

Abstract
Recent research in nutritional control of aging suggests that cytosolic increases in the reduced form of nicotinamide adenine dinucleotide and decreasing nicotinamide adenine dinucleotide metabolism plays a central role in controlling the longevity gene products sirtuin 1 (SIRT1), adenosine monophosphate‐activated protein kinase (AMPK) and forkhead box O3 (FOXO3). High nutrition conditions, such as the diabetic milieu, increase the ratio of reduced to oxidized forms of cytosolic nicotinamide adenine dinucleotide through cascades including the polyol pathway. This redox change is associated with insulin resistance and the development of diabetic complications, and might be counteracted by insulin C‐peptide. My research and others' suggest that the SIRT1–liver kinase B1–AMPK cascade creates positive feedback through nicotinamide adenine dinucleotide synthesis to help cells cope with metabolic stress. SIRT1 and AMPK can upregulate liver kinase B1 and FOXO3, key factors that help residential stem cells cope with oxidative stress. FOXO3 directly changes epigenetics around transcription start sites, maintaining the health of stem cells. ‘Diabetic memory’ is likely a result of epigenetic changes caused by high nutritional conditions, which disturb the quiescent state of residential stem cells and impair tissue repair. This could be prevented by restoring SIRT1–AMPK positive feedback through activating FOXO3.
Keywords: Adenosine monophosphate‐activated protein kinase, C‐peptide, Sirtuin 1
Introduction
The present review is based on my research experiences and written as a historical perspective review, rather than as a comprehensive review. I used this approach for several reasons, the first of which is that the biochemical consensus view of this topic has not changed in the past 10 years. Second, this format allows me to include more negative data than would otherwise be appropriate. Third, a historical perspective might be more interesting for new readers. For a comprehensive review focusing on the general biochemistry of diabetic complications, see Culcutt et al.1 Diabetic neuropathy was reviewed by Yagihashi et al.2, and a review of diabetic nephropathy from the perspective of cell biology was recently written by Maezawa et al.3
Polyol Pathway and the Pseudo‐Hypoxia Hypothesis
When I started PhD courses at Hirosaki University, Hirosaki, Japan, under Professor Yagihashi in pathology, the hottest topic in the field of diabetic complications was the polyol pathway4, 5. This might be the first theory that mechanistically explains how high‐glucose‐induced changes in metabolism lead to diabetic complications. The polyol (sorbitol) pathway consists of the two enzymatic reactions: (i) glucose + reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) ⇒ sorbitol + oxidized form of nicotinamide adenine dinucleotide phosphate (NADP+); and (ii) sorbitol + oxidized form of nicotinamide adenine dinucleotide (NAD+) ⇒ fructose + nicotinamide adenine dinucleotide (NADH). The first reaction is catalyzed by aldose reductase, and the second is catalyzed by sorbitol dehydrogenase. When intracellular free glucose increases, flux through the cascade increases. The development of aldose reductase inhibitors (ARI), a class of drugs that specifically targets diabetic complications, accelerated interest in this field and provided the first hope of stopping the development of complications in humans6, 7. Using animal models of diabetes, mostly rodents, more than one of these inhibitors has helped prevent retinopathy, nephropathy, neuropathy and vasculopathy7, 8, 9.
After receiving my PhD, I continued to study diabetic complications with Professor Joseph Williamson at Washington University in St. Louis, Missouri, USA. At that time, his laboratory had just developed a method to assess albumin vascular permeability and regional blood flow using radio‐isotopes10, 11. There, I developed the mathematical basis for the measurement of vascular permeability and glomerular filtration rate12, allowing us to more clearly monitor the nature of vascular dysfunction associated with diabetes or high glucose. In addition to using streptozotocin‐injected rats (hereafter referred to as STZ diabetic rats), his laboratory also developed a granulation tissue chamber model. Granulation tissue (connective tissue that is generated during wound healing) is grown inside small chambers (typically 2) mounted on the back of a rat. The initial study was carried out in diabetic rats, and found that newly‐formed vessels in granulation tissues were leakier than the ones in non‐diabetic rats. This leak could be prevented by aldose reductase inhibitors. Subsequently, they found that even in non‐diabetic rats, application of 25 mmol/L glucose to granulation tissue directly twice daily for 10 days caused a similar three‐ to fourfold increase in vascular permeability and blood flow13. This model was quite useful to screen compounds that could potentially ameliorate high‐glucose‐induced vascular dysfunction. As we published, inhibitors of aldose reductase13 and protein kinase C14, 15, aminoguanidine16, acetyl‐carnitine17 and insulin C‐peptide18 were found to suppress these changes caused by high glucose. In addition to glucose, lactate and non‐esterified fatty acids (palmitate) induced similar dysfunction.
Regarding ARI, we hypothesized that ARI not only inhibit the first step of the polyol pathway (glucose to sorbitol), but also decrease flux to the second step (sorbitol to fructose). The second step, which produces NADH from NAD+, is catalyzed by sorbitol dehydrogenase. It has been known since the 1960s that various tissues from diabetic rats19, including lens4 and liver20, 21, show an increase in the ratio of NADH to NAD+ compared with non‐diabetic rats. In non‐diabetic conditions, this is a typical sign of hypoperfusion hypoxia (low blood flow), which is caused by impaired utilization of NADH in mitochondria and accumulation of lactate. In diabetes, tissue hypoxia has been implicated in the pathogenesis of complications, retinopathy and neuropathy in particular. However, as glucose increased blood flow in the non‐diabetic rats of our tissue chamber model, the vascular dysfunction was not caused by hypoxia. To test whether vascular dysfunction was causally linked to increased cytosolic NADH/NAD+ ratios, 1 mmol/L of pyruvate was added with high glucose. Pyruvate converts NADH to NAD+, and produces lactate, thus decreasing the NADH/NAD+ ratio. Pyruvate treatment reduced vascular dysfunction. In addition, pyruvate also prevented vascular dysfunction caused by 10 mmol/L lactate with 5 mmol/L glucose19. During this experiment, we found that lactate‐induced dysfunction could be seen in blood flow just 5 h after incubation without affecting vascular permeability in tissue chambers22, and by bolus injection to the rats. This suggests that blood flow controlled by redox state is an acute process that could be more proximal than a change in permeability. Indeed, we showed that increased regional blood flow by physiological stimuli (muscle blood flow by electrical muscle contraction23, and increased retinal and visual cortex blood flow by visual stimulation24) are the result of similar NADH/NAD+ redox changes. These changes in blood flow could also be augmented by lactate infusion and suppressed by pyruvate infusion. This phenomenon was also observed in the human brain25. Experiments with longer duration of diabetes in STZ diabetic rats showed different results in blood flow. Increased regional blood flow could be observed up to 8 weeks. However, from 10 to 28 weeks of STZ diabetes, blood flow was no longer different from non‐diabetic controls, whereas albumin permeation and NADH/NAD+ redox change were still present from 6 to 28 weeks of diabetes. This showed that in longer durations of diabetes, the mechanisms to increase blood flow (likely affecting nitric oxide bioactivity) become inoperative.
This series of studies led us to postulate the ‘pseudohypoxia hypothesis’ (increased cytosolic NADH levels without real hypoxia) as a pathogenic mechanism of diabetic complications19, 22, 26, 27, 28. The original theory focused on high‐glucose‐induced mechanisms (polyol pathway). Later studies showed that pseudohypoxia could be caused by hyperlactatemia (obesity) and high non‐esterified fatty acids conditions (type 2 diabetes), which cause similar vascular dysfunctions29. Although the theory is old, the essence of this hypothesis is still valid, and its implications for current cell biology are beginning to unfold. The increased NADH/NAD+ ratio affects sirtuin 1 (SIRT1) and adenosine monophosphate‐activated protein kinase (AMPK) activity, as discussed later. Increasing NADH should also mediate epigenetic regulation through C‐terminal binding protein29, whose genome‐wide profiles were recently characterized in breast cancer30. Activation of C‐terminal binding protein by NADH causes the epithelial‐to‐mesenchymal transition and genome instability, whereas C‐terminal binding protein depletion or caloric restriction (decreasing NADH) reverses the gene repression and increases DNA repair. These factors are likely related to cancer risk or progression in diabetes31.
Brief Note on the Controversial Changes in Blood Flow in Early STZ Diabetic Rats
In humans, angiopathy is a late diabetic complication that impairs blood perfusion. However, there were conflicting reports in rats of how retinal and sciatic nerve blood flow were affected by early STZ diabetes. Professor Williamson's lab used the microsphere method, which is still the ‘gold standard’32, and found increased blood flow in the retina, which contradicted results from Dr King's group reporting a decreased blood flow as assessed by video fluorescein angiography33. We could not resolve this discrepancy. However, in human diabetes, dilation of the retinal arterioles was found, leading to speculation that loss of myogenic response and subsequent increased arterial diameter would permit physiologically high blood flow to reach the capillaries in the retina34.
Sciatic nerve blood flow in STZ diabetes is decreased in other researchers' reports, as shown by the hydrogen clearance and laser‐Doppler methods, which also contradicts our observation. However, we showed that exposing the sciatic nerve alone, as occurs during the hydrogen clearance/laser‐Doppler method, increased blood flow in control rats, but not diabetic rats. Thus, blood flow appears lower in diabetic rats, and this was corrected by treatment with an ARI35. In human diabetes, it was suggested that a change in vascular reactivity apparently predates the development of neuropathy36. Therefore, a method that measures absolute blood flow might produce a different result than a method that measures changes in blood flow in these tissues.
Insulin C‐Peptide
In addition to studying redox effects on the vasculature, I also studied the insulin‐connecting peptide (C‐peptide). Initially, C‐peptide was considered a mere by‐product of insulin maturation, a cleavage product without biological activity. However, there were some reports as early as 1975 showing biological actions37, 38, including a report that synthetic C‐peptide increased exogenous insulin effects in alloxan diabetic rats39. In the 1990s, Professor Wahren and other researchers showed more clearly that C‐peptide had beneficial effects. C‐peptide increased Na‐K‐adenosine triphosphatase (Na‐K‐ATPase) activity in kidney tubulus40, glucose uptake in rat skeletal muscle41 and decreased high glomerular filtration rate in diabetes patients42. We investigated C‐peptide actions in our systems18: the tissue chamber model and STZ diabetes. In the tissue chamber model, full‐length human and rat C‐peptide ameliorated high‐glucose‐induced vascular dysfunction in a dose response fashion. Both human and rat C‐peptide was fully effective at 100 nmol/L. A total of 10 amino acids in the middle of the sequence were critical for its function. In the STZ diabetic rat model, 400 μg/kg C‐peptide injected subcutaneously twice a day prevented vascular dysfunction in the retina, kidney, sciatic nerve, and aorta at 6 weeks and 3 months' duration of diabetes. In addition, it prevented diabetes‐induced decreases in nerve conduction velocity and nerve Na‐K‐ATPase activity in diabetes. Other authors43, 44 also observed the beneficial effects of C‐peptide on peripheral neuropathy in STZ‐rats.
C‐peptide distribution and kinetics in vivo was investigated in both non‐diabetic and STZ diabetic rats using 111In or 125I‐labeled C‐peptide (unpublished data). Time‐dependent distribution showed no clear preferential location for C‐peptide accumulation, and the majority of radioactive C‐peptide was cleared from the kidney. Non‐compartmental analysis showed that compared with non‐diabetic rats, diabetic rats had twofold C‐peptide whole‐body distribution volume and twofold delayed clearance, suggesting that C‐peptide distribution and clearance are affected by diabetes.
Although rat C‐peptide was slightly more effective at lower concentrations than human C‐peptide in the tissue chamber model, the concentration required for correction of vascular dysfunction (10–100 nmol/L) appeared to be more than the physiological concentration of C‐peptide (several hundred pmol/L). Peak human C‐peptide levels were approximately 9 nmol/L at 10 and 30 min after single subcutaneous injection at the dose of 400 μg/kg in the rats. Professor Wahren's group suggested that C‐peptide treatment is only beneficial in type 1 diabetes, in which endogenous C‐peptide levels drop below the physiological levels necessary to maintain normal functions45. As our tissue chamber model is made on non‐diabetic rats, the microvessels of granulation tissue are exposed to normal levels of endogenous C‐peptide. Therefore, the present results showed that an additional dose (pharmacological rather than physiological dose) of C‐peptide was required to prevent high‐glucose‐mediated insults.
We further tested this hypothesis in an acute glucose infusion model. Intravenous glucose infusion achieving 350–450 mg/dL plasma glucose, into non‐diabetic rats for 5 h causes an increase in regional blood flow similar to that observed in diabetic rats. Glucose infusion induces secretion of insulin and C‐peptide from pancreatic β‐cells, therefore this model is not C‐peptide deficient, but has excess C‐peptide during glucose infusion. We found that co‐infusion of C‐peptide did not prevent glucose‐induced changes in blood flow in eye tissues (retina, anterior uvea, posterior uvea), the kidney and sciatic nerve (Figure 1). Similarly, C‐peptide subcutaneous injection or infusion 1 day before glucose infusion had no effect (data not shown). However, C‐peptide fully prevented glucose‐induced increases in blood flow if 400 μg/kg C‐peptide was injected twice daily for 2 days before glucose infusion (Figure 1). These results suggest that C‐peptide action in preventing vascular dysfunction is not acute, and the high concentrations of C‐peptide might require eliciting this effect. C‐peptide itself does not reverse the redox change caused by high glucose18, yet twice daily injections for 2 days completely prevented blood flow increases by lactate infusion (unpublished data). This suggests that C‐peptide counteracts the downstream events caused by redox changes. As C‐peptide also worked in vascular permeability and peripheral nerve dysfunctions, the point(s) at which C‐peptide targets are very close to the redox change, perhaps mediated by transcription factors and changes in gene expression.
Figure 1.
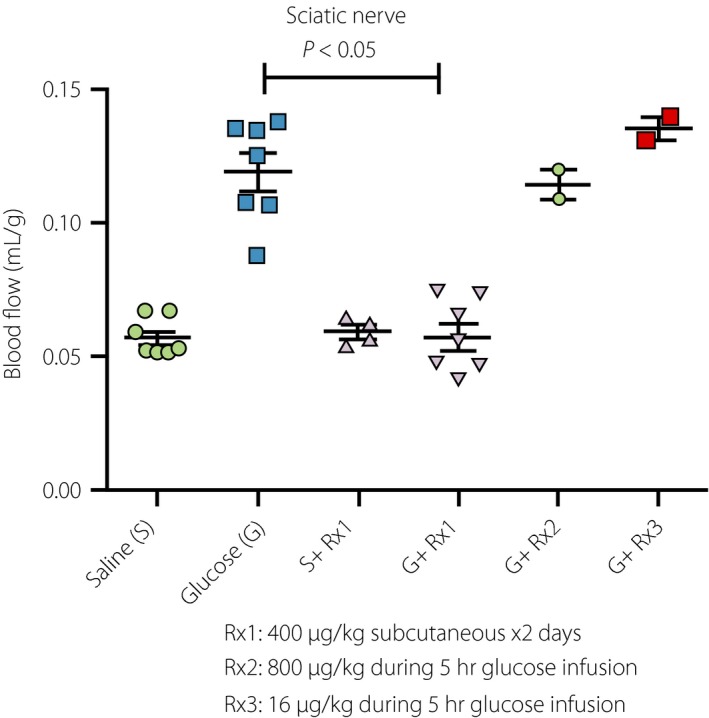
Effects of C‐peptide on high‐glucose‐induced increases in blood flow. Non‐diabetic Sprague–Dawley rats weighing 25–300 g received a 25% glucose infusion to maintain blood glucose levels at 400–500 mg/dL for 5 h: glucose infusion group (G). The control group received the same amount of saline (S). Blood flow was assessed at the end of the 5‐h infusion. Regional blood flows in the sciatic nerve, retina and kidney (not shown) were significantly increased by glucose infusion. Co‐infusion of human C‐peptide at the doses of 800 μg/kg (G + R × 2) and 16 mg/kg (G + R × 3) during the 5‐h infusion period had no effect on increased blood flow. In contrast, 400 μg/kg subcutaneous injection of C‐peptide for 2 days before glucose infusion (G + R × 1) completely prevented the effects of glucose on increased blood flow. The same regimen of C‐peptide had no effect on blood flow in the saline group (S + R × 1). Similar results were obtained by lactate infusion (not shown), suggesting that C‐peptide counteracts the effects of redox changes.
Clinical studies of C‐peptide have been mainly carried out by Professor Wahren, but they have not yet shown a beneficial effect. The receptor(s) for C‐peptide has not been identified, despite high‐affinity binding shown by surface plasmon resonance46. Discrepancies in reports of C‐peptide‐mediated signal transduction also make it difficult to understand its beneficial actions in the context of diabetes47. However, it has been reported that low nmol/L levels of C‐peptide activate AMPK48. I also independently found this phenomenon using 100 nmol/L C‐peptide when I began to study AMPK in 2001 in cultured human umbilical vein endothelial cells (although the activation was modest compared with the AMPK activator metformin). C‐peptide also attenuated 25 mmol/L glucose‐induced apoptosis; however, the effect was not impressive compared with AICAR (an AMPK activator). Interestingly, although D‐amino C‐peptide could also prevent vascular dysfunction in vivo 18, it was completely ineffective in preventing apoptosis (unpublished data). Therefore, activation of AMPK might not mediate the effect of C‐peptide on vascular function in rats. Activation of AMPK by native C‐peptide was acute (within 15 min), suggesting that it was likely mediated by calcium/calmodulin‐dependent protein kinase kinase beta activation. This phenomenon echoes a recent report that C‐peptide binds to a class of G‐protein‐coupled receptors49, analogous to the activation of AMPK by glucagon‐like peptide‐1, as we reported in endothelial cells50. However, in contrast to glucagon‐like peptide‐1, an early report suggests that C‐peptide might suppress insulin secretion, as shown by Toyota et al.37 Activation of AMPK could explain another early report that C‐peptide increased glucose uptake41 that appeared to be insulin‐independent, and activated nitric oxide synthase51. However, the report that C‐peptide increases Na‐K‐ATPase activity in kidney tubules is not compatible with observations that AMPK activation decreases Na‐K‐ATPase activity by inducing its internalization of Na‐K‐ATPase52, 53. In our studies using STZ diabetic rats, C‐peptide did not change blood glucose levels or bodyweight with/without a low dose of insulin18, which also does not support AMPK activation as a major action.
During our analysis of C‐peptide sequence and function, we noticed that effective C‐peptide sequences were very flexible. Reverse sequence and D‐amino acid sequence were also functional18. In addition to the middle part of the peptide, the C‐terminus penta‐peptide54 (which can be cleaved‐off in vivo) was also functional (unpublished data). However, its randomized sequence was not functional. This indicates that C‐peptide action appears to be mediated by at least two different parts of the peptide. Three‐dimensional structures of the reverse sequence and D‐amino acid sequence should be quite different, and therefore they must be recognized as random structures. The requirement for high concentrations suggests that these parts bind to low‐affinity receptors, which might be the reason why it could not be identified by radiolabeling. In addition, this scenario explains the diversity of C‐peptide actions, which might strongly depend on the receptors expressed. Alternatively, as suggested by Professor Kahn who commented on our paper, the effect we saw might not be mediated by its receptor(s).
In summary, beneficial effects of C‐peptide supplementation have not been confirmed in patients with type 1 diabetes. This could be because C‐peptide elicits its beneficial effect at concentrations much higher than physiological concentrations. If so, it might also work in type 2 diabetes. Mechanistically, C‐peptide counteracts downstream events caused by increased NADH/NAD+, possibly through changing gene expression.
Targeting Insulin Resistance as a General Mechanism for Cell Vulnerability in Diabetes
On the retirement of Professor Williamson, I began working with Dr Neil Ruderman at Boston University. I continued to study diabetic vascular complications, but decided to shift my focus. At that time, my hypothesis was that insulin resistance could be a fundamental cell biological problem in diabetes and its complications. In vivo, insulin resistance can be a result (type 1 diabetes) and cause (type 2 diabetes) of hyperglycemia. The insulin receptor is found in almost every cell type. A major signaling event of insulin is activation of Akt that can mediate anti‐apoptotic signaling. Thus, my hypothesis was that insulin resistance could increase the vulnerability of cells to high‐glucose‐induced stress. Based on observations that lactate infusion alone causes insulin resistance in the heart and some skeletal muscle in rats55, I proposed that high‐glucose‐induced redox changes could be related to insulin resistance.
AMP‐Activated Protein Kinase and Oxidative Stress
When I started studying AMPK, it was already known that AMPK activation ameliorates insulin resistance56; however, its presence and function were not known in endothelial cells. I first established the AMPK signaling cascade in endothelial cells in culture57, 58. I then established a cell culture model to induce apoptosis by high glucose, and examined whether AMPK activation prevented apoptosis by increasing Akt activity59. The result was positive, and served as the earliest indication that AMPK could be a target for treating diabetic vascular disease. As nuclear factor (NF)‐κB activation is one pathway of particular importance in retinopathy60, I evaluated whether AMPK activation could attenuate NF‐κB signaling. I found that AMPK activation prevented palmitate and tumor necrosis factor‐β‐mediated NF‐κB transactivation61. Palmitate is a substrate of ceramide de novo synthesis that was already known to cause insulin resistance in skeletal muscle56. In endothelial cells, both high glucose and high palmitate suppressed AMPK activation, and caused insulin resistance. We further investigated this phenomenon in cultured retinal pericytes. Similar to glucose, but at a much faster rate, palmitate induced apoptosis. This was mediated by increases in ceramide, nicotinamide adenine dinucleotide phosphate oxidase‐mediated oxidative stress and activation of NF‐κB62. Apoptosis, as well as palmitate‐induced endoplasmic reticulum stress, could be prevented by AMPK activation63. In the latter study, activation of AMPK did not prevent oxidative stress, as measured by 2′, 7′‐dichlorofluorescein fluorescence or electron spin resonance spectroscopy. In addition, 0.1 mmol/L palmitate produced peak levels of 8‐isoprostanes without causing apoptosis, whereas oleate produced more 8‐isoprostates than palmitate, but did not induce apoptosis63. These results suggest that oxidative stress is necessary, but not sufficient to produce cell damage in this model.
In 2000, Brownlee et al. showed that incubation of bovine aortic endothelial cells with high glucose induced oxidative stress from mitochondria64. We repeatedly tried to reproduce these results. However, we were not able to see any increase in oxidative stress by high glucose alone in aortic endothelial cells from human or bovine, human microvascular, or umbilical vein endothelial cells. In humans, anti‐oxidant treatment has not been shown to be effective in any disease conditions, suggesting that although oxidant levels might be increased, the levels themselves might not be enough to cause pathology. On the contrary, it was reported that anti‐oxidants prevent health‐promoting effects of physical exercise in humans65, suggesting that physiological levels of oxidative stress have an important role in inducing adaptive responses to stress as described later through forkhead box O3 (FOXO3).
Brownlee's study created a lot of interest in the role of mitochondria in diabetes and its associated conditions. There is no question of its importance. However, compared with the time when they published, research focus has shifted from metabolic and biochemical‐orientated studies (which examine metabolites and proteins) to cell biology‐oriented researches (which examine organelles and cell dynamics). From a cell biology stand‐point, sustained mitochondrial dysfunctions are likely to be as a result of problems of mitochondrial quality control mechanisms (mitochondriogenesis, autophagy [mitophagy] and fission–fusion cycle). In this respect, AMPK was shown to increase mitochondriogenesis by activating PGC‐1 and autophagy by phosphorylating Ulk‐1 and inhibiting mechanistic target of rapamycin signaling. In recent review articles about diabetic complications, AMPK has been postulated as a potential target in nephropathy66, retinopathy67, and neuropathy68 for its putative actions on mitochondria.
SIRT1, Senescence and Longevity Gene Products
Around 2004, my study interest became to include understanding diabetes, and its complications within the context of aging, senescence and epigenetics. In their early articles on sirtuins in Caenorhabditis elegans and other lower organisms, Drs Guarante and Sinclair69 listed longevity gene products including Amp‐1 (Ampk), Sir2, Tor, Foxo, p66Shc, Clock, Klotho and Catalase. For myself and other researchers studying AMPK, it was clear some of them (AMPK, SIRT1 [Sir2 ortholog], mechanistic target of rapamycin, FOXO, p66SHC and CATALASE) were interrelated even at that time and more clearly nowadays (Figure 2). However, the connection between AMPK and SIRT1 was not clear, so I decided to study this relationship. Together with Dr Fan Lan, we showed for the first time that sirtuin activators, such as resveratrol and other polyphenols, activate AMPK through liver kinase B1 (LKB1)70, 71. After a number of experiments, we concluded that SIRT1 deacetylates LKB1 at K48, which induces its translocation to the cytosol where it becomes activated72. This cascade was confirmed by Sinclair's group73, and is cited as one of the main connections between these two signaling pathways. We also showed that this activation cascade operates in aortic endothelial cells during exercise74, which might explain its beneficial effects.
Figure 2.
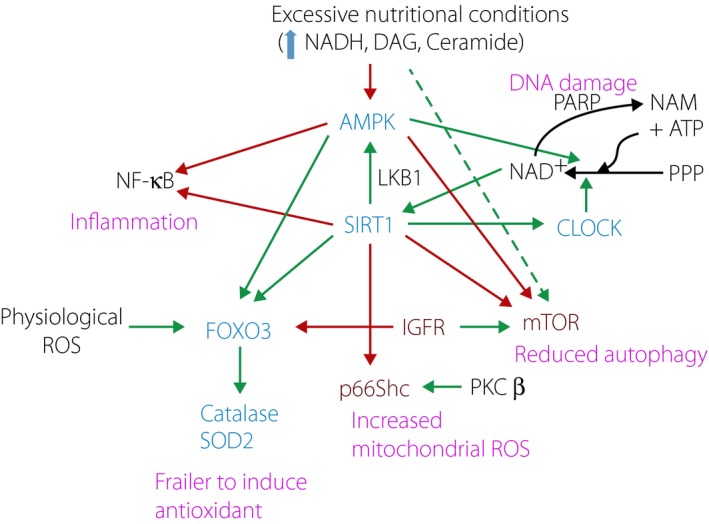
Relationships between longevity genes affected by the diabetic milieu. Blue: positive effectors for longevity. Red: negative effectors for longevity. Black: key regulators. Green arrow: positive effect. Red arrow: negative effect. Purple: changed in diabetes. Physiological stresses (exercise and starvation) under normal circadian rhythm activate adenosine monophosphate‐activated protein kinase (AMPK), sirtuin 1 (SIRT1), CLOCK, and forkhead box O3 (FOXO3) to increase catalase and superoxide dismutase 2, mitochondrial (SOD2) as an adaptive response. SIRT1 also maintains healthy mitochondria function and mass by increasing auto(mito)phagy through suppression of mechanistic target of rapamycin and controlling oxidant production through suppression of p66Shc. AMPK and SIRT1 suppress pro‐inflammatory signaling through nuclear factor (NF)‐κB by reducing transcriptional activation and de‐acetylation of RelA. Physiologically, SIRT1 de‐acetylates substrate proteins by consuming nicotinamide adenine dinucleotide (NAD+) to produce nicotinamide (NAM). This can be regulated by NAD+ synthesis by CLOCK and energy‐sensing AMPK activity. NAD+ is also consumed by the deoxyribonucleic acid (DNA) repair enzyme poly ADP ribose polymerase (PARP) that produces NAM, a physiological inhibitor of SIRT1. SIRT1 positively regulates AMPK through liver kinase B1 (LKB1) and CLOCK activity. The diabetic milieu increases the production of cytosolic nicotinamide adenine dinucleotide (NADH), diacylglycerol (DAG) and ceramide. These molecules suppress AMPK and possibly SIRT1. High nutritional conditions along with physical inactivity and problems in circadian rhythm induce a cascade of events that is observed in diabetes. First, suppression of FOXO3 activity leads to a failure to induce anti‐oxidant enzymes. This, in addition to activation of p66Shc by protein kinase C (PKC)‐β, leads to oxidative stress. DNA damage caused by oxidative stress activates PARP, and increases NAD+ catabolism and production of NAM that further decreases SIRT1 and AMPK activity. Decline of the activities of AMPK and SIRT1 decreases NAD+ synthesis. Oxidative stress and reduced activities of SIRT1 and AMPK favor NF‐κB activation. Reduction of autophagy or mitophagy induces endoplasmic reticulum stress and mitochondrial dysfunction. Mitochondrial biogenesis is inhibited by reduction of AMPK activity. When mitochondrial damage is strong enough, cells cannot maintain adenosine triphosphate (ATP) levels for synthesis of NAD+ from a substrate derived from the pentose phosphate pathway (PPP), further decreasing SIRT1 activity. ROS, reactive oxygen species.
Around the same time when we published the relationship between AMPK and SIRT1, another group suggested that AMPK might activate sirtuins by increasing NAD+ through activation of the key enzyme, nicotinamide phosphoribosyltransferease75, to synthesize NAD+ from nicotinamide76. The reaction would increase NAD+ and decrease nicotinamide, an endogenous SIRT1 inhibitor. We also found that there seemed to be concurrent regulation of AMPK and SIRT1 by NADH/NAD+ redox change induced by the addition of pyruvate77. Similar Sir2 regulation by redox was reported earlier78. These observations suggested an interrelationship between AMPK and SIRT179. This cascade together with our cascade would create a positive feedback loop (Figure 3). However, as the synthesis of NAD+ requires two ATP, this cascade might be operative only when cellular ATP levels are relatively normal. Indeed, the final reaction nicotinamide mononucleotide + ATP ⇔ NAD+ + pyrophosphate by nicotinamide mononucleotide adenyltransferase is reversible80, thus it might work for both nicotinamide and NAD+ synthesis. Consistent with this, it was reported that cytosolic NAD+ content is determined by ATP concentration81. Therefore, if ATP production is compromised by mitochondria dysfunction, the cascade from AMPK–NAD+–SIRT1 might not be operative. Currently, I believe that this positive feedback loop (Figure 2) is the key to maintaining homeostasis. Interestingly, a recent publication by Sinclair82 explained that a declining NAD+ induces a ‘pseudohypoxic state’, as it accumulates hypoxia‐inducible factor‐1α.
Figure 3.
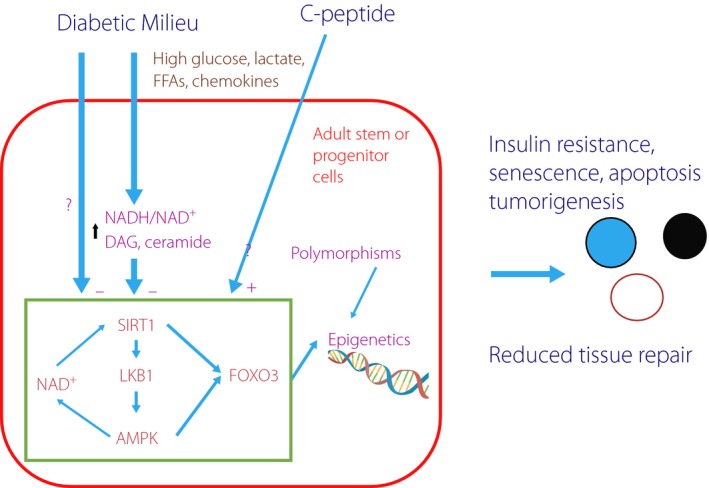
Proposed mechanisms for diabetes‐mediated tissue damage. Residential adult stem cells serve to replace existing cells to maintain tissue function. Sirtuin 1 (SIRT1), liver kinase B1 (LKB1) and adenosine monophosphate‐activated protein kinase (AMPK) form a positive feedback loop through the production of nicotinamide adenine dinucleotide (NAD+). This cascade and forkhead box O3 (FOXO3) maintain the health of these cells by increasing their ability to cope with various stresses, such as oxidative stress. The diabetic milieu, characterized by high nutritional states (glucose, FFA [FFA], lactate), causes increased nicotinamide adenine dinucleotide (NADH)/NAD+ redox, diacylglycerol (DAG) and ceramide accumulation, which negatively affect the SIRT1–AMPK cascade, resulting in epigenetic changes and gene instability. Polymorphisms also determine susceptibility for these epigenetic changes. These changes reduce tissue regeneration capability, increase vulnerability to insulin resistance, cellular senescence, apoptosis and tumorigenesis. C‐peptide counteracts redox‐mediated changes.
The SIRT1 activator, resveratrol, has been studied in human subjects and showed some beneficial effects, although there are also reports indicating that it has no effect. At this point (2015), I'm not aware of pharmaceutical companies pursuing the development of a SIRT1 activator. However, the structures of SIRT1 and of its activators became available this year83, which might raise industrial interest again.
Recent Development in Ageing Research and Implications for Diabetic Complications
Research on aging shown by heterochronic parabiosis experiments suggests that: (i) keeping a healthy population of quiescent residential (or adult) stem cells (which have both the ability to self‐renew and differentiate) are key for tissue regeneration84, 85, 86; and (ii) there are factors in blood that define age‐related tissue regeneration capacity87. As differentiated cells have a limited lifespan, stem cells, rather than differentiated cells already affected by disease, should be the targets for treatment. Nerve Schwann cells, retinal pericytes, glomerular parietal epithelial cells for podocytes and residential endothelial cells are examples of such potential targets for treating diabetic complications.
Longevity gene products (AMPK, SIRT1, etc.) act on these residential stem cells rather than on already differentiated cells, as I observed in human skin keratinocytes and mouse 3T3L1 pre‐adiopocytes while investigating the causes of cellular senescence. Contrary to its name, senescent cells are metabolically active, and produce a number of cytokines and chemokines that induce inflammatory signals to surrounding cells88. Thus, elimination of senescent cells in vivo delays age‐associated disorders89. Human skin keratinocytes are adult stem cells that renew indefinitely in vivo. Knockout of insulin receptor in keratinocytes in mice resulted in thinner skin90, and interestingly, glucose intolerance. I reported that resveratrol prevented oxidative stress‐induced cellular senescence91, and restored insulin action in cell proliferation92. In these capacities, the SIRT1–LKB1–AMPK–FOXO3 cascade played a significant role. Pre‐adipocytes are also believed to have stem cell characteristics. They supply additional adipocytes during progression of obesity, and a recent study suggested that proliferated pre‐adipocytes become senescent93. We found that Sirt1 knockdown in mouse 3T3L1 cells become senescent after differentiation, and express IL‐8Rb and transforming growth factor‐β. These phenotypes were prevented by expression of LKB1K48R, suggesting that LKB1 activity could play a significant role in senescence94. Consistent with our observations, LKB1 and FOXO were found to be the main factors that enable stem cells to cope with stress86.
Roles of FOXO3 on Epigenetics
The recent advancement of epigenetics and deep sequencing techniques is simply remarkable. The human genome project is a reference map of human genes and structures, and subsequent efforts to compile polymorphisms will reveal genetic influences on disease progression. Epigenetic differences among individuals can be the result of both genetic variability (polymorphisms) and environmental history to which the cells are exposed. In the case of diabetes, this would include high glucose and fat, oxidants, hormones, cytokines, and so on. It is known that diabetes is a condition that impairs tissue repair. Thus, it is likely that the epigenetics of these stem cells are not normal, which could cause ‘diabetic memory,’ and this might result in sustained tissue damage. If diabetic complications are the result of cell‐specific changes in epigenetics, what are they and which transcription factors are involved? This question has not been answered yet. The proteins that recognize DNA sequences are primarily transcription factors. FOXO3 is the only transcription factor so far identified as a longevity gene product. Genotypes of FOXO3 polymorphisms (rs2802292 and rs10457180)95 and possibly FOXO1 are associated with longevity. FOXO3 directly modifies epigenetic patterns around transcription start sites to enhance gene expression96. Perhaps FOXO3 is an aging master regulator. The fresh water creature, Hydra, is famous for its immortality. FOXO mediates immortality by maintaining self‐renewal capacity of its three stem cell lineage97. FOXO3 activity can be upregulated by SIRT1 and AMPK, and acutely by oxidative stress98. In keratinocytes, resveratrol enhances insulin action for proliferation only in the presence of low levels of oxidative stress92. Thus, it might work as a center for adaptive responses to stress along or together with nuclear factor (erythroid‐derived)‐like 2. FOXO3 plays a central role in controlling oxidative stress and insulin signaling95, and FOXO3 polymorphisms are associated with insulin sensitivity95. Thus, failure to respond properly to oxidative stress might be the feature of diabetes.
Summary and the Future Directions for the Study of Diabetic Complications
Diabetes research, in particular research in of its complications, enters the stage in cell biology and molecular medicine from just physiology and biochemistry. To address the ‘diabetic memory’ problem, we should focus on a treatment strategy targeting the epigenetics of progenitor or adult stem cells responsible for tissue regeneration. Ideally, this should be carried out in cultured human cells under the ‘diabetic milieu,’ and apply those results to diabetic animal models, followed by correlation studies in human samples. The ‘diabetic milieu’ is typically described as high glucose; however, serum factors of diabetes other than nutrition need to be explored further. From the heterochronic parabiosis experiments, it is clear that aging factors and possibly juvenile factors (GDF11 might not be the one) are present in serum. Although aging research helps to understand some aspect of diabetes, diabetic complications are unique in their pathology, thus different factor(s) or cascade(s) are likely involved. Once a significant genome‐wide association of polymorphisms for diabetic complications is established, and cascade‐wise polymorphisms (such as the AKR1B1 microsatellite allele [Z‐2] and C‐106T99) are more readily available, we might be able to use the cells derived from those individuals to differentiate target cell types by induced pluripotent stem cell or transdifferentiation techniques and test the effects of the diabetic milieu on epigenetics. It might also be feasible to carry out drug studies targeting individuals with these polymorphisms prone to diabetic complications (tailored‐made medicine).
LKB1 is required for adult stem cells to cope with stress conditions. The Schwan cell‐specific LKB1 knockout mouse was created recently100, and shows peripheral neuropathy preferential to sensory nerves and small fibers, resembling diabetic peripheral neuropathy. It is characterized by progressive non‐inflammatory axonopathy, and also by increased NADH/NAD+ ratio and an increased AMP/ATP ratio. Similar to the way in which C‐peptide antagonizes the effects of changes in redox through a seemingly AMPK‐independent mechanism, the effects of LKB1 on redox are AMPK independent. My current research focuses on how LKB1 activation could help diabetes‐related pathology, and explores novel ways to measure its activity in situ and on treatment with activators. With advanced molecular technologies, we now have new ways to address problems of diabetic complications.
Disclosure
The author declares no conflict of interest.
Acknowledgment
I thank Dr Karen Weikel and Dr Fan Lan for critical reading and corrections of this manuscripts. I also thank Kilo Diabetes Vascular Foundation for long‐term support for my previous research.
J Diabetes Investig 2016; 7: 448–458
References
- 1. Calcutt NA, Cooper ME, Kern TS, et al Therapies for hyperglycaemia‐induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov 2009; 8: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yagihashi S, Mizukami H, Sugimoto K. Mechanism of diabetic neuropathy: where are we now and where to go? J Diabetes Investig 2011; 2: 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maezawa Y, Takemoto M, Yokote K. Cell biology of diabetic nephropathy: roles of endothelial cells, tubulointerstitial cells and podocytes. J Diabetes Investig 2015; 6: 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varma SD, Kinoshita JH. Sorbitol pathway in diabetic and galactosemic rat lens. Biochim Biophys Acta 1974; 338: 632–640. [Google Scholar]
- 5. Hotta N. New concepts and insights on pathogenesis and treatment of diabetic complications: polyol pathway and its inhibition. Nagoya J Med Sci 1997; 60: 89–100. [PubMed] [Google Scholar]
- 6. Kikkawa R, Hatanaka I, Yasuda H, et al Effect of a new aldose reductase inhibitor, (E)‐3‐carboxymethyl‐5‐[(2E)‐methyl‐3‐phenylpropenylidene]rhodanine (ONO‐2235) on peripheral nerve disorders in streptozotocin‐diabetic rats. Diabetologia 1983; 24: 290–292. [DOI] [PubMed] [Google Scholar]
- 7. Hotta N, Akanuma Y, Kawamori R, et al Long‐term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3‐year, multicenter, comparative Aldose Reductase Inhibitor‐Diabetes Complications Trial. Diabetes Care 2006; 29: 1538–1544. [DOI] [PubMed] [Google Scholar]
- 8. Yagihashi S, Kamijo M, Ido Y, et al Effects of long‐term aldose reductase inhibition on development of experimental diabetic neuropathy. Ultrastructural and morphometric studies of sural nerve in streptozocin‐induced diabetic rats. Diabetes 1990; 39: 690–696. [DOI] [PubMed] [Google Scholar]
- 9. Oates PJ, Mylari BL. Aldose reductase inhibitors: therapeutic implications for diabetic complications. Expert Opin Investig Drugs 1999; 8: 2095–2119. [DOI] [PubMed] [Google Scholar]
- 10. Kilzer P, Chang K, Marvel J, et al Albumin permeation of new vessels is increased in diabetic rats. Diabetes 1985; 34: 333–336. [DOI] [PubMed] [Google Scholar]
- 11. Tilton RG, Chang K, Pugliese G, et al Prevention of hemodynamic and vascular albumin filtration changes in diabetic rats by aldose reductase inhibitors. Diabetes 1989; 38: 1258–1270. [DOI] [PubMed] [Google Scholar]
- 12. Ido Y, Tilton RG, Chang K, et al Rapid measurement of glomerular filtration rate in small animals. Kidney Int 1992; 41: 435–439. [DOI] [PubMed] [Google Scholar]
- 13. Williamson JR, Ostrow E, Eades D, et al Glucose‐induced microvascular functional changes in nondiabetic rats are stereospecific and are prevented by an aldose reductase inhibitor. J Clin Invest 1990; 85: 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolf BA, Williamson JR, Easom RA, et al Diacylglycerol accumulation and microvascular abnormalities induced by elevated glucose levels. J Clin Invest 1991; 87: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ido Y, Chang KC, Lejeune WS, et al Vascular dysfunction induced by AGE is mediated by VEGF via mechanisms involving reactive oxygen species, guanylate cyclase, and protein kinase C. Microcirculation 2001; 8: 251–263. [DOI] [PubMed] [Google Scholar]
- 16. Corbett JA, Tilton RG, Chang K, et al Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes 1992; 41: 552–556. [DOI] [PubMed] [Google Scholar]
- 17. Ido Y, McHowat J, Chang KC, et al Neural dysfunction and metabolic imbalances in diabetic rats. Prevention by acetyl‐L‐carnitine. Diabetes 1994; 43: 1469–1477. [DOI] [PubMed] [Google Scholar]
- 18. Ido Y, Vindigni A, Chang K, et al Prevention of vascular and neural dysfunction in diabetic rats by C‐peptide. Science 1997; 277: 563–566. [DOI] [PubMed] [Google Scholar]
- 19. Williamson JR, Chang K, Frangos M, et al Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993; 42: 801–813. [DOI] [PubMed] [Google Scholar]
- 20. Krebs HA. The redox state of nicotinamide adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Adv Enzyme Regul 1967; 5: 409–434. [DOI] [PubMed] [Google Scholar]
- 21. Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide‐adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 1967; 103: 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ido Y, Kilo C, Williamson JR. Interactions between the sorbitol pathway, non‐enzymatic glycation, and diabetic vascular dysfunction. Nephrol Dial Transplant 1996; 11(Suppl 5): 72–75. [DOI] [PubMed] [Google Scholar]
- 23. Ido Y, Chang K, Woolsey TA, et al NADH: sensor of blood flow need in brain, muscle, and other tissues. FASEB J 2001; 15: 1419–1421. [DOI] [PubMed] [Google Scholar]
- 24. Ido Y, Chang K, Williamson JR. NADH augments blood flow in physiologically activated retina and visual cortex. Proc Natl Acad Sci USA 2004; 101: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mintun MA, Vlassenko AG, Rundle MM, et al Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proc Natl Acad Sci USA 2004; 101: 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ido Y, Kilo C, Williamson JR. Cytosolic NADH/NAD+, free radicals, and vascular dysfunction in early diabetes mellitus. Diabetologia 1997; 40(Suppl 2): S115–S117. [DOI] [PubMed] [Google Scholar]
- 27. Williamson JR, Kilo C, Ido Y. The role of cytosolic reductive stress in oxidant formation and diabetic complications. Diabetes Res Clin Pract 1999; 45: 81–82. [DOI] [PubMed] [Google Scholar]
- 28. Nyengaard JR, Ido Y, Kilo C, et al Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes 2004; 53: 2931–2938. [DOI] [PubMed] [Google Scholar]
- 29. Ido Y. Pyridine nucleotide redox abnormalities in diabetes. Antioxid Redox Signal 2007; 9: 931–942. [DOI] [PubMed] [Google Scholar]
- 30. Di LJ, Byun JS, Wong MM, et al Genome‐wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun 2013; 4: 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gallagher EJ, LeRoith D. Obesity and diabetes: the increased risk of cancer and cancer‐related mortality. Physiol Rev 2015; 95: 727–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Visscher G, Haseldonckx M, Flameng W. Fluorescent microsphere technique to measure cerebral blood flow in the rat. Nat Protoc 2006; 1: 2162–2170. [DOI] [PubMed] [Google Scholar]
- 33. Bursell SE, Clermont AC, Shiba T, et al Evaluating retinal circulation using video fluorescein angiography in control and diabetic rats. Curr Eye Res 1992; 11: 287–295. [DOI] [PubMed] [Google Scholar]
- 34. Ikram MK, Cheung CY, Lorenzi M, et al Retinal vascular caliber as a biomarker for diabetes microvascular complications. Diabetes Care 2013; 36: 750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ido Y, Chang K, LeJeune W, et al Diabetes impairs sciatic nerve hyperemia induced by surgical trauma: implications for diabetic neuropathy. Am J Physiol 1997; 273: E174–E184. [DOI] [PubMed] [Google Scholar]
- 36. Ostergaard L, Finnerup NB, Terkelsen AJ, et al The effects of capillary dysfunction on oxygen and glucose extraction in diabetic neuropathy. Diabetologia 2015; 58: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toyota T, Abe K, Kudo M, et al Inhibitory effects of synthetic rat C‐peptide 1 on insulin secretion in the isolated perfused rat pancreas. Tohoku J Exp Med 1975; 117: 79–83. [DOI] [PubMed] [Google Scholar]
- 38. Dryburgh JR, Hampton SM, Marks V. Endocrine pancreatic control of the release of gastric inhibitory polypeptide. A possible physiological role for C‐peptide. Diabetologia 1980; 19: 397–401. [DOI] [PubMed] [Google Scholar]
- 39. Wojcikowski C, Maier V, Dominiak K, et al Effects of synthetic rat C‐peptide in normal and diabetic rats. Diabetologia 1983; 25: 288–290. [DOI] [PubMed] [Google Scholar]
- 40. Wahren J, Johansson BL, Wallberg‐Henriksson H, et al C‐peptide revisited–new physiological effects and therapeutic implications. J Intern Med 1996; 240: 115–124. [DOI] [PubMed] [Google Scholar]
- 41. Zierath JR, Handberg A, Tally M, et al C‐peptide stimulates glucose transport in isolated human skeletal muscle independent of insulin receptor and tyrosine kinase activation. Diabetologia 1996; 39: 306–313. [DOI] [PubMed] [Google Scholar]
- 42. Johansson BL, Sjoberg S, Wahren J. The influence of human C‐peptide on renal function and glucose utilization in type 1 (insulin‐dependent) diabetic patients. Diabetologia 1992; 35: 121–128. [DOI] [PubMed] [Google Scholar]
- 43. Zhang W, Kamiya H, Ekberg K, et al C‐peptide improves neuropathy in type 1 diabetic BB/Wor‐rats. Diabetes Metab Res Rev 2007; 23: 63–70. [DOI] [PubMed] [Google Scholar]
- 44. Jolivalt CG, Rodriguez M, Wahren J, et al Efficacy of a long‐acting C‐peptide analogue against peripheral neuropathy in streptozotocin‐diabetic mice. Diabetes Obes Metab 2015; 17: 781–788. [DOI] [PubMed] [Google Scholar]
- 45. Wahren J, Larsson C. C‐peptide: new findings and therapeutic possibilities. Diabetes Res Clin Pract 2015; 107: 309–319. [DOI] [PubMed] [Google Scholar]
- 46. Melles E, Bergman T, Stahlberg M, et al Large‐surface biosensor technology for enhanced recovery in protein characterization. J Biomol Tech 2005; 16: 392–397. [PMC free article] [PubMed] [Google Scholar]
- 47. Wahren J, Kallas A, Sima AA. The clinical potential of C‐peptide replacement in type 1 diabetes. Diabetes 2012; 61: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhatt MP, Lim YC, Kim YM, et al C‐peptide activates AMPKalpha and prevents ROS‐mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes 2013; 62: 3851–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yosten GL, Kolar GR, Redlinger LJ, et al Evidence for an interaction between proinsulin C‐peptide and GPR146. J Endocrinol 2013; 218: B1–B8. [DOI] [PubMed] [Google Scholar]
- 50. Krasner NM, Ido Y, Ruderman NB, et al Glucagon‐like peptide‐1 (GLP‐1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE 2014; 9: e97554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Forst T, De La Tour DD, Kunt T, et al Effects of proinsulin C‐peptide on nitric oxide, microvascular blood flow and erythrocyte Na+, K+‐ATPase activity in diabetes mellitus type I. Clin Sci (Lond) 2000; 98: 283–290. [PubMed] [Google Scholar]
- 52. Benziane B, Bjornholm M, Lantier L, et al AMP‐activated protein kinase activator A‐769662 is an inhibitor of the Na(+)‐K(+)‐ATPase. Am J Physiol Cell Physiol 2009; 297: C1554–C1566. [DOI] [PubMed] [Google Scholar]
- 53. Seo‐Mayer PW, Thulin G, Zhang L, et al Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am J Physiol Renal Physiol 2011; 301: F1346–F1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sato Y, Oshida Y, Han YQ, et al C‐peptide fragments stimulate glucose utilization in diabetic rats. Cell Mol Life Sci 2004; 61: 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vettor R, Lombardi AM, Fabris R, et al Lactate infusion in anesthetized rats produces insulin resistance in heart and skeletal muscles. Metabolism 1997; 46: 684–690. [DOI] [PubMed] [Google Scholar]
- 56. Tomas E, Lin YS, Dagher Z, et al Hyperglycemia and insulin resistance: possible mechanisms. Ann N Y Acad Sci 2002; 967: 43–51. [DOI] [PubMed] [Google Scholar]
- 57. Dagher Z, Ruderman N, Tornheim K, et al The effect of AMP‐activated protein kinase and its activator AICAR on the metabolism of human umbilical vein endothelial cells. Biochem Biophys Res Commun 1999; 265: 112–115. [DOI] [PubMed] [Google Scholar]
- 58. Dagher Z, Ruderman N, Tornheim K, et al Acute regulation of fatty acid oxidation and amp‐activated protein kinase in human umbilical vein endothelial cells. Circ Res 2001; 88: 1276–1282. [DOI] [PubMed] [Google Scholar]
- 59. Ido Y, Carling D, Ruderman N. Hyperglycemia‐induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP‐activated protein kinase activation. Diabetes 2002; 51: 159–167. [DOI] [PubMed] [Google Scholar]
- 60. Romeo G, Liu WH, Asnaghi V, et al Activation of nuclear factor‐kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 2002; 51: 2241–2248. [DOI] [PubMed] [Google Scholar]
- 61. Cacicedo JM, Yagihashi N, Keaney JF Jr, et al AMPK inhibits fatty acid‐induced increases in NF‐kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 2004; 324: 1204–1209. [DOI] [PubMed] [Google Scholar]
- 62. Cacicedo JM, Benjachareowong S, Chou E, et al Palmitate‐induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 2005; 54: 1838–1845. [DOI] [PubMed] [Google Scholar]
- 63. Cacicedo JM, Benjachareonwong S, Chou E, et al Activation of AMP‐activated protein kinase prevents lipotoxicity in retinal pericytes. Invest Ophthalmol Vis Sci 2011; 52: 3630–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nishikawa T, Edelstein D, Du XL, et al Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 65. Ristow M, Zarse K, Oberbach A, et al Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 2009; 106: 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tanaka Y, Kume S, Kitada M, et al Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res 2012; 2012: 628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kishikawa H, Araki E, Nishikawa T, et al Risk factors of diabetic retinopathy and their treatment. Nihon Rinsho 2010; 68(Suppl 9): 298–303. [PubMed] [Google Scholar]
- 68. Kammouni W, Hasan L, Saleh A, et al Role of nuclear factor‐kappaB in oxidative stress associated with rabies virus infection of adult rat dorsal root ganglion neurons. J Virol 2012; 86: 8139–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sinclair DA, Guarente L. Unlocking the secrets of longevity genes. Sci Am 2006; 294: 48–51, 54–57. [DOI] [PubMed] [Google Scholar]
- 70. Lan F, Cacicedo J, Ido Y. Activation of AMPKK‐AMP cascade by Silent Information Regulator 2 (Sir2). Diabetes 2005; 54(Suppl 1): A383. [Google Scholar]
- 71. Hou X, Xu S, Maitland‐Toolan KA, et al SIRT1 regulates hepatocyte lipid metabolism through activating AMP‐activated protein kinase. J Biol Chem 2008; 283: 20015–20026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lan F, Cacicedo JM, Ruderman N, et al SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP‐activated protein kinase activation. J Biol Chem 2008; 283: 27628–27635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Price NL, Gomes AP, Ling AJ, et al SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 2012; 15: 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cacicedo JM, Gauthier MS, Lebrasseur NK, et al Acute exercise activates AMPK and eNOS in the mouse aorta. Am J Physiol Heart Circ Physiol 2011; 301: H1255–H1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004; 279: 50754–50763. [DOI] [PubMed] [Google Scholar]
- 76. Fulco M, Cen Y, Zhao P, et al Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK‐mediated regulation of Nampt. Dev Cell 2008; 14: 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Suchankova G, Nelson LE, Gerhart‐Hines Z, et al Concurrent regulation of AMP‐activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun 2009; 378: 836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fulco M, Schiltz RL, Iezzi S, et al Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell 2003; 12: 51–62. [DOI] [PubMed] [Google Scholar]
- 79. Ruderman NB, Xu XJ, Nelson L, et al AMPK and SIRT1: a long‐standing partnership? Am J Physiol Endocrinol Metab 2010; 298: E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Berger F, Lau C, Dahlmann M, et al Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 2005; 280: 36334–36341. [DOI] [PubMed] [Google Scholar]
- 81. Devin A, Guerin B, Rigoulet M. Cytosolic NAD+ content strictly depends on ATP concentration in isolated liver cells. FEBS Lett 1997; 410: 329–332. [DOI] [PubMed] [Google Scholar]
- 82. Gomes AP, Price NL, Ling AJ, et al Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 2013; 155: 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dai H, Case AW, Riera TV, et al Crystallographic structure of a small molecule SIRT1 activator‐enzyme complex. Nat Commun 2015; 6: 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Villeda SA, Luo J, Mosher KI, et al The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011; 477: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat Cell Biol 2011; 13: 506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 2013; 14: 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Scudellari M. Ageing research: blood to blood. Nature 2015; 517: 426–429. [DOI] [PubMed] [Google Scholar]
- 88. Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal 2009; 11: 59–98. [DOI] [PubMed] [Google Scholar]
- 89. Baker DJ, Wijshake T, Tchkonia T, et al Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 2011; 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wertheimer E, Trebicz M, Eldar T, et al Differential roles of insulin receptor and insulin‐like growth factor‐1 receptor in differentiation of murine skin keratinocytes. J Invest Dermatol 2000; 115: 24–29. [DOI] [PubMed] [Google Scholar]
- 91. Ido Y, Duranton A, Lan F, et al Acute activation of AMP‐activated protein kinase prevents H2O2‐induced premature senescence in primary human keratinocytes. PLoS ONE 2012; 7: e35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ido Y, Duranton A, Lan F, et al Resveratrol prevents oxidative stress‐induced senescence and proliferative dysfunction by activating the AMPK‐FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015; 10: e0115341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tchkonia T, Morbeck DE, Von Zglinicki T, et al Fat tissue, aging, and cellular senescence. Aging Cell 2010; 9: 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lan F, Cacicedo J, Ruderman N, et al Sirt1 Knockdown promotes senescence‐like phenotype in 3T3L1 adipocyte, that is prevented by SIRT1‐independent active LKB1 expression. Diabetes 2010; 59(Suppl 1): A397. [Google Scholar]
- 95. Morris BJ, Willcox DC, Donlon TA, et al FOXO3: a major gene for human longevity – a mini‐review. Gerontology 2015; 61: 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Eijkelenboom A, Mokry M, Smits LM, et al FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep 2013; 5: 1664–1678. [DOI] [PubMed] [Google Scholar]
- 97. Boehm AM, Khalturin K, Anton‐Erxleben F, et al FoxO is a critical regulator of stem cell maintenance in immortal Hydra. Proc Natl Acad Sci USA 2012; 109: 19697–19702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Brunet A, Sweeney LB, Sturgill JF, et al Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303: 2011–2015. [DOI] [PubMed] [Google Scholar]
- 99. So WY, Wang Y, Ng MC, et al Aldose reductase genotypes and cardiorenal complications: an 8‐year prospective analysis of 1,074 type 2 diabetic patients. Diabetes Care 2008; 31: 2148–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Beirowski B, Babetto E, Golden JP, et al Metabolic regulator LKB1 is crucial for Schwann cell‐mediated axon maintenance. Nat Neurosci 2014; 17: 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]