

Abstract
Recently, it was reported that the role of mitochondria-reactive oxygen species (ROS) generating pathway in cisplatin-induced apoptosis is remarkable. Since a variety of molecules are involved in the pathway, a comprehensive approach to delineate the biological interactions of the molecules is required. However, quantitative modeling of the mitochondria-ROS generating pathway based on experiment and systemic analysis using the model have not been attempted so far. Thus, we conducted experiments to measure the concentration changes of critical molecules associated with mitochondrial apoptosis in both human mesothelioma H2052 and their ρ0 cells lacking mitochondrial DNA (mtDNA). Based on the experiments, a novel mathematical model that can represent the essential dynamics of the mitochondrial apoptotic pathway induced by cisplatin was developed. The kinetic parameter values of the mathematical model were estimated from the experimental data. Then, we have investigated the dynamical properties of this model and predicted the apoptosis levels for various concentrations of cisplatin beyond the range of experiments. From parametric perturbation analysis, we further found that apoptosis will reach its saturation level beyond a certain critical cisplatin concentration.
Keywords: Apoptosis, Cisplatin, Mitochondria-dependent pathway, Simulation, Systems approach
INTRODUCTION
Cisplatin is an effective chemotherapeutic agent being widely used as a first-line therapy for solid tumors, but it has been known that it can also induce the apoptosis of healthy normal cells [1,2,3]. There are three major signaling pathways involved in cisplatin-induced apoptosis [4,5,6,7]: i) death receptor pathway, ii) mitochondria--reactive oxygen species (ROS) generating pathway, and iii) endoplasmic reticulum (ER)-stress pathway. Mitochondrial signaling is known to tightly control the cell fate determination through cell survival or apoptotic cell death processes. In addition, a key feature of the apoptosis cascades is the disruption of mitochondrial transmembrane potential and apoptogenic protein release caused by the opening of the permeability transition pore [8]. In this respect, the mitochondria-ROS generating pathway plays a key role in apoptosis processes induced by cisplatin. Recently, it was reported that the role of mitochondria-ROS generating pathway in cisplatin-induced apoptosis is remarkable [9].
In spite of numerous studies of apoptosis caused by cisplatin, the detailed mechanism of the mitochondrial apoptotic pathway has not been completely understood. In particular, because a variety of critical molecules are involved in mitochondrial apoptotic pathway, a system-level understanding of the biological interactions between the molecules remained as a challenging task. Therefore, a comprehensive approach to delineate the biological interactions of the molecules is essential. However, quantitative modeling of the mitochondria-ROS generating pathway induced by cisplatin has not been based on the systemic analysis incorporating both wet experiment and computer simulation.
In this study, we conducted experiments to measure the concentration changes of critical molecules associated with mitochondrial apoptosis in both human mesothelioma H2052 and their ρ0 cells lacking mitochondrial DNA (mtDNA). In addition, a novel mathematical model that can represent the essential dynamics of the mitochondrial apoptotic pathway induced by cisplatin was developed. The kinetic parameter values of the mathematical model were estimated based on the experimental data. To validate our model, such model predictions were compared to experimental data. With this model, we performed parametric sensitivity analysis to investigate critical variables related to mitochondrial apoptotic pathway. By analyzing the experimentally validated mathematical model, we found varying apoptosis levels for an extensive range of cisplatin concentration, which could have not been observable from experimental study alone.
METHODS
Cells and reagents
Human mesothelioma H2052 cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured at 37℃ under 5% CO2 in Dulbecco's modified Eagle's medium or RPMI medium 1640 supplemented with 10% heatinactivated fetal bovine serum and antibiotics (Nacalai Tesque, Kyoto, Japan). Mitochondrial DNA-depleted ρ0 cells were established through long-term treatment of H2052 cells with ethidium bromide (25 ng/ml), as described previously (King and Attardi, 1989). Cisplatin was purchased from Sigma-Aldrich (St. Louis, MO). Other reagents used were of the highest grade commercially available.
Evaluation of cell death
Cell viability was evaluated using the trypan blue exclusion test. After treatment of cells with cisplatin for 24 h, both floating cells and adherent cells collected by trypsinization were centrifuged at 1500 rpm for 5 min. The cell pellets were resuspended in 50 µl of either Dulbecco's modified Eagle's medium or RPMI medium 1640, and 50 µl of 0.4% trypan blue was added. Dead cells were counted at four different areas using a hemocytometer.
Analysis of ROS generation
To analyze mitochondrial generation of ROS, cells were loaded with dihydroethidium (DHE; Wako Pure Chemicals) or MitoSOX (Invitrogen). DHE reacts with ROS to form fluorescent ethidium, which then intercalates into mtDNA. MitoSOX selectively accumulates in mitochondria and reacts with ROS to form red fluorescent mtDNA adducts. After incubation of cells with cisplatin, cells were collected and incubated in medium containing 5 µM of DHE or MitoSOX at 37℃ for 30 min in the dark. ROS generation was detected using a FACSCalibur flow cytometer (Becton Dickinson Japan, Tokyo, Japan).
Analysis of mitochondrial membrane potential
Cells treated with various anticancer agents were incubated with 5 µM JC-1 (Invitrogen) at 37℃ for 10 min and mitochondrial potential (Δψm) was analyzed by flow cytometry. Cationic JC-1 accumulates in the mitochondria and changes the fluorescence spectrum from orange to green depending on the decrease in membrane potential.
Western blot analysis
After incubating cells with cisplatin, cells were collected, lysed in a RIPA buffer with protease inhibitors (Nacalai Tesque), and subjected to 10~15% SDS-PAGE. The electrophoresed proteins were transferred onto a polyvinyidene difluoride membrane (Millipore, Billerica, MA) using a semidry blot system (2 mA/cm2 for 0.5 hr in 0.2 M Tris-glycine buffer, pH 8.8). Caspase-3 and -9 activation was analyzed using specific antibodies (Cell Signaling Technology, Beverly, MA). After incubation of the sheets in TBS solution (140 mM NaCl, 50 mM Tris-HCl, pH 7.2) containing 0.1% Tween 20 (TBST) and 5% low-fat milk powder at 25℃ for 1 hr, they were treated with anti-cleaved caspase-3 and -9 antibodies, as well as anti-β-actin antibody (Sigma) at 25℃ for 1 hr. The incubated membranes were washed five times with TBST for 5 min to eliminate nonspecific binding of the antibodies. After incubation with horseradish peroxidase-conjugated anti-IgG antibody (1:1,000) (Dako, Glostrup, Denmark) at 25℃ for 1 hr, immunoreactive spots were detected using ImmunoStar reagents (Wako) with the LAS-3000 instrument (FUJIFILM, Tokyo, Japan).
Analysis of released cytochrome c
To estimate the amount of cytochrome c released from the mitochondria to the cytosol, the harvested cells were incubated in an ice-cold cell lysis buffer (250 mM sucrose, 70 mM KCl, and 200 µM/mL digitonin in PBS) for 5 min, as described previously [10]. The supernatant and pellet fractions containing cytosolic and mitochondrial proteins, respectively, were subjected to SDS-PAGE and analyzed using anti-cytochrome c antibody (BD Pharmingen, San Diego, CA).
Statistical analysis
All experiments were repeated at least three times with similar results. Data were analyzed using Student's t-test (two-tailed), and p<0.05 was considered statistically significant. Data are expressed as means±SD.
Mathematical modeling, parameter estimation, and sensitivity analysis
In this study, we considered only key processes related to the mitochondria signaling pathway of cisplatin-induced apoptosis. A schematic representation of the mitochondria-dependent pathway of the model is shown in Fig. 1. Cisplatin enters into cells by passive diffusion or transportation [1]. The model begins with the uptake of cisplatin into a cell. After entering into the cell, cisplatin induces apoptosis by damaging mtDNA [3]. Cisplatin (which interacts with mtDNA) selectively enhances the generation of reactive oxygen species (ROS) in mitochondria, and the overproduction of ROS induces the opening of the mitochondrial permeability transition pore (MPTP) [11]. Intermembrane proteins such as cytochrome c (Cyt c), second mitochondria activator of caspase (Smac), and apoptosis-inducing factor (AIF) are released from MPTP, and the leaked Smac binds to several inhibitors of apoptosis proteins (IAP) which bind to active caspases and prevent accidental propagation of the apoptotic signaling cascade [12]. Cytochrome c in the cytosol activates caspase-9 and -3, resulting in the induction of apoptosis [13].
Fig. 1. Cellular responses to cisplatin-induced DNA damage.

MtDNA damage increases ROS generation, decreases mitochondrial membrane potential, and releases signals for cell death, including cytochrome c (Cyt c), second mitochondria activator of caspase (Smac) and apoptosis-inducing factor (AIF). Once released into the cytosol, cytochrome c triggers procaspase-9 activation and induces cell death. Solid arrows denote chemical reactions or upregulation; those terminated by a bar denote inhibition or down-regulation.
The signal transduction network of apoptotic reactions was modeled by ordinary differential equations (ODEs) based on the law of mass-action kinetics. The model consists of a set of 23 ODEs with 13 reaction partners (Tables 1 and 2). The ODEs were solved using MATLAB library functions and C++ in Visual Studio 2010. The model parameters in Table 1 were obtained by the iterative trials to match the model prediction with the experiment.
Table 1. The model parameters used in this study.
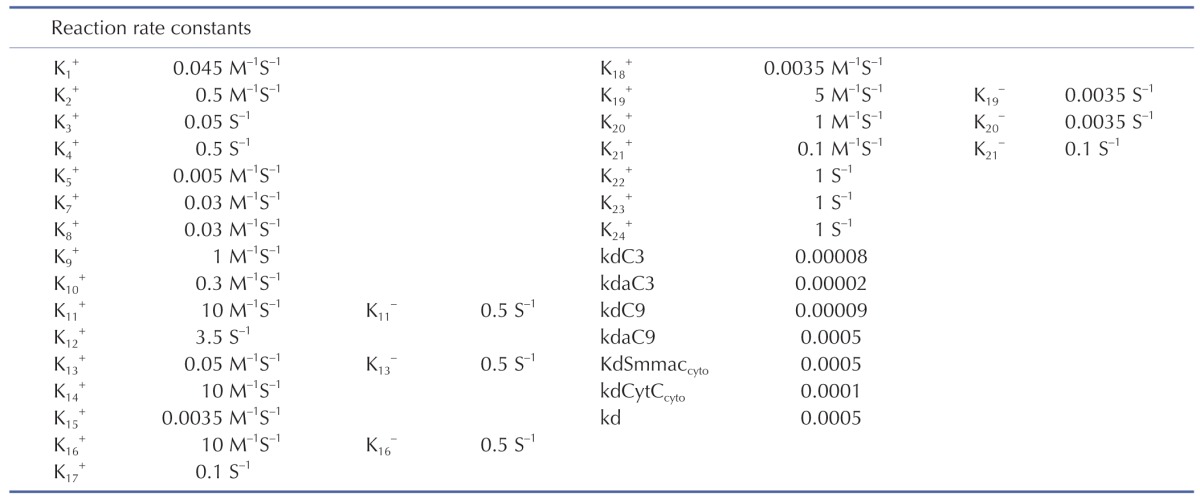
Table 2. Ordinary differential equations.

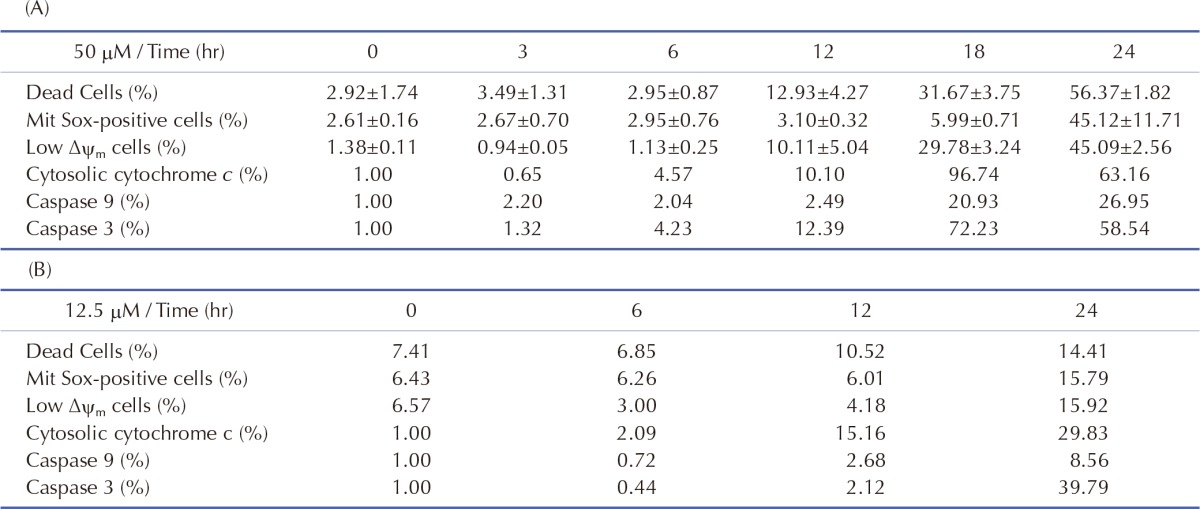
Initial conditions of the model were obtained from previous studies [14,15], as summarized in Table 3. To estimate the model parameters in Table 4, we performed iterative simulations using a genetic algorithm (GA) method in MATLAB with differential elimination of the Rosenfeld–Gröbner algorithm [16]. We then selected an optimal model parameter set by fitting the model simulation results to the experimental data for 50 µM cisplatin (Table 5). H2052 cells were incubated with 50 µM (Table 5A) and 12.5 µM cisplatin (Table 5B). Tables 3 and 4 show the initial conditions of the components at time zero and the kinetic parameters of the reactions (reaction rate constants), respectively [14,15].
Table 3. The initial concentrations [14,15].
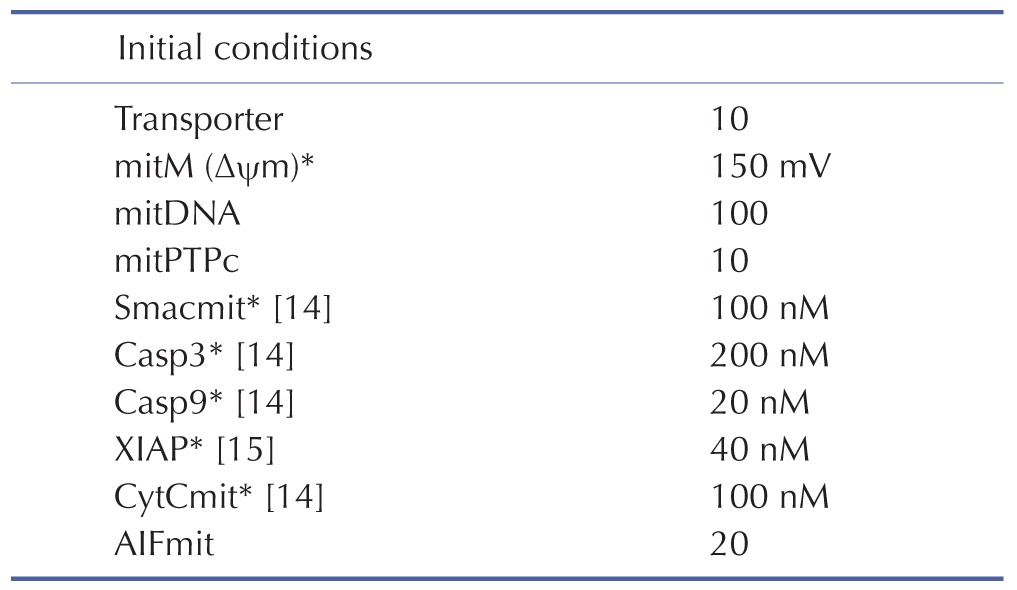
Table 4. The model parameters.
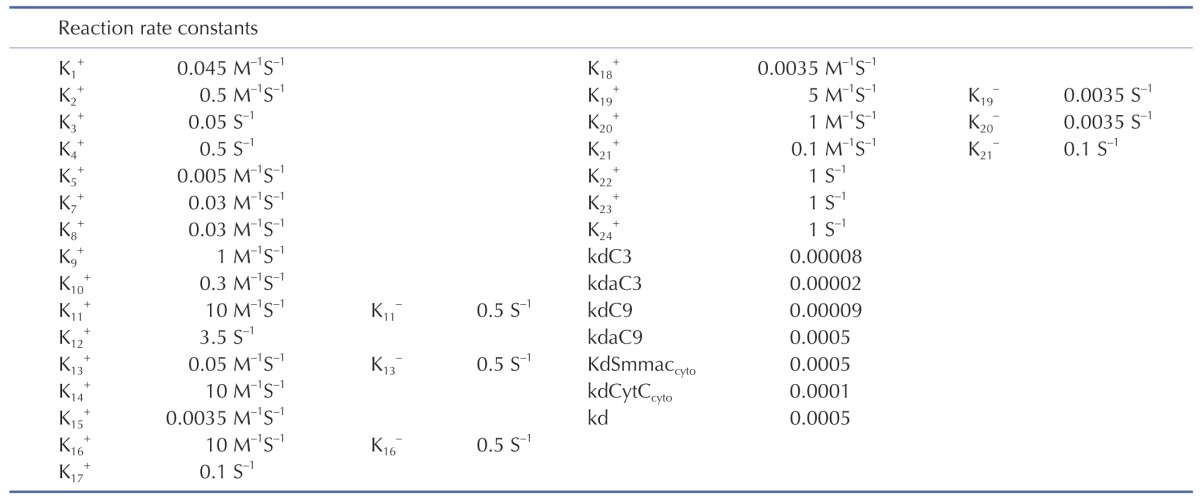
Table 5. Effect of cisplatin on the mitochondria-dependent apoptosis.
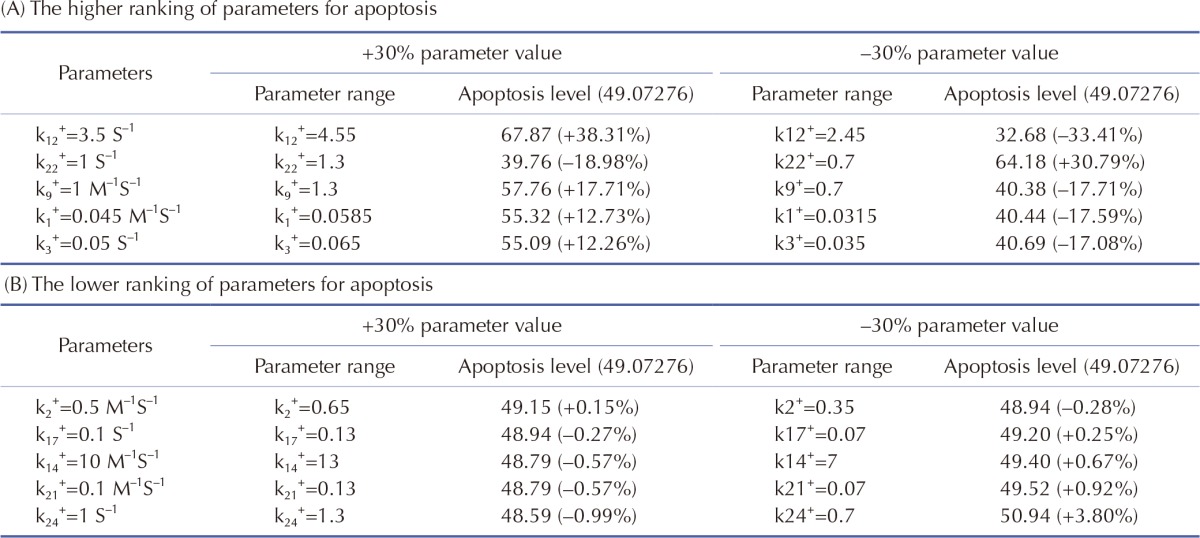
We performed sensitivity analysis of the model parameters using SimBiology in MATLAB. In biological simulation, sensitivity analysis is frequently used to evaluate how uncertainty in the output of a mathematical model can be attributed to different sources of uncertainty in the model input. Fig. 2 shows the sensitivities of rate constants for the apoptosis (A) and the whole model (B), respectively. For rate constants that had relatively high sensitivities, we tested the robustness of the parameters by perturbing the value within +/– 30%. The results of the perturbation test in all cases are shown in Table 6. For the higher ranking parameters (Table 6A), we found that k1, k3 and k9 affected apoptosis. In particular, k1 and k3 were associated with the concentration of cisplatin and mitROS generation, respectively. In addition, k12 and k22 affected apoptosis and major upstream signaling factors (activated caspase-9 and -3). However, the lower ranked parameters (Table 6B) were indicative of the overall robustness of the simulation results of the whole model. For the other parameters between a higher ranking and a lower ranking group, the level of apoptosis changed slightly within +/–10% (data not shown). Because Smacmit showed high sensitivity for k1, 3, 4, and 10 (Fig. 2B), we also performed a perturbation test. The apoptotic level was not affected by varying the parameters or Smacmit.
Fig. 2. Sensitivity analysis of rate constants for the model.

(A) Sensitivity of each rate constant for apoptosis. (B) Sensitivity of each rate constant for each initial condition.
Table 6. Effects of model parameters on apoptosis.
RESULTS
Mitochondrial-dependent apoptosis in H2052 cells induced by cisplatin
mtDNA-damaging agents (such as cisplatin) are potent inducers of cell death triggering apoptosis [1,3]. ROS promotes the mitochondrial membrane permeability transition (MPT), an early event leading to apoptosis, and cytochrome c is released from mitochondria into the cytosol through MPT pores [13,17]. As ROS are important in the induction of apoptosis [6], we analyzed the effects of cisplatin on ROS generation and cell death using H2052 cells (Fig. 3, Table 5). When ROS generation was enhanced, mitochondrial membrane potential decreased. Thus, apoptosis showed a time-dependent increase in the presence of cisplatin (Fig. 4A, Fig. 5F, and Fig. 6F). We then evaluated the degree of cytochrome c release as well as activated caspase-9 and -3 levels in cisplatin-treated cells using Western blot analysis (Fig. 3, Table 5). Western blot analysis revealed a significant increase in cytochrome c release and activated caspase-9 and -3 in H2052 cells. However, in mtDNA-depleted ρ0 cells, the cisplatin-induced apoptosis was significantly inhibited (Table 7A). These results suggest that cisplatin induces mitochondria-dependent cell death.
Fig. 3. Effect of cisplatin on cytochrome c localization and caspase activation.

Cytosolic cytochrome c and activated caspases were analyzed by SDS-PAGE and Western blotting after incubation with (A) 50 µM and (B) 12.5 µM cisplatin. The levels of cytosolic cytochrome c, activated caspase-9, -3, and β-actin were quantified by densitometric analysis.
Fig. 4. Model predictions and experimental validation.

(A) Apoptosis levels with increasing concentrations of cisplatin for 24 hr. (B) Cellular generation of ROS for 24 hr in the presence of 50 µM cisplatin.
Fig. 5. Comparisons between simulated and experimental obser vations in 50 µM cisplatin.

H2052 cells were cultured for 24 hr in the presence of 50 µM cisplatin, and the mitochondria-dependent apoptotic mechanisms were explored. (A) Mitochondrial generation of ROS was analyzed using MitoSOX and flow cytometry. (B) Mitochondrial membrane potential (Δψm) was assessed using JC-1 and flow cytometry. Cytosolic cytochrome c (C), and activated caspase-9 (D) and -3 (E) were quantified by densitometric analysis. (F) Apoptosis was analyzed using the trypan blue exclusion test.
Fig. 6. Comparisons between simulated and experimental ob servations at 12.5 µM cisplatin con centration.

Table 7. Effect of cisplatin on cellular generation of ROS and apoptosis in H2052 and their ρ0 cells.
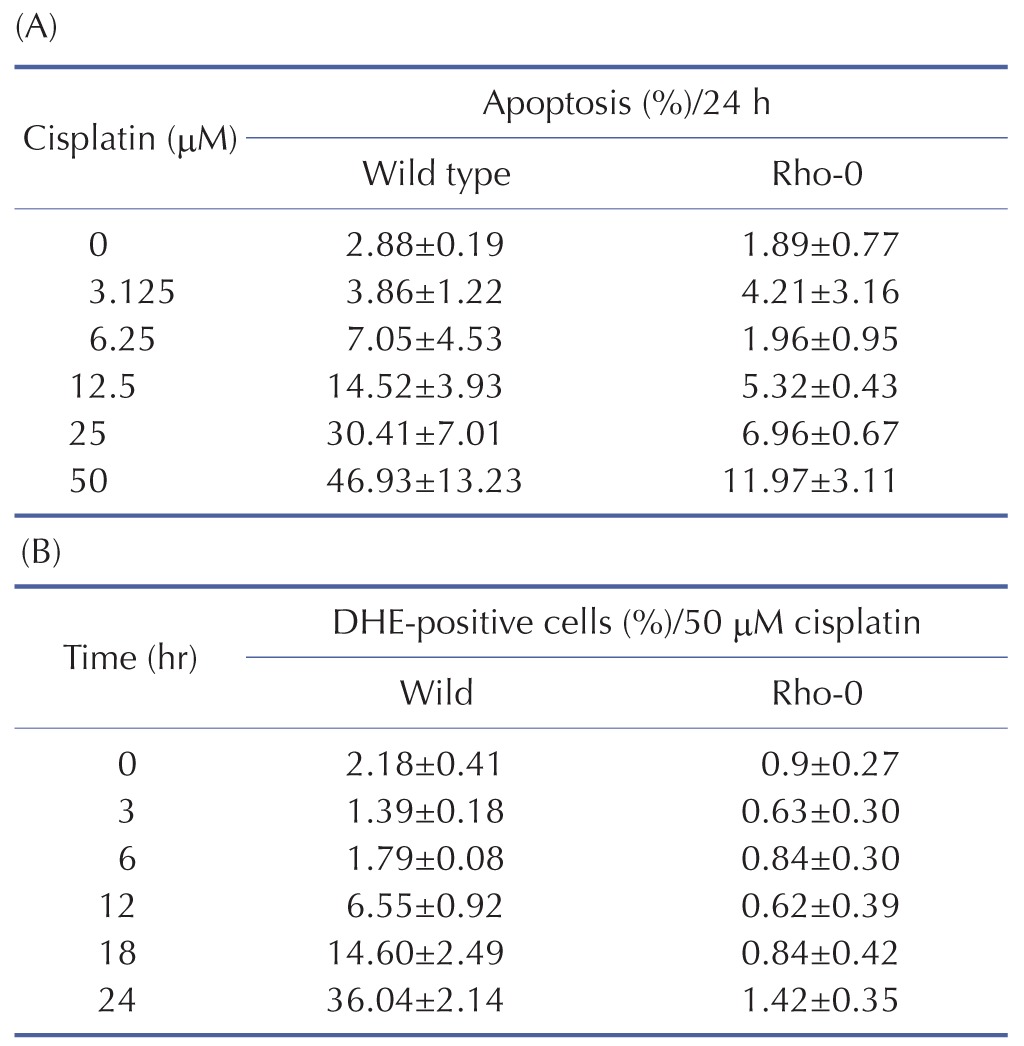
To examine the mechanism of mitochondria-dependent apoptosis, we compared the viability of H2052 cells and their ρ0 cells during their incubation with cisplatin (Table 7A, B, Fig. 4A, B). Here, H2052 and their ρ0 cells were incubated with increasing concentrations of cisplatin for 24 hr and analyzed their viability by trypan blue exclusion (Table 7A). After incubation of H2052 and their ρ0 cells with 50 µM cisplatin, ROS generation was analyzed by using DHE and flow cytometry (Table 7B). Cisplatin killed H2052 cells in a dose- and time-dependent manner, but not ρ0 cells (Fig. 4A). The apoptosis level in H2052 cells was fourfold higher than that in ρ0 cells in the presence of 50 µM cisplatin. Regarding ROS generation, mitochondrial generation of ROS (MitoSOX-positive cells) and cellular generation of ROS (DHE-positive cells) increased in a time-dependent way in the presence of cisplatin, whereas they did not increase in ρ0 cells (Fig. 5A, Fig. 4B). Our experiments showed that mitochondrial density is a key factor in cisplatin-induced apoptosis (Fig. 4A, B).
Simulation of cisplatin-induced apoptosis
In this study, the model parameters were estimated to fit the experimental results at 50 µM cisplatin concentrations. The comparison between experimental results and simulation results at 50 µM cisplatin infusion is shown in Fig. 5. The experimental and simulation results were also compared at 12.5 µM cisplatin concentration (Fig. 6). Excluding the transient percentage change in mitochondrial ROS production (Fig. 6A), all conditions were similar, suggesting that the model simulation was sufficiently robust to reproduce the apoptotic processes, regardless of the cisplatin concentration.
Apoptosis was enhanced in a time- and dose-dependent manner by cisplatin (Table 7B, Fig. 4A). Simulated apoptosis levels at 50 µM cisplatin concentration was similar to the experimental results in wild-type cells as shown in Fig. 4A, with nearly linear variation in apoptosis according to the cisplatin concentration. Furthermore, we performed a parametric perturbation study to delineate the mitochondria-dependent apoptosis processes induced by cisplatin. In vitro experiments with cisplatin concentrations over 100 µM have been carried out and the data were acquisited. Here, we numerically delineated the effect of cisplatin concentration on apoptosis at a wide range of cisplatin concentrations (from 0 to 400 µM) (Fig. 7A). Interestingly, the percentage of apoptosis after 24 hours reached saturation above 100 µM cisplatin concentration.
Fig. 7. Model predictions and experimental validation.

(A) Apoptosis levels with increasing concentrations of cisplatin for 24 hr. (B) Interaction between apoptosis and mitochondrial ROS. mitROS, ROS at baseline value; mitROS × 3, ROS increased by three fold; mitROS × 0.5, ROS decreased by two fold; mitROS × 0.1, ROS decreased by ten fold; mitROS=0. (C) Interaction between apoptosis and the amount of cytochrome c in the mitochondria. CytCmit × 2, amount of cytochrome c in the mitochondria increased by two fold; CytCmit=0. (D) Interaction between apoptosis and the amount of cytochrome c in the mitochondria or cytosol.
To examine the effects of mitochondrial density on apoptosis by cisplatin, we simulated various conditions of mitochondrial ROS (ROSmit), mitochondrial cytochrome c (Cytc mit), and leaked cytochrome c (Cytc cyto) using our model (Fig. 7B, C, D). The result was concordant with the observation that the amount of cellular mitochondria was positively correlated with cisplatin-induced apoptosis [18,19]. Because the release of cytochrome c from mitochondria to cytosol activates the mitochondria-dependent pathway leading to apoptosis, lower densities of both mitochondria and cytochrome c in ρ0 cells appeared to decrease the incidence of cell death. These observations suggest that the mitochondria-ROS generating pathway is most critical in cisplatin-induced apoptosis, which confirms our model predictions.
DISCUSSION
Mitochondria-ROS generating pathway plays a key role in the cellular apoptosis induced by a variety of drugs such as cisplatin, gentamicin, doxorubicin, cyclosporin, oxalate, etc. [20,21,22,23]. For in silico evaluation of the toxicity effect by such drugs, there has been a pressing need to develop a concise mathematical model of mitochondria apoptotic pathway validated by wet experiments. In this study, we aimed to quantitatively model the mitochondrial signaling pathway for cisplatin-induced apoptosis. To establish such a compact but well-representing model of the mitochondria-dependent apoptosis, we experimentally measured the concentration changes of critical molecules associated with mitochondrial apoptosis in both human mesothelioma H2052 and ρ0 cells lacking mitochondrial DNA, and used the data for the development of our mathematical model. For parameter estimation, we estimated the parameter values by fitting the simulated results to experimental results (Fig. 5) and finding a best fit between simulations and experiments.
ROS generation is involved in cisplatin-induced cell injury [24,25]. Cisplatin generates a significant amount of ROS and impairs the mitochondrial respiratory chain [26,27]. Uncontrollable production of ROS triggers the opening of MPT pore and induces apoptosis and/or necrosis [28]. Both our model predictions and experiments showed time- and dose-dependent increases of ROS generation and cell death in the presence of cisplatin (Fig. 4A, B, Fig. 5A, F, Fig. 7A). With respect to ROS generation, the cellular generation of ROS (DHE-positive cells) increased as an early event, but mitochondrial ROS (MitoSOX-positive cells) increased later than the cellular generation of ROS (Fig. 4B, Fig. 5A). It is possible that the sensitivity of MitoSOX is lower than that of DHE because we could not identify DHE-positive cells in ρ0 cells (Fig. 4B). We observed a complete inhibition of ROS generation in ρ0 cells, which was in accord with our simulation results. Hara et al. [18] also reported that cisplatin-induced cell death was inhibited by Mn-TBAP, a membrane-permeable superoxide scavenger. Together, these suggest that the cellular generation of ROS (DHE-positive) is mitochondria-derived, which can explain cisplatin-induced apoptosis.
A best fit between our model simulations and experiments was achieved. For this purpose, we estimated the parameter values by fitting the simulated results to experimental data at 50 µM cisplatin concentrations using MATLAB (Fig. 5). To evaluate the model, we changed the concentration of cisplatin from 50 µM to 12.5 µM and performed simulations. As shown in Fig. 6, the experimental results at 12.5 µM cisplatin concentrations were reproduced successfully using model simulations. In addition, our model predicted the apoptosis levels for various concentrations of cisplatin beyond the range of experiments. According to the simulation results, the apoptosis level reached its saturation over a critical cisplatin concentration. Thus, our model can predict detailed sequential events of the apoptotic signaling, allowing us to examine the effects of protein dysregulation by pharmacological intervention.
The present study has several limitations. First, in the present minimal model for the mitochondrial pathway in cisplatin-induced apoptosis, no other pathways were tested and their relative importance or cross-talk was not analyzed. Second, due to the minimal nature of the present model, there are no feedback loops to influence potentially important sequence of events in cisplatin-induced apoptosis. Third, the present model didn't take into consideration the release of the ROS accumulated in mitochondrion to cytosol and their subsequent uptake by other mitochondria, as observed in the previous study [29]. And ROS neutralization mechanism within mitochondria was neglected. However, these limitations were not expected to greatly alter the main findings of this study.
In summary, to develop a compact but well-representing mathematical model of the mitochondrial apoptotic pathway induced by cisplatin, we experimentally measured the concentration changes of critical molecules associated with mitochondrial apoptosis and used the data for the development of a mathematical model. To validate our model, we compared the simulated results with experimental observations. The parametric perturbation study of apoptosis by varying cisplatin indicated that the apoptosis level was nearly saturated beyond a certain critical cisplatin concentration. We also found that the mitochondria-ROS generating pathway is the most important pathway in cisplatin-induced apoptosis.
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government, the Ministry of Science, ICT and Future Planning (MSIP) (2012-0003065 and 2015M3A9A7067220, 2015R1A2A1A01007744) and by 2013 Research Grant from Kangwon National University (No. 120131352).
Footnotes
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
Author contributions: J.Y.H. performed the model design, experimental design and simulations, and wrote the manuscript. K.H. and E.F.S. performed experiments for data. J.K. assisted modeling, simulations and writing of the manuscript. E.B.S. supervised the work and reviewed the manuscript. K.H.C. assisted modeling and simulation design and writing of the manuscript.
References
- 1.Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 2010 doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of Cisplatin nephrotoxicity. Toxins (Basel) 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohno K, Wang KY, Takahashi M, Kurita T, Yoshida Y, Hirakawa M, Harada Y, Kuma A, Izumi H, Matsumoto S. Mitochondrial transcription factor A and mitochondrial genome as molecular targets for Cisplatin-based cancer chemotherapy. Int J Mol Sci. 2015;16:19836–19850. doi: 10.3390/ijms160819836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 5.Chen M, Wang J. Initiator caspases in apoptosis signaling pathways. Apoptosis. 2002;7:313–319. doi: 10.1023/a:1016167228059. [DOI] [PubMed] [Google Scholar]
- 6.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 7.Mayer B, Oberbauer R. Mitochondrial regulation of apoptosis. News Physiol Sci. 2003;18:89–94. doi: 10.1152/nips.01433.2002. [DOI] [PubMed] [Google Scholar]
- 8.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ROS crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong JY, Kim GH, Kim JW, Kwon SS, Sato EF, Cho KH, Shim EB. Computational modeling of apoptotic signaling pathways induced by cisplatin. BMC Syst Biol. 2012;6:122. doi: 10.1186/1752-0509-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 12.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 13.Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp. 1999;66:167–179. doi: 10.1042/bss0660167. [DOI] [PubMed] [Google Scholar]
- 14.Hua F, Cornejo MG, Cardone MH, Stokes CL, Lauffenburger DA. Effects of Bcl-2 levels on Fas signaling-induced caspase-3 activation: molecular genetic tests of computational model predictions. J Immunol. 2005;175:985–995. doi: 10.4049/jimmunol.175.2.985. [DOI] [PubMed] [Google Scholar]
- 15.Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem. 2002;277:11345–11351. doi: 10.1074/jbc.M109893200. [DOI] [PubMed] [Google Scholar]
- 16.Nakatsui M, Horimoto K, Okamoto M, Tokumoto Y, Miyake J. Parameter optimization by using differential elimination: a general approach for introducing constraints into objective functions. BMC Syst Biol. 2010;4(Suppl 2):S9. doi: 10.1186/1752-0509-4-S2-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 18.Hara K, Kasahara E, Takahashi N, Konishi M, Inoue J, Jikumaru M, Kubo S, Okamura H, Sato E, Inoue M. Mitochondria determine the efficacy of anticancer agents that interact with DNA but not the cytoskeleton. J Pharmacol Exp Ther. 2011;337:838–845. doi: 10.1124/jpet.111.179473. [DOI] [PubMed] [Google Scholar]
- 19.Qian W, Nishikawa M, Haque AM, Hirose M, Mashimo M, Sato E, Inoue M. Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am J Physiol Cell Physiol. 2005;289:C1466–C1475. doi: 10.1152/ajpcell.00265.2005. [DOI] [PubMed] [Google Scholar]
- 20.Cao LC, Honeyman TW, Cooney R, Kennington L, Scheid CR, Jonassen JA. Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney Int. 2004;66:1890–1900. doi: 10.1111/j.1523-1755.2004.00963.x. [DOI] [PubMed] [Google Scholar]
- 21.de Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol Lett. 2013;218:30–38. doi: 10.1016/j.toxlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Morales AI, Detaille D, Prieto M, Puente A, Briones E, Arévalo M, Leverve X, López-Novoa JM, El-Mir MY. Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int. 2010;77:861–869. doi: 10.1038/ki.2010.11. [DOI] [PubMed] [Google Scholar]
- 23.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93:105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 24.Biroccio A, Benassi B, Amodei S, Gabellini C, Del Bufalo D, Zupi G. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol Pharmacol. 2010;60:174–182. doi: 10.1124/mol.60.1.174. [DOI] [PubMed] [Google Scholar]
- 25.Miyajima A, Nakashima J, Yoshioka K, Tachibana M, Tazaki H, Murai M. Role of reactive oxygen species in cis-dichlorodiammineplatinum-induced cytotoxicity on bladder cancer cells. Br J Cancer. 1997;76:206–210. doi: 10.1038/bjc.1997.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kruidering M, Van de Water B, de Heer E, Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther. 1997;280:638–649. [PubMed] [Google Scholar]
- 27.Masuda H, Tanaka T, Takahama U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem Biophys Res Commun. 1994;203:1175–1180. doi: 10.1006/bbrc.1994.2306. [DOI] [PubMed] [Google Scholar]
- 28.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 29.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]