

Abstract
The most frequent epidermal growth factor receptor (EGFR) mutations found by traditional or comprehensive molecular profiling of lung adenocarcinomas include indels of exon 19 (the exon 19 deletion delE746_A750 being the most common) and the exon 21 L858R point mutation. The current approval labels for first line palliative gefitinib 250 mg/day, erlotinib 150 mg/day and afatinib 40 mg/day for advanced lung cancers require the presence of the aforementioned classical/sensitizing EGFR mutations. Other gefitinib, erlotinib and afatinib sensitizing mutations include exon 18 indels, G719X, exon 19 insertions, A763_Y764insFQEA, S768I and L861Q; for which off-label EGFR kinase inhibitor use is generally agreed upon by thoracic oncologists. The main biological mechanism of resistance to approved first line EGFR inhibitors is the selection/acquisition of EGFR-T790M that in itself can be inhibited by osimertinib 80 mg/day, a 3rd generation EGFR inhibitor that is bypassed by EGFR-C797X mutations. Another class of de novo inhibitor insensitive mutation includes EGFR exon 20 insertions. More recently, the dichotomy of only point mutations or indels explaining aberrant kinase activation of EGFR plus inhibitor response has been shattered by the discovery of uncommon (<0.5% of all EGFR mutations) genomic events involving exon 18–25 kinase domain duplications (KDD) and rearrangements (EGFR-RAD51 or EGFR-PURB). The latter lead to oncogene addiction, enhanced sensitivity to kinase inhibitors in vitro and clinical responses to approved EGFR inhibitors. The enhanced landscape of EGFR inhibitor-responsive genotypes highlights that comprehensive molecular profiling may be necessary to maximize the identification of all cases that can benefit from precision oncology.
Keywords: Epidermal growth factor receptor (EGFR), exon 18–25 duplication, rearrangement, exon 19, L858R, L861Q, G719X, exon 18, exon 20, T790M, C797S
Epidermal growth factor receptor (EGFR) mutations were first identified as driver oncogenes in non-small-cell lung cancers (NSCLCs) in 2004 by three separate independent groups (1-3), and originally thought to consistent of only in-frame deletions, insertions (i.e., indels) or point mutations within exons 18 to 21 of the kinase domain of EGFR (4). The most abundant EGFR mutations are deletions/indels (around amino-acid residues 747 to 752) of exon 19 (these account for ~45% of all EGFR mutations, with the most common delE746_A750) and the exon 21 point mutation L858R mutation (~35% of all EGFR mutations). Inhibition of mutant EGFR in preclinical models through tyrosine kinase inhibitors (TKIs) unsettles the intracellular signaling cascade, generating cell cycle arrest and apoptosis (5). In the clinic, the 1st generation EGFR TKIs gefitinib and erlotinib, both reversible ATP mimetics with a favorable therapeutic window in relation to the wild-type (WT) EGFR (4,6), induce overall response rate (ORR), progression-free survival (PFS) and quality of life (QoL) improvements that exceed platinum-doublet cytotoxic chemotherapies in advanced EGFR mutated NSCLCs (7,8). The 2nd generation irreversible EGFR TKI afatinib, with a narrower therapeutic window due to its exceedingly potent inhibition of WT EGFR, also improves ORR, PFS and QoL when compared to cytotoxic agents (9). Exceedingly high ORRs of >70% have been observed for EGFR-exon 19 deletion mutated NSCLCs treated with gefitinib 250 mg/day, erlotinib 150 mg/day or afatinib 40 mg/day (7-9). The ORR of EGFR-L858R mutated tumors seems to be slightly lower than 70% with afatinib 40 mg/day, while only at around 50–60% with gefitinib 250 mg/day and intermediate with erlotinib 150 mg/day (7-9). Indeed, a head-to-head phase II trial (LUX-Lung7) of afatinib 40 mg/day versus gefitinib 250 mg/day showed that the ORRs were 66% vs. 42% and median PFSs of 10.9 vs. 10.8 months (HR 0.71), respectively, for the 133 EGFR-L858R mutated NSCLCs (10). The ORRs were 73% vs. 66% and median PFSs of 12.7 vs. 11.0 months (HR 0.71), respectively, for the 186 EGFR-exon 19 deletion mutated NSCLCs (10). The improved predictive and prognostic impact of tumor EGFR-exon 19 deletions versus EGFR-L858R in TKI-treated patients are well known since 2006 (11,12) and confirmed in all randomized clinical trials of EGFR TKI versus chemotherapy (13). All three—gefitinib, erlotinib and afatinib—Food and Drug Administration (FDA) approved EGFR TKIs continue to be prescribed worldwide without a clear “go-to” drug in view of their different biological doses, toxicities (afatinib with higher rates of mucositis and diarrhea, erlotinib of rash, and gefitinib of liver dysfunction) and provider-patient preferences. As afatinib is the more toxic of the approved first line EGFR TKIs, one must take into consideration its reported higher ORR and PFS rates together with the increased rates of adverse events plus dose reductions required with this agent (9,10).
The third most common type of EGFR mutations (>7% of all EGFR mutations) consist of in-frame insertions and indels following/encompassing the regulatory C-helix amino-acids of exon 20 (14,15). In preclinical models, these mutations lead to auto-phosphorylation of EGFR and engagement of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinases (PI3K) cascades; concurrent with oncogene addiction (15). However, these mutant EGFRs at the structural and biological level do not have a favorable therapeutic window in relation to WT EGFR. The later realization explains why gefitinib (16), erlotinib (15) and afatinib (17) have limited activity (near 0% ORRs and short PFSs) in EGFR exon 20 insertion mutated NSCLCs (14). Grippingly, near identical exon 20 insertion mutations can be found on the erb-b2 receptor tyrosine kinase 2 (ERBB2) gene and the resulting encoded proteins are also not particularly sensitive to standard dosing schemes of dual EGFR/ERBB2 TKIs (18). The development of TKIs for these recalcitrant variants in EGFR and ERBB2 continues to be an unmet medical need for the management of NSCLC.
Certain other clinically-relevant kinase domain EGFR mutations, named by others as uncommon or atypical mutations, seem to be EGFR TKI sensitive in preclinical models (where they are transforming and activate the MAPK/PI3K signaling cascades) and in available published clinical reports (4,16,17). These mutations encompass EGFR-exon 18 indels/E709X (<0.5% of EGFR mutations), exon 18 G719X (~3% of EGFR mutations), exon 19 insertions (<0.5% of EGFR mutations), exon 20 A763_Y764insFQEA (<0.5% of EGFR mutations), exon 20 S768I (<1.5% of EGFR mutations) and the exon 21 L861Q (~3% of EGFR mutations); either alone or compound with other EGFR mutations (19). It is interesting to note that in preclinical models, the inhibitory concentrations of 1st generations EGFR TKIs are usually 10–200 times higher for EGFR-exon 18 indels/E709X (20,21), exon 18 G719X (20), exon 19 insertions (22), exon 20 A763_Y764insFQEA (15), exon 20 S768I (23) and the exon 21 L861Q (23) when compared to EGFR-exon 19 deletion mutants. These observations may explain why the ORRs in the clinic seldom exceed 55% for tumors that harbor these mutations types in patients treated with gefitinib or erlotinib (15,16). The same preclinical models show slightly higher relative potency for the 2nd generation EGFR TKI afatinib, specifically for EGFR exon 18 mutations (20). Indeed, the ORRs to afatinib 40 mg/day seem to be higher than 55% for tumors harboring EGFR-G719X, L861Q or S768I mutations (17).
Despite initial rapid and sometimes prolonged responses to gefitinib, erlotinib and afatinib for lung cancers with the aforementioned EGFR TKI-sensitizing mutations, acquired resistance to EGFR TKIs is inevitable for most tumors due to biological (on-target mutations, bypass tracks or histological transformation) and pharmacokinetic mechanisms (24). The most common abnormality identified on rebiopsy specimens is the EGFR-T790M (within the gatekeeper position of exon 20) mutation in >50–60% of progressing lesions (6,25). EGFR-T790M is most commonly identified in EGFR-exon 19 deletion mutated tumors but has also been reported in conjunction with L858R, L861Q, and S768I among others (26). Germline EGFR-T790M has also been described as a rare (<1%) high relative risk susceptibility allele in families with lung cancers independent of smoking risk (27,28). Eloquent structural and biochemical experiments have irrefutably defined that the addition of EGFR-T790M to a sensitizing mutant alters the kinetics of inhibitor binding of gefitinib, erlotinib and afatinib (29,30); leading to resistance to achievable clinical doses of these EGFR TKIs. However, 3rd generation EGFR TKIs that were selected on the basis of their covalent binding to EGFR-C797, plus their mutation over WT EGFR sensitivity, can inhibit EGFR-T790M bearing cancers (6,31). The most advanced of the clinical candidate 3rd generation EGFR TKIs is osimertinib given at 80 mg/day (32). The drug is exceedingly active against tumors with acquired resistance to gefitinib, erlotinib or afatinib when EGFR-T790M is present, with reported ORRs of >55% (26). Osimertinib was FDA-approved in 2015. Unfortunately, resistance to osimertinib monotherapy seems again to be inevitable with a predominance of on-target mutation events (including EGFR-C797S) in progressing tumors or circulating tumor DNA (33). The Thoracic Oncology community awaits a new generation of EGFR TKIs and of anti-cancer therapy combinations with EGFR TKIs to prevent and/or treat resistance to 3rd generation EGFR TKIs.
Just as the field of EGFR mutated NSCLC seemed to restricted to point mutations and indels that congregated in the kinase domain (as reviewed above and summarized in Table 1), two new reports led by investigators of the commercial comprehensive genomic profiling company Foundation Medicine and of Vanderbilt University School of Medicine have broadened our horizon to rare genomic events that also activate the kinase domain of EGFR: EGFR-exon 18–25 kinase domain duplication (EGFR-KDD) and EGFR rearrangements (34,35). It seems the frequency of these changes does not exceed individually 0.5% of all EGFR mutation events (Table 1). In the 1,510 EGFR mutated tumor cohort described from 10,097 analyzed cases using FoundationOne’s comprehensive genomic profiling (35), the frequency of EGFR-KDD was 0.2% and of EGFR rearrangements was 0.3% (Figure 1). These changes had not been reported previously because most traditional EGFR sequencing strategies used in day-to-day clinical care (Sanger sequencing, allele-specific PCR-based or focused next generation sequencing panels) are unable to identify these rare genomic variants.
Table 1. Types, frequency and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor sensitivity of EGFR kinase domain mutations in lung cancer.
| EGFR mutation | Approximate frequency (%) | EGFR TKI [in vitro sensitivity and expected overall response rate (ORR)] | ||
|---|---|---|---|---|
| EGFR TKI sensitivity type | 1st generation | 2nd generation | 3rd generation | |
| Gefitinib 250 mg Erlotinib 150 mg |
Afatinib 40 mg | Osimertinib 80 mg | ||
| Sensitizing | ||||
| Exon 19 deletion | 45.0 | ++++ (ORR >70%) | ++++ (ORR >75%) | ++++ (ORR >70%) |
| L858R | 35.0 | ++++ (ORR >60%) | ++++ (ORR >70%) | ++++ (ORR >60%) |
| G719X | 3.0 | ++ (ORR >55%) | +++ (ORR >65%) | ++ (ORR ?) |
| L861Q | 3.0 | ++ (ORR >55%) | ++ (ORR >55%) | ++ (ORR ?) |
| S768I | <1.5 | + (ORR >45%) | ++ (ORR >55%) | ? (ORR ?) |
| Exon 18 indel/E709X | <0.5 | ++ (ORR >55%) | +++ (ORR >65%) | ++ (ORR ?) |
| Exon 19 insertion | <0.5 | ++ (ORR >55%) | ++ (ORR ?) | ++ (ORR ?) |
| A763_Y764insFQEA | <0.5 | ++ (ORR >55%) | ++ (ORR ?) | ++ (ORR ?) |
| Exon 18–25 duplication (EGFR-KDD) | <0.5 | ++ (ORR >55%) | +++ (ORR >65%) | ++ (ORR ?) |
| Rearrangement (EGFR-RAD51) | <0.5 | ++ (ORR >55%) | +++ (ORR ?) | ++ (ORR ?) |
| Insensitizing | ||||
| Exon 20 insertion | >7.0 | – (ORR <5%) | – (ORR <10%) | – (ORR ?) |
| T790M inherited | <1.0 | – (ORR ~0%) | – (ORR ~0%) | ++++ (ORR >60%) |
| Others | >2.0 | ? (ORR ?) | ? (ORR ?) | ? (ORR ?) |
| Acquired resistance | ||||
| T790M + sens. | >50.0 (1st/2nd gen. TKI) | – (ORR ~0%) | – (ORR <5%) | ++++ (ORR >60%) |
| C797X + T790M + sens. | <50.0 (osimertinib) | – (ORR ~0%) | – (ORR ~0%) | – (ORR ~0%) |
++++, maximum inhibition; +++, moderate inhibition; ++, adequate inhibition; +, minimal inhibition; –, no significant inhibition beyond the therapeutic window of wild-type EGFR; EGFR, epidermal growth factor receptor; TKI, tyrosine kinase inhibitor; ?, unknown; sens, sensitizing mutation; gen., generation.
Figure 1.
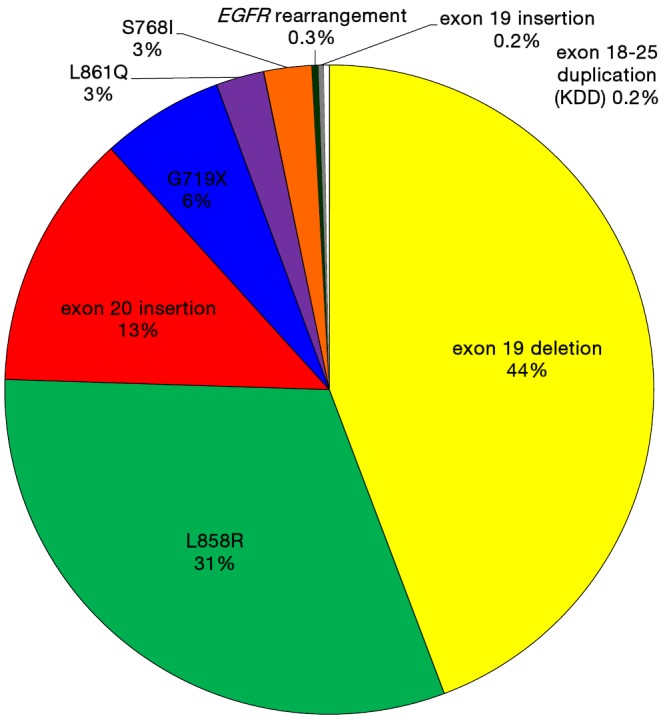
Pie chart display of epidermal growth factor receptor (EGFR) genomic aberrations identified by a single commercial vendor (Foundation Medicine) using the FoundationOne comprehensive genomic profiling that can be identify indels, point mutations, copy number changes, kinase domain duplications (KDD) and rearrangements. The data was obtained from (35).
The EGFR-KDD alteration consists of an intragenic alteration in EGFR, resulting in the tandem duplication of exons 18 to 25 (34). As these exons encompass the tyrosine kinase domain, this duplication generates an in-frame kinase domain duplication at the protein level. This type of EGFR-KDD had only been previously reported in rare cases of glioma (36) and was additionally found to occur in sarcomas, peritoneal carcinomas and Wilms’ tumors (34). In preclinical and computational models, the resulting EGFR-KDD protein is transforming, may generate EGFR intramolecular asymmetric activated dimers, and is hypersensitive to 1st, 2nd and 3rd generation EGFR TKIs (34). The same report also describes a case of advanced chemotherapy-progressive EGFR-KDD mutated lung adenocarcinoma with a 7-month partial response to afatinib (doses not provided) and subsequent progression due to amplification of the EGFR-KDD allele (34). Another case report of a prolonged multi-year response to gefitinib and then erlotinib has been described for advanced EGFR-KDD mutated lung adenocarcinoma (37). Therefore, it seems these variants are responsive to 1st and 2nd generation EGFR TKIs in the clinic.
EGFR rearrangements were for the first time described in 2016, with rearrangements following the kinase domain of EGFR (at exon 25) with other partners. The two reported partners include the C-terminal portion of the RAD51 recombinase (RAD51) or purine-rich element binding protein B (PURB) genes (35). The resulting N-terminal EGFR-RAD51 C-terminal fusion protein retains an important regulatory auto-phosphorylation site (Y845) of EGFR (35). In preclinical models, EGFR-RAD51 is transforming, activates downstream signaling pathways, may form activation dimers, and is hypersensitive to 1st, 2nd and 3rd generation EGFR TKIs (35). Of most interest, three patients with EGFR-RAD51 and one patient with EGFR-PURB rearranged NSCLCs had between 5- to 20-month periods of partial response to standard clinical doses of erlotinib (35); confirming that EGFR fusion proteins are TKI-sensitive variants. Other type of EGFR genomic aberrations outside the kinase domain of EGFR—including extracellular domain in-frame deletions (such as the truncated EGFR-vIII deletion), extracellular domain point mutations and C-terminal activating exon 25-26 deletions—have also been described in whole genome sequencing cohorts of lung adenocarcinoma (38). The prevalence and clinical significance of the latter genomic changes remains to be elucidated in the clinical care of NSCLC with off-label use of FDA-approved EGFR TKIs.
In summary, the enhanced landscape of EGFR TKI-responsive genotypes (including exon 19 deletions, L858R, exon 18 indels, G719X, exon 19 insertions, A763_Y764insFQEA, S768I, L861Q, KDD and rearrangements to gefitinib, erlotinib or afatinib; and T790M to osimertinib) highlights that comprehensive molecular profiling may be necessary to maximize the identification of all cases that can benefit from precision oncology when dealing with EGFR mutated NSCLC. It also demonstrates that we have not yet identified all genomic variants that are actionable and/or clinically-relevant in NSCLC (39-50).
Acknowledgements
Funding: This work was funded in part through a Lung Cancer Foundation of America-International Association for the Study of Lung Cancer grant (to the author), an American Cancer Society grant RSG 11-186 (to the author), and National Cancer Institute grants CA090578 (to the author).
Footnotes
Conflicts of Interest: The author has received consulting fees from Pfizer Inc., Boehringer Ingelheim and Ariad. The author also conducts unremunerated clinical trials using afatinib (Boehringer Ingelheim), erlotinib (Astellas), osimertinib (AstraZeneca) and rociletinib (Clovis Oncology).
References
- 1.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306-11. 10.1073/pnas.0405220101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500. 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39. 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- 4.Jorge SE, Kobayashi SS, Costa DB. Epidermal growth factor receptor (EGFR) mutations in lung cancer: preclinical and clinical data. Braz J Med Biol Res 2014;47:929-39. 10.1590/1414-431X20144099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 2007;4:1669-79; discussion 1680. [DOI] [PMC free article] [PubMed]
- 6.Costa DB, Kobayashi SS. Whacking a mole-cule: clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl Lung Cancer Res 2015;4:809-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57. 10.1056/NEJMoa0810699 [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46. 10.1016/S1470-2045(11)70393-X [DOI] [PubMed] [Google Scholar]
- 9.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327-34. 10.1200/JCO.2012.44.2806 [DOI] [PubMed] [Google Scholar]
- 10.Park K, Tan EH, O'Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol 2016;17:577-89. 10.1016/S1470-2045(16)30033-X [DOI] [PubMed] [Google Scholar]
- 11.Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res 2006;12:3908-14. 10.1158/1078-0432.CCR-06-0462 [DOI] [PubMed] [Google Scholar]
- 12.Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res 2006;12:839-44. 10.1158/1078-0432.CCR-05-1846 [DOI] [PubMed] [Google Scholar]
- 13.Lee CK, Wu YL, Ding PN, et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes After Treatment With EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR-Mutant Lung Cancer: A Meta-Analysis. J Clin Oncol 2015;33:1958-65. 10.1200/JCO.2014.58.1736 [DOI] [PubMed] [Google Scholar]
- 14.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13:e23-31. 10.1016/S1470-2045(11)70129-2 [DOI] [PubMed] [Google Scholar]
- 15.Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013;5:216ra177. 10.1126/scitranslmed.3007205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on "uncommon" epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 2011;17:3812-21. 10.1158/1078-0432.CCR-10-3408 [DOI] [PubMed] [Google Scholar]
- 17.Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015;16:830-8. 10.1016/S1470-2045(15)00026-1 [DOI] [PubMed] [Google Scholar]
- 18.Costa DB, Jorge SE, Moran JP, et al. Pulse Afatinib for ERBB2 Exon 20 Insertion-Mutated Lung Adenocarcinomas. J Thorac Oncol 2016;11:918-23. 10.1016/j.jtho.2016.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi S, Canepa HM, Bailey AS, et al. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol 2013;8:45-51. 10.1097/JTO.0b013e3182781e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi Y, Togashi Y, Yatabe Y, et al. EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin Cancer Res 2015;21:5305-13. 10.1158/1078-0432.CCR-15-1046 [DOI] [PubMed] [Google Scholar]
- 21.Ackerman A, Goldstein MA, Kobayashi S, et al. EGFR delE709_T710insD: a rare but potentially EGFR inhibitor responsive mutation in non-small-cell lung cancer. J Thorac Oncol 2012;7:e19-20. 10.1097/JTO.0b013e3182635ab4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He M, Capelletti M, Nafa K, et al. EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res 2012;18:1790-7. 10.1158/1078-0432.CCR-11-2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kancha RK, von Bubnoff N, Peschel C, et al. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin Cancer Res 2009;15:460-7. 10.1158/1078-0432.CCR-08-1757 [DOI] [PubMed] [Google Scholar]
- 24.Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 2009;10:281-9. 10.3816/CLC.2009.n.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786-92. 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- 26.Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015;372:1689-99. 10.1056/NEJMoa1411817 [DOI] [PubMed] [Google Scholar]
- 27.Oxnard GR, Nguyen KS, Costa DB. Germline mutations in driver oncogenes and inherited lung cancer risk independent of smoking history. J Natl Cancer Inst 2014;106:djt361. 10.1093/jnci/djt361 [DOI] [PubMed] [Google Scholar]
- 28.Oxnard GR, Miller VA, Robson ME, et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol 2012;7:1049-52. 10.1097/JTO.0b013e318250ed9d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eck MJ, Yun CH. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta 2010;1804:559-66. [DOI] [PMC free article] [PubMed]
- 30.Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res 2005;65:7096-101. 10.1158/0008-5472.CAN-05-1346 [DOI] [PubMed] [Google Scholar]
- 31.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009;462:1070-4. 10.1038/nature08622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X, Le X, Costa DB. The safety and efficacy of osimertinib for the treatment of EGFR T790M mutation positive non-small-cell lung cancer. Expert Rev Anticancer Ther 2016;16:383-90. 10.1586/14737140.2016.1162103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560-2. 10.1038/nm.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallant JN, Sheehan JH, Shaver TM, et al. EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer That Is Clinically Responsive to Afatinib. Cancer Discov 2015;5:1155-63. 10.1158/2159-8290.CD-15-0654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konduri K, Gallant JN, Chae YK, et al. EGFR Fusions as Novel Therapeutic Targets in Lung Cancer. Cancer Discov 2016;6:601-11. 10.1158/2159-8290.CD-16-0075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciesielski MJ, Fenstermaker RA. Oncogenic epidermal growth factor receptor mutants with tandem duplication: gene structure and effects on receptor function. Oncogene 2000;19:810-20. 10.1038/sj.onc.1203409 [DOI] [PubMed] [Google Scholar]
- 37.Baik CS, Wu D, Smith C, et al. Durable Response to Tyrosine Kinase Inhibitor Therapy in a Lung Cancer Patient Harboring Epidermal Growth Factor Receptor Tandem Kinase Domain Duplication. J Thorac Oncol 2015;10:e97-9. 10.1097/JTO.0000000000000586 [DOI] [PubMed] [Google Scholar]
- 38.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107-20. 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shea M, Huberman MS, Costa DB. Lazarus-Type Response to Crizotinib in a Patient with Poor Performance Status and Advanced MET Exon 14 Skipping Mutation-Positive Lung Adenocarcinoma. J Thorac Oncol 2016;11:e81-2. 10.1016/j.jtho.2016.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703. 10.1056/NEJMoa1006448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasuda H, de Figueiredo-Pontes LL, Kobayashi S, et al. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol 2012;7:1086-90. 10.1097/JTO.0b013e3182570919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jorge SE, Schulman S, Freed JA, et al. Responses to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and co-occurring mutations in lung adenocarcinomas with MET amplification or MET exon 14 skipping mutation. Lung Cancer 2015;90:369-74. 10.1016/j.lungcan.2015.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costa DB, Shaw AT, Ou SH, et al. Clinical Experience With Crizotinib in Patients With Advanced ALK-Rearranged Non-Small-Cell Lung Cancer and Brain Metastases. J Clin Oncol 2015;33:1881-8. 10.1200/JCO.2014.59.0539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963-71. 10.1056/NEJMoa1406766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Figueiredo-Pontes LL, Wong DW, Tin VP, et al. Identification and characterization of ALK kinase splicing isoforms in non-small-cell lung cancer. J Thorac Oncol 2014;9:248-53. 10.1097/JTO.0000000000000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shea M, Costa DB, Rangachari D. Management of advanced non-small cell lung cancers with known mutations or rearrangements: latest evidence and treatment approaches. Ther Adv Respir Dis 2016;10:113-29. 10.1177/1753465815617871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rangachari D, VanderLaan PA, Le X, et al. Experience with targeted next generation sequencing for the care of lung cancer: insights into promises and limitations of genomic oncology in day-to-day practice. Cancer Treat Commun 2015;4:174-81. 10.1016/j.ctrc.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanderlaan PA, Yamaguchi N, Folch E, et al. Success and failure rates of tumor genotyping techniques in routine pathological samples with non-small-cell lung cancer. Lung Cancer 2014;84:39-44. 10.1016/j.lungcan.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerber DE, Gandhi L, Costa DB. Management and future directions in non-small cell lung cancer with known activating mutations. Am Soc Clin Oncol Educ Book 2014:e353-65. 10.14694/EdBook_AM.2014.34.e353 [DOI] [PubMed] [Google Scholar]