

Abstract
Most human genes generate multiple protein isoforms through alternative pre-mRNA splicing, but the mechanisms controlling alternative splicing choices by RNA binding proteins are not well understood. These proteins can have multiple paralogs expressed in different cell types and exhibiting different splicing activities on target exons. We examined the paralogous polypyrimidine tract binding proteins PTBP1 and PTBP2 to understand how PTBP1 can exhibit greater splicing repression activity on certain exons. Using both an in vivo coexpression assay and an in vitro splicing assay, we show that PTBP1 is more repressive than PTBP2 per unit protein on a target exon. Constructing chimeras of PTBP1 and 2 to determine amino acid features that contribute to their differential activity, we find that multiple segments of PTBP1 increase the repressive activity of PTBP2. Notably, when either RRM1 of PTBP2 or the linker peptide separating RRM2 and RRM3 are replaced with the equivalent PTBP1 sequences, the resulting chimeras are highly active for splicing repression. These segments are distinct from the known region of interaction for the PTBP1 cofactors Raver1 and Matrin3 in RRM2. We find that RRM2 of PTBP1 also increases the repression activity of an otherwise PTBP2 sequence, and that this is potentially explained by stronger binding by Raver1. These results indicate that multiple features over the length of the two proteins affect their ability to repress an exon.
Keywords: alternative splicing, RNA binding proteins
INTRODUCTION
Alternative pre-mRNA splicing allows a single pre-mRNA transcript to produce more than one spliced mRNA and protein isoform. RNA binding proteins control these splicing choices by binding to cis-acting regulatory elements on the pre-mRNA and altering the assembly of the spliceosome at adjacent splice sites (Braunschweig et al. 2013; Han et al. 2014; Lee and Rio 2014). Differences in the concentration and activity of these regulators determine the expression of specific protein isoforms. Most RNA binding proteins are members of gene families, and paralogs within a family can share high sequence identity and domain structure but can have distinct tissue-specific expression patterns and direct different splicing events (Topp et al. 2008; Keppetipola et al. 2012). How these related members can exert different effects on a splicing pattern is not well understood.
The polypyrimidine tract binding proteins, PTBP1 and PTBP2, are paralogous RNA binding proteins encoded by related genes (Kafasla et al. 2012; Keppetipola et al. 2012). There is also a third family member, PTBP3 (Rod1). PTBP1 and PTBP2 share 74% sequence identity and a similar domain organization: Four RRM-type RNA binding domains joined by linker regions and an amino-terminal region containing a nuclear localization signal. PTBP1 and 2 most often function as splicing repressors, but can also enhance splicing of exons (Wollerton et al. 2001; Spellman et al. 2005; Boutz et al. 2007; Xue et al. 2009; Llorian et al. 2010; Licatalosi et al. 2012; Li et al. 2014). Each gene can produce more than one protein through alternative splicing. For PTBP1, the PTBP1.4 isoform has higher splicing repression activity than PTBP1.1 on some target exons (Wollerton et al. 2001; Gueroussov et al. 2015). Detailed studies of splicing repression by PTBP1 have examined several exons including the N1-exon of the Src pre-mRNA, exon 9 of GABAA receptor-γ2 subunit, and exon SM of α-actinin (Ashiya and Grabowski 1997; Chan and Black 1997; Southby et al. 1999). The PTBP2 target exons overlap with those of PTBP1, but are not identical (Markovtsov et al. 2000; Boutz et al. 2007; Spellman et al. 2007; Llorian et al. 2010; Tang et al. 2011; Zheng et al. 2012; Linares et al. 2015), and the two proteins show differential targeting of the Src N1 exon, CACNA1C exon 8, and other exons (Chan and Black 1997; Modafferi and Black 1999; Amir-Ahmady et al. 2005). The mechanistic basis for this difference in activity is not known.
PTBP1 and PTBP2 have distinct expression patterns. PTBP1 is widely expressed but is absent from neurons and muscle cells. PTBP2 is expressed in neurons, testis, and certain other cells. PTBP1 is expressed in neuronal progenitor cells but is replaced by PTBP2 during early neuronal differentiation (Polydorides et al. 2000; Boutz et al. 2007; Makeyev et al. 2007; Licatalosi et al. 2012; Zheng et al. 2012; Gueroussov et al. 2015; Linares et al. 2015). The switch from PTBP1 to PTBP2 alters the splicing of exons that are more sensitive to PTBP1, thus reprogramming neuronal splicing. The expression of PTBP2 is later reduced, leading to a second switch in the splicing program as neurons mature. The changes in the levels of these two regulators lead to changes in the expression and structure of many proteins playing critical roles in neuronal differentiation and maturation (Licatalosi et al. 2012; Zheng et al. 2012; Xue et al. 2013; Li et al. 2014; Linares et al. 2015).
Solution structures of each PTBP1 RRM bound to RNA identified residues important for RNA recognition and binding (Conte et al. 2000; Simpson et al. 2004; Oberstrass et al. 2005; Petoukhov et al. 2006; Auweter and Allain 2008). PTBP2 has the same residues at these positions of RNA contact except for one lysine to arginine and two phenylalanine to tyrosine substitutions (Fig. 1A). A carboxy-terminal fragment of PTBP2, containing RRM3, linker 3, and RRM4, is very similar in tertiary structure to the same region of PTBP1 (Joshi et al. 2014). The two proteins also share residues on the α helical face of RRM2 that mediate interaction with the corepressor protein Raver1 (Fig. 1A).
FIGURE 1.

(A, top) The domain structure of the PTB proteins indicating the segments defined in this study. The percent amino acid sequence identity between PTBP1.1 and PTBP2 is indicated below. (Bottom) Aligned amino acid sequences of human PTBP1.1, PTBP1.4, and PTBP2. Gaps in the alignment are indicated as dashes. Residues identical to PTBP1.4 are shown as dots. RNA recognition motifs (RRMs) are shaded gray (Oberstrass et al. 2005). PTBP1.4 residues that interact with Raver1 are shaded light pink (Joshi et al. 2011). Vertical lines above the sequence indicate PTBP1.4 residues that interact with RNA (Oberstrass et al. 2005). Arrowheads below the sequence indicate RNA interacting residues that are different in PTBP2. (B) Maps of PTB proteins and chimeras. The RRM domains and linker regions of PTBP1 are colored blue and yellow, respectively. The RRMs and linker regions of PTBP2 are colored plum and green, respectively. The positions of the amino-terminal Flag-tag and NLS are indicated. The color-coding indicates the combination of PTBP1.4 and PTBP2 regions in each chimera with the repression index measured for each at the right. Chimera 10 was unstable in cells and not expressed. A repression index was not determined for chimera 12, but in other assays this protein exhibited similar repression activity to PTBP1.4. Measurement of the splicing repression index. (C) Immunoblot of Flag-PTBP1.4 protein in cell lysates after transfection with increasing concentrations (0.25, 0.5, 1.0. 1.25, or 1.5 µg) of Flag- PTBP1.4 plasmid DNA. Protein levels were quantified and normalized to GAPDH. (D) PTBP1.4 represses splicing of test exon. Splicing reporter Dup 175-DS9 (0.5 µg) was cotransfected with empty expression vector or varying amounts (0.25–1.5 µg) of PTBP1.4 expression plasmid. RNA was harvested after 48 h, assayed by RT-PCR, and quantified. (E) Quantifying the level of repression per unit protein; Repression index. Percent-spliced-in (PSI) was plotted against normalized protein. The slope defines level of repression per unit protein (♦ = PTBP1.4, ▪ = PTBP2). The absolute value of the slope was defined as the repression index.
The NMR studies of the PTBP1 RRMs, as well as statistical modeling of PTBP1 and PTBP2 binding sites, indicated that the RRM domains preferentially bind to extended runs of alternating C and U, but that the protein could also bind sequences with interspersed G residues (Oberstrass et al. 2005; Han et al. 2014). Purified PTBP1 and PTBP2 bind extended pyrimidine rich RNA sequences with similar high affinity (Amir-Ahmady et al. 2005), but show differences in binding to shorter binding sites. When assayed in nuclear extracts the proteins differed in their assembly with other factors (Chou et al. 2000; Markovtsov et al. 2000). On some introns, PTBP1 interacts with the RNA binding proteins Raver1/2, Matrin 3, or MBNL to form higher-order RNP complexes (Gromak et al. 2003; Henneberg et al. 2010; Joshi et al. 2011; Gooding et al. 2013; Coelho et al. 2015). Of these, the PTBP1–Raver1 interaction is best characterized; the α helical surface of RRM2 interacts with a peptide motif within Raver1. PTBP2 was shown to bind this Raver1 peptide, but its interactions with full-length Raver1, or with Matrin 3 and MBNL are not known (Joshi et al. 2011). These studies indicate that there are differences in how PTBP1 and PTBP2 interact with cofactors.
To identify regions of primary structure that dictate their differences in splicing activity, we analyzed a series of PTBP1–PTBP2 chimeric proteins. Our data identify multiple new features contributing to the splicing properties of the two proteins.
RESULTS AND DISCUSSION
Primary structure features contributing to the differential splicing activity of PTBP1 and PTBP2
The high sequence identity between PTBP1 and PTBP2 allows their sequences to be precisely aligned and equivalent features defined. Based on the known structures of the RRM domains, the proteins were divided into eight segments: the amino-terminal NLS, RRM1, Linker 1, RRM2, Linker 2, RRM3, Linker 3, and RRM4 (Fig. 1A). A series of chimeric genes was constructed in the mammalian expression vector pcDNA 3.1 (+) driven by the CMV promoter. Each coding region included a portion of PTBP1 fused to the remaining regions of PTBP2 (Fig. 1B). Splicing repression activity of wild-type and chimeric genes was measured with the minigene reporter Dup 175-DS9. This minigene encodes a three exon pre-mRNA, where the central test exon is regulated by two high affinity PTBP1 binding sites, one upstream of the branch point sequence and the other within the test exon itself. Previous studies showed that this test exon is more strongly repressed by PTBP1 than by PTBP2 (Amir-Ahmady et al. 2005). To quantify this activity, each PTBP1–PTBP2 chimeric construct was titrated against a set amount of minigene reporter. Splicing repression activity was measured as a function of expressed protein. Flag-PTBP expression was quantified and normalized to the level of GAPDH (Fig. 1C). The percent of reporter mRNA containing the test exon (PSI) was measured by RT-PCR (Fig. 1D). Normalized protein levels were plotted against PSI. The repression index, representing the change in PSI per unit protein, was defined as the absolute value of the slope of these plots (Fig. 1E). Repression indices were determined for each chimeric protein (Fig. 1B).
Alternative splicing of PTBP1 exon 9 alters the length of linker 2 between RRMs 2 and 3. PTBP1 isoform 4 (PTBP1.4) includes exon 9 to produce a 557 amino acid protein. Skipping of exon 9 generates the shorter, 531 amino acid PTBP1 isoform 1 (PTBP1.1) (Gil et al. 1991; Ghetti et al. 1992; Wagner et al. 1999). A third isoform, PTBP1.2, resulting from the shortening of exon 9 was not tested. It was previously shown that PTBP1.4 was more active than PTBP1.1 for repression of tropomyosin and other exons, but showed equal repression activity for an exon in actinin (Wollerton et al. 2001; Gueroussov et al. 2015). When assayed for splicing of the Dup175-DS9 exon, PTBP1.1 (isoform 1) had a moderately lower repression index than PTBP1.4 (33.4 compared with 38.1), confirming that the linker 2 sequence can increase splicing repression activity. Compared with either PTBP1 isoform, PTBP2 had a dramatically lower repression index of 11.4, about threefold below PTBP1.1. These results confirm that PTBP2 is less repressive for certain exons than PTBP1 and that this activity can be quantified. Since linker 2 in PTBP1.1 is similar in length to PTBP2, additional features of PTBP1 must play a role in its higher activity. We tested a series of chimeras to map features on the two proteins that determine this difference in activity.
Chimera 1 is a fusion of the amino terminus and RRM1 of PTBP1 to the rest of PTBP2. Chimera 2 contains only RRM1 of PTBP1 in an otherwise PTBP2 protein. Both chimeras displayed high repression indices of 35.8 and 40.0, respectively (Fig. 1B), indicating that the RRM1 domain of PTBP1 can increase repression by an otherwise PTBP2 sequence. The PTBP2 RRM1 has 15 amino acid substitutions from PTBP1 (out of 82 total residues). Although many of these are conservative changes, at least one must alter the ability of the PTBP to repress splicing (Fig. 1A).
Chimera 3 contained RRM2, linker 2, and RRM3 of PTBP1.4 fused to the amino and carboxy termini of PTBP2 (Fig. 1B). (Unless otherwise noted, all chimeras used the PTBP1.4 isoform.) Chimera 3 yielded a repression index of 34.0, similar to PTBP1.1, but not as high as PTBP1.4. This construct is similarly repressive to Chimera 2, but shares no overlap of PTBP1 sequences. Later experiments found the converse protein to Chimera 3 (Chimera 12) containing the PTBP1 termini and the central PTBP2 RRMs and linker to have similar activity to Chimera 2 or full PTBP1.4 (data not shown). Thus, the determinants of PTBP repression activity are distributed over multiple segments of the protein.
Chimera 4 containing the L2 region of PTBP1.4 in an otherwise PTBP2 protein yielded a repression index of 35.2. This activity of L2 is consistent with the earlier comparisons of PTBP1.1 and PTBP1.4, although we found that other features of the central region of the protein can have similar effects. PTBP1 RRM2 with linker 2 in an otherwise PTBP2 protein (Chimera 5) yielded a repression index of 31.7, only slightly lower than PTBP1.1. Adding PTBP1 RRM3 (Chimera 6) increased the repression index of the protein to 37.6 even though it was lacking the PTBP1.4 L2 linker. These results are consistent with findings that an MS2 tethered fragment of PTBP1 containing RRM2 and L2 was sufficient to induce splicing repression of α tropomyosin exon 3 (Robinson and Smith 2006). The flexibility of the linker 2 region makes it a likely position for protein–protein interactions (Oldfield and Dunker 2014). Differences in activity between paralogous splicing regulators hnRNP L and hnRNP LL, mediated by regions outside the RRMs, were also identified by Shankarling and Lynch (2013).
Not all fragments of PTBP1 were found to increase the repression activity when substituted into a PTBP2 backbone. Replacing either RRM3 or RRM4 of PTBP2 with the equivalent PTBP1 domains yielded proteins (Chimeras 8 and 9) with low repression indices similar to wild-type PTBP2 itself (12.8 and 15.1, respectively). RRMs 3 and 4 form a single folded unit with a fixed orientation with respect to each other (Oberstrass et al. 2005; Vitali et al. 2006; Joshi et al. 2014). It is not clear whether this RRM3–RRM4 interaction is occurring in Chimeras 8 and 9. However, the L3 linkers of PTBP1 and PTBP2 are identical, and the residues mediating the inter-domain interactions are conserved between PTBP1 and PTBP2. Thus, other features of the RRM domains must cause these chimeras to behave differently. We note that fragments of PTBP1 and PTBP2 could exhibit different activities depending on what other portions of the protein are present with them (compare Chimeras 8 and 9 to Chimera 12).
Also intriguing was Chimera 10, which contained PTBP1 RRM3, L3, and RRM4 attached to the N-terminal domains of PTBP2. This protein was not expressed well and perhaps unstable, indicating that as yet unidentified features might affect the overall conformation of the protein. This will need further investigation.
Splicing repression activity of chimeric PTB proteins on the intact Src N1 exon
We next tested the chimeric proteins on a second reporter exon. The minigene Dup 4-5 contains the Src N1 exon with upstream and downstream intronic regulatory regions inserted between the flanking globin exons of Dup175 (Fig. 2A; Chan and Black 1995; Modafferi and Black 1999; Chou et al. 2000). The PTBP1/2 binding sites in Dup175-DS9 are derived from the upstream site of the Src N1 exon, and N1 was previously shown to be more strongly affected by PTBP1 than PTBP2 (Markovtsov et al. 2000). Wild-type and chimeric PTB proteins were cotransfected with Dup 4-5, with protein expression monitored by Western blot (Fig. 2B, top panel) and splicing by RT- PCR (Fig. 2B, bottom panel). The amount of protein expression plasmid was set at the middle of the previous titrations to allow observation of differences in protein activity. PSI values were measured relative to protein expression levels normalized to GAPDH and plotted in Figure 2C. By this measure, PTBP1.4 yielded a value of 1.2, indicating low inclusion of exon N1 or high repression activity. Similar to the results with Dup175-DS9, PTBP2 yielded a value of 5.2—about fourfold lower repression activity than PTBP1.4.
FIGURE 2.
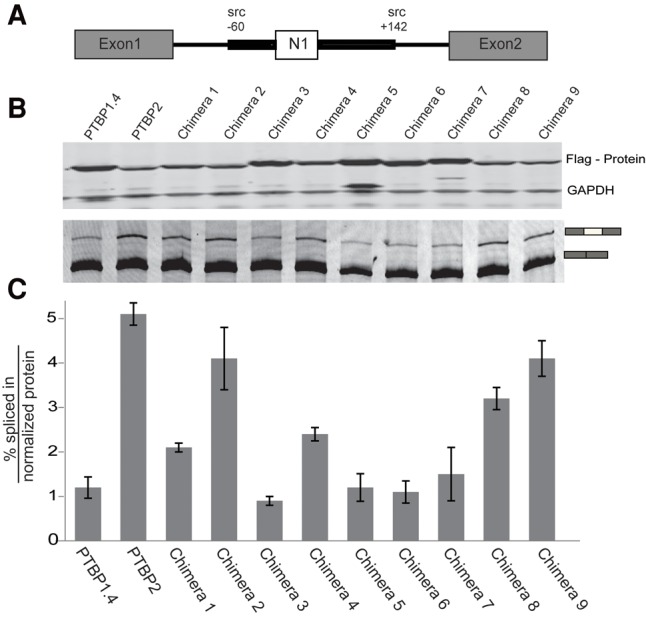
Splicing activity with minigene reporter plasmid Dup4-5. (A) The structure of the reporter mini gene Dup4-5, with β-globin exons 1 and 2 flanking the Src N1 exon and its intronic regulatory regions. (B) Western blots (top panel) and RT/PCR N1 exon splicing (bottom panel) in N2A cells after transient expression of the PTB chimeras and the Dup4-5 reporter. (C) Plot of the PSI per normalized protein for PTBP1.4, PTBP2, and the Chimeras. The bar height is the average of two experiments with the bars indicating the mean absolute error.
In general, chimeric proteins that strongly repressed the Dup175-DS9 exon similarly repressed Dup4-5 (see Chimeras 1, 3, 5, and 6). Conversely, chimeras with the weakest activity on Dup175-DS9 only weakly repressed Dup4-5 (e.g., Chimeras 8 and 9). However, there were interesting differences between the two exons in their responses to some chimeras. This was most notable for Chimera 2, which strongly repressed Dup175-DS9, but had little effect on Dup4-5. Quantitative differences were also observed. Chimera 3 was the strongest repressor of Dup4-5; although Chimera 3 is active on Dup175-DS9 (repression index = 34), several other chimeras had stronger effects on this exon. These results indicate that features in PTBP1 RRM2 were repressive for both exons, but features in RRM1 and Linker2 may affect the exons differently. These varying responses may arise from differences in the arrangement of the PTBP binding sites on the two exons, and differences in the other proteins assembling with the PTBPs. PTBP1 RRM1 makes contact with stem loop 4 of the U1 snRNA in repressing splicing of the Src N1 exon (Sharma et al. 2014). It would appear that the PTBP2 RRM1 should also make this contact. It will be interesting to examine both the protein and the RNA interactions of these two RRM1 domains within the N1 exon complex.
Protein segments exhibiting different activity in splicing could also be regulated through post-translational modification. Whole-cell proteomic approaches identify many modifications on PTBP1 and PTBP2 across multiple regions of the protein (Hornbeck et al. 2015). Another study found that PTBP1 and PTBP2 are sumoylated on RRM1 (Han et al. 2013). It will be interesting to examine how these modifications affect protein activity and whether PTBP1 and PTBP2 might be differentially modified.
PTBP-mediated repression of N1 exon splicing in vitro
We previously found that PTBP1 and PTBP2 exhibited different repression activities on the N1 exon in an in vitro splicing assay (Markovtsov et al. 2000). To examine whether chimeric proteins exhibited similar properties in vitro, we expressed recombinant Chimera 3 along with PTBP1.4 and PTBP2 in E. coli and purified them (Fig. 3A). A pre-mRNA transcript, BS713, was synthesized in vitro (Fig. 3B). This substrate contains the N1 exon flanked by its PTBP binding elements, as well as the downstream Src exon 4 with the intervening intron. PTBP1 was previously shown to strongly repress splicing of this intron in vitro. Splicing reactions containing BS713 in HeLa nuclear extract were incubated with increasing concentrations of PTBP1, PTBP2, and Chimera 3 protein (0.12–0.48 µM) (Fig. 3C). Splicing intermediates and products were progressively reduced with increasing concentrations of PTBP1, but not of PTBP2. Chimera 3 inhibited splicing similarly to PTBP1. Thus, Chimera 3 inhibits N1 splicing in both the in vivo and in vitro assays. An Adenovirus pre-mRNA substrate that lacks PTBP1 binding elements was only minimally repressed by these proteins (Fig. 3D). Thus, the features of RRM2 and L2 that mediate splicing repression in vivo make similar contacts in vitro.
FIGURE 3.

In vitro splicing assay of PTBP1.4, PTBP2, and Chimera 3. (A) Aliquots (5 µg) of recombinant PTBP1.4, PTBP2, and Chimera 3 were analyzed by SDS-PAGE and stained with Coomassie blue. The positions and sizes (in kDa) of marker polypeptides are indicated on the left. (B) Diagrams of the BS713 and adenovirus in vitro splicing substrates. Exons are indicated as boxes and introns as lines. Regulatory sequences upstream and downstream from the N1 exon are indicated as thick lines. (C) In vitro splicing activity of PTBP1.4, PTBP2, and Chimera 3. Reaction mixtures (25 µL) containing 15 µL of HeLa nuclear extract, 2.2 mM MgCl2, 0.4 mM ATP, 20 mM creatine phosphate, 1 U RNase out, 10,000cpm of 32-P UTP labeled BS713 and either no recombinant protein (lane 2) or increasing concentrations of PTBP1.4, PTBP2, or Chimera 3 (0.12, 0.24, or 0.48 µM) as indicated were incubated at 30°C for 90 min. The RNA splicing intermediates and products are diagrammed to the left. Bands showing repression by PTBP1 and PTBP2 are marked by black arrows to the right. (D) Splicing of adenovirus transcript in the presence of 0.48 µM PTBP1, PTBP2, and Chimera 3.
The RRM2 domains of PTBP1 and PTBP2 exhibit differences in binding of the Raver1 cofactor
We tested whether Raver1 exhibited differences in binding to PTBP1 and PTBP2, and whether this might contribute to the observed activity differences of the two proteins. We expressed Flag-tagged PTBP1 and PTBP2 in cells, as well as PTBP1 RRM2 or PTBP2 RRM2, each fused to Flag-mCherry. After cell lysis and nuclease digestion, we performed immunoprecipitation and immunoblot to examine the binding of endogenous Raver1 (Fig. 4). Flag-tagged RRM2 from PTBP1 pulled down substantial amounts of Raver1, confirming the earlier results (Rideau et al. 2006; Joshi et al. 2011). However, RRM2 from PTBP2 showed a much weaker interaction, with the level of Raver1 in the Flag-IP slightly above background. Similar differences in Raver1 binding were observed for full-length PTBP1 and PTBP2, although in this case PTBP2 exhibited greater binding than RRM2 alone, perhaps due to additional contacts outside of the RRM2 domains. The greater interaction of PTBP1 and its RRM2 domain may contribute to the increased splicing repression activity observed with chimeras containing this sequence.
FIGURE 4.
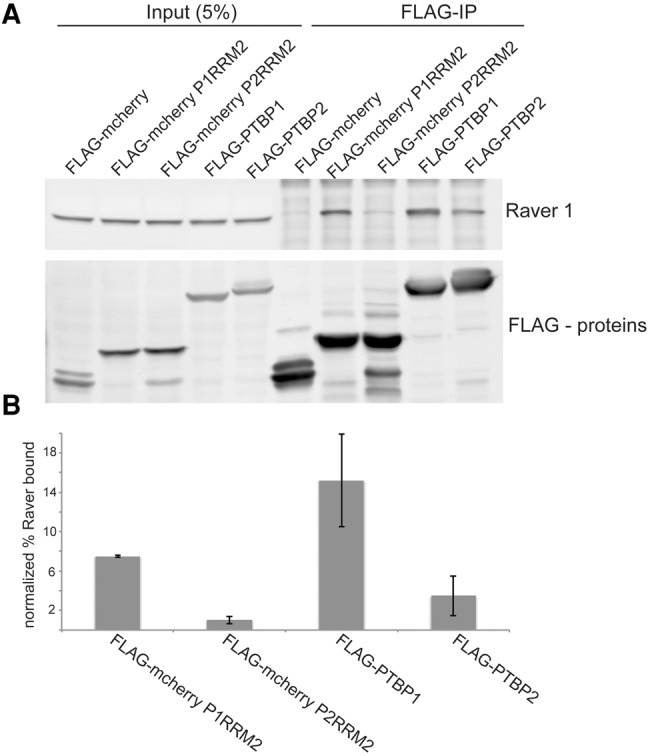
The interaction of Raver1 with PTB proteins (A) Flag-mCherry fusions and Flag-PTBP constructs were transfected into N2A cells. After cell lysis and nuclease digestion, tagged proteins were immunoprecipitated with Flag-agarose beads. Samples were assayed by immunoblot probed with anti-Raver 1 and anti-Flag antibodies as indicated to the right. Total protein in the transfected cells (left) and anti-flag immunoprecipitates (right) were assayed. (B) Raver1 protein in each immunoprecipitate was quantified and normalized against the input (5%). Bar heights are the averages of three separate experiments with the bars indicating mean absolute error.
Regulated exons assemble with multiple splicing regulatory proteins to form large RNP structures, and protein–protein interactions within these pre-mRNP complexes are important in determining the splicing pattern of the transcript (Rideau et al. 2006; Bonnal et al. 2008; Sharma et al. 2008, 2011; Chiou et al. 2013; Gooding et al. 2013; Lee and Rio 2014). The multiple features of PTBP1 that can confer higher repression activity on an otherwise PTBP2 sequence, including RRM1, RRM2, and the linker between RRM2 and RRM3, may mediate such protein–protein interactions. It is also possible that the sequences of PTBP1 and PTBP2 give them different conformational dynamics in solution or when bound to RNA. The RRMs of the cytoplasmic polyadenylation element binding proteins CPEB1 and CPEB4 undergo significant conformational changes upon RNA binding, with linker peptides mediating inter-RRM interactions (Afroz et al. 2014). Although NMR studies showed that linkers 1 and 2 were not rigid like linker 3, PTBP1 exhibited relatively little conformational flexibility in solution by low angle X-ray scattering measurements, suggesting some structuring of the linker 1 and linker 2 regions (Oberstrass et al. 2005; Petoukhov et al. 2006). It will be interesting to examine PTBP conformation upon RNA binding and how the structures of RNA-bound PTBP1 and PTBP2 are affected by the sequence determinants identified here.
MATERIALS AND METHODS
Cell culture and transfections
N2A cells were grown according to the American Type Culture Collection (ATCC) recommended protocols in DMEM (Fisher, MT-101-02CV) with 10% FBS (Omega scientific, FB-01). Transfections were done with Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Forty-eight hours after transfection, RNA and protein were isolated from the transfected cells.
Plasmid construction
Chimeras were constructed by two-step overlap extension PCR. Primers were designed to amplify regions of PTBP1 and PTBP2 carrying overlapping flanking sequences. The PCR fragments were cloned into pcDNA3.1 (+) (Life Technologies) using its BamH1 and EcoRV restriction sites. The inserts were sequenced completely to verify the absence of unwanted coding changes. Each expression plasmid carried an amino-terminal Flag-tag. The minigene reporter Dup 175-DS9 contained a 175-nt hybrid exon obtained from joining the 5′ end of β globin exon 2 to the 3′ end of β globin exon1 (Dominski and Kole 1991; Modafferi and Black 1997). The test exon is flanked by PTBP binding sites from Src and by wild-type β globin exons 1 and 2 (Amir-Ahmady et al. 2005). PQE80L-Chimera 3 was constructed by subcloning, using primers to introduce BamH1 and HindIII sites at the 5′ and 3′ end of the coding sequence, respectively.
Reverse-transcription PCR
RNA was isolated from N2A cells using the RNeasy Mini Kit (QIAGEN) and reverse transcribed with random hexamers and Superscript III (Life Technologies) according to the manufacturer's instructions. Spliced products were PCR amplified (17 cycles) using a 5′ primer (DUP-8 5′-GACACCATGCATGGTGCACCTG-3′) and FAM labeled 3′ primer (DUP-3) 5′-AACAGCATCAGGAGTGGACAGATCCC-3′ (Modafferi and Black 1997). The PCR products were separated by 8% polyacrylamide/7.5 M urea denaturing gels, visualized by a Typhoon Phosphorimager (Amersham Biosciences), and quantified using ImageQuant TL software (Amersham Biosciences).
Quantitative immunoblotting
Whole-cell lysates were separated by 12% SDS-PAGE, transferred to an Immobilon PVDF membrane, and probed with anti-Flag and GAPDH antibodies. Primary antibodies used were: Flag M2 1:3000 (Sigma), GAPDH 1:50,000 (Ambion), and Raver1 (a gift from Christopher Smith). After incubation with fluorescent-conjugated secondary antibodies, the blots were scanned using a Typhoon PhosphorImager. Western blot band intensities were quantified using Image Quant TL. Protein expression was quantified by normalizing the Flag-PTBP level to the level of GAPDH.
In vitro splicing assay
The BS713 and Adenovirus (pSPAd) constructs are described elsewhere (Chou et al. 2000; Sharma et al. 2005). The plasmids were linearized with Not1 and Sma1, respectively. BS713 was transcribed using T7 RNA polymerase and the Adeno template using SP6 RNA polymerase. Transcription reactions were carried out in the presence of cap analog and [α-32P] UTP as described previously (Chan and Black 1997). Nuclear extracts from HeLa cells were prepared as described previously (Chan and Black 1995). In vitro splicing was carried out in 25 µL reactions as previously described (Black 1992).
Expression and purification of recombinant protein
The His6-tagged PQE-PTB plasmids were transformed into E. coli BL21 (DE3). Five hundred milliliters of bacterial cultures were grown at 37°C in LB media containing 0.1 mg/mL ampicillin until the A600 reached ∼0.6. The cultures were adjusted to 0.4 mM IPTG and incubated at 37°C for 4 h with constant shaking. Cells were harvested by centrifugation, and the pellets were stored at −80°C. All subsequent procedures were performed at 4°C. Cells were resuspended in 25 mL Binding Buffer (50 mM NaPO4, pH 8.0, 300 mM NaCl, and 10 mM imidazole). Cell lysis was achieved by the addition of lysozyme to a final concentration of 1 mg/mL and sonicated to reduce viscosity. Insoluble material was removed by centrifugation. The soluble fractions were applied to 3-mL columns of Ni-NTA agarose beads that had been equilibrated with Binding Buffer. The columns were washed with 20 mL of Wash Buffer (50 mM NaPO4, pH 8.0, 300 mM NaCl, and 20 mM imidazole) and then eluted with 0.5 mL aliquots of Elution Buffer (50 mM NaPO4, pH 8.0, 300 mM NaCl, and 250 mM imidazole) containing 250 mM imidazole. Peak fractions were pooled and dialyzed against 2 L × 2 of buffer DG (20 mM HEPES-KOH pH 7.9, 80 mM K. glutamate, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, and 20% glycerol).
Immunoprecipitation
To achieve similar expression of the Flag-PTBP proteins, the amount of transfected DNA was adjusted according to their relative expression level from the pcDNA3.1+ vector. After 2 d incubation, N2A cells were collected and sonicated in Lysis Buffer (20 mM HEPES-KOH pH 7.9, 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.25% IGEPALCA-630, and protease inhibitor) followed by Benzonase (Sigma-Aldrich, E1014-25KU) treatment. Flag-PTBPs were immunopurified on anti-Flag M2 agarose affinity gel (Sigma, A2220). Flag-immunoprecipitates were eluted by boiling with SDS protein loading buffer and subjected to Western blot analysis.
ACKNOWLEDGMENTS
We thank Chris W. Smith (Cambridge University) for the Raver1 antibody. This work was supported in part by National Institutes of Health Ruth L. Kirschstein National Research Service Award (NIH-NRSA; 1F32GM093533), a CSUPERB New Investigator Award to N.M.K., and NIH grants R01GM049662 (to D.L.B.) and R21CA170786 (to S.S). K.H.Y. was supported by a fellowship from UCLA Broad Stem Cell Research Center's Training Program.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057505.116.
REFERENCES
- Afroz T, Skrisovska L, Belloc E, Guillén-Boixet J, Méndez R, Allain FHT. 2014. A fly trap mechanism provides sequence-specific RNA recognition by CPEB proteins. Genes Dev 28: 1498–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL. 2005. Exon repression by polypyrimidine tract binding protein. RNA 11: 699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiya M, Grabowski PJ. 1997. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA 9: 996–1015. [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Allain FHT. 2008. Structure-function relationships of the polypyrimidine tract binding protein. Cell Mol Life Sci 65: 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. 1992. Activation of c-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell 69: 795–807. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcarcel J. 2008. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell 32: 81–95. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Black DL. 2007. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21: 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. 2013. Dynamic integration of splicing within gene regulatory pathways. Cell 152: 1252–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL. 1995. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol Cell Biol 15: 6377–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL. 1997. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol 17: 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou NT, Shankarling G, Lynch KW. 2013. HnRNP L and HnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell 49: 972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MY, Underwood JG, Nikolic J, Luu MH, Black DL. 2000. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Molecular Cell 5: 949–957. [DOI] [PubMed] [Google Scholar]
- Coelho MB, Attig J, Bellora N, Konig J, Hallegger M, Kayikci M, Eyras E, Ule J, Smith CW. 2015. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J 34: 653–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte MR, Grune T, Ghuman J, Kelly G, Ladas A, Matthews S, Curry S. 2000. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J 19: 3132–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Kole R. 1991. Selection of splice sites in pre-mRNAs with short internal exons. Mol Cell Biol 11: 6075–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti A, Piñol-Roma S, Michael WM, Morandi C, Dreyfuss G. 1992. hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res 20: 3671–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A, Sharp PA, Jamison SF, Garcia-Blanco MA. 1991. Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev 5: 1224–1236. [DOI] [PubMed] [Google Scholar]
- Gooding C, Edge C, Lorenz M, Coelho MB, Winters M, Kaminski CF, Cherny D, Eperon IC, Smith CWJ. 2013. MBNL1 and PTB cooperate to repress splicing of Tpm1 exon 3. Nucleic Acids Res 41: 4765–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromak N, Rideau A, Southby J, Scadden ADJ, Gooding C, Hüttelmaier S, Singer RH, Smith CWJ. 2003. The PTB interacting protein raver1 regulates α-tropomyosin alternative splicing. EMBO J 22: 6356–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueroussov S, Gonatopoulos-Pournatzis T, Irimia M, Raj B, Lin Z, Gingras A, Blencowe B. 2015. An alternative splicing event amplifies evolutionary differences between vertebrates. Science 349: 868–873. [DOI] [PubMed] [Google Scholar]
- Han W, Wang L, Yin B, Peng X. 2013. Characterization of a novel posttranslational modification in polypyrimidine tract-binding proteins by SUMO1. BMB Rep 47: 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Stoilov P, Linares AJ, Zhou Y, Fu X-D, Black DL. 2014. De novo prediction of PTBP1 binding and splicing targets reveals unexpected features of its RNA recognition and function. PLoS Comput Biol 10: e1003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberg B, Swiniarski S, Sabine B, Illenberger S. 2010. A conserved peptide motif in Raver2 mediates its interaction with the polypyrimidine tract-binding protein. Exp Cell Res 316: 966–979. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. 2015. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Coelho MB, Kotik-Kogan O, Simpson PJ, Matthews SJ, Smith CWJ, Curry S. 2011. Crystallographic analysis of polypyrimidine tract-binding protein-raver1 interactions involved in regulation of alternative splicing. Structure 19: 1816–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Esteve V, Buckroyd A, Blatter M, Allain F, Curry S. 2014. Solution and crystal structures of a C-terminal fragment of the neuronal isoform of the polypyrimidine tract binding protein (nPTB). PeerJ 2: e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafasla P, Mickleburgh I, Llorian M, Coelho M, Gooding C, Cherny D, Smith CWJ. 2012. Defining the roles and interactions of PTB. Biochem Soc Trans 40: 815–820. [DOI] [PubMed] [Google Scholar]
- Keppetipola N, Sharma S, Li Q, Black DL. 2012. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol 47: 360–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Rio DC. 2014. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 84: 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zheng S, Han A, Lin C-H, Stoilov P, Fu X-D, Black DL. 2014. The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. eLife 3: e01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. 2012. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26: 1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares AJ, Lin C-H, Damianov A, Adams KL, Novitch BG, Black DL. 2015. The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. eLife 4: e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorian M, Schwartz S, Clark TA, Hollander D, Tan L-Y, Spellman R, Smith CWJ. 2010. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 17: 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. 2007. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27: 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovtsov V, Nikolic JM, Goldman JA, Turck CW, Chou MY, Black DL. 2000. Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol 20: 7463–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modafferi EF, Black DL. 1997. A complex intronic splicing enhancer from the c-src pre-mRNA activates inclusion of a heterologous exon. Mol Cell Biol 17: 6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modafferi EF, Black DL. 1999. Combinatorial control of a neuron-specific exon. RNA 5: 687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Allain FH-T. 2005. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309: 2054–2057. [DOI] [PubMed] [Google Scholar]
- Oldfield CJ, Dunker AK. 2014. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83: 553–584. [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Monie TP, Allain FH-T, Matthews S, Curry S, Svergun DI. 2006. Conformation of polypyrimidine tract binding protein in solution. Structure 14: 1021–1027. [DOI] [PubMed] [Google Scholar]
- Polydorides AD, Okano HJ, Yang YY, Stefani G, Darnell RB. 2000. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc Natl Acad Sci 97: 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideau AP, Gooding C, Simpson PJ, Monie TP, Lorenz M, Hüttelmaier S, Smith CWJ. 2006. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat Struct Mol Biol 13: 839–848. [DOI] [PubMed] [Google Scholar]
- Robinson F, Smith CWJ. 2006. A splicing repressor domain in polypyrimidine tract-binding protein. J Biol Chem 281: 800–806. [DOI] [PubMed] [Google Scholar]
- Shankarling G, Lynch KW. 2013. Minimal functional domains of paralogues hnRNP L and hnRNP LL exhibit mechanistic differences in exonic splicing repression. Biochem J 453: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Falick AM, Black DL. 2005. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell 19: 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. 2008. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol 15: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Maris C, Allain FHT, Black DL. 2011. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell 41: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Wongpalee SP, Vashisht A, Wohlschlegel JA, Black DL. 2014. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev 28: 2518–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PJ, Monie TP, Szendröi A, Davydova N, Tyzack JK, Conte MR, Matthews S. 2004. Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure 12: 1631–1643. [DOI] [PubMed] [Google Scholar]
- Southby J, Gooding C, Smith CW. 1999. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of α-actinin mutally exclusive exons. Mol Cell Biol 19: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R, Rideau A, Matlin A, Gooding C, Robinson F, McGlincy N, Smith CWJ. 2005. Regulation of alternative splicing by PTB and associated factors. Biochem Soc Trans 33: 457–460. [DOI] [PubMed] [Google Scholar]
- Spellman R, Llorian M, Smith CWJ. 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell 27: 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Sharma S, Zheng S, Chawla G, Nikolic J, Black DL. 2011. Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J Biol Chem 286: 10007–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp JD, Jackson J, Melton AA, Lynch KW. 2008. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA 14: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali F, Henning A, Oberstrass FC, Hargous Y, Auweter SD, Erat M, Allain FHT. 2006. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J 25: 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carstens RP, Garcia-Blanco MA. 1999. A novel isoform ratio switch of the polypyrimidine tract binding protein. Electrophoresis 20: 1082–1086. [DOI] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Robinson F, Brown EC, Jackson RJ, Smith CW. 2001. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 7: 819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon Y-S, Zhang Y. 2009. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Fu XD. 2013. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O'Dell TJ, Black DL. 2012. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci 15: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]