

Abstract
Accurate handling of stalled replication forks is crucial for the maintenance of genome stability. RAD51 defends stalled replication forks from nucleolytic attack, which otherwise can threaten genome stability. However, the identity of other factors that can collaborate with RAD51 in this task is poorly elucidated. Here, we establish that human Werner helicase interacting protein 1 (WRNIP1) is localized to stalled replication forks and cooperates with RAD51 to safeguard fork integrity. We show that WRNIP1 is directly involved in preventing uncontrolled MRE11‐mediated degradation of stalled replication forks by promoting RAD51 stabilization on ssDNA. We further demonstrate that replication fork protection does not require the ATPase activity of WRNIP1 that is however essential to achieve the recovery of perturbed replication forks. Loss of WRNIP1 or its catalytic activity causes extensive DNA damage and chromosomal aberrations. Intriguingly, downregulation of the anti‐recombinase FBH1 can compensate for loss of WRNIP1 activity, since it attenuates replication fork degradation and chromosomal aberrations in WRNIP1‐deficient cells. Therefore, these findings unveil a unique role for WRNIP1 as a replication fork‐protective factor in maintaining genome stability.
Keywords: genome instability, replication fork arrest, replication fork degradation, WRNIP1
Subject Categories: DNA Replication, Repair & Recombination
Introduction
The proper execution of DNA replication is an essential aspect of cellular life. Proliferating cells are constantly subjected to a wide variety of threats originating by the action of exogenous and endogenous agents that can hinder replication fork progression. Several studies have demonstrated that inaccurate handling of stalled replication forks can lead to genomic instability, a well‐known source of human diseases and cancer onset (Carr & Lambert, 2013; Magdalou et al, 2014). To minimize such a risk, cells have evolved sophisticated mechanisms to cope with perturbed replication forks (Branzei & Foiani, 2009, 2010; Yeeles et al, 2013; Zeman & Cimprich, 2014). The importance of stabilizing and restarting stalled replication forks is also evidenced by the increasing number of proteins identified as being part of these mechanisms. Accordingly, multiple pathways work in the recovery of replication stalling, and some homologous recombination (HR) proteins have been implicated in preserving the integrity of arrested replication forks (Petermann & Helleday, 2010; Costanzo, 2011). Indeed, a current model proposes that BRCA2 and RAD51 may act in preventing rather than repairing lesions at stalled replication forks, to protect nascent DNA strand from degradation mediated by the exonucleolytic activity of MRE11 (Hashimoto et al, 2010; Schlacher et al, 2011; Ying et al, 2012).
Among proteins participating in the maintenance of genome stability, whose function is still poorly characterized, is the human Werner helicase interacting protein 1 (WRNIP1). WRNIP1 was identified as a binding partner of the Werner protein (WRN) (Kawabe Kawabe et al, 2001, 2006), a member of the RecQ family of DNA helicases that plays a crucial role in response to replication stress, and significantly contributes to the recovery of stalled replication forks (Rossi et al, 2010; Franchitto & Pichierri, 2014). WRNIP1 belongs to the AAA+ class of ATPase family proteins that is evolutionary conserved (Kawabe Kawabe et al, 2001; Hishida et al, 2001). Although the yeast homolog of WRNIP1, MGS1, is required to prevent genome instability caused by replication arrest (Branzei et al, 2002), little is known about the function of human WRNIP1. However, in vitro studies support the possibility that the ATPase activity of WRNIP1 could stimulate DNA polymerase delta (Polδ) to re‐initiate DNA synthesis, for example after fork arrest, through a physical association with WRN and Polδ (Tsurimoto et al, 2005). Further in vitro investigations reveal that WRNIP1 binds in an ATP‐dependent manner to forked DNA that mimics stalled replication forks (Yoshimura et al, 2009). Furthermore, WRNIP1 foci overlap with replication factories, reinforcing the hypothesis of its function at replication forks (Crosetto et al, 2008).
In this study, we have identified an uncharacterized function of WRNIP1 at perturbed replication forks. Loss of WRNIP1 results in DNA damage accumulation due to the inability of cells to properly protect stalled replication forks from nucleolytic attack by MRE11. We demonstrate that WRNIP1 is recruited to stalled replication forks. We further show that WRNIP1 interacts with the BRCA2/RAD51 complex and contributes to the stabilization of RAD51 on ssDNA to preserve stalled fork integrity. Interestingly, we prove that blocking the removal of RAD51 from chromatin by depleting FBH1, the MRE11‐mediated degradation of stalled replication forks as well as chromosomal aberrations are counteracted in WRNIP1‐deficient cells. Furthermore, we establish that WRNIP1 is implicated in the stalled fork resumption through its ATPase activity. Altogether, our work suggests a molecular basis for the role of human WRNIP1 in safeguarding genome stability in response to replication stress.
Results
WRNIP1 is required for protection and restart of stalled forks upon replication stress
To investigate the function of human WRNIP1 during DNA replication, we monitored replication perturbation genomewide at single‐molecule level by performing DNA fibre assay. Firstly, we generated MRC5SV cells stably expressing WRNIP1‐targeting shRNA (shWRNIP1). Next, isogenic cell lines stably expressing the RNAi‐resistant full‐length wild‐type WRNIP1 (shWRNIP1WT) or its ATPase‐dead mutant form of WRNIP1 (shWRNIP1T294A) (Tsurimoto et al, 2005) were created using the shWRNIP1 cells (Fig 1A). To determine whether WRNIP1 affects replication under normal growth conditions (i.e. in the absence of any treatment), we measured the rate and symmetry of the replication fork progression in shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells. We sequentially labelled cells with the thymidine analogues 5‐chloro‐2′‐deoxyuridine (CldU) and 5‐iodo‐2′‐deoxyuridine (IdU) as described in the experimental scheme (Fig 1B). Under these conditions, shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells showed almost identical fork velocity with an average fork progression rate of about 1.0 kb per minute (Fig 1C). Moreover, the frequency of asymmetric replication tracks was similar in all cell lines (Fig 1D), confirming that no elongation defect is triggered when WRNIP1 or its enzymatic activity was lost.
Figure 1. Loss of WRNIP1 leads to nascent DNA strand degradation after HU‐induced replication stress.
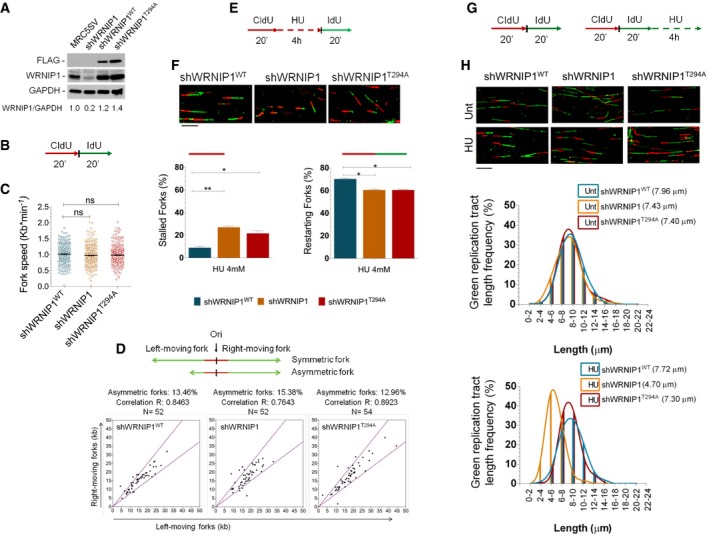
- Western blot analysis showing the expression of the WRNIP1 protein in wild‐type cells (shWRNIP1WT) and WRNIP1‐deficient (shWRNIP1) or mutant (shWRNIP1T294A) cells. MRC5SV fibroblasts were used as a positive control. The membrane was probed with an anti‐FLAG or anti‐WRNIP1. GAPDH was used as a loading control. Below each lane of the blot the ratio of WRNIP1 protein to total protein, then normalized to MRC5SV, is reported.
- Experimental scheme of dual labelling of DNA fibres in shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells. Cells were pulse‐labelled with CldU and then subjected to a pulse‐labelling with IdU.
- Analysis of replication fork velocity (fork speed) in the cells under unperturbed conditions. The length of the green tracks was measured. Mean values are represented as horizontal black lines (ns, not significant; Student's t‐test).
- Cells were treated as in (B). For each replication origin, the length of the right‐fork signal was measured and plotted against the length of the left‐fork signal. A schematic representation of symmetric and asymmetric forks is given. If the ratio between the left‐fork length and the right‐fork length deviated by more than 33% from 1 (that is, outside the violet dashed lines in the graphs), the fork was considered asymmetric. The percentage of asymmetric forks was calculated for all cell lines. N = number of forks counted for each cell line. R represents linear correlation coefficient.
- Experimental scheme of dual labelling of DNA fibres in shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells. Cells were pulse‐labelled with CldU, treated with 4 mM HU and then subjected to a pulse‐labelling with IdU.
- Graphs show the percentage of red (CldU) tracts (stalled forks) or red‐green (CldU‐IdU) contiguous tracts (restarting forks) in the cells. Means are shown, n = 3. Error bars represent standard error (*P < 0.05; **P < 0.01; Student's t‐test). Representative DNA fibre images are shown. Scale bar, 10 μm.
- Experimental scheme of dual labelling of DNA fibres in shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells. Cells were sequentially pulse‐labelled with CldU and IdU as indicated, then treated or not with 4 mM HU.
- Representative IdU tract length distributions in all cell lines under unperturbed conditions (top graph) or after HU treatment (bottom graph). Median tract lengths are given in parentheses. See also Appendix Tables S1 and S2 for details on the data sets and statistical test. Representative DNA fibre images are shown. Scale bar, 10 μm.
Source data are available online for this figure.
To obtain a deeper insight into the role of WRNIP1 in replication, we explored whether loss of WRNIP1 influences fork progression after HU‐induced replication stress. Thus, we pulse‐labelled shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells with CldU and IdU as reported (Fig 1E). DNA fibre analysis showed that WRNIP1 depletion resulted in a significant enhancement in the percentage of stalled forks induced by HU with respect to wild‐type cells (Fig 1F). Similarly, the expression of the mutant form of WRNIP1 greatly affected fork progression after HU (Fig 1F). Interestingly, comparing the percentage of restarting forks in all cell lines, we observed that loss of WRNIP1 reduced the ability of cells to resume replication after release from HU in the same extent as loss of its ATPase activity (Fig 1F). All other replication parameters were not significantly different among the cell lines (Appendix Fig S1A and B). These results implicate WRNIP1, through its ATPase activity, in restarting stalled forks.
We next verified whether WRNIP1 was involved in the protection of stalled forks, by examining the stability of nascent replication strands. To this aim, we changed the DNA labelling scheme. Thus, shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were sequentially pulse‐labelled with CldU and IdU to mark nascent replication tracts before fork stalling with HU (Fig 1G). The maintenance of the IdU label after HU treatment measures the extent of fork stability on the stretched DNA fibres. The analysis showed that IdU tract length remained unchanged with or without HU treatment in cells expressing wild‐type WRNIP1 (shWRNIP1WT) (7.72 and 7.96 μm, respectively; Fig 1H). On the contrary, in WRNIP1‐deficient cells (shWRNIP1), fork stalling led to a significant shortening of IdU tract length compared to unperturbed replication (4.70 and 7.43 μm, respectively; Fig 1H). Notably, in shWRNIP1T294A cells, IdU tract length was left unaffected after HU as in wild‐type cells, revealing that the ATPase activity is dispensable for protection of stalled forks (7.30 and 7.40 μm, with and without HU, respectively; Fig 1H). Since nascent IdU tracts are formed before treatment with HU, it is plausible that the decrease in length of the IdU tracts takes place during exposure to the drug, as previously demonstrated (Schlacher et al, 2011). Thus, we deduced that WRNIP1 is essential in avoiding degradation of nascent DNA strands at stalled forks.
To determine whether the phenotype of WRNIP1‐deficient cells is a general response to replication arrest, we pulse‐labelled shWRNIP1WT and shWRNIP1 cells with IdU, followed by exposure to a high dose of aphidicolin (Aph), a selective inhibitor of the replicative DNA polymerases (Appendix Fig S2A). Since we observed that Aph showed substantial similarity to HU in the ability to reduce IdU tract length in the absence of WRNIP1 (7.34 and 4.83 μm, shWRNIP1WT and shWRNIP1, respectively; Appendix Fig S2B), we concluded that replication stress caused by various agents needs WRNIP1 to protect stalled forks. Moreover, to ascertain whether the role of WRNIP1 is kept in other cell types, we tested HEK293T cells transfected with control siRNA (HEK293TsiCtrl) or WRNIP1 siRNA (HEK293TsiWRNIP1). After transfection, cells were pulse‐labelled with IdU and then exposed to HU (Appendix Fig S3A). Although similar IdU tract length was observed in both cell lines under unperturbed conditions, however, WRNIP1‐deficient cells (HEK293TsiWRNIP1) exhibited a defective maintenance of nascent length tracts after HU treatment as compared to the wild‐type cells (HEK293TsiCtrl) (4.42 and 7.34 μm, respectively; Appendix Fig S3B). This confirms that the fork‐protective role of WRNIP1 is independent from the cell lines.
Overall, our results suggest that, when replication is perturbed, WRNIP1 maintains the integrity of stalled forks and ensures their restart via its ATPase activity.
MRE11 nuclease activity is responsible for degradation of nascent DNA strand at stalled forks in the absence of WRNIP1
It has been reported that MRE11 activity is responsible for degradation of HU‐stalled forks in BRCA2‐defective cells (Schlacher et al, 2011; Ying et al, 2012). Since we proved that WRNIP1‐deficient cells show instability of stalled forks, which is reminiscent of that observed in the absence of BRCA2, we asked whether MRE11 nuclease could similarly promote fork degradation in our cells. To test this hypothesis, we double‐labelled shWRNIP1WT and shWRNIP1 cells, followed by treatment with HU and mirin, a chemical inhibitor of MRE11 activity (Dupré et al, 2008); then, we measured the length of the IdU tracts (Fig 2A). As expected, mirin had no effect on HU‐treated wild‐type cells (Fig 2B). However, we found that loss of MRE11 activity prevented IdU tract shortening by HU treatment in the absence of WRNIP1, reaching a value comparable to that of wild‐type cells (7.89 and 4.95 μm, with or without MRE11 inhibition, respectively; Fig 2B).
Figure 2. Inhibition of MRE11 exonuclease activity prevents nascent DNA strand degradation after replication stress.
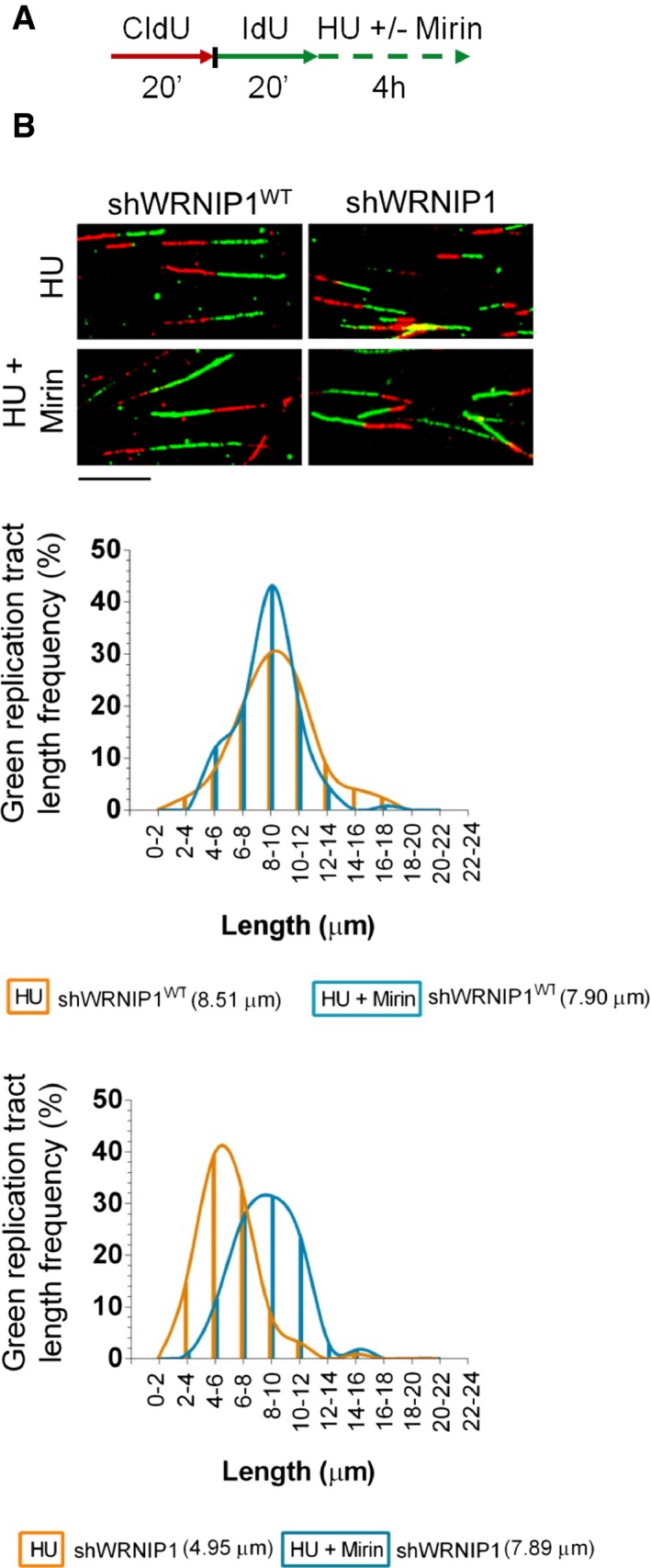
- Experimental scheme of dual labelling of DNA fibres in wild‐type cells (shWRNIP1WT) or WRNIP1‐deficient cells (shWRNIP1). Cells were sequentially pulse‐labelled with CldU and IdU as indicated, then left untreated or treated with 4 mM HU in combination or not with 50 μM mirin.
- Representative IdU tract length distributions in shWRNIP1WT (top graph) or shWRNIP1 cells (bottom graph) after treatment. Median tract lengths are given in parentheses. See Appendix Tables S1 and S2 for details on the data sets and statistical test. Representative DNA fibre images are shown. Scale bar, 10 μm.
Next, to exclude off‐target effects produced by the MRE11 inhibitor, shWRNIP1 cells were transfected with siRNAs directed against MRE11, then labelled with IdU and treated with HU (Appendix Fig S4A). Depletion of MRE11 resulted in a clear evidence of protection from nascent strand degradation during HU exposure, as IdU tract length was longer in HU‐treated cells in which MRE11 was abrogated (7.43 and 4.72 μm, with or without MRE11 knockdown, respectively; Appendix Fig S4B).
Therefore, we conclude that MRE11 nuclease activity degrades stalled forks in the absence of WRNIP1.
WRNIP1 depletion results in parental‐strand ssDNA accumulation and RAD51 destabilization after fork stalling
Next, we tested whether WRNIP1 depletion caused an increased parental‐strand ssDNA accumulation at replication forks due to degradation of nascent DNA strand. We specifically visualized ssDNA by immunofluorescence using an anti‐IdU antibody under non‐denaturing conditions. To this aim, shWRNIP1WT and shWRNIP1 cells were labelled with IdU for 24 h and then released into fresh culture medium for 2 h before stalling forks with HU (Fig 3A). Moreover, to assess the dependence of ssDNA formed on MRE11 activity, parallel samples were exposed to mirin (Fig 3A). Our analysis showed that WRNIP1‐deficient cells presented higher amount of ssDNA than wild‐type cells under unperturbed and HU‐treated conditions (Fig 3A). However, MRE11 inhibition substantially lowered the accumulation of ssDNA detected with or without fork stalling only in shWRNIP1 cells (Fig 3A). Experiments with HU‐treated shWRNIP1WT and shWRNIP1 cells, in which MRE11 activity was disrupted by RNAi, confirmed the nuclease‐dependent formation of ssDNA at parental strand in the absence of WRNIP1 (Appendix Fig S5). Then, to verify whether nascent strand became single‐stranded at stalled forks, shWRNIP1WT and shWRNIP1 cells were shortly labelled with IdU immediately before HU treatment (Appendix Fig S6A). Immunofluorescence analysis showed little, but similar, IdU labelling in both shWRNIP1WT and shWRNIP1 cells after HU treatment (Appendix Fig S6).
Figure 3. Analysis of parental ssDNA formation and RAD51 destabilization at stalled replication forks.
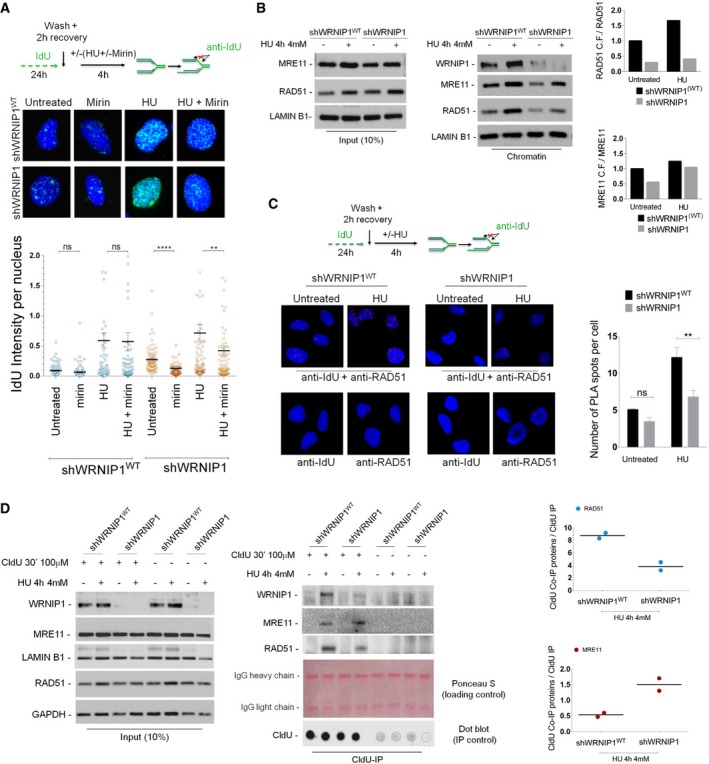
- Evaluation of ssDNA accumulation at parental‐strand by immunofluorescence analysis in wild‐type (shWRNIP1WT) or WRNIP1‐deficient (shWRNIP1) cells. Experimental design of ssDNA assay is shown. Cells were labelled with IdU for 24 h, as indicated, washed and left to recover for 2 h, then treated or not with 4 mM HU. In parallel samples, the MRE11 activity was chemically inhibited with 50 μM mirin, alone or in combination with HU‐induced replication stress. After treatment, cells were fixed and stained with an anti‐IdU antibody without denaturing the DNA to specifically detect parental ssDNA. Horizontal black lines and error bars represent the mean ± SE; n = 3 (ns, not significant; **P < 0.01; ****P < 0.0001; two‐tailed Student's t‐test). Representative images are shown. DNA was counterstained with DAPI (blue).
- Analysis of chromatin binding of MRE11 and RAD51 in shWRNIP1WT and shWRNIP1 cells. Chromatin fractions of cells, treated or not with 4 mM HU, were analysed by immunoblotting. The membrane was probed with the anti‐WRNIP1, anti‐MRE11 and anti‐RAD51 antibodies. Lamin B1 was used as a loading for the chromatin fraction. Total amount of RAD51 and MRE11 (input) in the cells was determined with the relevant antibodies. Lamin B1 was used as a loading control. In the graph, the fold increase with respect to the wild‐type untreated of the normalized ratio of the chromatin‐bound RAD51 (or MRE11)/total RAD51 (or MRE11) is reported for each cell line.
- Analysis of DNA–protein interactions between ssDNA and endogenous RAD51 in shWRNIP1WT and shWRNIP1 cells by in situ PLA assay. Experimental design used for the assay is given. Cells were labelled with IdU for 24 h, as indicated, washed and left to recover for 2 h, then treated or not with 4 mM HU for 4 h. Next, cells were fixed, stained with an anti‐IdU antibody without denaturing the DNA to specifically detect parental‐strand ssDNA and subjected to PLA assay as described in the Materials and Methods section. Antibodies raised against IdU or RAD51 were used to reveal ssDNA or endogenous RAD51, respectively. Each red spot represents a single interaction between ssDNA and RAD51. No spot has been revealed in cells stained with each single antibody (negative control). DNA was counterstained with DAPI (blue). Representative images of the PLA assay are given. Graph shows data presented as mean ± SE of the number of PLA spots per cell from three independent experiments (ns, not significant; **P < 0.01; two‐tailed Student's t‐test); n = 3.
- Localization of WRNIP1, MRE11 and RAD51 to stalled replication forks. Forks were isolated by CldU co‐immunoprecipitation (CldU‐IP). shWRNIP1WT or shWRNIP1 cells were pulse‐labelled with CldU, then fixed or treated with HU. Cells were cross‐linked, and the nuclear extracts were isolated (input) and subjected to CldU‐IP using an anti‐CldU antibody (CldU‐IP). The membranes were probed with the anti‐WRNIP1 or anti‐RAD51 antibodies. After stripping, the membranes were probed with an anti‐MRE11 antibody. Lamin B1 and GAPDH were used as loading controls (input). Ponceau S was used as a loading control of CldU‐IP. Dot blot analysis was performed to confirm that equal amounts of immunoprecipitated DNA from each sample. 10% of each IP was loaded on a nitrocellulose membrane. The membrane was probed with an anti‐CldU antibody. The graph shows the normalized ratio of the proteins co‐immunoprecipitated with CldU (CldU Co‐IP proteins)/the total of labelled DNA immunoprecipitated with CldU (CldU‐IP) for each cell line after replication stress from two independent experiments. The dots in the graph represent the individual data points from each single experiment. Horizontal black line represents the mean value from two replicates; n = 2.
Source data are available online for this figure.
Since RAD51‐ssDNA complex is functionally relevant in protecting stalled replication forks from degradation (Schlacher et al, 2011), we wondered whether the greater amount of ssDNA detected in WRNIP1‐deficient cells could correlate with a larger amount of RAD51 bound to chromatin. To address this point, we performed a Western blot analysis after cellular fractionation in shWRNIP1WT and shWRNIP1 cells treated or not with HU as indicated (Fig 3B). As shown in Fig 3B, the amount of chromatin‐bound RAD51 was lower in shWRNIP1 than in shWRNIP1WT cells under both unperturbed and fork‐stalling conditions. Furthermore, as expected, in wild‐type cells we observed an enhanced chromatin loading of MRE11 after fork stalling (Mirzoeva & Petrini, 2003); however, in WRNIP1‐deficient cells, we detected a greater increase (Fig 3B).
In agreement with our biochemical fractionation experiments, immunofluorescence detection of RAD51 relocalization in shWRNIP1WT and shWRNIP1 cells, treated or not with HU, showed a reduced percentage of RAD51 foci in the absence of WRNIP1 after fork stalling (Appendix Fig S7). We further confirmed the presence of low levels of RAD51 in WRNIP1‐deficient cells. Using a modification of the in situ proximity ligation assay (PLA), a fluorescence‐based improved method that makes possible to reveal physical protein–protein interaction (Söderberg et al, 2008), to detect protein/DNA association (Iannascoli et al, 2015), we next investigated the co‐localization of RAD51 at/near ssDNA. To this aim, shWRNIP1WT and shWRNIP1 cells were treated or not with HU (Fig 3C). We found that the co‐localization between ssDNA (anti‐IdU signal) and RAD51 significantly decreased in shWRNIP1 cells after replication stress (Fig 3C). Since high amount of ssDNA formation was revealed in shWRNIP1 cells (Fig 3A), and given that visualization of a red spot in the cell requires the presence of both ssDNA (anti‐IdU signal) and RAD51, the smaller number of PLA spots observed in the absence of WRNIP1 may correlate with the reduced levels of RAD51.
Finally, to exclude the possibility that, in shWRNIP1 cells, RAD51 was susceptible to proteasome‐mediated degradation, we examined the amount of RAD51 upon MG132 treatment alone or in combination with HU. We found that proteasomal inhibition led to accumulation of RAD51 in unperturbed shWRNIP1 cells, but not after fork stalling (Appendix Fig S8). Therefore, we concluded that, under replication stress, RAD51 is not degraded but likely not properly stabilized in the absence of WRNIP1.
Altogether these findings indicate that, when cells are depleted for WRNIP1, fork stalling results in a large enhancement of ssDNA at template DNA strand produced by the action of MRE11 nuclease activity, which does not lead to a greater amount of RAD51 bound to chromatin.
RAD51 and MRE11 are differently recruited to stalled replication forks in WRNIP1‐deficient cells
Our experiments suggest that loss of WRNIP1 results in reduced RAD51 loading to chromatin and MRE11‐dependent nascent strand degradation after fork stalling. Thus, we wanted to ascertain whether RAD51 and MRE11 were differently recruited to stalled replication forks in the absence of WRNIP1. To this end, shWRNIP1WT and shWRNIP1 cells were pulse‐labelled with CldU to mark newly replicated DNA and exposed or not to HU. Co‐immunoprecipitation of RAD51 or MRE11 with CldU‐labelled replication sites was performed from cross‐linked chromatin to detect DNA‐associated proteins at replication forks. Equal loading of proteins was evaluated by Ponceau S staining, and equal amounts of immunoprecipitated DNA from each sample were verified by dot blot analysis (Fig 3D). In line with previous studies (Petermann et al, 2010; Somyajit et al, 2015), our CldU‐IP experiments confirmed the loading of RAD51 at nascent strand in HU‐treated wild‐type cells (Fig 3D). Interestingly, although in shWRNIP1 cells, RAD51 was present at sites of stalled replication forks, the level was significantly lower than that in wild‐type cells (Fig 3D). On the contrary, the amount of MRE11 was higher in the absence of WRNIP1 as compared to wild‐type cells (Fig 3D). Moreover, and in accordance with a previous study (Dungrawala & Cortez, 2015), our experiments indicated that WRNIP1 co‐immunoprecipitated with CldU‐labelled replication sites after HU treatment in wild‐type cells, proving that WRNIP1 is associated with stalled replication forks (Fig 3D).
Consistently with the MRE11‐mediated nascent strand degradation, these results provide evidence for enhanced recruitment of MRE11, but reduced level of RAD51 at stalled replication forks in WRNIP1‐deficient cells.
RAD51 protects nascent DNA strand from degradation after replication stalling in WRNIP1‐deficient cells
The RAD51 recombinase is directly implicated in the protection of nascent strand from MRE11‐mediated degradation (Hashimoto et al, 2010; Schlacher et al, 2011), and BRCA2 stimulates RAD51 assembly on ssDNA (Jensen et al, 2010; Liu et al, 2010; Moynahan & Jasin, 2010). Since loss of WRNIP1 leads to a phenotype similar to that observed in BRCA2‐defective cells, to identify the pathway in which WRNIP1 functions under replication stress, we examined whether chemical inhibition of RAD51, which disrupts RAD51 binding to DNA (Huang & Mazina, 2012), could affect stabilization of stalled forks in WRNIP1‐deficient cells. To this end, shWRNIP1WT and shWRNIP1 cells were exposed to IdU and RAD51 inhibitor and treated with HU; then, the length of the IdU tracts was measured (Fig 4A). As expected, HU treatment resulted in IdU tract shortening in WRNIP1‐deficient cells, but not in wild‐type cells (5.31 and 7.40 μm, respectively; Fig 4B). Moreover, as previously demonstrated (Schlacher et al, 2011), in wild‐type cells treated with HU the inability to form RAD51 nucleoprotein filaments led to nascent strand degradation (Fig 4B). However, our analysis showed that concomitant loss of WRNIP1 and RAD51 activity did not have a synergistic effect on degradation of nascent strand after HU (5.36 and 5.72 μm, shWRNIP1 and shWRNIP1WT cells after HU+RAD51i, respectively; Fig 4B). Moreover, DNA fibre analysis executed in wild‐type and WRNIP1‐deficient cells, in which RAD51 was downregulated by RNA interference, was comparable to that deriving from chemical inhibition of RAD51 (Appendix Fig S9).
Figure 4. RAD51 protects nascent DNA strand from degradation after fork stalling in the absence of WRNIP1.
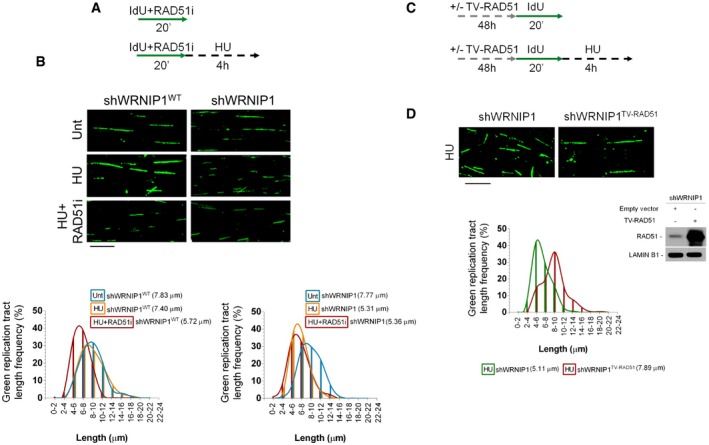
- Experimental scheme of pulse‐labelling of DNA fibres in wild‐type cells (shWRNIP1WT) or WRNIP1‐deficient cells (shWRNIP1). Cells were labelled with IdU and exposed or not to 25 μM RAD51 inhibitor, then treated or not with 4 mM HU.
- Representative IdU tract length distributions in shWRNIP1WT cells (left graph) or shWRNIP1 cells (right graph). Median tract lengths are reported in parentheses. See Appendix Tables S1 and S2 for details on the data sets and statistical test. Representative DNA fibre images are reported. Scale bar, 10 μm.
- Scheme of DNA fibre tract analysis in shWRNIP1 cells. Cells were transfected with an empty vector or a plasmid expressing a wild‐type human RAD51, and 48 h thereafter labelled with IdU and treated or not with 4 mM HU.
- Representative IdU tract length distributions in shWRNIP1 cells or shWRNIP1 cells expressing exogenous wild‐type RAD51 after HU exposure. Median tract lengths are given in parentheses. See Appendix Tables S1 and S2 for details on the data sets and statistical test. Representative DNA fibre images are given. Scale bar, 10 μm. Western blot shows the expression of the RAD51 protein in shWRNIP1 cells. The membrane was probed with an anti‐RAD51. Lamin B1 was used as a loading control.
Source data are available online for this figure.
Next, to directly test the requirement of RAD51 in the protection of stalled forks in the absence of WRNIP1, we over‐expressed the wild‐type human RAD51 in shWRNIP1 cells, and 48 h thereafter, we treated them with HU (Fig 4C). The over‐expression of RAD51 in WRNIP1‐deficient cells counteracted the shortening of IdU tracts upon HU (5.11 and 7.89 μm, with empty vector or wild‐type RAD51, respectively; Fig 4D).
Therefore, WRNIP1 protects stalled replication forks by effective loading or retention of RAD51.
WRNIP1 stabilizes RAD51 on stalled forks
To understand the functional correlation between WRNIP1 and RAD51, we investigated their possible interaction in vivo, by performing co‐IP studies. HEK293T cells were transfected with the FLAG‐tagged wild‐type WRNIP1 and treated or not with HU. Our co‐IP demonstrated that WRNIP1 associated with RAD51 both in the presence or absence of replication stress (Fig 5A). In addition, we found that WRNIP1 immunoprecipitated also BRCA2 (Fig 5A). To confirm the physical interaction of WRNIP1 with RAD51, we carried out the PLA analysis, a method allowing the detection of protein–protein interactions (Söderberg et al, 2008). To do this, shWRNIP1WT cells were treated or not with HU, then subjected to the PLA. As shown in Fig 5B, a fluorescent signal requiring the presence of both WRNIP1 and RAD51 was detected, showing their close localization in situ. Moreover, HU treatment increased the number of PLA spots per cell (Fig 5B). Interestingly, similar results were obtained in shWRNIP1T294A cells, suggesting that inhibition of catalytic activity of WRNIP1 does not hamper its interaction with RAD51 (Fig 5B).
Figure 5. WRNIP1 stabilizes RAD51 on stalled forks.
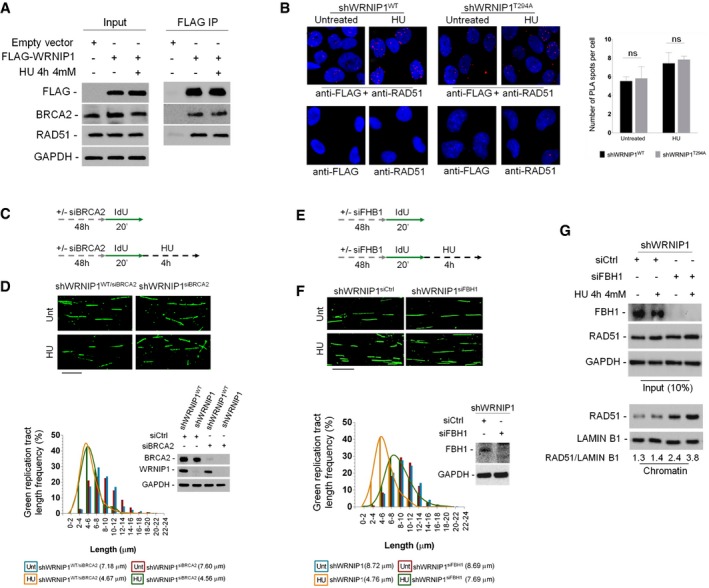
- Co‐immunoprecipitation experiments in HEK293T cells transfected with empty vector or FLAG‐WRNIP1 plasmid. Cells were treated or not with HU. After treatment, cell lysates were immunoprecipitated (FLAG IP) using anti‐FLAG antibody. The presence of WRNIP1, BRCA2 and RAD51 was assessed by immunoblotting using the anti‐FLAG, anti‐RAD51 and anti‐BRCA2 antibodies, respectively. Whole‐cell extracts were analysed (input). The membrane was probed with the same antibodies used for IP. GAPDH was used as a loading control.
- Analysis of protein–protein interactions between WRNIP1 and endogenous RAD51 in wild‐type (shWRNIP1WT) or WRNIP1‐mutant (shWRNIP1T294A) cells by in situ PLA assay. Cells were labelled with IdU for 24 h, washed and left to recover for 2 h, then treated or not with 4 mM HU. Antibodies raised against FLAG‐Tag and RAD51 were used to reveal FLAG‐WRNIP1 or endogenous RAD51, respectively. Each red spot represents a single interaction between WRNIP1 and RAD51. No spot has been revealed in cells stained with each single antibody (negative control). DNA was counterstained with DAPI (blue). Representative images of the PLA assay are shown. Graph shows the mean number of PLA spots per cell ± SE. Error bars represent standard error (ns, not significant; two‐tailed Student's t‐test); n = 3.
- Experimental scheme of pulse‐labelling of DNA fibres in wild‐type cells (shWRNIP1WT) or WRNIP1‐deficient cells (shWRNIP1). Cells were transfected with BRCA2 siRNA (siBRCA2), and 48 h thereafter labelled with IdU, then treated or not with 4 mM HU.
- Representative IdU tract length distributions in shWRNIP1WT/siBRCA2 or shWRNIP1siBRCA2 cells treated or not with HU. Median tract lengths are given in parentheses. See Appendix Tables S1 and S2 for details on the data sets and statistical test. Representative DNA fibre images are reported. Scale bar, 10 μm. Western blot shows BRCA2 depletion in shWRNIP1WT and shWRNIP1 cells. The membrane was probed with an anti‐BRCA2 or anti‐WRNIP1. GAPDH was used as a loading control.
- Experimental scheme of pulse‐labelling of DNA fibres in shWRNIP1 cells. Cells were transfected with control siRNA (shWRNIP1siCtrl) or FBH1 siRNA (shWRNIP1siFBH1), and 48 h thereafter labelled with IdU, then treated or not with 4 mM HU.
- Representative IdU tract length distributions in shWRNIP1siCtrl or shWRNIP1siFBH1 cells with or without HU treatment. Representative DNA fibre images are reported. Scale bar, 10 μm. Western blot shows FBH1 depletion in the cells. The membrane was probed with an anti‐FBH1. GAPDH was used as a loading control. Median tract lengths are given in parentheses. See Appendix Tables S1 and S2 for details on the data sets and statistical test.
- Analysis of chromatin binding of RAD51 in shWRNIP1 cells depleted for FBH1. Cells were transfected with control siRNA (shWRNIP1siCtrl) or FBH1 siRNA (shWRNIP1siFBH1), and 48 h treated or not with HU for 4 h. Chromatin fractions of cells were analysed by immunoblotting. The membrane was probed with the anti‐FBH1 and anti‐RAD51 antibodies. Lamin B1 was used as a loading for the chromatin fraction. Total amount of RAD51 (input) in the cells was determined with the relevant antibodies. GAPDH was used as a loading control. The ratio of the RAD51/Lamin B1 signal (chromatin) is reported below each lane.
Source data are available online for this figure.
To explore the link existing between WRNIP1 and the BRCA2/RAD51 complex in response to replication perturbation, DNA fibre assay was performed in HU‐treated shWRNIP1WT or shWRNIP1 cells, in which BRCA2 was downregulated by RNAi (Fig 5C). In agreement with the observation that inhibition of RAD51 did not enhance the level of fork degradation at HU‐stalled forks in WRNIP1‐deficient cells (Fig 4B), our analysis showed that concomitant depletion of WRNIP1 and BRCA2 did not result in further destabilization of stalled forks (4.56 and 4.67 μm, shWRNIP1WT/siBRCA2 and shWRNIP1siBRCA2 cells after HU, respectively; Fig 5D). This suggests that WRNIP1 and the BRCA2/RAD51 complex lie on a pathway involved in blocking degradation of newly synthesized DNA strand.
Since BRCA2 mediates RAD51 loading on ssDNA (Jensen et al, 2010; Liu et al, 2010; Moynahan & Jasin, 2010), we verified whether, in WRNIP1‐deficient cells, the low amount of chromatin‐bound RAD51 could depend on inefficient recruitment of BRCA2 upon replication stress. Immunofluorescence analysis showed no difference in the ability of shWRNIP1WT and shWRNIP1 cells to relocalize BRCA2 after HU (Appendix Fig S10). Thus, we asked whether the defective phenotype could derive from an uncontrolled translocase activity of the F‐box DNA helicase 1 (FBH1) (Simandlova et al, 2013), leading to disruption of RAD51 filaments in the absence of WRNIP1. To this aim, we examined the stability of nascent strand in shWRNIP1 cells depleted for FBH1 using siRNA and treated according to the scheme (Fig 5E). Interestingly, DNA fibre analysis showed that abrogation of FBH1 prevented the shortening of IdU tracts after HU in WRNIP1‐deficient cells (Fig 5F). In addition, in the absence of BRCA2, depletion of FBH1 results in nascent strand degradation after fork stalling, as it does when WRNIP1 is lacking (Appendix Fig S11).
FBH1 is involved in extracting RAD51 from chromatin (Simandlova et al, 2013), and we found that its depletion restores DNA fibre length in the absence of WRNIP1. To test whether this phenotypic reversion could relate to stabilization of RAD51, we then performed cellular fractionation experiments in shWRNIP1 cells, in which FBH1 was downregulated. Our analysis showed that loss of FBH1 was associated with an increase in the proportion of RAD51 that is chromatin‐bound under unperturbed and HU‐treated conditions (Fig 5G).
Altogether, these experiments allow us to conclude that WRNIP1 serves to stabilize than recruit RAD51 to stalled forks, protecting them from the MRE11‐dependent degradation.
Loss of WRNIP1 or its ATPase activity leads to DNA damage accumulation and cell death after replication stalling
We next sought to characterize the physiological consequences of the inability of WRNIP1‐deficient or mutant cells to preserve fork stability or promote fork restart after replication stress. We first examined the levels of DNA damage in wild‐type (shWRNIP1WT), WRNIP1‐deficient (shWRNIP1) or mutant cells (shWRNIP1T294A) under unperturbed cell growth conditions or upon HU‐induced replication stress. We measured DNA damage accumulation at the single‐cell level using anti‐phospho‐H2AX immunostaining. H2AX phosphorylation (γ‐H2AX) is considered an early sign of DNA damage induced by replication stalling (Ward & Chen, 2001). Thus, shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were treated or not with HU for 4 h, then immunostained with an anti‐γ‐H2AX antibody (Fig 6A). Our results showed that loss of WRNIP1 function or its ATPase activity resulted per se in about fivefold and threefold increase in the percentage of γ‐H2AX‐positive foci‐containing cells, respectively, relative to that of the control cells (Fig 6A). In contrast, HU treatment led to enhanced accumulation of γ‐H2AX‐positive nuclei in all cell lines (Fig 6A). However, the increase in γ‐H2AX foci formation appeared greater for shWRNIP1 and shWRNIP1T294A cells, about 40 and 34%, respectively, versus 24% of wild‐type cells (Fig 6A). Interestingly, flow cytometric analysis confirmed that the different percentages detected among cells lines were not due to differences in cell cycle distribution with or without HU treatment (Appendix Fig S12).
Figure 6. Loss of WRNIP1 or its ATPase activity results in DNA damage accumulation and enhanced chromosomal instability in response to fork stalling.
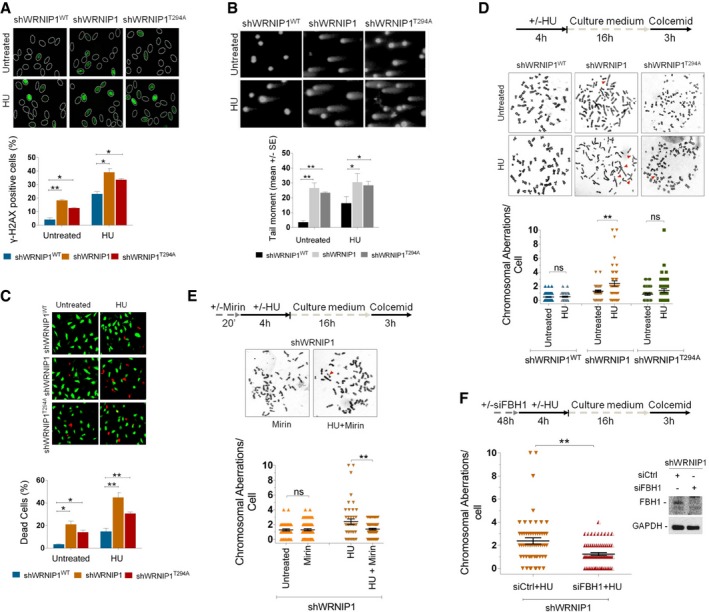
- Analysis of DNA damage accumulation. Wild‐type (shWRNIP1WT), WRNIP1‐deficient (shWRNIP1) or mutant (shWRNIP1T294A) cells were treated or not with 4 mM HU for 4 h, then subjected to γ‐H2AX immunofluorescence. Graph shows data presented as mean of γ‐H2AX‐positive cells ± SE from three independent experiments; n = 3 (*P < 0.1; **P < 0.01; two‐tailed Student's t‐test). Representative images of nuclei showing the different number of foci per nucleus are reported.
- Analysis of DNA breakage accumulation. shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were treated as in (A), then subjected to alkaline comet assay. Graph shows data presented as mean tail moment ± SE from three independent experiments; n = 3 (*P < 0.1; **P < 0.01; two‐tailed Student's t‐test). Representative images are shown.
- Evaluation of cell death. shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were treated or not with 4 mM HU for 16 h. Cell viability was evaluated by LIVE/DEAD fluorescent assay. Data are expressed as mean of dead cells ± SE from three independent experiments; n = 3 (*P < 0.1; **P < 0.01; two‐tailed Student's t‐test). Representative images of double‐staining of viable (green) and dead (red) cells are shown.
- Experimental scheme for evaluation of the chromosomal aberrations is shown. shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were treated or not with 4 mM HU, then left to recover for 16 h in drug‐free medium and metaphases collected with colcemid. Next, cells were fixed and processed as reported in Appendix Supplementary Materials and Methods. Dot plot shows the number of chromosomal aberrations per cell. Horizontal black lines and error bars represent the mean ± SE (ns, not significant; **P < 0.01; two‐tailed Student's t‐test). Representative Giemsa‐stained metaphases of cells treated or not with 4 mM HU. Arrows indicate chromosomal aberrations.
- Experimental scheme of the chromosomal aberration analysis is given. The experiment was carried out as in (D) but cells were pre‐treated or not with 50 μM mirin. Dot plot shows the effect of mirin exposure on the number of chromosome aberrations per cell in shWRNIP1 cells. Horizontal black lines and error bars represent the mean ± SE (ns, not significant; **P < 0.01; two‐tailed Student's t‐test). Representative Giemsa‐stained metaphases of shWRNIP1 cells treated with mirin alone or in combination with HU. Arrows indicate chromosomal aberrations.
- Experimental design of the chromosomal aberration assay is reported. shWRNIP1 cells were transfected with control siRNAs (siCtrl) or FBH1 siRNA (siFBH1). Forty‐eight hours thereafter, cells were treated or not with 4 mM HU and then left to recover for 16 h. Metaphases were collected with colcemid and prepared as reported in Appendix Supplementary Materials and Methods. Dot plot shows the number of chromosomal aberrations per cell. Western blot shows FBH1 depletion in the cells. The membrane was probed with an anti‐FBH1. GAPDH was used as a loading control. Horizontal black lines and error bars represent the mean ± SE (**P < 0.01; two‐tailed Student's t‐test).
Source data are available online for this figure.
As an alternative and sensitive method for the detection of DNA damage in individual cells, we used alkaline comet assay. The experiments were performed under the same conditions as for γ‐H2AX analysis. We found that loss of WRNIP1 or its ATPase activity increased the spontaneous level of DNA breakage compared with wild‐type cells, similar to what observed by fluorescent data (Fig 6B). Moreover, HU treatment caused a further enhancement of comet tail moment reaching values significantly higher in shWRNIP1 and shWRNIP1T294A cells than those in the control cells (Fig 6B). To verify whether double‐strand breaks (DSBs) were formed under our experimental conditions, we performed the neutral comet assay in parallel samples. Comparing the comet tail moment in the different cell lines tested, we did not notice appreciable amounts of DSBs (Appendix Fig S13).
Furthermore, evaluation of cell viability by the fluorescence‐based LIVE/DEAD assay confirmed that WRNIP1‐deficient cells were more sensitive to HU, as the percentage of dead cells was higher than that of wild‐type cells (Fig 6C). Similarly, expression of mutant form of WRNIP1 (shWRNIP1T294A) led to enhanced cell death after HU exposure respect to shWRNIP1WT cells (Fig 6C).
Thus, these experiments demonstrate that WRNIP1‐deficient and mutant cells exhibit high sensitivity to HU‐induced fork stalling, leading to DNA damage accumulation and cell death.
Unprotected stalled forks lead to chromosomal instability in WRNIP1‐deficient cells
To obtain further insights into the role of WRNIP1 in maintaining genome stability, we analysed the consequences of loss of WRNIP1 functions on chromosomal damage after HU‐induced replication stress. To this aim, shWRNIP1WT, shWRNIP1 and shWRNIP1T294A cells were exposed to HU for 5 h and released into drug‐free medium for 16 h prior to the addition of colcemid for 3 h to collect metaphase chromosomes (Fig 6D). Our analysis showed that WRNIP1‐deficient as well as mutant cells, displayed higher spontaneous levels of chromosomal aberrations respect to wild‐type cells (Fig 6D), suggesting that loss of WRNIP1 by itself or of its ATPase activity can cause genome instability. Moreover, HU treatment significantly increased the mean number of total chromosomal aberrations per cell in shWRNIP1 cells, whereas both shWRNIP1WT and shWRNIP1T294A cells did not produce a similar effect (Fig 6D). Next, we verified whether chromosomal damage formed after HU treatment in the absence of WRNIP1 could correlate with the MRE11‐dependent nascent strand degradation. To do this, we treated shWRNIP1 cells with HU and mirin or with mirin alone. Interestingly, we found that chemical inhibition of MRE11 activity during fork stalling led to attenuation of the level of chromosomal aberrations per cell in shWRNIP1 cells (Fig 6E). In addition, the same analysis performed in cells in which MRE11 was downregulated by RNA interference (siMRE11) was comparable to that resulting from chemical inhibition of MRE11 (Appendix Fig S14).
Finally, the impact of RAD51‐ssDNA filament stabilization by FBH1 depletion on chromosomal aberrations in shWRNIP1 cells was analysed. To this end, shWRNIP1 cells were depleted for FBH1 and treated as described in the scheme (Fig 6F). As shown in Fig 6F, inhibition of RAD51 dismantling from chromatin alleviated the level of chromosomal damage after HU treatment in WRNIP1‐deficient cells.
Therefore, our results suggest that loss of WRNIP1 as well as of its ATPase activity leads to a mild genomic instability. They also show that the WRNIP1‐mediated fork protection function, rather than the role in restarting stalled forks, is responsible for chromosomal instability arising after fork stalling.
Discussion
The ability to properly counteract replication stress is of paramount importance to ensure genome stability in living cells. Recently, it has emerged that some HR proteins, that is BRCA2 and RAD51, are essential components of a mechanism responsible for the defence against replication stress (Petermann & Helleday, 2010; Costanzo, 2011). Despite extensive research, it is still not completely understood how the HR proteins operate during the resolution of fork stalling, and which their partners are. In the present study, we have identified WRNIP1 as a factor working in conjunction with the RAD51 recombinase in response to replication stress.
Our experiments establish a function not previously described for WRNIP1 in maintaining the integrity of stalled forks, a behaviour conserved among human cells. So far, clear data showing an involvement of WRNIP1 in the dynamics of replication fork progression were missing. Our DNA fibre analysis demonstrates that loss of WRNIP1 results in impaired fork progression under stressful conditions. Moreover, it shows that nascent DNA tracts undergo destabilization due to the nucleolytic activity of MRE11, which in turn causes marked genome instability in the absence of WRNIP1. We observed that WRNIP1‐depleted cells exhibit increased fork degradation, envisaging a mechanism very similar to the pathological MRE11‐mediated degradation of stalled replication intermediates reported in the absence of BRCA2 (Schlacher et al, 2011; Ying et al, 2012). In keeping with this, combined depletion of WRNIP1 and BRCA2 has no additional effect on the destabilization of newly synthesized DNA tracts compared to loss of the single genes. This observation indicates that WRNIP1 may function within the same pathway of BRCA2 to preserve stalled fork integrity. Our data reveal that loss of WRNIP1 results in a large amount of MRE11‐dependent parental‐strand ssDNA, but little nascent strand ssDNA. It has been proposed that, in response to perturbed replication, MRE11 activity does not process parental DNA in eukaryotes, making it impossible to expose ssDNA at nascent strand (Hashimoto et al, 2010). Consistently, accumulation of parental‐strand ssDNA could derive from defects of the early stages of fork remodelling before regression, as probably occurs in the absence of BRCA2 (Schlacher et al, 2011, 2012; Ying et al, 2012). Alternatively, exposure of parental‐strand ssDNA may result from over‐processing of the extruded arm of a regressed fork, as opposed to the limited degradation reported in wild‐type cells (Thangavel et al, 2015; Zellweger et al, 2015). Interestingly, further supporting the hypothesis that WRNIP1 and BRCA2 can collaborate in a common pathway, WRNIP1 co‐immunoprecipitates with BRCA2 and RAD51 under both unaltered and replication perturbed conditions, and physically interacts with RAD51. The fact that WRNIP1 is associated with these HR proteins, even under unperturbed conditions, raises the possibility that they may exist in a single complex ready to safeguard the integrity of the forks whenever they arrested. Previous studies have shown that WRNIP1 binds to forked DNA, which resembles stalled forks (Yoshimura et al, 2009). Our CldU‐IP experiments reveal the association of WRNIP1 with replication forks upon replication stress, suggesting that WRNIP1 could be actually recruited to perturbed forks in vivo, also confirming recent observations from iPOND approaches (Dungrawala & Cortez, 2015). In contrast, and consistently with the increased MRE11‐mediated fork degradation, enhanced recruitment of MRE11 to chromatin and to stalled forks after replication stress is observed in WRNIP1‐deficient cells.
BRCA2 is required for preserving stalled fork stability after replication perturbation, and this function is achieved by its direct interaction with RAD51, which is loaded on ssDNA (Jensen et al, 2010; Moynahan & Jasin, 2010). The inability to form RAD51‐coated nucleofilament renders BRCA2‐deficient cells susceptible to MRE11 nucleolytic degradation (Schlacher et al, 2011, 2012; Ying et al, 2012). Given that WRNIP1 directly interacts with RAD51, loss of this interaction may interfere with efficient nucleation of RAD51 on ssDNA, thus undermining nascent strand integrity. Interestingly, WRNIP1‐deficient cells show increased accumulation of ssDNA, which is not accompanied by an excess of RAD51 loaded on chromatin. In addition, reduced fork recruitment and association between ssDNA and RAD51 is found in the absence of WRNIP1.
In line with this, co‐depletion of WRNIP1 and RAD51 does not alter the excessive degradation occurring at stalled forks, but RAD51 over‐expression effectively prevents the excessive fork destabilization in WRNIP1‐defective cells. Defective accumulation of RAD51 at stalled forks in WRNIP1‐deficient cells may be explained by the failure of proper relocalization of RAD51 on ssDNA. Indeed, WRNIP1 could act as assisting factor for docking RAD51 recruitment to ssDNA through its association with the BRCA2/RAD51 complex. Alternatively, loss of WRNIP1 could result in the inability to retain RAD51 on chromatin. Interestingly, depletion of FBH1, which is involved in the removal of RAD51 from chromatin (Simandlova et al, 2013), restores RAD51 levels in chromatin and reverts both the fork degradation and chromosome instability phenotypes of WRNIP1‐deficient cells. In contrast, downregulation of FBH1 in BRCA2‐deficient cells does not rescue fork degradation. Since BRCA2, which mediates RAD51 loading to chromatin, is recruited correctly in WRNIP1‐deficient cells, these results support the hypothesis of a role for WRNIP1 in stabilizing or retaining RAD51 at stalled forks.
As a member of the AAA+ family proteins, human WRNIP1 possesses an ATPase activity that is stimulated by association with template/primer DNA (Tsurimoto et al, 2005). Interestingly, the catalytic activity of WRNIP1 would not be involved in the protection of nascent strand. Indeed, loss of ATPase activity of WRNIP1 does not hinder its interaction with RAD51 and consistently does not compromise the stability of nascent strand. However, we demonstrate that the ATPase activity of WRNIP1 is needed for restart of stalled forks. In vitro studies have indicated that WRNIP1 is able to bind DNA structures resembling stalled forks and template/primer DNA (Tsurimoto et al, 2005; Yoshimura et al, 2009). Similarly to MGS1, WRNIP1 associates with DNA polymerase delta (Polδ) (Kanamori et al, 2011) and by its ATPase activity promotes the Polδ‐mediated DNA synthesis enhancing the frequency on template/primer DNA (Tsurimoto et al, 2005). Also WRN, a partner of WRNIP1 (Kawabe et al, 2006; Kawabe Kawabe et al, 2001), has shown the capacity to bind on template/primer DNA and to interact with Polδ increasing its activity in the elongation step of replication (Kamath‐Loeb et al, 2000; Szekely et al, 2000). Interestingly, WRN is involved in the stability and restart of perturbed replication forks (Sidorova, 2008; Ammazzalorso et al, 2010; Basile et al, 2014). Thus, it is possible that WRNIP1, WRN and Polδ could form a complex, acting under replication stress to promote reinitiation of DNA synthesis at stalled forks, as it has been proposed in vitro (Tsurimoto et al, 2005).
It is worth noting that both WRNIP1 deficiency and loss of ATPase activity result in comparable levels of DNA damage and chromosomal instability under unperturbed conditions. As MGS1, the yeast homolog of WRNIP1, is essential for Okazaki fragment processing preventing genome instability (Kim et al, 2005), then WRNIP1 could play a similar function. However, our results suggest that these phenotypes are not associated with any apparent impairment of normal replication. One possible explanation may be that specific replication defects are not detectable using our assays. Alternatively, the genome instability phenotype observed in untreated WRNIP1‐deficient and ATPase mutant cells could be due to non‐replicative events, such as the post‐replication gap repair. The enhanced alkaline tail moment detected in untreated WRNIP1‐deficient or ATPase mutant cells could support both possibilities, but further investigations are necessary to address this point.
Taken together, previous works and the present study allow us to draw a model to explain how WRNIP1 could participate in the replication stress response (Fig 7). Upon fork stalling, replication fork progression is arrested and extended ssDNA is generated. Thus, BRCA2 recruits RAD51, and WRNIP1 contributes to the stabilization of RAD51, in order to protect stalled forks and prevent their degradation by the nuclease MRE11. Once the reason of the stall is removed, the ATPase activity of WRNIP1, perhaps in association with other proteins, stimulates the restart of DNA synthesis, which can be completed, thus guaranteeing genome stability. However, when WRNIP1 or its ATPase activity is lost, cells undergo to a pathological process. The absence of WRNIP1 leads to extensive MRE11‐dependent degradation of nascent DNA strand and uncontrolled action of the translocase, FBH1, resulting in enhanced accumulation of chromosomal damage and cell death. On the other hand, inhibition of the ATPase activity may abolish the binding of WRNIP1 with Polδ, as seen for MGS1 (Hishida et al, 2001; Branzei et al, 2002), making difficult the resumption of stalled forks.
Figure 7. Schematic model for the role of WRNIP1 at stalled forks.
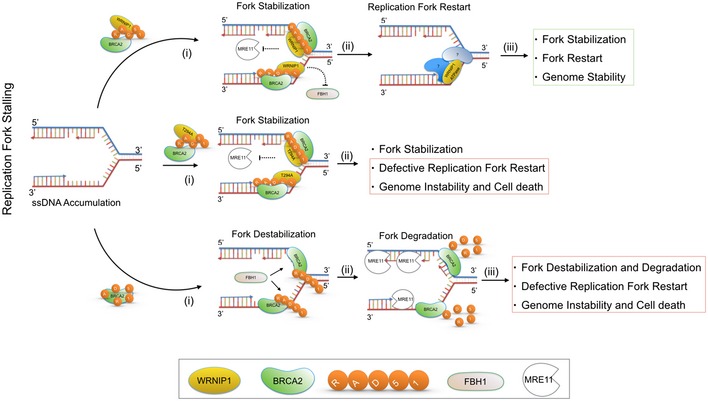
WRNIP1 interacts with the BRCA2/RAD51 complex and stabilizes RAD51 on ssDNA at stalled forks, counteracting the dissolution of the RAD51 filament by FBH1. After stalled fork stabilization, the ATPase activity of WRNIP1, in collaboration with other proteins, could be required for stimulating the restart of DNA synthesis, which ensures genome stability. Loss of WRNIP1 or its catalytic activity leads to DNA damage accumulation and enhanced chromosomal instability. See the text for more details.
Collectively, our data define a role for WRNIP1 in avoiding the pathological degradation of stalled forks and contribute to explain how DNA damage accumulates in the absence of WRNIP1 in human cells. Stabilization of RAD51 at stalled forks is emerging as an essential function to preserve genome integrity upon replication stress (Simandlova et al, 2013; Higgs et al, 2015). These findings expand our understanding of the pathway required for the stabilization of stalled forks, identifying WRNIP1 as a novel crucial factor to the RAD51 function. As genomic instability is often associated with cancer development, our study can help to clarify how downregulation of WRNIP1 gene could give rise to several human tumours (Lukk et al, 2010).
Materials and Methods
Cell lines and culture conditions
The SV40‐transformed MRC5 fibroblast cell line (MRC5SV) was a generous gift from Patricia Kannouche (IGR, Villejuif, France). MRC5SV was transduced with shRNA lentiviral transduction particles targeting the UTR region of the mRNA (Sigma‐Aldrich TRCN0000004526 PLKO.1‐puro) and selected on puromycin (5 μg/ml; Invitrogen) to create the stable shWRNIP1 cell line. Cells were cultured in the presence of puromycin (100 ng/ml; Invitrogen) to maintain selective pressure for shRNA expression. By using the Neon™ Transfection System Kit (Invitrogen) according to the manufacturer's instructions, shWRNIP1 cells were stably transfected with FLAG‐tagged full‐length cDNA encoding wild‐type WRNIP1 plasmid (shWRNIP1WT) or expressing a FLAG‐tagged full‐length WRNIP1 plasmid carrying Ala substitution at Thr294 site missense‐mutant form of WRNIP1 with inactive ATPase activity (WRNIP1T294A) (Tsurimoto et al, 2005). Cells were cultured in the presence of neomycin and puromycin (1 mg/ml and 100 ng/ml, respectively) to maintain selective pressure for expression. HEK293T cells were obtained from American Type Culture Collection (VA, USA). All cell lines were maintained in DMEM (Invitrogen) supplemented with 10% FBS (Boehringer Manheim) and incubated at 37°C in a humidified 5% CO2 atmosphere.
DNA fibre analysis
Cells were pulse‐labelled with 25 μM 5‐chloro‐2′‐deoxyuridine (CldU) and 250 μM 5‐iodo‐2′‐deoxyuridine (IdU) at specified times, with or without treatment as reported in the experimental schemes. Alternatively, cells were pulse‐labelled with 250 μM IdU for the indicated times and treated or not as indicated. DNA fibres were prepared and spread out as previously reported (Basile et al, 2014). For immunodetection of labelled tracks, the following primary antibodies were used: anti‐CldU (rat monoclonal anti‐BrdU/CldU; BU1/75 ICR1 Abcam, 1:100) and anti‐IdU (mouse monoclonal anti‐BrdU/IdU; clone b44 Becton Dickinson, 1:10). The secondary antibodies were goat anti‐mouse Alexa Fluor 488 or goat anti‐rabbit Alexa Fluor 594 (Molecular Probes, 1:200). The incubation with antibodies was accomplished in a humidified chamber for 1 h at RT.
Images were acquired randomly from fields with untangled fibres using Eclipse 80i Nikon Fluorescence Microscope, equipped with a Video Confocal (ViCo) system. The length of labelled tracks was measured using the Image‐Pro‐Plus 6.0 software, and values were converted into kilobases using the conversion factor 1 μm = 2.59 kb as reported (Basile et al, 2014). A minimum of 100 individual fibres was analysed for each experiment and the mean of at least three independent experiments presented. Statistics were calculated using GraphPad Prism Software (see Appendix Tables S1 and S2).
In situ PLA assay
The in situ proximity ligation assay (PLA; Olink, Bioscience) was performed according to the manufacturer's instructions. Exponential growing cells were seeded into 24‐multiwell plates at a density of 8 × 104 cells/well. After the indicated treatment, cells were permeabilized with 0.5% Triton X‐100 for 10 min at 4°C, fixed with 3% formaldehyde/2% sucrose solution for 10 min and then blocked in 3% BSA/PBS for 15 min. After washing with PBS, cells were incubated with the two relevant primary antibodies. Antibody staining was carried out in the standard immunofluorescence procedure. The primary antibodies used were as follows: mouse monoclonal anti‐FLAG (Sigma‐Aldrich, 1:1,000), rabbit polyclonal anti‐WRNIP1 (GeneTex, 1:1000), rabbit polyclonal anti‐RAD51 (Santa Cruz Biotechnology, 1:500) and anti‐IdU (mouse monoclonal anti‐BrdU/IdU; clone b44 Becton Dickinson, 1:10). The negative control consisted of using only one primary antibody. Samples were incubated with secondary antibodies conjugated with PLA probes MINUS and PLUS: the PLA probe anti‐mouse PLUS and anti‐rabbit MINUS (OLINK Bioscience). The incubation with all antibodies was accomplished in a humidified chamber for 1 h at 37°C. Next, the PLA probes MINUS and PLUS were ligated using two connecting oligonucleotides to produce a template for rolling‐cycle amplification. After amplification, the products were hybridized with red fluorescence‐labelled oligonucleotide. Samples were mounted in Prolong Gold anti‐fade reagent with DAPI (blue). Images were acquired randomly using Eclipse 80i Nikon Fluorescence Microscope, equipped with a Video Confocal (ViCo) system.
CldU co‐immunoprecipitation of proteins at replication forks
CldU co‐immunoprecipitation of proteins present at replication forks was carried out according to the protocol reported elsewhere (Bryant et al, 2009). Exponential growing cells were seeded into plates at a density of 3 × 106 cells/plate. The day after, cells were labelled with 100 μM CldU for 30 min and then subjected to either no treatment or treatment with 4 mM HU for 4 h. Cells were cross‐linked in 1% formaldehyde for 15 min at RT. The reaction was stopped by incubating cells with 125 mM glycine for 15 min at RT. Cells were washed twice with cold PBS and harvested in cold PBS using a cell scraper. The cytosolic protein fraction was removed by centrifugation (5 min, 1,500 g, 4°C) of cells, after incubation with hypotonic buffer (10 mM HEPES pH 7.5, 50 mM NaCl, 0.3 M sucrose, 0.5% TX‐100, supplemented with protease inhibitor cocktail (Thermo Scientific)) for 10 min on ice. Next, the nuclear soluble fraction was removed by centrifugation (2 min, 15,000 g, 4°C) of cells, after incubation with nuclear buffer (10 mM HEPES pH 7.0, 200 mM NaCl, 1 mM EDTA, 0.5% NP‐40, supplemented with protease inhibitor cocktail) for 10 min on ice. The pellets were resuspended in lysis buffer (10 mM HEPES pH 7.0, 500 mM NaCl, 1 mM EDTA, 1% NP‐40, supplemented with protease inhibitor cocktail), sonicated and centrifuged (30 s, 15,000 g, 4°C). The supernatant was then collected. Total protein concentration was determined using the standard Bradford assay (Bio‐Rad). A total of 300 μg protein was used for IP reaction and incubated with 6 μg of anti‐CldU antibody (rat monoclonal anti‐BrdU/CldU; BU1/75 ICR1 Abcam) and 25 μl of Dynabeads Protein G (Novex). The IP reaction was washed 3 times with nuclear buffer and then 3 times with washing buffer (10 mM HEPES pH 7.0, 0.1 mM EDTA, supplemented with protease inhibitor cocktail). The reaction was resuspended in 2× sample loading buffer (100 mM Tris/HCl pH 6.8, 100 mM DTT, 4% SDS, 0.2% bromophenol blue and 20% glycerol), boiled for 30 min at 90°C and then subjected to Western blot as described in Appendix Supplementary Materials and Methods.
Statistical analysis
Statistical differences in all case were determined by Student's t‐test, except for fork degradation, which was analysed by Mann–Whitney test (see Appendix Tables S1 and S2). In all cases: ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Author contributions
GL created cell lines, performed DNA fibre assay, fluorescence, DNA damage analysis and biochemical experiments. VM performed LIVE/DEAD assay, fluorescence and chromosomal aberrations analysis. GL and VM analysed data and contributed to design experiments. PP and AF designed experiments and analysed data. PP and AF wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
The authors thank Drs. Eugenia Dogliotti and Filomena Mazzei for critical reading of the manuscript, and all members of our laboratories for stimulating discussion. The authors are grateful to Drs. Patricia Kannuoche and Maria Spies for sharing research materials. This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) IG #15410 to A.F.
The EMBO Journal (2016) 35: 1437–1451
References
- Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P (2010) ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J 29: 3156–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile G, Leuzzi G, Pichierri P, Franchitto A (2014) Checkpoint‐dependent and independent roles of the Werner syndrome protein in preserving genome integrity in response to mild replication stress. Nucleic Acids Res 42: 12628–12639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Seki M, Onoda F, Yagi H, Kawabe Y, Enomoto T (2002) Characterization of the slow‐growth phenotype of S. cerevisiae Whip/Mgs1 Sgs1 double deletion mutants. DNA Repair (Amst) 1: 671–682 [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2009) The checkpoint response to replication stress. DNA Repair (Amst) 8: 1038–1046 [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219 [DOI] [PubMed] [Google Scholar]
- Bryant HE, Petermann E, Schultz N, Jemth A‐S, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T (2009) PARP is activated at stalled forks to mediate Mre11‐dependent replication restart and recombination. EMBO J 28: 2601–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM, Lambert S (2013) Replication stress‐induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425: 4733–4744 [DOI] [PubMed] [Google Scholar]
- Costanzo V (2011) Brca2, Rad51 and Mre11: performing balancing acts on replication forks. DNA Repair (Amst) 10: 1060–1065 [DOI] [PubMed] [Google Scholar]
- Crosetto N, Bienko M, Hibbert RG, Perica T, Ambrogio C, Kensche T, Hofmann K, Sixma TK, Dikic I (2008) Human Wrnip1 is localized in replication factories in a ubiquitin‐binding zinc finger‐dependent manner. J Biol Chem 283: 35173–35185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Cortez D (2015) Purification of proteins on newly synthesized DNA using iPOND. Methods Mol Biol 1228: 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré A, Boyer‐Chatenet L, Sattler RM, Modi AP, Lee J‐H, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J (2008) A forward chemical genetic screen reveals an inhibitor of the Mre11‐Rad50‐Nbs1 complex. Nat Chem Biol 4: 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pichierri P (2014) Replication fork recovery and regulation of common fragile sites stability. Cell Mol Life Sci 71: 4507–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, Stankovic T, Boulton SJ, Niedzwiedz W, Stewart GS (2015) BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell 59: 462–477 [DOI] [PubMed] [Google Scholar]
- Hishida T, Iwasaki H, Ohno T, Morishita T, Shinagawa H (2001) A yeast gene, MGS1, encoding a DNA‐dependent AAA(+) ATPase is required to maintain genome stability. Proc Natl Acad Sci USA 98: 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Mazina O (2012) Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J Med Chem 55: 3011–3020 [DOI] [PubMed] [Google Scholar]
- Iannascoli C, Palermo V, Murfuni I, Franchitto A, Pichierri P (2015) The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1‐dependent degradation. Nucleic Acids Res 43: 9788–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51‐mediated recombination. Nature 467: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath‐Loeb AS, Johansson E, Burgers PM, Loeb LA (2000) Functional interaction between the Werner Syndrome protein and DNA polymerase delta. Proc Natl Acad Sci USA 97: 4603–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M, Seki M, Yoshimura A, Tsurimoto T, Tada S, Enomoto T (2011) Werner interacting protein 1 promotes binding of Werner protein to template‐primer DNA. Biol Pharm Bull 34: 1314–1318 [DOI] [PubMed] [Google Scholar]
- Kawabe Y, Branzei D, Hayashi T, Suzuki H, Masuko T, Onoda F, Heo SJ, Ikeda H, Shimamoto A, Furuichi Y, Seki M Enomoto T (2001) A novel protein interacts with the Werner's syndrome gene product physically and functionally. J Biol Chem 276: 20364–20369 [DOI] [PubMed] [Google Scholar]
- Kawabe Y, Seki M, Yoshimura A, Nishino K, Hayashi T, Takeuchi T, Iguchi S, Kusa Y, Ohtsuki M, Tsuyama T, Imamura O, Matsumoto T, Furuichi Y, Tada S, Enomoto T (2006) Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Repair (Amst) 5: 816–828 [DOI] [PubMed] [Google Scholar]
- Kim J‐H, Kang Y‐H, Kang H‐J, Kim D‐H, Ryu G‐H, Kang M‐J, Seo Y‐S (2005) In vivo and in vitro studies of Mgs1 suggest a link between genome instability and Okazaki fragment processing. Nucleic Acids Res 33: 6137–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, Heyer W‐D (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA‐covered single‐stranded DNA. Nat Struct Mol Biol 17: 1260–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukk M, Kapushesky M, Nikkilä J, Parkinson H, Goncalves A, Huber W, Ukkonen E, Brazma A (2010) A global map of human gene expression. Nat Biotechnol 28: 322–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdalou I, Lopez BS, Pasero P, Lambert SAE (2014) The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 30: 154–164 [DOI] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JHJ (2003) DNA replication‐dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res 1: 207–218 [PubMed] [Google Scholar]
- Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11: 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Helleday T (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11: 683–687 [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Bohr VA (2010) Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 9: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51‐BRCA1/2. Cancer Cell 22: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM (2008) Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair (Amst) 7: 1776–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, Chatterjee S, Reid DA, Liu Y, Janscak P, Rothenberg E, Hickson ID (2013) FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem 288: 34168–34180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45: 227–232 [DOI] [PubMed] [Google Scholar]
- Somyajit K, Saxena S, Babu S, Mishra A, Nagaraju G (2015) Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res 43: 9835–9855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely AM, Chen Y, Zhang C, Oshima J, Weissman SM (2000) Werner protein recruits DNA polymerase delta to the nucleolus. Proc Natl Acad Sci USA 97: 11365–11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, Vindigni A (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurimoto T, Shinozaki A, Yano M, Seki M, Enomoto T (2005) Human Werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase delta. Genes Cells 10: 13–22 [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR‐dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Yeeles JTP, Poli J, Marians KJ, Pasero P (2013) Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5: a012815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T (2012) Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821 [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Seki M, Kanamori M, Tateishi S, Tsurimoto T, Tada S, Enomoto T (2009) Physical and functional interaction between WRNIP1 and RAD18. Genes Genet Syst 84: 171–178 [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6