

Abstract
Crizotinib is an oral inhibitor of anaplastic lymphoma kinase (ALK) with remarkable clinical activity in patients suffering from ALK-rearranged non-small cell lung cancer (NSCLC), accounting to its superiority compared to chemotherapy. Unfortunately, virtually all ALK-rearranged tumors acquire resistance to crizotinib, frequently within one year since the treatment initiation. To date, therapeutic strategies to overcome crizotinib resistance have focused on the use of more potent and structurally different compounds. Second-generation ALK inhibitors such as ceritinib (LDK378), alectinib (CH5424802/RO5424802) and brigatinib (AP26113) have shown relevant clinical activity, consequently fostering their rapid clinical development and their approval by health agencies. The third-generation inhibitor lorlatinib (PF-06463922), selectively active against ALK and ROS1, harbors impressive biological potency; its efficacy in reversing resistance to crizotinib and to other ALK inhibitors is being proven by early clinical trials. The NTRK1-3 and ROS1 inhibitor entrectinib (RXDX-101) has been reported to act against NSCLC harboring ALK fusion proteins too. Despite the quick development of these novel agents, several issues remain to be discussed in the treatment of patients suffering from ALK-rearranged NSCLC. This position paper will discuss the development, the current evidence and approvals, as long as the future perspectives of new ALK inhibitors beyond crizotinib. Clinical behaviors of ALK-rearranged NSCLC vary significantly among patients and differential molecular events responsible of crizotinib resistance account for the most important quote of this heterogeneity. The precious availability of a wide range of active anti-ALK compounds should be approached in a critical and careful perspective, in order to develop treatment strategies tailored on the disease evolution of every single patient.
Keywords: Non-small-cell lung cancer (NSCLC), EML4-ALK rearrangement, crizotinib, anaplastic lymphoma kinase inhibitors (ALK inhibitors)
Introduction
Lung cancer remains one of the deadliest neoplasms worldwide, with less than 18% of patients alive five years after diagnosis (1); non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers (2). Notably, NSCLC is not a single pathological entity, but it is rather a mixture of malignancies different in terms of histology and molecular patterns, differentially impacting upon disease outcomes. Several genetic events lead to the expression of molecularly altered tyrosine kinase receptors (TKRs) and signaling proteins that, while conferring decisive oncogenic potential to malignant cells, represent suitable and crucial therapeutic targets. In addition to mutations occurring in EGFR, KRAS, BRAF and other genes (3), recent interest has been conferred to gene fusions determining the aberrant expression of proteins, which generates similar profiles of oncogenic addiction (4).
Among these, the anaplastic lymphoma kinase (ALK) protein, encoded by the ALK gene on chromosome 2p, is a transmembrane TKR. ALK protein is important for fetal development, but its expression is lost in all adult tissues, with exception of the brain. Rearrangements in ALK gene release it from the negative control exerted by silencing promoters, giving rise thus far to ALK fusion transcripts and active proteins. The latter are the crucial and funding events in ALK-rearranged pathognomonic anaplastic large cell lymphoma (ALCL), inflammatory myofibroblastic tumors (IMT) and NSCLC (5). Concerning lung tumors, inversions within chromosome 2p are the most frequent genetic alterations leading to the fusion of ALK with the partner gene echinoderm microtubule associated protein like 4 (EML4) (6). Other mechanisms of gene rearrangement and different partner genes on different chromosomes are possible. The loss of ALK transmembrane domain alters its localization, addressing the fusion protein almost invariably to the cytoplasmic compartment (4). Importantly, the kinase domain of ALK is always conserved and constitutively active, transmitting mitogenic, survival and anti-apoptotic signals to the nucleus through intracellular pathways.
Taking into account the estimation of 224,390 new diagnoses of lung cancers in 2016 in USA (1) and the large proportion of advanced NSCLC, the absolute number of ALK-rearranged tumors is expected to be between 2,670 and 10,680, considering its relative incidence of 2–8% (6-9). In Europe, about 410,000 new lung cancer cases are diagnosed every year (10): between 5,000 and 19,000 cases of ALK-positive advanced NSCLC are thus far expected.
Crizotinib, initially developed as a cMET inhibitor, is an oral inhibitor of ALK that exert its activity competing with ATP binding in the kinase domain of the enzyme. Crizotinib showed impressive response rates (RR) and prolonged progression-free survival (PFS) compared with chemotherapy in ALK-positive patients in both pretreated and first-line settings (11-13); PROFILE 1007, 1005 and 1014, respectively). This molecule is currently approved for patients whose tumors harbor ALK rearrangements. Unfortunately, almost every ALK-rearranged tumor eventually acquires resistance during crizotinib treatment, frequently within one year since the initiation of treatment (11). Mechanisms responsible of resistance to crizotinib include ALK mutations, ALK amplification and bypass of target signaling (14,15). Furthermore, different mutations may coexist in the same patient, increasing the difficulty of detecting every event conferring resistance to crizotinib (14). As brain is among the most frequent site of disease progression, pharmacokinetics issues concerning crizotinib ability to cross the blood-brain barrier have also been risen up (16).
Second- and third-generation ALK inhibitors have been developed to overcome acquired crizotinib resistance. These novel compounds are more potent than crizotinib and structurally distinct. Ceritinib (LDK378) and alectinib (CH5424802/RO5424802) are already approved by U.S. Food and Drug Administration (FDA) in crizotinib-resistant patients, while brigatinib (AP26113), lorlatinib (PF-06463922) and entrectinib (RXDX-101) are in different phases of clinical development.
Nevertheless, some of these new compounds are currently compared to the first generation molecule in crizotinib-naive patients, in order to evaluate if an upfront “stronger” ALK inhibition can control the disease longer than the sequential treatment. Moreover, also if almost all patients do respond to crizotinib, primary (de novo) resistance actually exists (17).
The rapid and continuous development of the new-generation ALK inhibitors requires a thorough and updated knowledge of their biological relevance, of their early and confirmed clinical activity, as long as their current approval and experimental status. Such availability provides incomparable opportunities for the treatment of ALK-rearranged NSCLC patients. Nevertheless, the careful analysis of published and presented data is of crucial importance, as long as the design of smart clinical trials, in order to define the best treatment strategies. This position paper reviews current clinical evidence and discusses the development and future perspectives of recently discovered ALK inhibitors.
Molecular pathology of ALK-rearranged NSCLC
ALK is a member of the insulin receptor super-family of receptor tyrosine kinases with unclear physiologic functions. In humans, ALK expression is limited to the adult brain, whereas no expression has been evidenced in normal lung tissue (18).
EML4-ALK rearranged NSCLC harbor a chimeric fusion gene involving ALK. This fusion results from the rearrangement within chromosome 2 [inv (2)(p21p23)] and fusion of the 5' portion of EML4 with the 3' portion of ALK (6). At least 14 variants of the EML4-ALK fusion gene have been reported thus far, encoding for the cytoplasmic portion of ALK protein and containing varying lengths of EML4 (19). Variants v1, v2, v3a and v3b of EML4-ALK fusion gene are the most commonly detected, together accounting for more than 90% of variants in some series (20). Although more uncommon, other ALK fusion partners have been identified as TFG (TRK-fused gene), KIF5B, PTPN3 and KLC1 (21-23). The different EML4 variants and the further partner genes do not seem to significantly impact on biology and sensitivity of ALK-rearranged malignant cells and tumors to specific inhibitors (24-26) although a putative role in conditioning response to treatments has been reported in vitro (27).
Retrospective and prospective screenings of ALK in NSCLC have consistently demonstrated that ALK-rearranged (ALK-positive) tumors are associated with adenocarcinoma histology, particularly when showing a solid, signet ring cell or mucinous cribriform pattern (7,28,29). In addition, ALK-positive NSCLC are associated with a lower age at diagnosis (median, 54 years) than that observed in other genetic subsets of lung adenocarcinoma, and never/light smoker status (6,7). ALK and ROS1 fusion-positive tumors seem to have a significantly shorter disease-free survival after adjusting for confounding factors (30); however, this does not necessarily translate into a short overall survival, since some patients present a favorable natural history of disease and may be candidate to several lines of therapy. ALK rearrangements in NSCLC initially appeared to be mutually exclusive with activation occurring in other oncogenic drivers (31); however, recent studies revealed concomitant EGFR or KRAS mutations and ALK rearrangements (32-34).
Testing for ALK rearrangements
Although the prevalence of tumors harboring ALK gene fusion is relatively low, the significant absolute number of patients diagnosed with NSCLC and the dramatic effect exerted by ALK inhibitors on disease courses make the identification of ALK-positive patients crucial (35). Albeit the screening for ALK-positive tumors in clinical trials with ALK inhibitors was based on fluorescence in situ hybridization (FISH) assay, the identification of ALK rearrangements may be performed by other diagnostic approaches, including immunohistochemistry (IHC) and reverse transcriptase-polymerase chain reaction (RT-PCR) (36). Beyond these more “traditional” techniques, the applicability of next-generation sequencing (NGS) technologies to the detection of gene fusions and their current widespread availability deserve mention. Diagnostic algorithms emerging from the integration of the cited analysis should lead to standardized procedures. The latters aim therefore to combine sensitivity and specificity, to generate reproducible data and to allow the best management of tumor tissue, always precious in lung malignancies, as frequently derived from small biopsies or cytological samples.
FISH
ALK FISH analysis relies on a break-apart probe provided with two fluorochromes, respectively labeling the 3' (telomeric) and 5' (centromeric) parts of the fusion breakpoint (Vysis LSI ALK dual-color, break-apart probe, Abbott Laboratories, Abbott Park, IL, USA). Superimposed signals indicate ALK wild-type status, while inversion and rearrangements generate signals that can be identified as split or isolated (36). This technique works appropriately either on archival paraffin-embedded tumor specimens and well-prepared, mono-layer, non-bloody smeared cytology (37,38). A cut-off of >15% rearranged tumor cells, counting at least 50 tumor cell nuclei, has been decided to quote ALK fusions by FISH (Figure 1) (26). As already stated, ALK rearrangement is the key mechanism of transformation and oncogenesis: virtually all cells therefore harbor at least one rearranged copy of the ALK gene. The threshold of 15% is thus far technique-dependent and it is not driven by tumor biology (39). This cut-off revealed as the best option to couple specificity (ALK positive cells can appear FISH negative due to nuclear chromatin status) and sensitivity (ALK negative cells can appear FISH positive because of stochastic and isolated genomic alterations or artifact) (36,39). Due to the lack of clear-cut discrimination nature of the cut-off >15%, rates comprised between 10% and 15%, occurring in the 8.5% of NSCLC (40), require a critical approach. In these specific cases more than in others, re-performing FISH analysis and addressing to further techniques of detection (see below) is necessary in order to “catch” every single NSCLC patient with true ALK rearrangements.
Figure 1.

Different ALK staining expression at IHC with clone 5A4 (A) and Ventana D5F3 CD assay (B). Insert in image (A) shows the ALK positivity in signet ring tumor cells with a thin rim of brownish-stained cytoplasm dislocated under the nuclear membrane. FISH analysis showing tumor cells with normal ALK set-up with one normal fusion signal and one single red/orange signal (C); ALK positivity by gene deletion with single 3' orange rearranged signals (deleted green signal) (D, arrowheads) and ALK positivity by inversion with “broken apart” signals, 2 or more signal diameters apart (E, arrows). IHC, immunohistochemistry; ALK, anaplastic lymphoma kinase; FISH, fluorescence in situ hybridization.
As mentioned, inversion in the chromosome 2p are the most frequent event leading to EML4-ALK fusions; this occurrence implies that the breakpoint in ALK gene generate a small physical separation between the 3' and 5' extremities, sometimes making the “break apart” challenging to identify (41).
Moreover, interpretation of FISH results can be very challenging, as it requires great expertise to interpret fluorescence microscope images when only a subset of tumor cells may be evaluable for split assessments. Another major issue with FISH derives from the correct recognition of tumor cells in dark-field microscopy among inflammatory and stromal cells or normal bronchus-associated salivary glands that may lead to false-negatives or false-positives results (36). FISH is also labor intensive and costly when adopted as a screening tool for thousands of patients (42).
IHC
Since ALK protein is not expressed in normal tissues, IHC demonstrating ALK expression is an alternative approach that enables analysis of paraffin-embedded tumor specimens in clinical practice. IHC is a routine procedure and well-known ancillary method in all pathology settings, is less labor intensive and more cost-effective than FISH, permits the identification of tumor cells at light morphology and therefore results as a good screening test (43). There is a consistent body of evidence demonstrating that IHC is comparable, if not superior, to FISH (44,45). In a recent study (46), the sensitivity and specificity were 43% and 98% for FISH and 100% and 98% for IHC, respectively. Relevant discrepancies between FISH and IHC results have indeed been reported (47-49). Importantly, patients responding to ALK inhibitors with an ALK IHC-positive but FISH-negative adenocarcinoma have been extensively described (49,50). The main issue with IHC is that ALK protein in ALK-rearranged lung NSCLC is restricted in the cytoplasm with/without membrane staining and it is expressed at lower level than those pathologists are used to observe in ALCL, where the t(2;5) translocation generates the nucleophosmin (NPM)-ALK fusion gene, leading to strong nuclear and cytoplasmic immunostaining with ALK antibody (51). There are at least four different commercially-available clones with different sensitivities, namely ALK1, 5A4, 1A4 and D5F3. Comparison studies have not evidenced any significant difference among them in detecting ALK-positive NSCLC (52). However, staining intensity may be quite different and intercalated antibody-enhanced polymer methods or amplification kits have been used to improve the sensitivity. ALK1 clone indeed, useful in addressing molecular diagnosis in ALCL and provided of good specificity, lacks of appropriate sensibility in NSCLC, due to the aforementioned lower expression of ALK transcripts (53).
While FDA approved the use of ALK inhibitor crizotinib employing the companion diagnostic Vysis probe for FISH, the EMA required the demonstration of ALK-positive status by any appropriate method, not only by FISH (43). Recently, both the FDA (June 2015) and the Italian Drug Agency (Agenzia Italiana del Farmaco, AIFA) (April 2015) approved the selection of ALK-positive NSCLC using IHC. While the FDA opened to IHC identification of ALK-positive patients only with Ventana (Ventana Medical Systems, Inc., Tucson, AZ, USA) ALK (D5F3) companion diagnostic assay (54), AIFA permitted the use of various clones adopting basic and clear rules based on the scoring staining and the antibody clone (Figure 2). D5F3 and 5A4 with an appropriate highly sensitive amplification kit are the best clones when using a platform different from Ventana/Roche, while the Ventana/Roche CDx D5F3 is the most adequate IHC system when a Ventana/Roche platform is available in the own lab (49). The possibility to have a dual ALK IHC/in-situ hybridization assay is realistic and appears promising (55,56).
Figure 2.
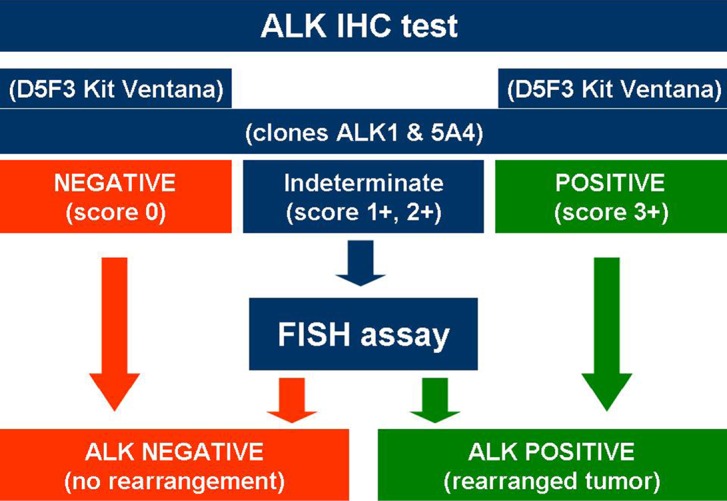
AIFA-based scheme in detecting ALK-positive NSCLC using an integrated approach with IHC and FISH. All antibody clones against ALK are accepted employing a scoring system, then requiring FISH confirmation in indeterminate cases at IHC (scores 1+ and 2+). AIFA, Agenzia Italiana del Farmaco; ALK, anaplastic lymphoma kinase; NSCLC, non-small cell lung cancer; IHC, immunohistochemistry; FISH, fluorescence in situ hybridization.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RT-PCR represents a valuable tool to detect ALK rearrangements in NSCLC (57). However, the high quality of RNA required by this analysis, not always provided by formalin-fixed, paraffin-embedded (FFPE) specimens, make the clinical applicability challenging. Most of the current guidelines do not include it ALK RT-PCR in their diagnostic algorithms, as the rate of failures and false-negative tests can be significant. Nevertheless, RT-PCR can find its best utility in samples not suitable for tissue blocking, mainly represented by bronchial washing fluid and pleural effusions (19,58). RT-PCR could represent a useful tool to detect fusion variants of the EML4-ALK fusion gene which otherwise, besides harboring unclear clinical significance (see above), can be currently revealed by next-generation genomic and transcriptomic analyses (see next paragraph).
Next-generation techniques
The current or upcoming diffuse possibility to perform deep genomic analysis on both DNA (targeted NGS, whole exome sequencing, comparative genomic hybridization) and RNA (RNA sequencing) allow to obtaining large amounts of information from a single specimen (59). Focusing on relevant fusions in NSCLC, molecular events involving ALK, ROS1, RET and NTRK1-3 genes would hopefully, in the next future, routinely been simultaneously sounded out (60), allowing the exploitation of small amounts of material. Moreover, deep sequencing analysis can be performed on FFPE samples and not necessarily on frozen tissues (61). With special regard to ALK rearrangement, NGS feasibility and reliability have been already proven (46).
Differently from FISH and IHC (and with overall better performances than RT-PCR), NGS analysis can provide information concerning ALK fusion partner gene and, in the most of the cases, the involved variant of EML4 gene. A similar scenario is depicted for rearrangements involving ROS1, which is strictly related, for phylogenetic and oncogenic homologies, to ALK (62).
The issue of acquired resistance to crizotinib
Once ALK rearrangements are detected in NSCLC patients, crizotinib can be usefully administered as front-line treatment (13) or after chemotherapy (11). Irrespective of the line of treatment, crizotinib exhaustion manifests after a median period of 7.7 to 10.9 months (11,13,63). As observed in other oncogene-addicted tumors such as EGFR-driven NSCLC, acquired resistance to crizotinib almost invariably emerges and leads to clinical progression.
Resistance to crizotinib derives from different mechanisms, that include ALK secondary mutations in approximately 20% to 40% of patients: the L1196M (gatekeeper) and G1269A are the more frequent, while the 1151T-ins, L1152R, C1156Y, F1174C/V, G1202R, D1203N, 1206Y and G1269A are less often observed (13,14,64-66). Moreover, the current availability of NGS methods allows the detection of further rare mutations in ALK kinase domain conferring either crizotinib sensitivity (67) or resistance, while allowing the inhibition exerted by second-generation inhibitors (68).
Amplification of rearranged ALK gene is reported in approximately 15% of crizotinib-refractory patients (14), while activation of EGFR, KRAS, c-KIT or IGF-1R signaling is reported in up to 30% of patients (14,15,69). Depending of the case series, the mechanisms underlying acquired resistance to crizotinib remain unknown in approximately 50% of patients.
The virtually inevitable occurrence of biological resistance to crizotinib accelerated, in the recent past years, the development of new ALK inhibitors provided with pharmacodynamics and pharmacokinetics improvements, whose best integration in treatment strategies is of pivotal interest.
Development program of new ALK inhibitors
Among second-generation ALK inhibitors, ceritinib received FDA and EMA approvals in April 2014 and in May 2015 respectively, in the setting of ALK-positive NSCLC patients showing crizotinib resistance or intolerance. Alectinib, available in Japan since July 2014, received FDA approval in December 2015 and is currently under evaluation by EMA. Brigatinib received FDA granted breakthrough-therapy designation for ALK-positive advanced NSCLC that have progressed to crizotinib. Lorlatinib and entrectinib, in less-advanced clinical development, are promising drugs belonging to this category. Table 1 resumes the activity of registered and experimental ALK-inhibitors driven from clinical trials.
Table 1. Activity of ALK-inhibitors emerged from completed and ongoing clinical trials.
| Drug | Study (reference) | Previous crizotinib | Crizotinib-naive | |||
|---|---|---|---|---|---|---|
| ORR | mPFS (months) | ORR | mPFS (months) | |||
| Crizotinib | Phase I (63) (PROFILE 1001) | 87/143 (60.8%) | 9.7 | |||
| Phase II (12) (PROFILE 1005) | 155/259 (59.8%) | 8.1 | ||||
| Phase III 2nd line (11) (PROFILE 1007) | 113/173 (65%) | 7.7 | ||||
| Phase III 1st line (13) (PROFILE 1014) | 128/172 (74%) | 10.9 | ||||
| Ceritinib (LDK378) | Phase I (70) (ASCEND-1) | 92/163 (56%) | 6.9 | 60/83 72% |
18.4 | |
| Phase II (71) (ASCEND-2) | 54/140 (38.6%) | 5.7 | ||||
| Phase II (72) (ASCEND-3) | 79/124 (63.7%) | 11.1 | ||||
| Alectinib (CH542480/RO5424802) | Phase I/II (73) (AF-001JP), 240/300 mg BID | 58/61 (95%) | NA | |||
| Phase I/II (74), dose escalation (AF-002JG) | 24/44 (55%) | NA | ||||
| Phase II (75) (NP28761) | 33/69 (48%) | NA | ||||
| Phase II (76) (NP28673) | 61/122 (50%) | 10.3 | ||||
| Brigatinib (AP26113) | Phase I/II (77), phase II portion (NCT01449461) | 50/70 (71%) | 13.4 | 8/8 (100%) | Not reached | |
| Lorlatinib (PF-06463922) | Phase I/II (78), dose escalation (NCT01970865) | 17/32 (53.1%) | NA | 2/2 (100%) | NA | |
| Entrectinib (RXDX-101) | Phase I/II (79), dose escalation (NCT02097810) | 2/4 (50%) | NA | |||
ALK, anaplastic lymphoma kinase; ORR, objective response rate; mPFS, median progression-free survival; NA, not available.
Ceritinib
Ceritinib is 20 times more potent than crizotinib against native ALK protein, as shown in enzymatic assays (80), and is capable of reconverting crizotinib resistance in cellular functional studies (66). Its intrinsic potency and chemical structure allow its interference with ATP within ALK kinase domains that have undergone several of the cited mutations (66).
In crizotinib-naive ALK-positive patients treated with ceritinib in the phase I ASCEND-1 trial, the overall RR was 62% when treated with at least 400 mg daily; 21/34 patients reported a partial response. In the same subgroup of patients, the median PFS was 10.4 months, with 18 patients (53%) censored and a median follow-up of 9.5 months (81). In the same study, 83 ALK-positive NSCLC patients who had progressed under crizotinib were enrolled in the dose-escalation or phases. Among them, the ORR was of 56% and median PFS was 6.9 months. Importantly, all the 19 cases for whom tumor material was available at crizotinib progression responded to ceritinib, regardless of the respective mechanism of acquired resistance, also when the precise molecular explanation was not uncovered (81). The updated analysis of this trial at median duration of follow-up of 11.1 months, showed a median PFS of 18.4 and of 6.9 months in ALK inhibitor naive and pre-treated patients that received ceritinib at the recommended 750 mg/day dose (70).
In patients with chemotherapy and crizotinib-refractory ALK-positive NSCLC, a phase II trial with ceritinib at the dose of 750 mg daily was conducted (ASCEND-2; Clinicaltrials.gov identifier: NCT01685060) enrolling a total of 140 patients (71). Patients should have received cytotoxic therapy (1–3 lines, including 1 platinum doublet) and progressed on crizotinib as the last treatment prior to study entry. The overall response rate (ORR) by investigators was 38.6% (35.7% by blinded independent review committee), with a PFS of 5.7 months.
ASCEND 3 (Clinicaltrials.gov identifier: NCT01685138) is a one-arm, phase II trial in 124 ALK-positive NSCLC patients who had not received prior treatment with ALK inhibitors (98.4% had received at least 1 line of prior chemotherapy and 25% of patients had received ≥3 prior antineoplastic regimens) (72). The study met its primary end-point, with a whole-body ORR of 63.7% (by blinded independent review committee of 58.9%); responses were durable, leading to a median PFS of 11.1 months with a median follow-up of 8.3 months (72).
The results of two phase III trials comparing ceritinib with standard chemotherapy are awaited. In ASCEND 4 (Clinicaltrials.gov identifier: NCT01828099) patients suffering by ALK-positive advanced NSCLC, not previously exposed to crizotinib, are randomized to receive a first line chemotherapy with platinum-pemetrexed or ceritinib. In ASCEND-5 (Clinicaltrials.gov identifier: NCT01828112) patients progressing after crizotinib and chemotherapy are randomized to receive either a cytotoxic treatment with pemetrexed or docetaxel (per investigator choice) or ceritinib.
A recent retrospective multicenter analysis estimated at 17.4 months the median combined PFS to sequential crizotinib and ceritinib treatment in 73 ALK-positive patients (82). Considering the results obtained by ceritinib in crizotinib-naïve patients (70), current trials evaluating ceritinib treatment in ALK-TKI-naive patients (ASCEND 3 and 4) should aim to confirm the latter data and to reach a comprehensive duration of disease control at least overlapping with the one reported with two compounds in sequential treatment.
Although preclinical and clinical research mainly focuses on acquired resistance to crizotinib, absolute lack of response to the inhibitor can be distinctly observed in 5% to 7% of ALK-positive patients (11,13,63). Albeit most of the primary resistance cases can be attributed to misdiagnoses of ALK-rearrangements (48), “true” biological de novo resistance can be driven by co-occurring K-RAS mutations (83) or by unknown mechanisms (17). Although the eventual role of ceritinib in the first scenario is not clear, its intrinsic greater potency can explicate the complete inhibition of ALK signaling when the second circumstances occur (17).
Alectinib
Alectinib is a potent and selective ALK inhibitor (84). The initial phase I dose-escalation portion of the Japanese AF-001JP study included 24 crizotinib-naive patients with recurrent/relapsed ALK-positive NSCLC. Alectinib was administered at doses ranging from 20 to 300 mg twice daily. In the phase II portion of the trial, 46 ALK inhibitor-naïve patients were treated with 300 mg twice daily of alectinib. This drug displayed an excellent RR in both moments of the study (93.5% in the phase II cohort), with long response durations and an acceptable toxicity profile (73). Two complete responses and 41 partial responses were achieved among the 46 evaluable patients, and the two-year PFS rate was 76%. Published data were too early to deduce robust information concerning median PFS in the phase II portion with the recommended posology of alectinib; with a median follow-up of 12 months, the mean duration of treatment was 11.8 months within the escalation-dose phase (73).
Alectinib demonstrated promising antitumor activity also in patients with ALK-rearranged NSCLC resistant to crizotinib, including those with central nervous system (CNS) metastases. In the dose-finding portion of the AF-002JG trial, a phase I/II study performed in the United States in ALK-positive NSCLC patients previously treated with crizotinib, alectinib allowed a RR of 55% (74). In a global phase II study testing alectinib in crizotinib-refractory ALK-rearranged NSCLC (NP28673), the RR was 50% in the whole study population (including both patients pretreated with chemotherapy and chemotherapy-naive) and 45% in the subgroup of patients pretreated with chemotherapy (76). In an ongoing phase II study enrolling 87 ALK-positive patients who had progressed on crizotinib (NP28761), at the time of the primary analysis (median follow-up 4.8 months), 33 out of 69 patients (48%) with measurable disease at baseline had a confirmed partial response with alectinib (75).
On the basis of these results, ALEX, a phase III randomized trial, has been initiated to compare alectinib with crizotinib in treatment-naïve, ALK-positive, advanced NSCLC patients (ClinicalTrials.gov identifier NCT02075840).
Importantly, during the final preparation of this manuscript, the study sponsor of ALEX trial released a press communication reporting that among a Japanese population of ALK-rearranged NSCLC patients, the front-line administration of alectinib versus crizotinib achieved an impressive benefit in mPFS, quantified with a Hazard Ratio of 0.34 (85). The anticipated outstanding activity of alectinib will be extensively presented at the upcoming 2016 ASCO meeting (Abstract #9008).
Brigatinib
Brigatinib is a tyrosine kinase inhibitor that showed preclinical activity against ALK and 9 clinically-identified crizotinib-resistant mutants (86,87). Moreover, this compound exerts in vitro activity against both ALK and EGFR (88), making it a suitable therapeutic option for patients progressing to crizotinib with activation of EGFR pathway as the mechanism of resistance (14,15,66). Indeed, in an ongoing phase I/II study of brigatinib both in ALK-rearranged and EGFR-mutated NSCLC patients, brigatinib is demonstrating significant antitumor activity in ALK-positive NSCLC patients with and without prior crizotinib, including patients with brain metastases. After the dose-escalating portion of the study, patients were divided into three cohorts that received 90, 90–180 (escalating after 7 days) or 180 mg of the drug daily (77,89). According to the latest updates concerning those 78 evaluable ALK-positive NSCLC patients treated in the phase II portion of the study, 50 (71%) responded: 50/70 (71%) pretreated with crizotinib and 8/8 crizotinib-naive patients (with three complete responses in this last sub-cohort) (77). The overall median PFS was not reached in the crizotinib-naive sub-group and corresponded to 13.4 months in patients already exposed to crizotinib; one-year OS rates were 100% and 81% for the two just mentioned population, respectively (77).
The pivotal phase II ALTA (ALK in Lung cancer Trial of AP26113) trial in patients with ALK-positive NSCLC patients pretreated with crizotinib has recently terminated enrolment (ClinicalTrials.gov identifier: NCT02094573) and primary outcome evaluations are expected in November 2016. As reported for ceritinib and alectinib, a head-to-head comparison of crizotinib versus brigatinib will soon begin (ClinicalTrials.gov identifier: NCT02737501; ALTA-1L trial). Finally and interestingly, brigatinib will be soon evaluated after the exhaustion of other second-generation ALK-inhibitors (namely ceritinib or alectinib) in a phase II study (ClinicalTrials.gov identifier: NCT02706626). It would be noteworthy to assess the clinical response to this compounds in patients affected by tumors harboring G1202R and I1171N/S/T mutations, not overcome by ceritinib and alectinib respectively (66,90,91).
Lorlatinib
Lorlatinib is an extremely selective and potent third-generation ALK/ROS1 inhibitor, with sub-nanomolar activity against EML4-ALK enzyme and almost all its mutant forms, including the ones driving resistance to ceritinib and alectinib (92,93). Preclinical activity in cellular assays and in mouse models (93) have been recently confirmed in the dose-escalation portion on a phase I/II study (78). Recommended phase II dose was set at 100 mg once daily. Among the 43 ALK/ROS1-positive NSCLC patients evaluable for radiological responses, one complete and 19 partial response were observed, with seven, 14 and two cases accounting for disease stability, progression and indeterminate response, respectively (78). Treatment efficacy was observed regardless of the number of prior specific inhibitors assumption; lorlatinib confirmed clinical activity also against G1202R mutation, which confers the highest degree of resistance to both crizotinib and ceritinib (66).
As described for EGFR-mutated NSCLC, progressing to the third-generation inhibitor AZD9291 because of the emergence of C797S mutation, sensible to first-generation compounds (94), ALK substitution L1198F drives resistance to ceritinib, alectinib, brigatinib and lorlatinib, while re-sensitizing tumor cells to crizotinib (95).
Entrectinib
Entrectinib, initially envisaged as an ALK inhibitor (96), currently represents the best antagonist of NTRK1-3 fusion proteins, an emerging reality in oncogenic-driven NSCLC (97), while expressing activity against ROS1-rearranged cancer cells too (98). Concerning specific ALK inhibition, entrectinib has shown remarkable in vitro and in vivo activity in preclinical models also against L1196M and CC1156Y crizotinib-resistance mutations (99). Patients affected by a wide range of tumor histologies harboring the cited fusion oncogenes were treated with entrectinib in two different cohorts (the Italian ALKA-372-001 and the international STARTRK-1), flowing into the same phase I study (ClinicalTrials.gov identifier: NCT02097810). Updated data of the study were recently presented (79); the recommended phase II dose was defined as 600 mg once daily in a continuous schedule. Among the 24 patients who had not previously undergone specific targeted inhibition and who were evaluable for treatment response, four suffered from ALK-rearranged NSCLC; in this small sub-cohort, two partial responses according to RECIST criteria were observed. Lack of previous specific molecular therapy is a main inclusion criteria in the STARTRK-2 trial (ClinicalTrials.gov identifier: NCT02568267), a phase II basket trial currently addressing to entrectinib treatment patients screened, by means of NGS techniques, for NTRK, ROS1 and ALK gene rearrangements.
Efficacy of ALK inhibitors on brain metastases
ALK-positive NSCLC are associated with a relevant incidence of CNS metastases, affecting approximately 35–50% of patients (11,81,100). It is actually not clear whether ALK-positive patients have an increased risk of developing CNS metastases independently from therapy received (as an expression of the natural disease course) or if this higher risk may be related to treatment with ALK inhibitors. In fact, high tumor activity of these compounds with prolonged control of extra-cranial disease could make emerge CNS as relevant site of progression. This is particularly true for crizotinib, which may inefficiently cross the blood brain barrier thus reaching limited concentration in the cerebrospinal fluid (CSF) (16,101,102). CNS involvement resulted the first site of progression in 46% of ALK-positive patients treated with crizotinib, with 85% of these lacking systemic progression (103).
A retrospective analysis of crizotinib-treated patients enrolled in the PROFILE 1005 (12) and 1007 (11) trials found that 20% of patients without brain metastases at the time of therapy initiation subsequently experienced brain progression; in patients with known brain metastases, CNS was site of progression in 70% of cases (104). Therefore, in ALK-positive patients CNS represents a frequent site of disease and the main site of progression during crizotinib.
Although CSF concentrations of crizotinib are modest, this agent is associated with intracranial disease control in TKI-naïve ALK-positive patients (104-106). The intracranial activity of crizotinib and chemotherapy in ALK-positive NSCLC patients included in PROFILE 1014, not exposed to systemic therapy but who had already undergone radiotherapy for brain disease, have been recently and comprehensively reported (107). Crizotinib administration allowed an intracranial disease control rate (IDCR) in 85% and 56% at 12 and 24 weeks, respectively (33 and 22 out of 39 patients, respectively); in the chemotherapy arm IDCR was achieved in 18 (45%) out of 40 patients at 12 weeks and maintained in 10 (25%) at 24 weeks.
According to the National Comprehensive Cancer Network (NCCN) guidelines, radiotherapy to cerebral sites of progression, in association with crizotinib continuation, represents an accepted treatment strategy (108). Although no randomized studies are available, when ALK-positive patients with isolated CNS progression on TKIs received brain radiotherapy without crizotinib withdrawal, further progression occurred approximately 12 months after the initiation of this strategy (109).
The greatest promise in the treatment of ALK-positive NSCLC patients with CNS involvement comes from new ALK inhibitors, expressly ceritinib, alectinib, brigatinib and lorlatinib. All these new agents showed promising activity against CNS metastases (Table 2), both in TKI-naive and in crizotinib-progressing patients. The improved CNS activity likely reflects the higher potency in ALK inhibition and improved CNS penetration (93). Although these studies were not specifically designed to evaluate patients with brain metastases, there is now enough evidence to support the use of these agents in clinical practice and, potentially, to delay of brain radiotherapy in favor of a further novel ALK inhibitor in case of asymptomatic brain lesions.
Table 2. Activity of ALK inhibitors on brain metastases.
| Drug | Study (reference) | Pts with brain mts/total treated | Pts with previous RT | iORR | iDCR | |||
|---|---|---|---|---|---|---|---|---|
| Untreated brain mts1 | Treated brain mts2 | Untreated brain mts1 | Treated brain mts2 | |||||
| Crizotinib | Phase II–III (104) (PROFILE 1005–1007) | 275/888 | 166 | 18% | 33% | 56% | 62% | |
| Phase III (107) (PROFILE 1014) | 39/172 | 39 | NA | 12-w: 85%; 24-w: 56% |
||||
| Ceritinib (LDK378), 750 mg/die | Phase I (70) (ASCEND-1) | 124/246 | 83 | 63%# | 36%# | 79%* | 65%* | |
| Phase II (71) (ASCEND-2) | 100/140 | 72 | 39% by IRC | 85% by IRC | ||||
| Phase II (72) (ASCEND-3) | 50/124 | 27 | 59% by IRC | 82% by IRC | ||||
| Alectinib (CH542480/RO5424802) | Phase I/II (74), dose escalation (AF-002JG) | 21/47 | 17 | 75%* by IRC | 47%* by IRC | 100%* by IRC | 88% by IRC | |
| Phase II (75) (NP28761) | 52/87 | 34 | 67%* | 26.5%* | 94.5%* | NA | ||
| Phase II (76) (NP28673) | 84/138 | 61 | 52%* | 39%* | 74%* | 87%* | ||
| Brigatinib (88) (AP26113) | Phase I/II, phase II portion (NCT01449461) | 52/79 | 27 | 52%* | 28%* | 88%* | ||
| Lorlatinib (PF-06463922) | Phase I/II (78), dose escalation (NCT01970865) | 30Φ/50 | NA | 33%Φ,* | 63%Φ,* | |||
1, untreated brain metastasis: in case of ceritinib, data are relative to ALK-TKI naive patients; in other drugs are relative to no previous brain RT; 2, treated brain metastasis: in case of ceritinib, data are relative to ALK-TKI pre-treated patients; in other drugs are relative to previous brain RT; #, considering 36 patients with measurable disease; *, considering patients with measurable and non-measurable CNS disease; Φ, including ALK- and ROS1-positive patients. ALK, anaplastic lymphoma kinase; Pts, patients; mts, metastases; RT, brain radiotherapy; iORR, intracranial objective response rate; iDCR, intracranial disease control rate; NA, not available; 12-w, at 12 weeks; 24-w, at 24 weeks; IRC, independent review committee.
Regarding the activity of ceritinib in CNS disease, patients with controlled or asymptomatic brain metastases were eligible in the ASCEND-1 trial (70,81). Of 124 ALK-positive NSCLC patients with brain metastases at baseline, 94 had brain scans evaluated by central review. In ALK-TKI naive (n=19) and ALK-TKI pre-treated patients (n=75), disease-control rate (DCR) was 79% and 65%, respectively; whole body disease-control rate and duration were similar to what observed in the overall population (70). The median time required to achieve intracranial responses (6.1 weeks) overlapped with what observed for extracranial disease. 23 patients (67%) had previously received radiotherapy to the brain; no difference in brain responses was observed between patients who had undergone or nor radiotherapy before ceritinib administration. Thirty-six patients (eight TKI-naive and 28 previously exposed to crizotinib) had measurable brain metastases: among them, intracranial ORR was 63% and 36% in naïve and pre-treated patients, respectively. The median ceritinib exposure was 49.6 weeks in ALK-TKI-naïve patients and 40.6 weeks in pre-treated patients.
The ASCEND-2 preliminary results support those reported for previously ALK inhibitor-treated patients in ASCEND-1 trial (71). Among patients with brain metastases at study entry, 20 had investigator-assessed brain lesions selected as target lesions at baseline, for whom ORR and DCR was 45% and 80%, respectively. According to the Independent Review Committee, 33 patients had brain lesions selected as target lesions at baseline, for whom ORR and DCR were 39% and 85%, respectively. In the ASCEND-3 trial, 124 patients with ALK-positive NSCLC who had not received prior treatment with ALK inhibitor were enrolled (72). Among subjects with brain metastases at study entry, 10 patients had investigator-assessed brain lesions selected as target lesions at baseline, for whom intracranial ORR (iORR) and iDCR were 20% and 80%, respectively. According to the independent review committee, 17 patients had brain lesions selected as target lesions at baseline; ORR and DCR were 59% and 82%, respectively. An international prospective phase II open-label study evaluating the antitumor activity of ceritinib in patients with ALK-positive NSCLC metastatic to the brain or leptomeninges is ongoing (ASCEND-7, ClinicalTrials.gov identifier: NCT02336451).
Initial data concerning intracranial activity of alectinib derived from a phase I/II trial enrolling patients who progressed on or were intolerant to crizotinib (74). Among the 21 patients with CNS metastases at baseline treated in the dose-escalation portion of the study, 11 (52%) achieved a brain objective response (6 complete and 5 partial responses) and eight (38%) patients had stable disease; brain responses were observed at different drug doses. Results of a phase II trial carried out in USA were recently published; 87 patients were enrolled and treated with alectinib at 600 mg twice daily (75). In patients with baseline-measurable CNS disease (n=16), ORR and DCR stated by an independent review committee were 75% and 100%, respectively, with notably four complete intracranial responses. When both patients with baseline-measurable and non-measurable CNS disease were included (n=52), ORR and DCR were 40% and 89%, respectively.
At the 2015 ASCO meeting, updated data of the 46 crizotinib-naive patients enrolled in the phase II part of Japanese trial AF-001JP were presented (73,110). At a median follow-up >30 months, Ohe et al. showed that seven out of 14 patients with baseline brain metastases were still in the study without CNS or systemic progression; PFS of patients with or without brain metastases was similar. Moreover, in the recently published phase II NP28763 trial (76), among the 84 crizotinib-resistant patients with baseline CNS metastases, 23 patients obtained a CNS complete response (27%); the overall intracranial DCR was 83% and the median duration of CNS response was 10.3 months. Considering only the 35 patients with measurable brain lesions at baseline, CNS objective RR was 57%, including achievement of seven CNS complete responses. High RR and DCR were confirmed also stratifying patients according to previous radiotherapy. At the 2015 World Conference on Lung Cancer, a pooled analysis of CNS data from two phase II multicenter studies on patients previously treated with crizotinib (NP28761, NP28673) showed a complete RR of 22%, an ORR of 39% and a DCR of 85% in patients with CNS disease, irrespective of prior radiotherapy (111). The CNS response was sustained for a duration similar to the systemic response, suggesting that alectinib could provide an effective treatment for patients with ALK-positive NSCLC while actively targeting CNS metastases.
In the phase II portion of an ongoing trial of brigatinib, 46 out of 50 patients with intracranial metastases were evaluable (77). The 8/15 (53%) patients with measurable disease had a brain response and 11/31 (35%) patients with only non-measurable lesions showed disappearance of all lesions. Brigatinib treatment leads to a median intracranial PFS of 15.6 months in the 46 evaluable patients (77).
Lorlatinib shown impressive activity results in mouse models of CNS metastases from NSCLC tumor cells harboring EML4-ALK gene fusion, with or without the gatekeeper L1196M mutation (93). Preclinical data were confirmed by recent reports (78) testifying an intracranial disease control in 19 out of 30 ALK/ROS1-positive evaluable patients, who had previously undergone systemic treatments with at least one ALK inhibitor. Four complete and six CNS partial responses were observed, with one leptomeningeal disease resolution obtained after sequential crizotinib, ceritinib and alectinib administration (78).
For future clinical trials with new ALK-TKIs, it will be important to consider dedicated cohorts with CNS disease and to focus on patients with previously not irradiated brain metastases or those with brain metastases progressing after irradiation.
Safety profile of ALK inhibitors
The wide majority of available data concerning safety of second-generation ALK inhibitors have been obtained within studies including patients, who had already undergone crizotinib treatment (Table 3). Thus, the populations analyzed for safety are mixed and many data derive from phase I studies or their expansions cohorts. Anyway, ALK inhibitors are globally characterized by very good safety and tolerability profiles, without real substantial differences among compounds, combining thus far significant activity with the lack of frequent relevant adverse events.
Table 3. Safety profile of ALK inhibitors.
| Drug | Study | Grade 3–4 toxicities | Most common all-grade adverse event |
|---|---|---|---|
| Crizotinib | Phase I (63) (PROFILE-1001), 149 patients | ALT: 6 pts (4%); AST: 5 pts (3%) | Visual disorders: 96 pts (64%) |
| Phase II (12) (PROFILE-1005), 901 patients | Neutropenia: 72 pts (8%); ALT: 55 pts (6.1%); fatigue: 22 pts (2.5%) | Visual disorders: 571 pts (63.3%) | |
| Phase III (11) (PROFILE-1007), 173 patients | ALT/AST: 27 pts (16%); dyspnea: (7 patients, 4%) |
Visual disorders: 103 pts (60%) | |
| Phase III (13) (PROFILE-1014), 172 patients | ALT/AST: 24 pts (14%) | Visual disorders: 122 pts (71%) | |
| Ceritinib (LDK378), 750 mg/die | Phase I (70) (ASCEND-1), 255 patients | ALT: 73 pts (30%); AST: 25 pts (10%); diarrhea: 15 pts (6%); nausea: 15 pts (6%); lipase: 16 pts (6%); hyperglycaemia: 15 pts (6%) |
Diarrhea: 213 pts (86%) |
| Phase II (71) (ASCEND-2), 140 patients | ALT: 24 pts (17.1%), nausea: 9 pts (6.4%), diarrhea: 9 pts (6.4%), fatigue: 9 pts (6.4%), dyspnea: 8 pts (5.7%), AST: 7 pts (5%) |
Nausea: 114 pts (81.4%) | |
| Phase II (72) (ASCEND-3), 124 patients | Gamma-GT: 23 pts (18.5%); ALT: 19 pts (15.3%); AST: 9 pts (7.3%) |
Diarrhea: 102 pts (82.3%) | |
| Alectinib (CH5424802, RO5424802) | Phase I/II (73) (AF-001JP), phase II portion, 46 patients | Neutropenia: 2 pts (4%); CPK: 2 pts (4%) | Dysgeusia: 14 pts (30%) |
| Phase I/II (74) (AF-002JG), dose escalation, 47 patients | Gamma-GT: 2 pts (4%); neutropenia: 2 pts (4%); hypophosphatemia: 2 pts (4%) | Fatigue: 14 patients (30%) | |
| Phase II (75) (NP28761), 86 patients | CPK: 7 pts (8%); ALT: 5 pts (6%); AST: 4 patients (5%) | Constipation: 31 pts (36%) | |
| Phase II (76) (NP28673), 136 patients | Dyspnea: 4 pts, 3%; ALT: 2 pts (2%); AST: 2 pts (2%) | Constipation: 45 pts (33%) | |
| Brigatinib (AP26113) | Phase I/II (77) (NCT01449461), 137 patients | Lipase: 12 pts (9%); dyspnea: 9 pts (7%); pneumonia: 8 pts (6%); fatigue: 6 pts (4%); hypoxia: 7 pts (5%); amylase: 6 pts (4%) |
Nausea: 71 pts (52%) |
| Lorlatinib (PF-06463922) | Phase I/II (78), dose escalation (NCT01970865), 45 patients | Hypercholesterolemia: 5 pts (11%) | Hypercholesterolemia: 26 pts (59%) |
| Entrectinib (RXDX-101) | Phase I (112) (ALKA-372-001), 38 patients | Asthenia (DLT): 2 pts (5.3%); muscular weakness (DLT): 2 pts (5.3%) | Paresthesia: 16 pts (42%) |
| Phase I/IIa (113) (STARTRK-1), 27 patients | Neutropenia (DLT): 3 pts (11%); fatigue (DLT): 2 pts (7%); cognitive impairment (DLT): 2 pts (7%) | Fatigue: 9 pts (33%) |
ALK, anaplastic lymphoma kinase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; gamma-GT, gamma-glutamyl-transpeptidase; CPK, creatine-phosphokinase; DLT, dose-limiting toxicity.
With respect to ceritinib, in the expansion cohort of the phase I trial (ASCEND-1) the most frequent all-grade toxicity was diarrhea, which occurred in 213 out of 255 patients (86%); nausea was reported in 205 cases (83%) (70). The most common grade 3–4 laboratory abnormalities were increased alanine aminotransferase [73 (30%) patients] and increased aspartate aminotransferase [25 (10%)] (70). Concerning the phase II study ASCEND-2, conducted in patients who underwent chemotherapy and crizotinib, the most common all-grade toxicity was nausea, occurring in 114 patients (81.4%); increased ALT (24 patients, 17.1%), nausea (9 patients, 6.4%), diarrhea (9 patients, 6.4%), fatigue (9 patients, 6.4%), dyspnea (8 patients, 5.7%), and AST elevation (7 patients, 5%) were the most frequent grade 3–4 events (70). Diarrhea (102 patients, 82.3%) was the most common all-grade toxicity in the phase II study ASCEND-3, conducted in patients naive from ALK inhibitors; in this trial, elevated gamma-glutamyl-transpeptidase (gamma-GT, 23 patients, 18.5%), ALT (19 patients, 15.3%) and AST (9 patients, 7.3%) were the most frequent grade 3–4 events (72).
In the phase I study of alectinib carried out in United States, fatigue (occurring in 14 patients, 30%) was the most common all-grade toxicity, while elevated gamma-GT (2 patients, 4%), neutropenia (2 patients, 4%), and hypophosphataemia (2 patients, 4%) were the most frequent grade 3–4 events (74). In the USA phase II study NP28761, constipation (31 patients, 36%) represented the most common all-grade toxicity, and increase in creatine-phosphokinase (CPK; 7 patients, 8%), ALT (5 patients, 6%), and AST (4 patients, 5%) levels were the most frequent grade 3–4 events (75). In the global phase II study NP28763, constipation (45 patients, 33%) represented the most common all-grade toxicity; dyspnea (4 patients, 3%); ALT (2 patients, 2%), and AST (2 patients, 2%) elevations were the most frequent grade 3–4 events (76).
With regards to brigatinib, the most common treatment-emergent, all-grade adverse events, included: nausea (52%, 71/137 patients), diarrhea (42%), fatigue (42%), headache (33%), cough (32%) (77). Early-onset pulmonary events, observed ≤7 days after starting treatment, included dyspnea, hypoxia, or new pulmonary opacities on chest computed tomography suggestive of pneumonia or pneumonitis and occurred in 11/137 (8%) patients (77). The occurrence of pulmonary events was remarkably less frequent with lower starting doses of the drug. As these early lung features are unique to brigatinib, they represent a drug- and not a class-related toxicity, requiring clinical attention to be detected and treated as soon as possible.
In the phase I/dose escalation study of lorlatinib, hypercholesterolemia represented the most common all-grade (32 patients, 64%) and grade 3–4 (5 patients, 10%) adverse event (78). Although almost completely accounting for grade 1–2 toxicities, CNS effects (neurologic and psychiatric disorders affecting the state of consciousness) and peripheral neuropathy, occurring in 28% to 36% of patients, require a prompt management, which allows symptoms regression (78).
With regard to entrectinib, paresthesia (16 patients, 42%) represented the most common all-grade toxicity in the phase I study ALKA-372-001 conducted in solid tumors in general, while asthenia, the dose-limiting toxicity (DLT) in 2 patients (5.3%) and muscular weakness (DLT in 2 patients, 5.3%) were the most frequent grade 3–4 events (112). In the twin study STARTRK-1, fatigue (nine patients, 33%) was the most common all-grade toxicity, with neutropenia (DLT in three patients, 11%), fatigue (DLT in two patients, 7%) and cognitive impairment (DLT in two patients, 7%) being the most frequent grade 3–4 events (113).
Positioning in clinical practice
A few years ago, the availability of crizotinib made ALK-positive patients the second subgroup of advanced NSCLC eligible for treatment with a targeted drug in clinical practice, following the introduction of Epidermal Growth Factor receptor (EGFR) tyrosine kinase inhibitors for EGFR-mutations positive cases. Despite the major clinical improvement determined by the introduction of crizotinib, all treated patients, after a variable duration of clinical benefit, experience disease progression and need further treatment approaches.
Even before the results of the PROFILE 1014 trial, which demonstrated the superiority of crizotinib compared with platinum-based chemotherapy as first-line treatment for ALK-positive patients (13), most experts were convinced of the opportunity of using crizotinib as first-line treatment. Regardless of the line of treatment crizotinib is administered, the most challenging therapeutic decisions come when progression to the compound occurs, in terms of both the correct definition of crizotinib failure and of the selection of proper treatment. When other targeted agents were not yet available, patients with disease progression on crizotinib, if eligible for further treatment, could remain on crizotinib “beyond progression”, or were switched to chemotherapy (114). In patients with frank disease progression, this latter strategy was a clinically sound approach. On the other hand, for patients experiencing progression in a single site or in a few disease sites (the so called “oligo-progressive diseases”), continuation of crizotinib, alone or associated with local therapy (surgery and/or ablative treatments), has been commonly proposed as a reasonable option (103,109,115).
In the last years, many patients treated with crizotinib have received this drug beyond progression. In the phase I trial, 56.5% of progressive patients were still obtaining clinical benefit according to investigators’ opinion and continued crizotinib, in some cases for a long period (63). Similarly, in the phase III trial comparing crizotinib versus chemotherapy (docetaxel or pemetrexed) as second-line treatment (11), many patients continued crizotinib beyond documented progression, with a median duration of further treatment of 16 weeks (range, 3–73 weeks). In a retrospective analysis of two single-arm trials, in which patients who developed disease progression according to RECIST criteria were allowed to continue treatment with crizotinib if they were still obtaining clinical benefit, overall survival from progression was significantly longer for patients who continued crizotinib than for those who stopped the drug (16.4 vs. 3.9 months; HR 0.27, 95% CI, 0.17–0.42; P<0.0001) (116). Based on these data, the possibility of continuing crizotinib is discussed in NCCN guidelines as an option for patients with asymptomatic progression but also, together with local therapy, for patients with symptomatic brain lesions (isolated or multiple) or with isolated systemic lesions (108). However, it should be considered that the results discussed above with the “beyond progression” strategy are clearly affected by selection bias. Although supporting the feasibility and the acceptable tolerability of a prolonged administration of crizotinib, they do not provide robust evidence of the efficacy of this treatment strategy.
Of note, the South West Oncology Group is conducting a randomized phase II trial (SWOG 1300; ClinicalTrials.gov identifier: NCT02134912) testing the role of continuing crizotinib beyond progression in addition to chemotherapy. In this trial, patients assigned to the control arm receive pemetrexed alone, while patients assigned to the experimental arm receive crizotinib plus pemetrexed. This trial likely will not add evidence on the efficacy of pemetrexed in this setting, given that both arms receive this drug, but will add prospective, randomized evidence about the use of crizotinib beyond progression.
The availability of second- and third-generation ALK inhibitors in clinical practice has and will hopefully assume a relevant impact in the therapeutic choices for patients experiencing disease progression on crizotinib as well as for crizotinib-naive patients. For the former population of patients, the treatment algorithm is now clearly enriched by the development of novel-generation ALK inhibitors. The use of these new agents is associated with high clinical activity in this setting. To date, ceritinib is the only treatment approved both by FDA and EMA, as another line of treatment to overcome crizotinib-resistant tumors.
Among the different clinical presentation of patients with ALK rearrangement, the activity of ceritinib (and, more in general, of second-generation ALK inhibitors) is particularly appealing in patients with brain metastases, frequently present either at diagnosis of advanced disease or as lesions accounting for disease progression. Although crizotinib is active also in these patients, progression of preexisting intracranial lesions, or development of new lesions while on treatment with crizotinib is very common. In these subjects, the availability of a treatment that has shown a good activity on brain metastases assumes a great clinical value.
It must be observed that the onset of ALK resistance is a heterogeneous process: target mutations, amplifications and bypasses are proven mechanisms of resistance (14,15). For this reason, the rational choice of the optimal ALK inhibitor after progression to crizotinib (the same concept is applicable to progression to novel compounds) is of crucial importance and should be guided by the particular molecular events detected at disease relapse when re-biopsies are envisageable. Treatment can be oriented by preclinical evidence of drug activity when mutations in the ALK kinase domain occur (66,93). As explained for brigatinib, its double activity upon ALK and EGFR signaling could be useful when the second is involved in resistance mechanisms. The recently reported activation of c-MET, HER-3 and IGF-1R pathways in alectinib-resistant ALK-rearranged cells lines rise the issue of combined molecular treatments addressing to tumor-tailored biologic alterations (117). The latter concept has already been approached in in vitro pharmacological screens (118) and could be applicable in ALK-positive tumors where KRAS activation is the putative responsible of treatment resistance and clinical progression, with the putative double blockade (ALK and MEK inhibition) in a melanoma-oriented fashion.
Sequential or combinatorial treatments with ALK inhibitors and immune checkpoints inhibitors, targeting the programmed cell death receptor and its ligand (PD-1 and PD-L1, respectively) as well as the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) are in current clinical study (ClinicalTrials.gov identifiers: NCT01998126, NCT02393625, NCT02584634). Nevertheless, the substantial lack of high mutational rates in ALK-rearranged NSCLC, due also to the scarce tobacco exposure of ALK-positive patients, makes less probable a substantial benefit derivable from immune-releasing compounds (119), as seen for EGFR-mutated tumors (120,121) and emerged from retrospective series (122).
Heat shock protein 90 (HSP90) inhibitors could have a potential role in treating crizotinib-resistant patients, but mono-therapy has been studied with results indicating inferior ORR and less-tolerable toxicity profile, as well as poor CNS activity compared with ALK inhibitors. These drugs are currently under evaluation in combination with ALK inhibitors, postulating the synergistic effect of direct kinase inhibition and disruption of cell signaling (123,124). Nevertheless, the outstanding activity of novel ALK inhibitors, as well as their sub-optimal toxicity profiles, does not allow to allocate HSP90 combinations among the plausible future breakthrough for the treatment of ALK-positive patients.
An intriguing combination, suggested by the reported interplays between ALK signaling and hypoxia-inducible factors, is on the other and represented by combination of alectinib and the anti-angiogenic antibody bevacizumab (ClinicalTrials.gov identifier: NCT02521051).
With respect to the possibility to use second-generation ALK inhibitors, like ceritinib and alectinib, in crizotinib-naïve patients, it can be argued that these novel agents generally show more than 10-fold higher potency against the ALK-rearranged protein with a wild-type ALK kinase domain (66,84,86,93) and display increased affinity for second-site mutated ALK. Potentially, second-generation ALK inhibitors, compared with crizotinib, may offer an improved duration of response with a delay in the onset of ALK resistance and a treatment option available for secondary mutation-dependent resistance. These two concepts should be translated in new treatment paradigms. In fact, taking into consideration that crizotinib is a relatively weak ALK-inhibitor in NSCLC lines carrying an EML4-ALK fusion gene compared to novel compounds, the prescription of the new molecules in crizotinib-naïve patients can be envisaged. However, in the next future, the administration of crizotinib as first-line treatment (which was the “backbone” of all the therapeutic decisions discussed above) could be challenged by the results of the trials currently testing second-generation ALK inhibitors in the same setting. If the randomized phase III trial comparing ceritinib and cisplatin or carboplatin plus pemetrexed (ClinicalTrials.gov identifier: NCT01828099) will lead to positive results, ceritinib could represent an alternative to crizotinib as first-line option. The study completion date is estimated in June 2018. The selection among different drugs will be likely based on indirect comparison of activity and toxicity. Furthermore, also alectinib is currently tested as first-line treatment, in a head-to-head comparison versus crizotinib (ClinicalTrials.gov identifier: NCT02075840): the estimated study completion date is December 2017. The anticipation of the impressive activity of front-line alectinib in the Japanese cohort has already been mentioned (85) and, if preliminary results will be confirmed, the scenario of ALK inhibitions in NSCLC will ostensibly change.
As previously approached, the results of these trials will require a critical interpretation beyond the assumption of their eventual positivity. If crizotinib treatment can be usefully followed by next-generation molecules, the inverse therapeutic sequence cannot be envisaged, with the exception of only one case reported worldwide so far (95). Novel molecules administered as upfront ALK inhibitors should therefore allow disease control periods at least comparable to experienced crizotinib-based sequences. Moreover, mechanisms of resistance to the second- and third-generation ALK inhibitor are up to now clearly defined and seem to differ from events leading to crizotinib exhaustion (95,125). From this point of view, the administration of the novel compounds as the first ALK-directed approach could leave physicians “molecularly unarmed” at the moment of disease progression. Besides the precious contributions of trails evaluating novel ALK inhibitors in ALK-TKI naïve patients, studies prospectively comparing sequences of targeted treatments versus next-generation compounds upfront could be particularly useful.
The wide range of ALK inhibitors, an extremely precious resource, helps to emphasize the concept that ALK-positive patients should be maintained under specific targeted inhibition as long as possible. Beyond-progression strategies, local treatment for oligometastatic disease and SNC progression, eventual tailored-inhibition re-challenges, as well as optimal treatment sequences based on the mechanism of biological resistance, should be optimally integrated in every single patient.
Acknowledgements
None.
Footnotes
Conflicts of Interest: Dr. Marcello Tiseo declares advisory boards and speakers’ fee for Astra-Zeneca, Pfizer, Eli-Lilly, BMS, Novartis, Roche MSD, Boehringer Ingelheim, Otsuka, Pierre Fabre. Pr. Massimo Di Maio acted as consultant and received honoraria from AstraZeneca, Bayer, Eli Lilly, Boehringer Ingelheim, Novartis, MSD. Pr. Paolo Graziano has attended advisory boards and has received speakers’ fee—Astra-Zeneca, Pfizer, Eli-Lilly, Novartis, Roche, Boehringer Ingelheim. Dr. Giulio Rossi declares advisory boards and speakers’ fee: Astra-Zeneca, Pfizer, Eli-Lilly, Novartis, Roche, MSD Oncology, Boehringer Ingelheim. Pr. Silvia Novello attened speaker bureau of Eli Lilly, BMS, MSD, Roche, Astra Zeneca, Pfizer. Other authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7-30. 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2.Wood SL, Pernemalm M, Crosbie PA, et al. Molecular histology of lung cancer:from targets to treatments. Cancer Treat Rev 2015;41:361-75. 10.1016/j.ctrv.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 3.Carper MB, Claudio PP. Clinical potential of gene mutations in lung cancer. Clin Transl Med 2015;4:33. 10.1186/s40169-015-0074-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Hsu PP, Awad MM, et al. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013;13:772-87. 10.1038/nrc3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer:a paradigm for precision cancer medicine. Clin Cancer Res 2015;21:2227-35. 10.1158/1078-0432.CCR-14-2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6. 10.1038/nature05945 [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247-53. 10.1200/JCO.2009.22.6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiseo M, Gelsomino F, Bartolotti M, et al. Anaplastic lymphoma kinase as a new target for the treatment of non-small-cell lung cancer. Expert Rev Anticancer Ther 2011;11:1677-87. 10.1586/era.11.157 [DOI] [PubMed] [Google Scholar]
- 9.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998-2006. 10.1001/jama.2014.3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe:estimates for 40 countries in 2012. Eur J Cancer 2013;49:1374-403. 10.1016/j.ejca.2012.12.027 [DOI] [PubMed] [Google Scholar]
- 11.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385-94. 10.1056/NEJMoa1214886 [DOI] [PubMed] [Google Scholar]
- 12.Kim DW, Ahn MJ, Shi Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol 2012;30:abstr 7533.
- 13.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167-77. 10.1056/NEJMoa1408440 [DOI] [PubMed] [Google Scholar]
- 14.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18:1472-82. 10.1158/1078-0432.CCR-11-2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med 2012;4:120ra17. 10.1126/scitranslmed.3003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443-5. 10.1200/JCO.2010.34.1313 [DOI] [PubMed] [Google Scholar]
- 17.Facchinetti F, Caramella C, Auger N, et al. Crizotinib primary resistance overcome by ceritinib in a patient with ALK-rearranged non-small cell lung cancer. Tumori 2016. [Epub ahead of print]. 10.5301/tj.5000520 [DOI] [PubMed] [Google Scholar]
- 18.Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res 2010;16:1561-71. 10.1158/1078-0432.CCR-09-2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soda M, Isobe K, Inoue A, et al. A prospective PCR-based screening for the EML4-ALK oncogene in non-small cell lung cancer. Clin Cancer Res 2012;18:5682-9. 10.1158/1078-0432.CCR-11-2947 [DOI] [PubMed] [Google Scholar]
- 20.Li T, Maus MK, Desai SJ, et al. Large-scale screening and molecular characterization of EML4-ALK fusion variants in archival non-small-cell lung cancer tumor specimens using quantitative reverse transcription polymerase chain reaction assays. J Thorac Oncol 2014;9:18-25. 10.1097/JTO.0000000000000030 [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009:15:3143-9. 10.1158/1078-0432.CCR-08-3248 [DOI] [PubMed] [Google Scholar]
- 22.Jung Y, Kim P, Jung Y, et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosomes Cancer 2012;51:590-7. 10.1002/gcc.21945 [DOI] [PubMed] [Google Scholar]
- 23.Togashi Y, Soda M, Sakata S, et al. KLC1-ALK:a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One 2012;7:e31323. 10.1371/journal.pone.0031323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nanjo S, Nakagawa T, Takeuchi S, et al. In vivo imaging models of bone and brain metastases and pleural carcinomatosis with a novel human EML4-ALK lung cancer cell line. Cancer Sci 2015;106:244-52. 10.1111/cas.12600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontana D, Ceccon M, Gambacorti-Passerini C, et al. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK Cancer Med 2015;4:953-65. 10.1002/cam4.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703. 10.1056/NEJMoa1006448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res 2012;18:4682-90. 10.1158/1078-0432.CCR-11-3260 [DOI] [PubMed] [Google Scholar]
- 28.Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol 2009;22:508-15. 10.1038/modpathol.2009.2 [DOI] [PubMed] [Google Scholar]
- 29.Yoshida A, Tsuta K, Nakamura H, et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am J Surg Pathol 2011;35:1226-34. 10.1097/PAS.0b013e3182233e06 [DOI] [PubMed] [Google Scholar]
- 30.Kim MH, Shim HS, Kang DR, et al. Clinical and prognostic implications of ALK and ROS1 rearrangements in never-smokers with surgically resected lung adenocarcinoma. Lung Cancer 2014;83:389-95. 10.1016/j.lungcan.2014.01.003 [DOI] [PubMed] [Google Scholar]
- 31.Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS:an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 2013;19:4273-81. 10.1158/1078-0432.CCR-13-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang JJ, Zhang XC, Su J, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements:diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res 2014;20:1383-92. 10.1158/1078-0432.CCR-13-0699 [DOI] [PubMed] [Google Scholar]
- 33.Won JK, Keam B, Koh J, et al. Concomitant ALK translocation and EGFR mutation in lung cancer:a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol 2015;26:348-54. 10.1093/annonc/mdu530 [DOI] [PubMed] [Google Scholar]
- 34.Ulivi P, Chiadini E, Dazzi C, et al. Nonsquamous, non-small-cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS:frequency, clinical-pathological characteristics, and response to therapy. Clin Lung Cancer 2015. [Epub ahead of print]. 10.1016/j.cllc.2015.11.004 [DOI] [PubMed] [Google Scholar]
- 35.Boyd N, Dancey JE, Gilks CB, et al. Rare cancers:a sea of opportunity. Lancet Oncol 2016;17:e52-61. 10.1016/S1470-2045(15)00386-1 [DOI] [PubMed] [Google Scholar]
- 36.Tsao MS, Hirsch FR, Yatabe Y. IASLC Atlas of ALK testing in lung cancer. International Association for the Study of Lung Cancer, Colorado, USA, 2013. [Google Scholar]
- 37.Bozzetti C, Nizzoli R, Tiseo M, et al. ALK and ROS1 rearrangements tested by fluorescence in situ hybridization in cytological smears from advanced non-small cell lung cancer patients. Diagn Cytopathol 2015;43:941-6. 10.1002/dc.23318 [DOI] [PubMed] [Google Scholar]
- 38.Rosenblum F, Hutchinson LM, Garver J, et al. Cytology specimens offer an effective alternative to formalin-fixed tissue as demonstrated by novel automated detection for ALK break-apart FISH testing and immunohistochemistry in lung adenocarcinoma. Cancer Cytopathol 2014;122:810-21. 10.1002/cncy.21467 [DOI] [PubMed] [Google Scholar]
- 39.Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res 2010;16:5581-90. 10.1158/1078-0432.CCR-10-0851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camidge DR, Skokan M, Kiatsimkul P, et al. Native and rearranged ALK copy number and rearranged cell count in non-small cell lung cancer:implications for ALK inhibitor therapy. Cancer 2013;119:3968-75. 10.1002/cncr.28311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerr KM. ALK testing in non-small cell lung carcinoma:what now? J Thorac Oncol 2014;9:593-5. 10.1097/JTO.0000000000000171 [DOI] [PubMed] [Google Scholar]
- 42.Atherly AJ, Camidge DR. The cost-effectiveness of screening lung cancer patients for targeted drug sensitivity markers. Br J Cancer 2012;106:1100-6. 10.1038/bjc.2012.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelsomino F, Rossi G, Tiseo M. Clinical implications and future perspectives in testing non-small cell lung cancer (NSCLC) for anaplastic lymphoma kinase (ALK) gene rearrangements. J Thorac Dis 2015;7:220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossi G, Ragazzi M, Tamagnini I, et al. Does immunohistochemistry represent a robust alternative technique in determining drugable predictive gene alterations in non-small cell lung cancer? Curr Drug Targets 2015. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 45.Bavieri M, Tiseo M, Lantuejoul S, et al. Fishing for ALK with immunohistochemistry may predict response to crizotinib. Tumori 2013;99:e229-32. [DOI] [PubMed] [Google Scholar]
- 46.Pekar-Zlotin M, Hirsch FR, Soussan-Gutman L, et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. Oncologist 2015;20:316-22. 10.1634/theoncologist.2014-0389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cabillic F, Gros A, Dugay F. Parallel FISH and immunohistochemical studies of ALK status in 3244 non-small-cell lung cancers reveal major discordances. J Thorac Oncol 2014;9:295-306. 10.1097/JTO.0000000000000072 [DOI] [PubMed] [Google Scholar]
- 48.Ilie MI, Bence C, Hofman V, et al. Discrepancies between FISH and immunohistochemistry for assessment of the ALK status are associated with ALK 'borderline'-positive rearrangements or a high copy number:a potential major issue for anti-ALK therapeutic strategies. Ann Oncol 2015;26:238-44. 10.1093/annonc/mdu484 [DOI] [PubMed] [Google Scholar]
- 49.Marchetti A, Di Lorito A, Pace MV, et al. ALK protein analysis by IHC staining after recent regulatory changes:a comparison of two widely used approaches, revision of the literature, and a new testing algorithm. J Thorac Oncol 2016;11:487-95. 10.1016/j.jtho.2015.12.111 [DOI] [PubMed] [Google Scholar]
- 50.Ren S, Hirsch FR, Varella-Garcia M, et al. Atypical negative ALK break-apart FISH harboring a crizotinib-responsive ALK rearrangement in non-small-cell lung cancer. J Thorac Oncol 2014;9:e21-3 10.1097/JTO.0000000000000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cataldo KA, Jalal SM, Law ME, et al. Detection of t(2;5) in anaplastic large cell lymphoma:comparison of immunohistochemical studies, FISH, and RT-PCR in paraffin-embedded tissue. Am J Surg Pathol 1999;23:1386-92. 10.1097/00000478-199911000-00009 [DOI] [PubMed] [Google Scholar]
- 52.Selinger CI, Rogers TM, Russell PA, et al. Testing for ALK rearrangement in lung adenocarcinoma:a multicenter comparison of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol 2013;26:1545-53. 10.1038/modpathol.2013.87 [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Pan Y, Wang R, et al. ALK-rearranged lung cancer in Chinese:a comprehensive assessment of clinicopathology, IHC, FISH and RT-PCR. PLoS One 2013;8:e69016. 10.1371/journal.pone.0069016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wynes MW, Sholl LM, Dietel M, et al. An international interpretation study using the ALK IHC antibody D5F3 and a sensitive detection kit demonstrates high concordance between ALK IHC and ALK FISH and between evaluators. J Thorac Oncol 2014;9:631-8. 10.1097/JTO.0000000000000115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nitta H, Tsuta K, Yoshida A, et al. New methods for ALK status diagnosis in non-small-cell lung cancer:an improved ALK immunohistochemical assay and a new, Brightfield, dual ALK IHC-in situ hybridization assay. J Thorac Oncol 2013;8:1019-31. 10.1097/JTO.0b013e31829ebb4d [DOI] [PubMed] [Google Scholar]
- 56.Shen Q, Wang X, Yu B, et al. Comparing four different ALK antibodies with manual immunohistochemistry (IHC) to screen for ALK-rearranged non-small cell lung cancer (NSCLC). Lung Cancer 2015;90:492-8. 10.1016/j.lungcan.2015.10.002 [DOI] [PubMed] [Google Scholar]
- 57.Wang R, Pan Y, Li C, et al. The use of quantitative real-time reverse transcriptase PCR for 5' and 3' portions of ALK transcripts to detect ALK rearrangements in lung cancers. Clin Cancer Res 2012;18:4725-32. 10.1158/1078-0432.CCR-12-0677 [DOI] [PubMed] [Google Scholar]
- 58.Liu L, Zhan P, Zhou X, et al. Detection of EML4-ALK in lung adenocarcinoma using pleural effusion with FISH, IHC, and RT-PCR methods. PLoS One 2015;10:e0117032. 10.1371/journal.pone.0117032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim SM, Kim EY, Kim HR, et al. Genomic profiling of lung adenocarcinoma patients reveals therapeutic targets and confers clinical benefit when standard molecular testing is negative. Oncotarget 2016. [Epub ahead of print]. 10.18632/oncotarget.8138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao X, Wang A, Walter V, et al. Combined targeted DNA sequencing in non-small cell lung cancer (NSCLC) using UNCseq and NGScopy, and RNA sequencing using UNCqer for the detection of genetic aberrations in NSCLC. PLoS One 2015;10:e0129280. 10.1371/journal.pone.0129280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20:1479-84. 10.1038/nm.3729 [DOI] [PubMed] [Google Scholar]
- 62.Ou SH, Tan J, Yen Y, et al. ROS1 as a 'druggable' receptor tyrosine kinase:lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 2012;12:447-56. 10.1586/era.12.17 [DOI] [PubMed] [Google Scholar]
- 63.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer:updated results from a phase 1 study. Lancet Oncol 2012;13:1011-9. 10.1016/S1470-2045(12)70344-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734-9. 10.1056/NEJMoa1007478 [DOI] [PubMed] [Google Scholar]
- 65.Camidge DR, Doebele RC.Treating ALK-positive lung cancer--early successes and future challenges. Nat Rev Clin Oncol 2012;9:268-77. 10.1038/nrclinonc.2012.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 2014;4:662-73. 10.1158/2159-8290.CD-13-0846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Togashi Y, Mizuuchi H, Kobayashi Y, et al. An activating ALK gene mutation in ALK IHC-positive/FISH-negative nonsmall-cell lung cancer. Ann Oncol 2015;26:1800-1. 10.1093/annonc/mdv240 [DOI] [PubMed] [Google Scholar]
- 68.Kodityal S, Elvin JA, Squillace R, et al. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016;92:19-21. 10.1016/j.lungcan.2015.11.023 [DOI] [PubMed] [Google Scholar]
- 69.Lovly CM, McDonald NT, Chen H, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med 2014;20:1027-34. 10.1038/nm.3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim DW, Mehra R, Tan DS, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol 2016;17:452-63. 10.1016/S1470-2045(15)00614-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mok T, Spigel D, Felip E, et al. ASCEND-2:A single-arm, open-label, multicenter phase 2 study of ceritinib in adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (crz). J Clin Oncol 2015;33:abstr 8059.
- 72.Felip E, Orlov S, Park K, et al. ASCEND-3:A single-arm, open-label, multicenter phase 2 study of ceritinib in ALKi-naive adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC). J Clin Oncol 2015;33:abstr 8060.
- 73.Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study):A single-arm, open-label, phase 1-2 study. Lancet Oncol 2013;14:590-8. 10.1016/S1470-2045(13)70142-6 [DOI] [PubMed] [Google Scholar]
- 74.Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-smallcell lung cancer (AF-002JG):results from the dose-finding portion of a phase1/2 study. Lancet Oncol 2014;15:1119-28. 10.1016/S1470-2045(14)70362-6 [DOI] [PubMed] [Google Scholar]
- 75.Shaw AT, Gandhi L, Gadgeel S, et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer:a single-group, multicentre, phase 2 trial. Lancet Oncol 2016;17:234-42. 10.1016/S1470-2045(15)00488-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ou SH, Ahn JS, De Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol 2016;34:661-8. 10.1200/JCO.2015.63.9443 [DOI] [PubMed] [Google Scholar]
- 77.Rosell R, Gettinger SN, Bazhenova LA, et al. Brigatinib efficacy and safety in patients with Anaplastic Lymphoma Kinase-positive non-small cell lung cancer in a phase 1/2 trial. 2016 European Lung Cancer Conference. Geneva, Switzerland, 2016:abstr 1330. [Google Scholar]
- 78.Bauer T, Solomon BJ, Besse B, et al. Clinical activity and safety of the ALK/ROS1 TKI inhibitor PF-06463922 in advanced NSCLC. J Thor Oncol 2015;10:S239. [Google Scholar]
- 79.Drilon A, De Braud FG, Siena S, et al. Entrectinib, an oral pan-Trk, ROS1, and ALK inhibitor in TKI-naïve patients with advanced solid tumors harboring gene rearrangements. 2016 AACR Annual Meeting. New Orleans, USA, 2016:abstr 2136. [Google Scholar]
- 80.Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675-90. 10.1021/jm400402q [DOI] [PubMed] [Google Scholar]
- 81.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK- rearranged non-small-cell lung cancer. N Engl J Med 2014;370:1189-97. 10.1056/NEJMoa1311107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gainor JF, Tan DS, De Pas T, et al. Progression-free and overall survival in ALK-positive NSCLC patients treated with sequential crizotinib and ceritinib. Clin Cancer Res 2015;21:2745-52. 10.1158/1078-0432.CCR-14-3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mengoli MC, Barbieri F, Bertolini F, et al. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung:Report of two cases and review of the literature. Lung Cancer 2016;93:55-8. 10.1016/j.lungcan.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 84.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. Ch5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679-90. 10.1016/j.ccr.2011.04.004 [DOI] [PubMed] [Google Scholar]
- 85.Media Release. Available online: http://www.roche.com/media/store/releases/med-cor-2016-05-19b.htm
- 86.Zhang S, Wang F, Keats J, et al. AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066 (PF1066). Cancer Res, 2010;70:abstr LB-298.
- 87.Squillace RM, Anjum R, Miller D, et al. AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. Cancer Res 2013;73:abstr 5655.
- 88.Rivera VM, Wang F, Anjum R, et al. AP26113 is a dual ALK/EGFR inhibitor:Characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res 2012;72;abstr 1794.
- 89.Camidge DR, Bazhenova L, Salgia R, et al. Safety and efficacy of brigatinib (AP26113) in advanced malignancies, including ALK+ non-small cell lung cancer (NSCLC). J Clin Oncol 2015;33:abstr 8062.
- 90.Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res 2014;20:5686-96. 10.1158/1078-0432.CCR-14-1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ou SH, Klempner SJ, Greenbowe JR, et al. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J Thorac Oncol 2014;9:1821-5. 10.1097/JTO.0000000000000368 [DOI] [PubMed] [Google Scholar]
- 92.Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem 2014;57:4720-44. 10.1021/jm500261q [DOI] [PubMed] [Google Scholar]
- 93.Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015;28:70-81. 10.1016/j.ccell.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res 2015;21:3924-33. 10.1158/1078-0432.CCR-15-0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med 2016;374:54-61. 10.1056/NEJMoa1508887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ardini E, Menichincheri M, De Ponti C, et al. Characterization of NMS-E628, a small molecule inhibitor of anaplastic lymphoma kinase with antitumor efficacy in ALK-dependent lymphoma and non-small cell lung cancer models. Mol Cancer Ther 2009;8:abstr 244.
- 97.Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol 2015;10:1670-4. 10.1097/01.JTO.0000473485.38553.f0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ardini E, Menichincheri M, Banfi P, et al. The ALK inhibitor NMS-E628 also potently inhibits ROS1 and induces tumor regression in ROS-driven models. Cancer Res 2013;73:abstr 2092.
- 99.Ardini E, Menichincheri M, Banfi P, et al. In vitro and in vivo activity of NMS-E628 against ALK mutations resistant to Xalkori. Mol Cancer Ther 2011;10:abstr 232.
- 100.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement:a retrospective analysis. Lancet Oncol 2011;12:1004-12. 10.1016/S1470-2045(11)70232-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chun SG, Choe KS, Iyengar P, et al. Isolated central nervous system progression on Crizotinib:an Achilles heel of non-small cell lung cancer with EML4-ALK translocation? Cancer Biol Ther 2012;13:1376-83. 10.4161/cbt.22255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Metro G, Lunardi G, Floridi P, et al. CSF concentration of crizotinib in two ALK-positive non-small-cell lung cancer patients with CNS metastases deriving clinical benefit from treatment. J Thorac Oncol 2015;10:e26-7. 10.1097/JTO.0000000000000468 [DOI] [PubMed] [Google Scholar]
- 103.Weickhardt AJ, Scheier B, Burke JM, et al. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol 2012;7:1807-14. 10.1097/JTO.0b013e3182745948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Costa DB, Shaw AT, Ou SH, et al. clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol 2015;33:1881-8. 10.1200/JCO.2014.59.0539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaneda H, Okamoto I, Nakagawa K. Rapid response of brain metastasis to crizotinib in a patient with ALK rearrangement-positive non-small-cell lung cancer. J Thorac Oncol 2013;8:e32-3. 10.1097/JTO.0b013e3182843771 [DOI] [PubMed] [Google Scholar]
- 106.Camidge DR. Taking aim at ALK across the blood-brain barrier. J Thorac Oncol 2013;8:389-90. 10.1097/JTO.0b013e3182864e7c [DOI] [PubMed] [Google Scholar]
- 107.Solomon BJ, Cappuzzo F, Felip E, et al. Intracranial Efficacy of Crizotinib Versus Chemotherapy in Patients With Advanced ALK-Positive Non-Small-Cell Lung Cancer: Results From PROFILE 1014. J Clin Oncol 2016. [Epub ahead of print]. 10.1200/JCO.2015.63.5888 [DOI] [PubMed] [Google Scholar]
- 108.NCCN guidelines, version 7.2015. Available online: www.nccn.org. Last accessed 8th December 2015.
- 109.Johung KL, Yeh N, Desai NB, et al. Extended survival and prognostic factors for patients with ALK-rearranged non-small-cell lung cancer and brain metastasis. J Clin Oncol 2016;34:123-9. 10.1200/JCO.2015.62.0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ohe Y, Nishio N, Kiura K, et al. A phase I/II study with a CNS-penetrant, selective ALK inhibitor alectinib in ALK-rearranged non-small cell lung cancer (ALK+ NSCLC) patients (pts):Updates on progression free survival (PFS) and safety results from AF-001JP. J Clin Oncol 2015;33:abstr 8061.
- 111.Gadgeel S, Shaw A, Ramaswamy G, et al. Pooled analysis of CNS response to alectinib in two studies of pre-treated ALK+ NSCLC. J Thorac Oncol 2015;10:S238. [Google Scholar]
- 112.De Braud F, Niger M, Damian S, et al. Alka-372-001:First-in-human, phase I study of entrectinib - an oral pan-trk, ROS1, and ALK inhibitor - in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol 2015;33:abstr 2517.
- 113.Patel MR, Bauer TM, Liu SV, et al. STARTRK-1:Phase 1/2a study of entrectinib, an oral Pan-Trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol 2015;33:abstr 2596.
- 114.Di Maio M, De Marinis F, Hirsch FR, et al. Diagnostic and therapeutic issues for patients with advanced non-small cell lung cancer harboring anaplastic lymphoma kinase rearrangement:European vs. US perspective (review). Int J Oncol 2014;45:509-15. [DOI] [PubMed] [Google Scholar]
- 115.Gan GN, Weickhardt AJ, Scheier B, et al. Stereotactic radiation therapy can safely and durably control sites of extra-central nervous system oligoprogressive disease in anaplastic lymphoma kinase-positive lung cancer patients receiving crizotinib. Int J Radiat Oncol Biol Phys 2014;88:892-8. 10.1016/j.ijrobp.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ou SH, Jänne PA, Bartlett CH, et al. Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol 2014;25:415-22. 10.1093/annonc/mdt572 [DOI] [PubMed] [Google Scholar]
- 117.Isozaki H, Ichihara E, Takigawa N, et al. Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res 2016;76:1506-16. 10.1158/0008-5472.CAN-15-1010 [DOI] [PubMed] [Google Scholar]
- 118.Crystal AS, Shaw AT, Sequist LV, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346:1480-6. 10.1126/science.1254721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015;373:1627-39. 10.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010):a randomised controlled trial. Lancet 2016;387:1540-50. 10.1016/S0140-6736(15)01281-7 [DOI] [PubMed] [Google Scholar]
- 122.Gainor JF, Shaw AT, Sequist LV, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer (NSCLC):A Retrospective Analysis. Clin Cancer Res 2016. [Epub ahead of print]. 10.1158/1078-0432.CCR-15-3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sang J, Acquaviva J, Friedland JC, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discovery 2013;3:430-43. 10.1158/2159-8290.CD-12-0440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Awad MM, Shaw AT. ALK Inhibitors in non-small cell lung cancer:crizotinib and beyond. Clin Adv Hematol Oncol 2014;12:429-39. [PMC free article] [PubMed] [Google Scholar]
- 125.Fujita S, Masago K, Katakami N, et al. Transformation to SCLC after treatment with the ALK inhibitor alectinib. J Thorac Oncol 2016;11:e67-72. 10.1016/j.jtho.2015.12.105 [DOI] [PubMed] [Google Scholar]