

Abstract
Aims/Introduction
The involvement of glucose‐dependent insulinotropic polypeptide (GIP) on inflammation was explored in atherosclerosis and adipose tissue. Periodontal disease is a chronic inflammatory disease, and is considered one of the diabetic complications. In the present study, to examine the effect of GIP on periodontitis, we induced experimental periodontitis in glucose‐dependent insulinotropic polypeptide receptor‐knockout mice (GIPRKO). We also investigated the anti‐inflammatory effect of GIP in a culture system.
Materials and Methods
Experimental periodontitis was induced by ligature wire in GIPRKO and C57BL/C mice. Two weeks after the ligature, immunohistological evaluation and inflammatory messenger ribonucleic acid expression in the gingiva was examined. To elucidate the role of GIP in inflammation, the effects of GIP on lipopolysaccharide‐induced gene expressions in THP‐1 cells were evaluated.
Results
Periodontitis increased inflammatory cell infiltration, macrophage accumulation and tumor necrosis factor‐α and nitric oxide synthase gene expressions in the gingiva. Periodontitis in GIPRKO showed a marked increase of inflammatory cells in the gingivomucosal tissue. Mac‐1‐positive macrophages and the inflammatory gene expressions were significantly increased in periodontitis in GIPRKO compared with C57BL/C mice periodontitis. Immunohistochemical staining confirmed that GIP receptors were expressed in residual and infiltrated Mac‐1‐positive macrophages. The in vitro study showed that GIP suppressed lipopolysaccharide‐induced tumor necrosis factor‐α and nitric oxide synthase gene expression in a dose‐dependent manner. Furthermore, the inhibitory effect of GIP on lipopolysaccharide‐induced inflammatory gene expressions was at least partially through cyclic adenosine monophosphate/protein kinase A pathway.
Conclusions
These results suggest the beneficial effects of GIP on periodontal disease. In diabetic patients, GIP is expected to have a direct anti‐inflammatory effect on periodontitis in addition to its glucose‐lowering effect.
Keywords: Glucose‐dependent insulinotropic polypeptide, Inflammation, Periodontal disease
Introduction
Periodontal disease is a chronic inflammatory disease, and the periodontium is destroyed by existing bacterial infection in the periodontal pocket. The periodontopathic bacteria, which is Gram‐negative anaerobic bacterium that possesses lipopolysaccharide (LPS), aggravates the inflammatory reaction in the periodontal tissue and finally results in resorption of the alveolar bone.
Because of its high morbidity and severity in diabetic patients, periodontal disease is considered to be one of the diabetic complications1. Hyperinflammatory response in diabetes is one of the candidates in the aggravation of periodontitis2. Furthermore, emerging epidemiological studies showed the bidirectional relationship between diabetes and periodontal disease3. In both type 1 and type 2 diabetic patients, periodontal disease has a higher incidence and severity compared with healthy subjects4, 5, and poor glycemic control aggravates periodontal disease6. In contrast, the existence of severe periodontitis increases the development of diabetes7, 8, 9. A meta‐analysis suggested the possibility that the treatment of periodontitis might improve blood glucose control10, 11. It was also reported that Pima Indians with severe periodontitis had a higher risk of death of diabetic nephropathy and ischemic heart disease12. The chronic inflammation of periodontal disease might be involved in the development of diabetes and diabetic complications.
The incretins, glucagon‐like polypeptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP; also called gastric inhibitory polypeptide), are secreted during intake of a meal by the gastrointestinal tract, and stimulate pancreas β‐cells to secrete insulin secretion. There are many investigations about the extrapancreatic effects of incretins13. As the receptor expression patterns are different between GLP‐1 and GIP, it is believed that GLP‐1 and GIP have distinct effects in organs14.
Glucose‐dependent insulinotropic polypeptide is secreted from K‐cells of the small intestine, and exerts its effects through a specific receptor, the GIP receptor (GIPR)14, 15. In pancreatic β‐cells, GIP binds to GIPR and increases intracellular cyclic adenosine monophosphate (cAMP), which leads to insulin secretion. In addition to pancreatic cells, GIPR is expressed in many other organs, such as the nervous system, eyes, adipose tissue and bone, as well as immune cells13, 16. Glucose‐dependent insulinotropic polypeptide increases fat deposition in adipose tissue and promotes bone formation in bone17, 18. The effects of GIP on inflammation are controversial. The administration of long‐acting GIP analog reduced adipose tissue inflammation and circulating inflammatory cells19. Glucose‐dependent insulinotropic polypeptide also suppresses atherosclerosis by inhibiting macrophage infiltration, suggesting an anti‐inflammatory effect of GIP16, 20. In contrast, GIP increased inflammatory cytokine expressions in GIPR‐overexpressing adipocytes21.
The aim of the present study was to investigate whether GIP ameliorates periodontitis using GIPR knockout mice (GIPRKO), and to elucidate the mechanisms. We thus induced experimental periodontitis in GIPRKO and wild‐type mice (WT). Furthermore, to explore the role of GIP in inflammatory monocytes/macrophages, we investigated the effects of GIP on the lipopolysaccharide (LPS)‐stimulated inflammatory response in a human monocyte/macrophage cell line, THP‐1 cells.
Materials and Methods
Animals
The generation of GIPRKO was previously described22. Male C57BL/C mice (WT) were obtained from Chubu Kagakushizai (Nagoya, Japan). All mice were housed in individual cages under controlled temperature (24 ± 1.0°C), on a 12‐h light/dark cycle and given standard laboratory mouse chow with water ad libitum. The Institutional Animal Care and Use Committees of Aichi Gakuin University approved all experimental protocols (AGUD 190).
Induction of ligature‐induced experimental periodontitis
In half of the male WT and GIPRKO aged 8‐weeks‐old, a ligature wire (0.10 mm®; Nilaco, Tokyo, Japan) was placed on the second molar tooth (M2) between the left maxillary section to clog the plaque, which subsequently induced experimental periodontitis (Figure 1a)23. Mice without any ligation were used as the control mice. Two weeks after the ligature, the following assessments were carried out.
Figure 1.
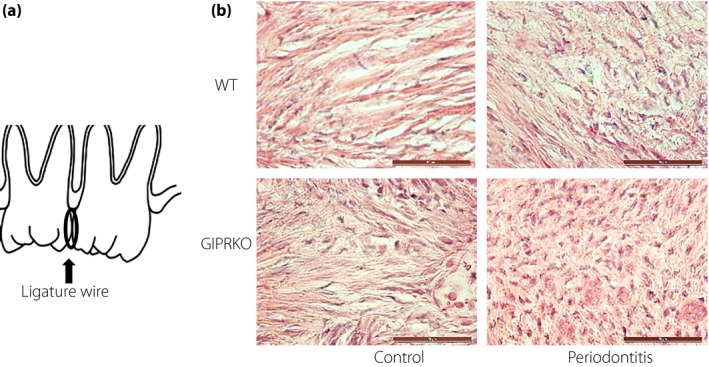
Histological evaluation of gingiva. (a) Periodontitis was induced by a ligature wire placed on the second molar tooth (M2) between the left section maxillary to clog the plaque, which subsequently induced experimental periodontitis. (b) Frozen sections of the periodontal tissues between the teeth were subjected to evaluation of the inflammatory cell infiltration by hematoxylin–eosin staining. Scale bar, 50 μm. GIPRKO, glucose‐dependent insulinotropic polypeptide receptor‐knockout mice; WT, wild‐type mice.
Measurements of plasma insulin and GIP levels
After 12‐h fasting, blood samples were taken from the vein. For the GIP measurements, a dipeptidyl peptidase‐4 (DPP‐4) inhibitor (Millipore, Billerica, MA, USA) was immediately added to the blood samples. Plasma insulin levels were measured by using an enzyme‐linked immunosorbent assay kit (Morinaga, Yokohama, Japan), and total GIP levels were measured by using an enzyme‐linked immunosorbent assay kit (Yanaihara Institute Inc., Shizuoka, Japan) according to the manufacturer's instructions.
Tissue collection
Two weeks after the ligation, WT and GIPRKO were killed with an overdose of pentobarbital (150 mg/kg). The maxillary were excised and separated from the surrounding tissues. For messenger ribonucleic acid (mRNA) analyses, a gingival tissue strip from between the teeth of the M2 was excised and immersed in RNAlater RNA stabilization reagent (Qiagen, Hilden, Germany) and kept at −80°C until use. For immunohistological study, maxillary bones with the attached gingival tissue were fixed in 4% paraformaldehyde.
Quantitative polymerase chain reaction for mRNA expressions
Total RNA was extracted from samples of gingiva or THP‐1 cells using RNeasy (Qiagen) and complementary deoxyribonucleic acid was synthesized from 500 ng of RNA using ReverTra Ace (Toyobo, Osaka, Japan) according to the manufacturer's instructions. Primers for mouse tumor necrosis factor‐α (TNF‐α; Mm00443258_m1), mouse inducible nitric oxide synthase (iNOS; also known as Nos2; Mm00440502_m1), mouse interleukin‐1α (IL‐1α; Mm99999060_m1), mouse interleukin‐1β (IL‐1β; Mm01336189_m1), mouse β‐actin (4352341E‐1112017) and human β‐actin (4326315E‐19112022) were purchased from TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA). The primer sequences (Nippon Gene Material Co. Ltd., Toyama, Japan) were as follows: human TNF‐α (sense primer: 5′‐CACCTAGAAATTGACACAAG‐3′, antisense primer: 5′‐AGTGCAAACATAAATAGAGG‐3′, probe: 5′‐ACC1TAG GCCTTCCTCTCTCCA‐3′), human iNOS (sense primer: 5′‐GGATGACCTTCAGTATCAC‐3′, antisense primer: 5′‐CAGAGATTCTGGAGACTTC‐3′, probe: 5′‐TCAGCAAGCAGCAG AATGAGTCC‐3′). The following protocol was used: 1 min at 95°C, 1 min at 52°C and 30 s at 72°C, and it was repeated for a total of 40 cycles. Relative quantity was calculated by the ΔΔCT method using β‐actin as the endogenous control24. All reactions were run on an ABI7000 platform (Applied Biosystems).
Histological analysis
Maxillary bones with gingiva were fixed in 4% paraformaldehyde solution for 24 h, washed in tap water and immersed in a specific immersing solution containing liquid nitrogen and isopentane. After this procedure, frozen non‐deossified sections were made according to the Kawamoto method25. For hematoxylin–eosin staining and immunohistological staining with primary antibody, the second molars were cut serially into 5‐μm thick sections, with the sections mounted using adhesive film (Cryofilm type I; Leica Microsystems, Osaka, Japan) and mounting medium (SCMM‐R2; Leica Microsystems)26.
Immunohistochemical staining
Immunohistochemical analysis was carried out using anti‐Mac‐1 polyclonal antibody (1:1,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and anti‐GIPR antibody (1:1,000; Abcam Plc, Cambridge, UK). This was followed by incubation with the secondary antibodies labeled with Alexa Fluor 488, 594 or Texas Red (Molecular Probes Inc., Eugene, OR, USA) and with 4′,6‐diamidino‐2‐phenylindole (Sigma‐Aldrich, St. Louis, MO, USA) for 1 h at room temperature. Slides were investigated with a FU‐200 confocal system (Olympus, Tokyo, Japan) and Leica AF6000LX (Leica Microsystems, Wetzlar, Germany) inverted microscope. The numbers of Mac‐1‐immunopositive cells were counted, and the cell density was calculated as the cell count divided by the total area.
Cell culture
Human monocyte/macrophage cell line, THP‐1 cells, were purchased from American Type Culture Collection (Manassas, VA, USA). THP‐1 cells were cultured in RPMI 1640 (Invitrogen Life Technologies, Paisley, UK) supplemented with 10% fetal bovine serum (Life Technologies Corp., Carlsbad, CA, USA), 50 IU/mL penicillin and 50 IU/mL streptomycin (Invitrogen Life Technologies) under standard conditions (humidified atmosphere of 5% CO2 at 37°C).
Effect of GIP on LPS‐induced TNF‐α and iNOS mRNA expressions in THP‐1 cells
Cells were seeded in six‐well plates. Preconfluent cells were starved without serum for 24 h. Glucose‐dependent insulinotropic polypeptide (GIP1–42; PEPTIDE, Osaka, Japan) was added at the concentration of 10−7–10−9 mol/L 30 min before LPS (Sigma‐Aldrich; 100 ng/mL) administration. Four hours after LPS stimulation, cells were washed with phosphate buffer saline and total RNA was extracted using RNeasy (Qiagen).
To investigate the GIP signaling pathway, cells were incubated with 5 μmol/L of cis‐N‐(2‐phenylcyclopentyl)‐azacyclotridec‐1‐en‐2‐amine (MDL‐12330A; Sigma‐Aldrich), a specific adenylate cyclase inhibitor, 10 μmol/L of protein kinase inhibitor 14–22 (PKI[14–22]) amide or H‐89 (Sigma‐Aldrich), a cAMP‐dependent protein kinase A (PKA) inhibitor or 10 μmol/L of 8‐pCPT‐2′‐O‐Me‐cAMP (Sigma‐Aldrich), a selective activator against Epac, 30 min before adding GIP27.
Statistical analyses
Data are expressed as means ± standard error of the mean. Datasets were assessed by analysis of variance (one‐way anova) followed by the Bonferroni correction for multiple comparisons. Statistical tests were carried out with SPSS (IBM SPSS Statistics for Windows; Armonk, NY, USA). The differences were considered to be significant when P < 0.05.
Results
Characteristics and hormonal levels in WT and GIPRKO mice with or without periodontitis
Bodyweights, fasting blood glucose, and the levels of insulin and GIP were measured 2 weeks after the induction of periodontitis. As shown in Table 1, all parameters, except white blood cell count, were not significantly changed between WT and GIPRKO with or without periodontitis (Table 1), indicating that the existence of periodontitis did not affect fasting blood glucose, insulin levels or GIP levels in either WT or GIPRKO. In contrast, periodontitis significantly increased the number of white blood cells in GIPRKO. When compared with WT periodontitis and GIPRKO periodontitis, there were no significant differences in bodyweights, blood glucose, insulin/GIP levels and the number of white blood cells.
Table 1.
Bodyweights, blood glucose, hormone levels and white blood cells in wild‐type and glucose‐dependent insulinotropic polypeptide receptor‐knockout mice
| Variable | WT control | WT periodontitis | GIPRKO control | GIPRKO periodontitis |
|---|---|---|---|---|
| Bodyweight (g) | 21.1 ± 1.9 | 20.7 ± 2.8 | 21.7 ± 2.5 | 23.1 ± 2.0 |
| Blood glucose (mmol/L) | 3.6 ± 0.3 | 3.3 ± 0.1 | 3.2 ± 0.3 | 3.4 ± 0.3 |
| WBC (/μL) | 5,100 ± 750 | 6,180 ± 520 | 4,700 ± 520 | 6,770 ± 470* |
| GIP (pmol/L) | 38.1 ± 1.5 | 40.1 ± 1.9 | 39.5 ± 1.9 | 40.3 ± 2.0 |
| Insulin (pmol/L) | 49.7 ± 11.8 | 47.3 ± 8.6 | 42.5 ± 5.2 | 46.1 ± 7.6 |
Values are given as mean ± standard error of the mean (n = 9). *P < 0.05. GIP, glucose‐dependent insulinotropic polypeptide; GIPRKO, glucose‐dependent insulinotropic polypeptide receptor‐knockout mice; WBC, white blood cells; WT, wild‐type mice.
Inflammatory cell infiltration was increased by periodontitis in GIPRKO
Histological evaluation showed that the inflammatory cell infiltration increased on the periodontitis side in WT and GIPRKO (Figure 1b). In particular, periodontitis in GIPRKO showed a marked increase of inflammatory cells in the gingival connective tissue.
Macrophages in the gingival connective tissue were evaluated by staining with Mac‐1 antibody, a macrophage marker. As shown in Figure 2a,b, there were only a few Mac‐1‐positive macrophages in the periodontal tissue of the WT controls (25.0 ± 7.3/mm2). Macrophages were significantly increased by 6.4‐fold in the vicinity of the wire in WT periodontitis (159.4 ± 21.7/mm2, P < 0.05). In contrast, in the GIPRKO, macrophages already existed on the control side of the periodontal tissue, which increased by 4.5‐fold compared with the WT control (113.2 ± 13.9/mm2, P < 0.01). The induction of periodontitis in GIPRKO showed a marked increase of infiltrated macrophages in the gingivomucosal tissue near the wire (291.2 ± 29.7/mm2, P < 0.01 vs GIPRKO control). Macrophages in GIPRKO periodontitis showed a 1.8‐fold increase compared with those in WT periodontitis (P < 0.01).
Figure 2.
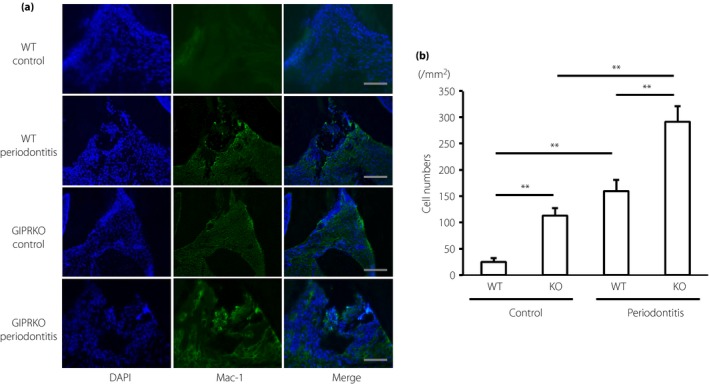
Macrophage accumulation in gingiva. (a) Macrophages were visualized by immunohistochemical staining with anti‐Mac‐1 polyclonal antibody. Scale bar, 100 μm. (b) Quantification of Mac‐1‐positive macrophages in gingiva. Results are expressed as mean ± standard error of the mean (n = 6). **P < 0.01. DAPI, 4′,6‐diamidino‐2‐phenylindole; GIPRKO, glucose‐dependent insulinotropic polypeptide receptor‐knockout mice; KO, knockout mice; WT, wild‐type mice.
Inflammatory mediators mRNA expressions in gingiva
Periodontitis significantly increased the inflammatory gene expressions of TNF‐α and iNOS in the gingiva of both WT and GIPRKO (Figure 3). In the comparison between WT periodontitis and GIPRKO periodontitis, gene expressions of TNF‐α and iNOS were significantly increased in GIPRKO by 1.6‐fold and 1.6‐fold, respectively (P < 0.01). IL‐1α and IL‐1β gene expressions tended to be increased by the induction of periodontitis, and periodontitis significantly increased IL‐1β gene expression in GIPRKO. However, IL‐1α and IL‐1β gene expressions did not differ between WT periodontitis and GIPRKO periodontitis.
Figure 3.
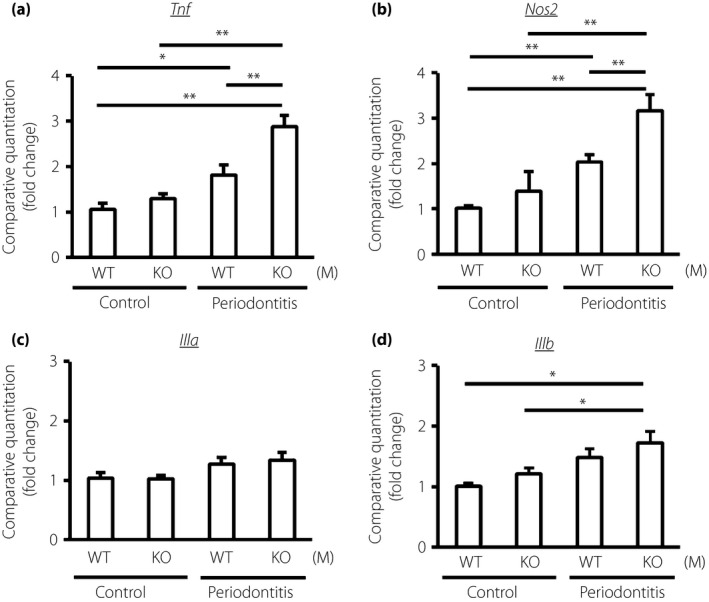
Messenger ribonucleic acid expressions of inflammatory cytokines, (a) tumor necrosis factor‐α (TNF‐α; Tnf), (b) inducible nitric oxide synthase (iNOS; Nos2), (c) interleukin‐1α (IL‐1α; Il1a) and (d) IL‐1β (Il1b), in the control and the periodontitis side of gingiva in the wild‐type mice (WT) and glucose‐dependent insulinotropic polypeptide receptor‐knockout mice (GIPRKO). Messenger ribonucleic acid expressions were determined by quantitative reverse transcription polymerase chain reaction. Results are expressed as mean ± standard error of the mean (n = 9). *P < 0.05; **P < 0.01.
Expressions of GIPR in gingiva
The immunohistochemical staining using anti‐GIPR antibody showed GIPR‐expressing cells in the gingiva in WT mouse (Figure 4). Glucose‐dependent insulinotropic polypeptide receptor‐expressing cells were increased in the periodontitis gingiva, suggesting that GIPR‐expressing cells in the gingival connective tissue are the infiltrated inflammatory cells. To confirm whether the residual and infiltrated macrophages by periodontitis expressed GIPR, we double‐stained using anti‐GIPR antibody and anti‐Mac‐1antibody in the gingiva. Mac‐1‐positive macrophages expressed GIPR in the gingival connective tissue in WT mice.
Figure 4.
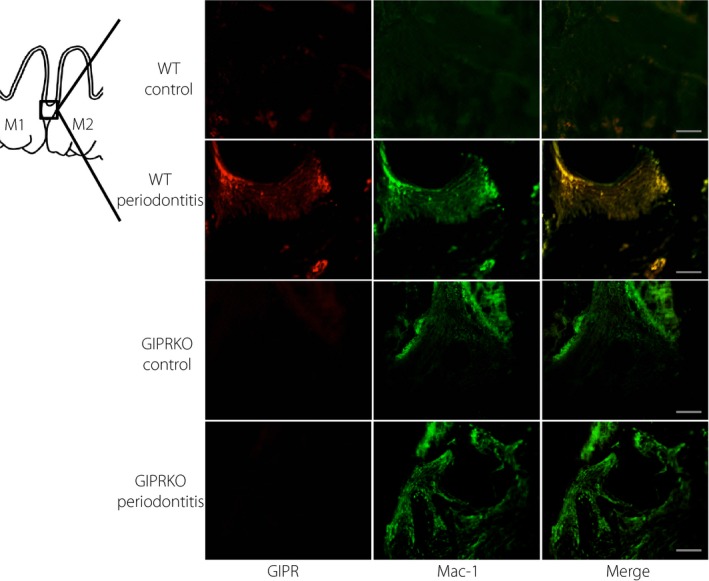
Expression of glucose‐dependent insulinotropic polypeptide receptor (GIPR) in gingiva. Immunohistochemical double staining was carried out using anti‐Mac‐1 polyclonal antibody and anti‐GIPR antibody in gingiva. Scale bar, 100 μm. GIPRKO, glucose‐dependent insulinotropic polypeptide receptor‐knockout mice; WT, wild‐type mice.
Effects of GIP on LPS‐induced TNF‐α and iNOS gene expressions in THP‐1 cells
TNF‐α and iNOS gene expressions in THP‐1 cells were significantly increased by LPS by 3.5‐fold and 3.0‐fold, respectively (P < 0.01; Figure 5). Glucose‐dependent insulinotropic polypeptide inhibited LPS‐induced TNF‐α and iNOS gene expressions in THP‐1 cells in a dose‐dependent manner.
Figure 5.
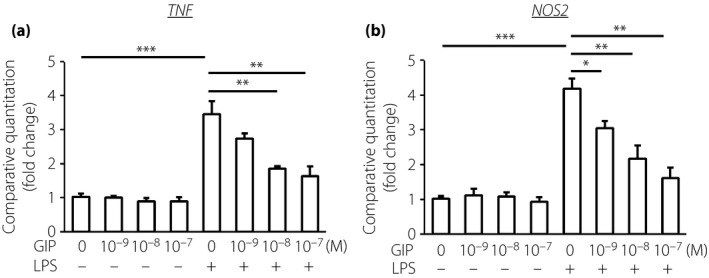
Effects of glucose‐dependent insulinotropic polypeptide (GIP) on lipopolysaccharide (LPS)‐stimulated gene expressions of (a) tumor necrosis factor‐α (TNF‐α; TNF) and (b) inducible nitric oxide synthase (iNOS;NOS2) in THP‐1 cells. After 30 min pre‐incubation with GIP (10−9–10−7 mol/L), cells were stimulated by LPS for 4 h. Messenger ribonucleic acid expressions were determined by quantitative reverse transcription polymerase chain reaction. Results are expressed as mean ± standard error of the mean (n = 6). *P < 0.05; **P < 0.01; ***P < 0.001.
To examine the mechanism of the inhibitory effects of GIP on the LPS‐induced inflammatory cytokine appearances in THP‐1 cells, THP‐1 cells were pre‐incubated with MDL‐12330A, a specific inhibitor of cAMP, PKI(14–22) amide or H‐89, a specific inhibitor of PKA, before GIP administration and then stimulated by LPS. As shown in Figure 6a, both MDL‐12330A and PKI(14–22) amide significantly neutralized the inhibitory effects of GIP on LPS‐induced TNF‐α and iNOS gene expressions in THP‐1 cells. The inhibition of PKA by H‐89 also abolished the reduction of LPS‐induced TNF‐α and iNOS gene expressions by GIP in THP‐1 cells (Figure S1). As there is another pathway, cAMP/Epac2 pathway in the downstream of GIPR, the effects of Epac2 activator 8‐pCPT‐2′‐O‐Me‐cAMP on LPS‐induced TNF‐α and iNOS gene expressions were investigated. As shown in Figure 6b, there were no effects of Epac pathway activator on TNF‐α and iNOS gene expressions in THP‐1 cells with or without LPS and GIP. These results showed that GIP suppressed the LPS‐induced inflammatory response at least in part through cAMP and PKA.
Figure 6.
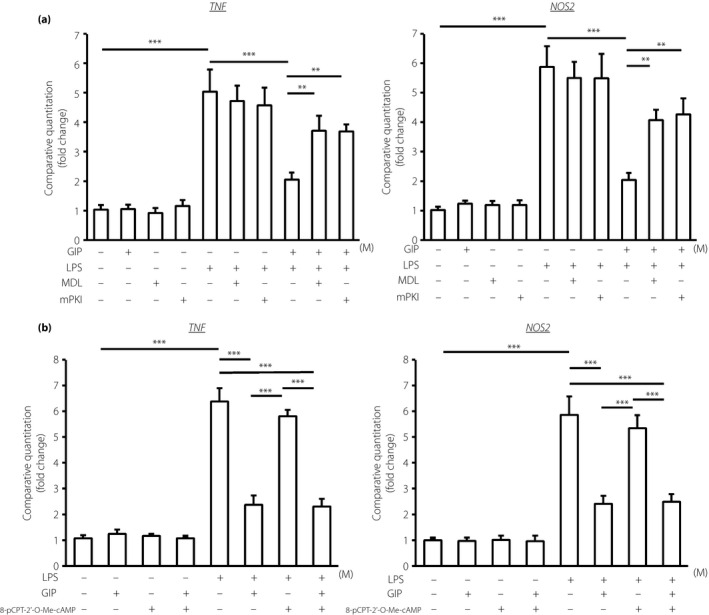
Pathway of the inhibitory effect of glucose‐dependent insulinotropic polypeptide (GIP) on lipopolysaccharide (LPS)‐induced inflammatory gene expressions, tumor necrosis factor‐α (TNF‐α; TNF) and inducible nitric oxide synthase (iNOS;NOS2), in THP‐1 cells. Messenger ribonucleic acid expressions were determined by quantitative reverse transcription polymerase chain reaction. (a) Involvement of cyclic adenosine monophosphate and protein kinase A. After 30‐min pre‐incubation of GIP with or without MDL‐12330A, a specific inhibitor of cyclic adenosine monophosphate, or protein kinase inhibitor 14–22 (PKI[14–22]) amide (mPKI), a specific inhibitor of protein kinase A, cells were stimulated by lipopolysaccharide (LPS) for 4 h. (b) Involvement of Epac2. After 30 min pre‐incubation of GIP with or without 8‐pCPT‐2′‐O‐Me‐cyclic adenosine monophosphate, a selective activator against Epac, cells were stimulated by LPS for 4 h. Results are expressed as mean ± standard error of the mean (n = 6). **P < 0.01; ***P < 0.001. MDL, cis‐N‐(2‐phenylcyclopentyl)‐azacyclotridec‐1‐en‐2‐amine.
Discussion
In the present study, we explored the inhibitory effects of GIP on periodontitis through GIPR. In the periodontitis, residential and infiltrated macrophages expressed GIPR. In vitro experiments using THP‐1 cells confirmed the suppressive effect of GIP on LPS‐induced inflammation at least partially through cAMP and PKA pathways.
We induced experimental periodontitis by wire ligation around a tooth23. This ligature‐induced periodontitis well mimics human periodontitis28, 29, 30. After the plaque accumulation around the placed wire, gingival and periodontal inflammation occurred. Using this experimental periodontitis model, we clearly showed that the deletion of GIP signal aggravated periodontitis. Experimental periodontitis in GIPRKO presented severe periodontitis with increased inflammatory cell infiltration in the gingival connective tissue around the wire. The gene expressions of gingival inflammatory cytokines, TNF‐α and iNOS, were significantly increased in the GIPRKO periodontitis compared with the WT periodontitis. These results suggest the anti‐inflammation activity of GIP in periodontitis.
Periodontitis is a chronic infectious disease initiated by a group of periodontopathic bacteria, such as Porphyromonas gingivalis and Aggregatobacter actinomycetemcomitans, which possess LPS31. LPS activates inflammatory cells, which secrete inflammatory cytokines. In the present study, we have shown that GIP inhibited LPS‐stimulated inflammatory cytokine gene expressions in THP‐1 cells in a dose‐dependent manner, suggesting an anti‐inflammatory role of GIP in bacterial infection. The expression of GIPR was confirmed in mouse macrophages and human monocytes, as well as THP‐1 cells16, 32. It is also shown that GIP increased intracellular cAMP levels in human monocytes, THP‐1 cells and RAW264.7 cells32. We further elucidated that GIP suppressed LPS‐induced TNF‐α and iNOS gene expressions, at least in part through cAMP and PKA. On the contrary, there were no effects of Epac pathway activator 8‐pCPT‐2′‐O‐Me‐cAMP on LPS‐induced inflammatory expressions in THP‐1 cells.
Glucose‐dependent insulinotropic polypeptide exerted anti‐atherogenic effects by suppressing macrophage foam cell formation through GIPR, followed by cAMP activation. Glucose‐dependent insulinotropic polypeptide suppressed the cholesteryl ester accumulation in exudate peritoneal macrophages, and reduced macrophage infiltrations in the atherosclerotic lesion in apolipoprotein E knockout mice16, 20. The contribution of GIP to adipose tissue inflammation is controversial. Glucose‐dependent insulinotropic polypeptide infusion increased inflammatory chemokine and cytokine gene networks, especially MCP‐1, in human subcutaneous adipose tissue from obese participants and mice adipose tissue from ob/ob mice32, 33. On the contrary, GIP did not increase MCP‐1 in misty mice adipocytes. Furthermore, Varol et al.19 showed that long‐acting GIP administration reduced monocytes infiltrations and inflammatory cytokine expressions in adipose tissue in obesity model mice. Glucose‐dependent insulinotropic polypeptide increased MCP‐1 transcripts in the co‐culture condition with human macrophages and adipocytes, whereas GIP did not lead to any significant increase of MCP‐1 gene expression in the single culture with human THP‐1 macrophages, primary human macrophages or human adipocytes32. As GIP increased inflammatory response in GIPR‐overexpressing adipocytes, but not in naive adipocytes21, the expression level of GIPR might be crucial to determine the inflammatory response. Further study is required to elucidate this issue.
Glucose‐dependent insulinotropic polypeptide receptor‐knockout mice impaired early insulin secretion after oral glucose ingestion, whereas the fasting glucose level was kept in the normal range22. In the present study, the induction of periodontitis did not affect fasting blood glucose or insulin in WT or GIPRKO. Because we only made one site of periodontitis of a second molar interproximal part in the whole mouth, the effect might have been more prominent if we had induced multi‐sites periodontitis or observed for a longer duration. Future study is required to explore this issue.
In conclusion, the induction of periodontitis in GIPRKO showed more severe periodontitis than that in WT, accompanied by increased accumulation of macrophages in the gingiva. These results suggest the beneficial effects of GIP on periodontal disease. In vitro study suggested that GIP suppressed LPS‐induced inflammatory gene expressions at least partially through the cAMP/PKA pathway. In diabetic patients, GIP is expected to have a direct anti‐inflammatory effect on periodontitis in addition to its glucose‐lowering effect.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1 ¦ Involvement of PKA in the inhibitory effect of GIP on LPS‐induced inflammatory gene expressions, TNF‐a(TNF) and iNOS(NOS2), in THP‐1 cells. mRNA expressions were determined by quantitative RT‐PCR. After 30‐min pre‐incubation of GIP with or without H89, a specific inhibitor of PKA, cells were stimulated by LPS for 4 hrs. Results are expressed as mean ± SEM (n = 6). ***P < 0.001.
Acknowledgments
We thank Mr Brent Bell for reading the manuscript. This research was supported, in part, by a Grant‐in‐Aid for Scientific Research (22592322) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and, in part, by the ‘Strategic Research AGU‐Platform Formation (2008–2012)’ Project for Private Universities: matching fund subsidy from MEXT of Japan.
J Diabetes Investig 2016; 7: 497–505
References
- 1. Loe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care 1993; 16: 329–334. [PubMed] [Google Scholar]
- 2. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol 2011; 7: 738–748. [DOI] [PubMed] [Google Scholar]
- 3. Chapple IL, Genco R. Diabetes and periodontal diseases: consensus report of the joint EFP/AAP workshop on periodontitis and systemic diseases. J Clin Periodontol 2013; 40(Suppl 14): S106–S112. [DOI] [PubMed] [Google Scholar]
- 4. Nelson RG, Shlossman M, Budding LM, et al Periodontal disease and NIDDM in Pima Indians. Diabetes Care 1990; 13: 836–840. [DOI] [PubMed] [Google Scholar]
- 5. Khader YS, Dauod AS, El‐Qaderi SS, et al Periodontal status of diabetics compared with nondiabetics: a meta‐analysis. J Diabetes Complications 2006; 20: 59–68. [DOI] [PubMed] [Google Scholar]
- 6. Demmer RT, Holtfreter B, Desvarieux M, et al The influence of type 1 and type 2 diabetes on periodontal disease progression: prospective results from the Study of Health in Pomerania (SHIP). Diabetes Care 2012; 35: 2036–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saito T, Shimazaki Y, Kiyohara Y, et al The severity of periodontal disease is associated with the development of glucose intolerance in non‐diabetics: the Hisayama study. J Dent Res 2004; 83: 485–490. [DOI] [PubMed] [Google Scholar]
- 8. Demmer RT, Jacobs DR, Desvarieux M. Periodontal disease and incident type 2 diabetes results from the first national health and nutrition examination survey and its epidemiologic follow‐up study. Diabetes Care 2008; 31: 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morita T, Yamazaki Y, Mita A, et al A cohort study on the association between periodontal disease and the development of metabolic syndrome. J Periodontol 2010; 81: 512–519. [DOI] [PubMed] [Google Scholar]
- 10. Teeuw WJ, Gerdes VE, Loos BG. Effect of periodontal treatment on glycemic control of diabetic patients: a systematic review and meta‐analysis. Diabetes Care 2010; 33: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corbella S, Francetti L, Taschieri S, et al Effect of periodontal treatment on glycemic control of patients with diabetes: a systematic review and meta‐analysis. J Diabetes Investig 2013; 4: 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saremi A, Nelson RG, Tulloch‐Reid M, et al Periodontal disease and mortality in type 2 diabetes. Diabetes Care 2005; 28: 27–32. [DOI] [PubMed] [Google Scholar]
- 13. Seino Y, Yabe D. Glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1: incretin actions beyond the pancreas. J Diabetes Investig 2013; 4: 108–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seino Y, Fukushima M, Yabe D. GIP and GLP1, the two incretin hormones: similarities and differences. J Diabetes Investig 2010; 1: 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inagaki N, Seino Y, Takeda J, et al Gastric inhibitory polypeptide: structure and chromosomal localization of the human gene. Mol Endocrinol 1989; 3: 1014–1021. [DOI] [PubMed] [Google Scholar]
- 16. Nogi Y, Nagashima M, Terasaki M, et al Glucose‐dependent insulinotropic polypeptide prevents the progression of macrophage‐driven atherosclerosis in diabetic apolipoprotein E‐null mice. PLoS One 2012; 7: e35683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyawaki K, Yamada Y, Ban N, et al Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742. [DOI] [PubMed] [Google Scholar]
- 18. Tsukiyama K, Yamada Y, Yamada C, et al Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol 2006; 20: 1644–1651. [DOI] [PubMed] [Google Scholar]
- 19. Varol C, Zvibel I, Spektor L, et al Long‐acting glucose‐dependent insulinotropic polypeptide ameliorates obesity‐induced adipose tissue inflammation. J Immunol 2014; 193: 4002–4009. [DOI] [PubMed] [Google Scholar]
- 20. Nagashima M, Watanabe T, Terasaki M, et al Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 2011; 54: 2649–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nie Y, Ma RC, Chan JC, et al Glucose‐dependent insulinotropic peptide impairs insulin signaling via inducing adipocyte inflammation in glucose‐dependent insulinotropic peptide receptor‐overexpressing adipocytes. FASEB J 2012; 26: 2383–2393. [DOI] [PubMed] [Google Scholar]
- 22. Miyawaki K, Yamada Y, Yano H, et al Glucose intolerance caused by a defect in the entero‐insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA 1999; 96: 14843–14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizuno M, Miyazawa K, Tabuchi M, et al A new experimental mouse model of periodontitis using an orthodontic ligature wire. J Hard Tissue Biol 2014; 23: 255–260. [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2− ΔΔCT method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 25. Kawamoto T. Use of a new adhesive film for the preparation of multi‐purpose fresh‐frozen sections from hard tissues, whole‐animals, insects and plants. Arch Histol Cytol 2003; 66: 123–143. [DOI] [PubMed] [Google Scholar]
- 26. Nishikawa T, Naruse K, Kobayashi Y, et al Involvement of nitrosative stress in experimental periodontitis in diabetic rats. J Clin Periodontol 2012; 39: 342–349. [DOI] [PubMed] [Google Scholar]
- 27. Arakawa M, Mita T, Azuma K, et al Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon‐like peptide‐1 receptor agonist, exendin‐4. Diabetes 2010; 59: 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kennedy JE, Polson AM. Experimental marginal periodontitis in squirrel monkeys. J Periodontol 1973; 44: 140–144. [DOI] [PubMed] [Google Scholar]
- 29. Rovin S, Costich ER, Gordon HA. The influence of bacteria and irritation in the initiation of periodontal disease in germfree and conventional rats. J Periodontal Res 1966; 1: 193–204. [DOI] [PubMed] [Google Scholar]
- 30. Takada T, Yoshinari N, Sugiishi S, et al Effect of restraint stress on the progression of experimental periodontitis in rats. J Periodontol 2004; 75: 306–315. [DOI] [PubMed] [Google Scholar]
- 31. Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts‐CD14 and Toll‐like receptors. Crit Rev Oral Biol Med 2002; 13: 132–142. [DOI] [PubMed] [Google Scholar]
- 32. Gögebakan Ö, Osterhoff MA, Schüler R, et al GIP increases adipose tissue expression and blood levels of MCP‐1 in humans and links high energy diets to inflammation: a randomised trial. Diabetologia 2015; 58: 1759–1768. [DOI] [PubMed] [Google Scholar]
- 33. Chen S, Okahara F, Osaki N, et al Increased GIP signaling induces adipose inflammation via a HIF‐1alpha‐dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2015; 308: E414–E425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ¦ Involvement of PKA in the inhibitory effect of GIP on LPS‐induced inflammatory gene expressions, TNF‐a(TNF) and iNOS(NOS2), in THP‐1 cells. mRNA expressions were determined by quantitative RT‐PCR. After 30‐min pre‐incubation of GIP with or without H89, a specific inhibitor of PKA, cells were stimulated by LPS for 4 hrs. Results are expressed as mean ± SEM (n = 6). ***P < 0.001.