

Abstract
The Gram-negative bacterial lipopolysaccharide (LPS) is a major component of the outer membrane that plays a key role in host–pathogen interactions with the innate immune system. During infection, bacteria are exposed to a host environment that is typically dominated by inflammatory cells and soluble factors, including antibiotics, which provide cues about regulation of gene expression. Bacterial adaptive changes including modulation of LPS synthesis and structure are a conserved theme in infections, irrespective of the type or bacteria or the site of infection. In general, these changes result in immune system evasion, persisting inflammation and increased antimicrobial resistance. Here, we review the modifications of LPS structure and biosynthetic pathways that occur upon adaptation of model opportunistic pathogens (Pseudomonas aeruginosa, Burkholderia cepacia complex bacteria, Helicobacter pylori and Salmonella enterica) to chronic infection in respiratory and gastrointestinal sites. We also discuss the molecular mechanisms of these variations and their role in the host–pathogen interaction.
Keywords: adaptive mutation, O antigen, lipid A, Pseudomonas aeruginosa, Burkholderia cenocepacia, cystic fibrosis, Helicobacter pylori, gastric ulcer
The authors review modifications of lipopolysaccharide structure and biosynthetic pathways that occur upon bacterial adaptation to chronic respiratory and gastrointestinal infections.
Graphical Abstract Figure.
The authors review modifications of lipopolysaccharide structure and biosynthetic pathways that occur upon bacterial adaptation to chronic respiratory and gastrointestinal infections.
INTRODUCTION
The lipopolysaccharide (LPS) is a central component of the outer membrane in Gram-negative bacteria and frequently plays a key role in pathogenesis (Fig. 1) (Whitfield and Trent 2014). LPS is the dominant glycolipid in the outer leaflet of the outer membrane, forming a layer that is stabilized by divalent cations and provides an effective permeability barrier against deleterious molecules such as antibiotics and cationic antimicrobial peptides (Nikaido 2003). The classical LPS molecule has a tripartite structure comprising (i) lipid A, the hydrophobic moiety that anchors LPS to the outer leaflet of the outer membrane; (ii) core oligosaccharide (herein core), which together with lipid A, contributes to maintain the integrity of the outer membrane; and (iii) O antigen polysaccharide or O antigen, which is connected to the core and consists of a polymer made of repeating oligosaccharide units in direct contact with the external milieu (Fig. 1) (Whitfield and Trent 2014). LPS molecules only including lipid A and core are generally referred to as ‘rough’ and often called lipooligosaccharides, while the complete LPS capped with O antigen is called ‘smooth’.
Figure 1.

Cell envelope organization of Gram-negative bacteria. The cell envelope of Gram-negative bacteria is characterized by the presence of two lipid bilayers: the outer membrane (OM) and the cytoplasmic membrane (CM), which are separated by the periplasm, containing hydrolytic enzymes, binding proteins, chemoreceptors and the peptidoglycan cell wall. The OM is an asymmetric lipid bilayer. The outer leaflet of the OM contains mainly LPS molecules, which form contacts with integral outer membrane proteins (OMPs). The inner layer of the OM and the lipid layers of the cytoplasmic membrane contain phospholipids and membrane proteins.
The lipid A is embedded in the outer membrane and composed of acyl chains linked to a backbone dimer of glucosamine by ester and/or amide linkages. The typically hexa-acylated lipid A elicits robust inflammatory responses upon recognition by the complex Toll-like receptor 4 and myeloid differentiation factor 2 (TLR4-MD2), which is predominantly found on macrophages, monocytes and dendritic cells (Park et al. 2009; Park and Lee 2013). Modification of the lipid A acylation patterns, or addition of positively charged substituents to the lipid A phosphate groups (Raetz et al. 2007), confers protection against host innate defenses by reducing even further the permeability of the outer membrane to antimicrobial peptides and dampening inflammatory responses by the host (Raetz et al. 2007; Needham and Trent 2013; Di Lorenzo et al. 2015a).
Lipid A is glycosylated at the 6′-position with two residues of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo); the inner Kdo serves as the point of attachment for the remaining core. Some bacterial species such as Burkholderia (Silipo et al. 2005, 2007) produce a modified Kdo, which is converted into d-glycero-d-talo-oct-2-ulosonic acid (Ko) by a unique Kdo-3 hydroxylase (Chung and Raetz 2011). The next sugars added to the lipid A-Kdo2 are typically two or more residues of l-glycero-d-manno-heptose, although in some species LPS molecules are devoid of heptose (Valvano, Messner and Kosma 2002). The rest of the core consists of a set of sugars that differs among species and even among strains of the same species (Mamat, Skurnik and Bengoechea 2011). Phosphorylation of the core sugars in Pseudomonas aeruginosa has been associated with increased membrane impermeability and resistance to antibiotics (Walsh et al. 2000), and is also required for the transport of LPS to the outer membrane (Delucia et al. 2011). The P. aeruginosa core may also be a ligand for the cystic fibrosis (CF) transmembrane conductance regulator protein displayed on the apical surface of epithelial cells (Schroeder et al. 2002).
O antigens comprise repeating oligosaccharide units that may be linear or branched (Whitfield and Trent 2014). The O-repeating unit is highly variable immunochemically giving rise to a vast number of different O-specific serotypes (Valvano, Patel and Furlong 2011; Whitfield and Trent 2014). The O antigen contributes to evasion of host immune defenses, particularly evasion of the complement cascade in Salmonella enterica serovar Typhimurium (Murray, Attridge and Morona 2006), delay of recognition and internalization in epithelial cells in Salmonella Typhimurium and Burkholderia cenocepacia (Duerr et al. 2009; Saldías, Ortega and Valvano 2009), enhanced intracellular survival in Shigella flexneri (West et al. 2005) and Brucella melitensis (Paixão et al. 2009), and protection against oxidative stress in Erwinia amylovora (Berry et al. 2009). O antigen also contributes to swimming and swarming motility in E. amylovora (Berry et al. 2009), B. cenocepacia (Coutinho et al. 2011a) and Pectobacterium atrosepticum (Bowden et al. 2013). The immunogenicity of the O antigen polysaccharide elicits a robust antibody response, which may cause selective pressure on bacteria to lose the ability to produce O antigen (King et al. 2009); this is particularly common for chronic P. aeruginosa strains infecting the lungs of patients with CF (Hancock et al. 1983). Conceivably, once the bacteria become mucoid (Govan and Deretic 1996), the nutrient burden is so high producing alginate and that the bacteria are replicating in a ‘protected’ niche in which O antigen becomes dispensable. However, this may not be a universal notion since other bacteria chronically infecting the CF lung, such as members of the B. cepacia complex, undergo different adaptive changes than those reported for P. aeruginosa (Zlosnik et al. 2014), including the observation of an inverse correlation between the quantity of mucoid exopolysaccharide production and the rate of decline in CF lung function (Zlosnik et al. 2011).
Most P. aeruginosa strains produce two types of O antigen molecules (‘A-band’ and ‘B-band’), which are structurally and serologically distinct and have different mechanisms of biosynthesis (King et al. 2009; Lam et al. 2011). The ‘A-band’ or ‘common polysaccharide antigen’ is a homopolymer of d-rhamnose that elicits a relatively weak antibody response (King et al. 2009). ‘B-band’ or ‘O-specific antigens’ are highly immunogenic heteropolymers composed of repetitive units of different sugars and form the basis for the AITS P. aeruginosa-serotyping scheme that includes 20 serotypes (Knirel et al. 2006). Structural data in several Pseudomonas serotype strains (Sadovskaya et al. 2000; Bystrova et al. 2006) and genetic experiments (Abeyrathne et al. 2005) demonstrate that both common and O-specific antigens are linked to the lipid A-core.
In this article, we review the literature on LPS variations occurring upon bacterial adaptation to chronic infection, with special emphasis on chronic respiratory infections in patients with CF and gastric infections. CF is a genetic disease that leads to ineffective mucociliary clearance of the airways, resulting in chronic airways infection by several Gram-negative bacterial opportunistic pathogens, such as P. aeruginosa, the Burkholderia cepacia complex (Bcc) and Achromobacter xylosoxidans (Ciofu et al. 2015; Cullen and McClean 2015; Parkins and Floto 2015). Chronic gastric infection by Helicobacter pylori leads to a pre-cancerous state associated with loss of acid-producing parietal cells, which results in increased gastric pH, and pepsinogen-producing zymogenic cells. The gastric environment changes during disease progression and as a result, infecting H. pylori strains must adapt to persist in a gastric habitat with increased pH and different cell composition (Skoglund et al. 2009; Rubin and Trent 2013; Malnick et al. 2014). Because both respiratory infections in patients with CF and gastric infections by H. pylori remain during the lifetime of the patient, they provide natural human models of disease progression and microbial adaptation to the host environment.
LPS BIOSYNTHESIS
Lipid A-core biosynthesis
The biosynthesis of LPS has been reviewed in detail elsewhere (Raetz et al. 2007; King et al. 2009; Lam et al. 2011; Greenfield and Whitfield 2012; Whitfield and Trent 2014; Valvano 2015). Briefly, the lipid A is synthesized on the cytoplasmic side of the inner membrane by a conserved pathway of nine enzymes catalyzing the sequential conversion of the precursor UDP-N-acetyl-glucosamine into lipid A-Kdo2, which is the acceptor for the rest of the core sugars that are added from nucleotide sugar precursors via sequential glycosyl transfer reactions (Fig. 2) (Mamat, Skurnik and Bengoechea 2011; Whitfield and Trent 2014). The complete lipid A-core is transported to the periplasmic face of the inner membrane by the ABC transporter MsbA (Whitfield and Trent 2014). Diverse covalent modifications of lipid A may occur during its transit from the periplasmic side of the inner membrane to the outer leaflet of the outer membrane (Raetz et al. 2007), which are important for niche adaptation and can influence the virulence of the pathogen (Needham and Trent 2013). In bacteria that produce O antigen, the O polysaccharide is assembled by a separate biosynthesis pathway (see the next section) and attached to the core at the periplasmic side of the inner membrane (Fig. 2).
Figure 2.

Simplified overview of the LPS biosynthesis. Lipid A-Kdo2 is synthesized on the cytoplasmic surface of the cytoplasmic membrane. The rest of the core is assembled to the lipid A-Kdo2 and MsbA flips the whole complex to the periplasmic side of the cytoplasmic membrane. The O antigen is synthesized by cytoplasmic membrane-associated enzyme complexes using C55 Und-P as an acceptor for chain assembly and is then flipped to the periplasmic face of the membrane by one of the three pathways: (1) Wzy dependent, (2) ABC transporter dependent or (3) synthase dependent. For simplicity, only the ABC-transporter pathway is represented. Once on the periplasmic side, the O antigen is linked to the lipid A-core by the WaaL ligase and the mature LPS molecule is then transported across the periplasm and inserted into the outer leaflet of the outer membrane by the Lpt (LPS transport) system, a complex that spans the Gram-negative cell envelope to deliver LPS to the outer membrane (E). OM, outer membrane; CM, cytoplasmic membrane.
O antigen biosynthesis
The O antigen is synthesized by cytoplasmic membrane-associated enzyme complexes and requires C55-undecaprenyl phosphate (Und-P), which serves as an acceptor for O antigen chain assembly (Valvano 2011). Chain assembly occurs by the action of diverse glycosyltransferases that synthesize the specific O antigen of each strain. Genes at the wb* (formerly rfb) locus encode most of the enzymes involved in O antigen assembly (Reeves et al. 1996). Because of the great diversity of O antigen structures, the wb* loci are highly polymorphic (Raetz and Whitfield 2002; Lam et al. 2011). The O antigen is initially assembled on the cytoplasmic side of the membrane and then translocated to the periplasmic side and ligated to lipid A-core (Valvano 2015) (Fig. 2). There are three pathways for O antigen biosynthesis and export: (1) Wzy dependent, (2) ABC transporter dependent and (3) synthase dependent (Keenleyside and Whitfield 1996; Lam et al. 2011; Greenfield and Whitfield 2012; Valvano 2015). The mature LPS molecule is then transported across the periplasm and inserted into the outer leaflet of the outer membrane by the conserved Lpt (LPS transport) pathway (May et al. 2015; Simpson et al. 2015). Lpt proteins form a complex that traverses the Gram-negative cell envelope to deliver LPS to the outer membrane and include an ABC protein complex (LptBFG) that uses energy from ATP hydrolysis to extract LPS from the periplasmic face of the inner membrane, several proteins that dock and promote the transfer of LPS across the periplasm (LptCA and YhjD) and a complex of proteins on the outer membrane (LptDE, YtfN, YfgH and YceK), responsible for the correct insertion of LPS in the outer leaflet (Babu et al. 2011; Sperandeo, Dehò and Polissi 2011; Sperandeo et al. 2011; May et al. 2015; Simpson et al. 2015). The Lpt system has not been investigated in Gram-negative pathogens other than Escherichia coli and sequence homology between E. coli and P. aeruginosa genes is low, with the exception of LptB (66% sequence identity). Recently, it was shown that P. aeruginosa LptA has a dimeric structure, unlike the oligomeric structure of E. coli LptA (Shapiro, Gu and Gao 2014).
In P. aeruginosa, the common polysaccharide and the O-specific antigens are synthesized via the ABC-transporter-dependent pathway and the Wzy-dependent pathway, respectively (King et al. 2009; Lam et al. 2011). In both the synthesis is initiated by the same glycosyltransferase, WbpL (homologous to the E. coli WecA), resulting in the formation of an Und-P-P-sugar intermediate (King et al. 2009; Lam et al. 2011). Four enzymes are required for the biosynthesis of GDP-d-rhamnose, the nucleotide sugar precursor for the common polysaccharide antigen: WbpW, AlgC, Gmd and Rmd (King et al. 2009; Lam et al. 2011). The glycosyltransferases WbpX, WbpY and WbpZ are involved in the synthesis of the common polysaccharide antigen (King et al. 2009; Lam et al. 2011), while genes pa54-55pa5459 have been suggested to encode proteins that play a role in controlling chain length (Hao et al. 2013). Once the common polysaccharide antigen is linked to the Und-P carrier, the complex is exported across the membrane by the ABC-transport system Wzm-Wzt (King et al. 2009; Lam et al. 2011). While the genes for the synthesis and assembly of the common polysaccharide are conserved, different set of genes are responsible for the biosynthesis of the O-specific antigen in each serotype strain. These genes are in a cluster flanked by the highly conserved genes himD/ihfB and wbpM (King et al. 2009; Lam et al. 2011). While the P. aeruginosa O5, O6 and O11 O antigen clusters were studied to some extent, very little experimental work was conducted into the functions of genes in the remaining O antigen loci (Lam et al. 2011). The synthesized Und-PP-linked O-repeat units are translocated to the periplasmic side of the membrane and polymerized. The proteins Wzy, Wzz and Wzx are required for this process, acting as polymerase, chain-length regulator and flippase, respectively (Lam et al. 2011). Once on the periplasmic side, both the common polysaccharide antigen and the O-specific antigen are independently linked to the lipid A-core complex by the WaaL ligase (Fig. 2) (Abeyrathne et al. 2005; Valvano 2011; Ruan et al. 2012).
LPS VARIATION DURING CHRONIC RESPIRATORY INFECTIONS IN PATIENTS WITH CF
Pseudomonas aeruginosa infection
Pseudomonas aeruginosa is the most common pathogen isolated from the respiratory tract of adult patients with CF (Lipuma 2010; Hauser et al. 2011). Chronic airway infections caused by P. aeruginosa are found in up to 80% of adult patients with CF (Aaron et al. 2010; Lipuma 2010) and are associated with increased morbidity and mortality (Hauser et al. 2011). Phenotypic changes suggesting P. aeruginosa adaptation to the CF lung have been reported in several studies (Hogardt and Heesemann 2010). They include loss of motility associated with growth in microcolony (Sriramulu et al. 2005), reduced expression of virulence factors, which is presumably an adaptive strategy to escape detection by the host immune system (Smith, Buckley and Wu 2006), increased activity of efflux pumps associated with antibiotic resistance, especially against those antibiotics used clinically (Poole 2005) and a switch from non-mucoid to mucoid phenotypes (Ciofu et al. 2010; Hogardt and Heesemann 2010). The phenotypic changes reflect point mutations accumulating in P. aeruginosa lineages that persist in CF airways (Lorè et al. 2012), and include mutations in alginate biosynthesis regulator genes (Bragonzi et al. 2006) and genes involved in the LPS modification (Cigana et al. 2009), motility (Mahenthiralingam, Campbell and Speert 1994), quorum-sensing regulation (D'Argenio et al. 2007; Hoffman et al. 2009), type 3 secretion system biosynthesis (Jain et al. 2004), multidrug-efflux pumps and mutator genes (Oliver et al. 2000).
The longitudinal course of chronic airway infection with P. aeruginosa in CF has been followed in various studies (Smith, Buckley and Wu 2006; Cigana et al. 2009; Cramer et al. 2011; Mowat et al. 2011; Warren et al. 2011; Yang et al. 2011; Lorè et al. 2012; Dettman et al. 2013). A study investigating over 1700 serial isolates obtained from 10 patients infected with the same strain showed that within-patient diversity made the largest contribution to the overall variation in the population and also that population compositions fluctuated over time (Mowat et al. 2011). The authors suggested that extensive diversity within the P. aeruginosa population during chronic infection has the potential to provide a reservoir for antibiotic-resistant mutations and mutations in other virulence traits (Mowat et al. 2011). Despite these differences, certain traits were overrepresented in all isolates, most of which include properties regulated by quorum sensing (Mowat et al. 2011). In silico simulations reveal that virulence factor expression declines towards the end of chronic infections and adaptive mutations that tend to improve metabolic fitness, which would optimize growth over the more energetically expensive virulence factor production (Oberhardt et al. 2010). Pseudomonas aeruginosa LPS modifications appear to be an important factor in the adaptation of this pathogen to chronic infection (Cigana et al. 2009). Indeed, chronic P. aeruginosa CF isolates have rough colony phenotypes and contain few, short or no O side chains, becoming non-typeable (Hancock et al. 1983). O-antigen-deficient isolates are sensitive to in vitro killing by serum complement and become more tolerant to the antibiotic gentamicin (Kadurugamuwa, Lam and Beveridge 1993). Analysis of sequential variants of P. aeruginosa shows O antigen loss (Lee et al. 2005) and lipid A modifications (Cigana et al. 2009). Whole-genome analysis of two clinical P. aeruginosa variants recovered from a patient with chronic CF after 6 and 96 months of infection also revealed non-synonymous mutations in the O antigen biosynthetic genes wbpA and pa5238 in the latter variant (Smith, Buckley and Wu 2006). Another study of genes responsible for modifying lipid A revealed one mutation in pagL in late variants, which abolish PagL expression and leads to reduced TLR4-MD2-signaling (Cigana et al. 2009). Thus, initial lipid A modifications by addition of palmitate to the lipid A of P. aeruginosa make the LPS more proinflammatory, but the subsequent modification through the loss of PagL activity decreases its proinflammatory activity. Together, the results of these studies suggest that reduced LPS immunostimulatory potential contributes to immune system evasion and survival over the course of the chronic P. aeruginosa infection. Experimental data support this hypothesis since a comparison of the pathogenicity of nine P. aeruginosa sequential clonal variants in the infection models Caenorhabditis elegans, Galleria mellonella, Drosophila melanogaster and two different mice backgrounds (C57Bl/6NCrl and BALB/cAnNCrl) show that early P. aeruginosa variants were lethal in all infection models tested, while late strains exhibited reduced or no virulence (Lorè et al. 2012).
A microevolution analysis based on whole-genome sequencing of sequential P. aeruginosa variants recovered from patients with CF for more than 20 years (Cramer et al. 2011) identified codon changes in genes for lipid A biosynthesis (lpxC, lpxO2 and yciK), core biosynthesis (rfaD and wapP) and common polysaccharide antigen biosynthesis (wbpZ) (Cramer et al. 2011). Another genomic analysis taken over 200 000 bacterial generations of 12 selected P. aeruginosa DK2 variants recovered from six patients with CF identified a total of 234 non-synonymous single nucleotide polymorphisms (SNPs) among the genomes in relation to their common ancestor strain, suggesting that an initial period of rapid adaptation is followed by a period of genetic drift in this lineage (Yang et al. 2011). Three of the non-synonymous SNPs occurred in genes needed for lipid A biosynthesis and modification (pagL and lpxO2) and O-specific antigen synthesis (wzz) (Yang et al. 2011). A recent study analyzing whole-genome sequence data from P. aeruginosa clinical isolates sampled from the sputum of 32 different patients reported that the O antigen ligase waaL is one of the few hotspots of gene polymorphisms (Dettman et al. 2013). To gain insight into the role of mutator genes for generating adaptive variation, Warren et al. (2011) analyzed the genomes of two series of isolates recovered from two patients, similar in duration but different in mutator incidence, and identified 15 LPS genes that lacked in multiple members both in mutator and non-mutator series. All the identified genes are involved in the synthesis of serogroup O2/O5/O26/O18/O20 O antigen (wbpA, wbpB, wbpC, wbpD, wbpE, wbpG, wbpH, wbpI, wbpJ, wbpK, wbpL, wzx, wzy, wzz and pa1385) (Warren et al. 2011).
In addition to changes in O antigen, adaption of P. aeruginosa to chronic lung infection in patients with CF involves the synthesis of various lipid A structures (Fig. 3) (Ernst et al. 2007), which result in alteration of host innate immune responses and promote bacterial persistence (Moskowitz and Ernst 2010). These modifications involve deacylation of the lipid A resulting in the loss of an acyl chain from the 3-position, which is catalyzed by PagL (Fig. 3) (Trent et al. 2001; Geurtsen et al. 2005; Ernst et al. 2006). Underacylation of lipid A has been associated with low inflammatory activity (Moskowitz and Ernst 2010; Di Lorenzo et al. 2015b) and modulation of TLR4-MD2 receptor recognition (Ernst et al. 2003). Also, P. aeruginosa lipid A can acquire a secondary acyl chain into the 3′-position, which is catalyzed by a divergent palmitoyltransferase functionally analogous to the Salmonella and E. coli PagP enzyme (Fig. 3) (Thaipisuttikul et al. 2014). Further modifications involve the addition of secondary acyl chains to the chains present at the 2- and 2′-positions, which is catalyzed by HtrB and LpxO, respectively (Fig. 3), as well as the incorporation of 4-amino-4-deoxy-l-arabinopyranose (Arap4N) to phosphate groups at the 1- and 4′-positions by the two-component regulatory system PmrAB (Fig. 3) (Moskowitz, Ernst and Miller 2004). These lipid A modifications contribute to P. aeruginosa adaptation to the CF airway (Moskowitz and Ernst 2010). The addition of phosphoethanolamine to the P. aeruginosa lipid A via the ColRS two-component system (Fig. 3) in a Zn2+-dependent manner was recently reported (Nowicki et al. 2015), but the role of this modification in vivo is not clear.
Figure 3.

Lipid A modifications occurring in P. aeruginosa during adaptation to long-term chronic infection. The basic tetra-acylated lipid A structure can be modified by: deacylation by PagL; palmitoylation by PagP; acylation by HtrB; acylation by LpxO; addition of Arap4N by PmrAB on position 1 or 4′; and addition of phosphoethanolamine by ColRS on position 1 or 4′.
Collectively, the studies described above support the notion that chronically infecting bacteria adapt to host immune responses by producing LPS lacking O antigen and by introducing lipid A modifications in isolates recovered in late stages of CF chronic infection (Table 1) (Lyczak, Cannon and Pier 2002; Lee et al. 2005; Smith, Buckley and Wu 2006; Cigana et al. 2009; Moskowitz and Ernst 2010; Cramer et al. 2011; Yang et al. 2011; Dettman et al. 2013). This conclusion is also supported from comparative studies using various host models demonstrating that adaptation of different P. aeruginosa lineages within CF lungs selects populations with reduced pathogenic potential in acute infections (Lorè et al. 2012).
Table 1.
LPS genes altered in P. aeruginosa during chronic infections.
| LPS metabolism | Genes | Reference |
|---|---|---|
| Lipid A biosynthesis and modification | lpxO2 | Cramer et al. (2011); Yang et al. (2011) |
| lpxC, yciK | Cramer et al. (2011) | |
| pagL | Cigana et al. (2009); Yang et al. (2011) | |
| Core biosynthesis and modification | rfaD, wapP | Cramer et al. (2011) |
| Common polysaccharide antigen biosynthesis | wbpZ | Cramer et al. (2011) |
| O-specific antigen biosynthesis | wbpA, pa5238 | Smith, Buckley and Wu (2006) |
| wzz | Yang et al. (2011) | |
| wbpA, wbpB, wbpC, wbpD, wbpE, wbpG, wbpH, wbpI, wbpJ, wbpK, wbpL, wzx, wzy, wzz, pa1385 | Warren et al. (2011) | |
| O antigen ligase | waaL | Dettman et al. (2013) |
Chronic infections by other Gram-negative CF pathogens
Bacteria from the Bcc emerged as significant CF pathogens in the early 1980s, when a minority of infected patients exhibited rapid clinical deterioration, resulting in early death (Mahenthiralingam, Urban and Goldberg 2005; Loutet and Valvano 2010). Respiratory infections with Bcc bacteria in patients with CF generally lead to faster decline in lung function and, in some cases to cepacia syndrome, a fatal necrotizing pneumonia frequently accompanied by septicemia (Mahenthiralingam, Urban and Goldberg 2005; Coutinho et al. 2011b). Further, Bcc bacteria are transmissible through social contacts and are intrinsically resistant to most clinically used antibiotics, which renders their eradication from the CF lung very difficult, if not virtually impossible (Mahenthiralingam, Urban and Goldberg 2005; Drevinek and Mahenthiralingam 2010; Coutinho et al. 2011b). Although transient infection of the respiratory tract may occur in some patients, acquisition of Bcc most typically results in chronic infection (Mahenthiralingam, Urban and Goldberg 2005; Coutinho et al. 2011b). The same level of adaptation is not so clear cut in B. cenocepacia infections, as studies using the various infection models (C. elegans, G. mellonella, alfalfa, mice and rats) reported that most virulence factors are specific for one infection model only and rarely essential for pathogenicity in multiple hosts (Uehlinger et al. 2009; Lorè et al. 2012). Furthermore, less is known about Burkholderia adaptation during CF chronic infection; however, there has been an effort to characterize the evolution of Burkholderia populations in the lung, including phenotyping (Coutinho et al. 2011a; Moreira et al. 2014) and genotyping of serial isolates (Lieberman et al. 2011; Traverse et al. 2013), and comparative expression profiling of the transcriptome (Mira et al. 2011) and the proteome (Madeira et al. 2011, 2013).
Lieberman et al. (2011) sequenced the genomes of 112 clinical B. dolosa isolates that resulted from the evolution of a single strain in 14 patients with CF over 16 years of epidemic spread and discovered that genes involved in oxygen regulation, antibiotic resistance, outer membrane synthesis and secretion have recurrent mutation patterns Interestingly, recurrent mutations in the same amino acid of the glycosyltransferase WbaD were observed in nine patients, which resulted in production of O-unit repeats that were absent in the ancestral phenotype (Lieberman et al. 2011). The ancestral B. dolosa genotype encodes a stop codon at this locus that prevents O antigen synthesis. In some variants, two different mutations affecting the same amino acid were detected, both of them restoring the full-length WbaD protein and leading to O antigen production (Table 2) (Lieberman et al. 2011). Although this gain-of-function mutation does not follow the loss of O antigen tendency described in P. aeruginosa, these results underpin the importance of the O antigen switch mechanism during chronic infection. Another metagenomic analysis of six lineages evolved in biofilm mode of growth revealed an extraordinary mutational parallelism, including genes known to affect LPS biosynthesis, transcription, galactose metabolism, tricarboxylic acid cycle enzymes and altered metabolism of cyclic diguanosine monophosphate (Traverse et al. 2013). One commonly mutated locus, showing 20 independent mutations in both B. dolosa and B. cenocepacia, was manC, encoding a nucleotide mannose biosynthesis protein presumably involved in surface polysaccharide biosynthesis that could be either an exopolysaccharide or LPS (Traverse et al. 2013). Interestingly, these authors showed that complementation of one of the manC mutations dramatically reduced biofilm formation, and they speculated that the loss of polysaccharide may be required for efficient biofilm formation rather than immune evasion (Traverse et al. 2013).
Table 2.
LPS genes altered in B. dolosa and B. cenocepacia during chronic infections. The homologous gene in P. aeruginosa is also indicated.
| Homologous gene in | ||
|---|---|---|
| Gene or locus | P. aeruginosa PAO1 | Reference |
| wbaD | – | Lieberman et al. (2011) |
| YP_834517 | rmlB | Traverse et al. (2013) |
| YP_834518 | rmlA | Traverse et al. (2013) |
| YP_834524 | migA | Traverse et al. (2013) |
| YP_834525 | wbpW | Traverse et al. (2013) |
| YP_834526 | gmd | Traverse et al. (2013) |
| YP_834528 | – | Traverse et al. (2013) |
| YP_834530 | wapR | Traverse et al. (2013) |
| YP_834532 | wbpL | Traverse et al. (2013) |
| YP_834533 | wbpM | Traverse et al. (2013) |
A comparison of the transcriptome and the proteome of three B. cenocepacia isolates recovered at the beginning of the infection and later during the progress of the disease suggests that the expression from genes involved in LPS biosynthesis is altered during chronic infection (Madeira et al. 2011, 2013; Mira et al. 2011), in particular of those required for O antigen biosynthesis. Indeed, recent analysis of the LPS structure of these isolates revealed that although the early-stage isolate has a complete LPS with the O-chain moiety, the late-stage variants have a rough-type LPS, lacking O antigen (Maldonado et al. unpublished data).
Several studies at genome, transcriptome and proteome levels have contributed to a better understanding of Bcc bacteria genome-wide adaptive mechanisms during chronic infections. Together, they suggest that there is a high selective pressure on the O antigen locus leading to alterations both at the structural, sequence and regulatory levels. Given the exceptional parallelism found among the relatively few studies dedicated to Bcc bacteria and P. aeruginosa, the LPS seems to play an important role during chronic infection, both in immune system evasion and biofilm adaptation. Moreover, lack of O antigen in B. cenocepacia leads to increased internalization into macrophages upon phagocytosis (Saldías, Ortega and Valvano 2009), which may explain the higher invasiveness of epidemic strains, such as J2315, which do not produce O antigen. O antigen loss could therefore facilitate access of Bcc bacteria to macrophages, where intracellular bacteria could find a niche to persist, in agreement with a recent study showing that in human lungs, Bcc bacteria but not P. aeruginosa are found mainly inside macrophages (Schwab et al. 2014). Other Gram-negative opportunistic pathogens that cause CF chronic infections include Stenotrophomonas maltophilia, Achromobacter xylosoxidans and Haemophilus influenza. Recently, some studies characterizing the adaptive traits of sequential isolates of S. maltophilia (Vidigal et al. 2014), A. xylosoxidans (Trancassini et al. 2014) and H. influenza (Watson, Burns and Smith 2004) recovered from patients with CF have been published; however, the LPS characterization of these clinical isolates is still lacking.
LPS VARIATION DURING CHRONIC GASTRIC INFECTION
The human gastric pathogen H. pylori is usually acquired during childhood by colonizing the human gastric mucosa and producing a superficial gastritis, which may remain asymptomatic during the lifetime of colonized individuals or eventually lead to gastric ulcer and atrophic gastritis (Linz et al. 2013; Otero, Ruiz and Perez Perez 2014). This geographically widespread bacteria infects more than half of the human population and is one of the most genetically diverse bacterial species, being also one of the most ubiquitous infectious organisms (Linz et al. 2013). The genetic diversity of H. pylori is caused by a high mutation rate, presumably due to the lack of several mutation repair genes (Kang and Blaser 2006). Chronic infection with H. pylori is recognized as the most common cause of gastric and duodenal ulcers (Brown 2000). Helicobacter pylori chronic infection is also associated with the development of gastric adenocarcinoma and lymphoma of mucosa-associated lymphoid tissue (Otero, Ruiz and Perez Perez 2014; Mégraud, Bessède and Varon 2015), for which this bacterium is considered to be a class 1 carcinogen (WHO 1994).
Helicobacter pylori produces several virulence factors of which the vacuolating toxin A (VacA), the cytotoxin-associated gene A (CagA) and LPS play major roles in immunomodulation and contribute to maintain chronic infection (Posselt, Backert and Wessler 2013; Rubin and Trent 2013; Chmiela, Miszczyk and Rudnicka 2014; de Bernard and Josenhans 2014; Hatakeyama 2014). These factors contribute to maintain the infection by preventing the clearance of H. pylori from the gastric mucosa and interfering with innate and adaptive immune responses. Structural modifications of the lipid A result in reduced endotoxicity, while expression and variation of Lewis determinants exposed on the bacterial cell surface as a terminal O-specific oligosaccharide (Aspinall et al. 1996; Monteiro et al. 1998) mimic host components expressed on the human gastric epithelium (Moran, Prendergast and Appelmelk 1996; Moran 2008) and reduce detection by the immune system. Helicobacter pylori lipid A presents a unique structure and shows remarkably lower biological activity compared with lipid A from other bacteria (Muotiala et al. 1992; Moran and Aspinall 1998). Structural analysis revealed that the lipid A acyl chains are longer (16 to 18 carbons) than those present in enterobacterial lipid A (Moran, Lindner and Walsh 1997). The predominant form is tetra-acylated lipid A, which is also underphosphorylated (Moran, Lindner and Walsh 1997; Cullen et al. 2011). Underphosphorylation and underacylation of H. pylori lipid A are responsible for reduced endotoxicity (Ljungh, Moran and Wadström 1996), as determined by its low reactivity against anti-lipid A antibodies (Mattsby-Baltzer et al. 1992), reduced ability to induce the production of cytokines, nitric oxide and prostaglandin E2 (Pérez-Pérez et al. 1995), and E-selectin expression (Darveau et al. 1995), as well as reduced activation of leukocytes (Baker et al. 1994; Semeraro et al. 1996). Lipid A remodeling in H. pylori occurs mainly on the periplasmic side of the inner membrane. A first set of modifications involves removal of the 1-phosphate group by LpxE and the addition of a phosphorylethanolamine in its place by EptA (Tran et al. 2004, 2006). These modifications increase bacterial resistance to antimicrobial peptides (Tran et al. 2006). Second, a two-protein Kdo-hydrolase complex removes the terminal Kdo sugar, a modification that is critical to allow the ligation of the O-specific oligosaccharides to the lipid A core (Stead et al. 2010). Third, LpxF catalyzes the removal of the 4′-phosphate group (Cullen et al. 2011). After ligation of the O-specific oligosaccharide (see below), the complete LPS molecule is transported and displayed on the surface of the bacterial outer membrane. Once in the outer membrane, the lipid A undergoes a final modification that consist on the removal of the 3′-linked acyl chains by LpxR, producing the characteristic tetra-acylated lipid A structure (Stead et al. 2008).
The H. pylori O-specific oligosaccharide is initially formed as a lipid-linked oligosaccharide resulting from the addition of monosaccharides, but does not form a repeating oligosaccharide unit (Berg et al. 1997; Rubin and Trent 2013). The O-specific oligosaccharide has a common backbone that is further modified by fucosyltransferases generating structures that mimic human Lewis antigen molecules and other related blood group antigens such as LeX; LeY, Lea, Leb, sialyl-LeX, H-1 antigen, and blood groups A and B antigens (Rubin and Trent 2013) (Fig. 4) This lipid-linked fucosylated oligosaccharide is translocated across the inner membrane by Wzk, an ABC-transporter protein homologous to PglK from Campylobacter jejuni, and subsequently ligated to the lipid A-core by the WaaL ligase (Hug et al. 2010).
Figure 4.
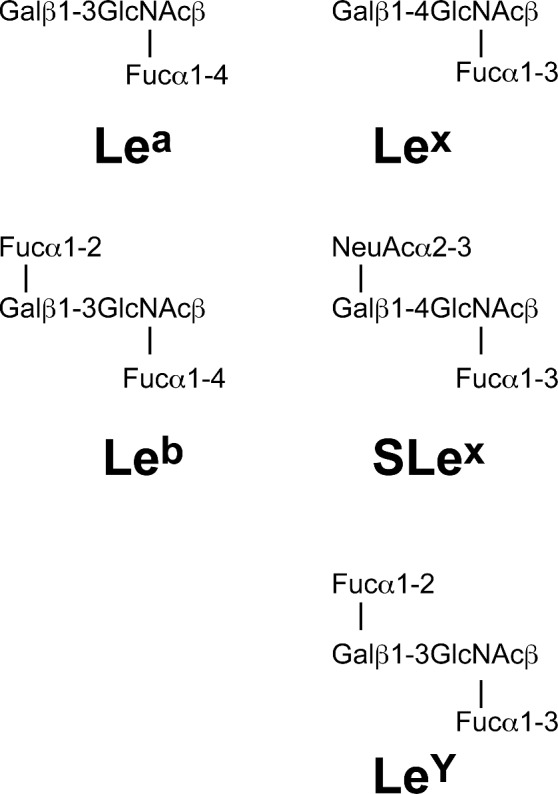
Lewis antigen structures. Helicobacter pylori can produce type 1 (based on a β-(1,3)-linked galactose-GlcNAc sugar backbone) and type 2 (based on a β-(1,4)-linked galactose-GlcNAc sugar backbone) Lewis antigens. Lea and Lex are built by addition of a fucose residue to the GlcNAc sugar of the type 1 and type 2 backbone, through α-(1,4) or α-(1,3) linkages, respectively. Leb and LeY are built by addition of a fucose residue through α-(1,2) linkage to Lea and Lex structures, respectively. Sialyl-Lex (SLex) is built by addition of a sialyl group to the Lex antigen by a α-(2,3) linkage.
The presence of terminal fucosylated sugars on the outer surface of the bacterium, in particular the most common LeX and LeY structures, is critical for colonization in mice models (Logan et al. 2000; Moran et al. 2000). However, the diversity of Lewis antigen expression in H. pylori hampers efforts to clearly define the role of these molecules in infection and disease progression. In humans, LeX H. pylori O-specific oligosaccharide is recognized by galectin-3, a β-galactoside-binding lectin that serves as a gastric receptor (Fowler et al. 2006). However, the main role attributed to the Lewis antigens is that of molecular mimicry, which could be manifested is several ways. For example, H. pylori can change its Lewis antigens in response to those present in the host, as demonstrated with Leb-transgenic mice infected with LeX-expressing H. pylori, which over time switched on Leb expression (Pohl et al. 2009). This change allowed better bacterial colonization than in the transgenic mice lacking Leb expression, suggesting that Leb H. pylori could survive better in a self-tolerant Leb host (Pohl et al. 2009). Alternatively, H. pylori expressing different Lewis antigens than those in the host can induce production of autoantibodies that recognize gastric parietal cells leading to disease (Negrini et al. 1996; Faller et al. 1997). Further, Lewis antigens can also dampen host immune responses to H. pylori through interactions with the C-type lectin DC-SIGN on the surface of gastric dendritic cells, which lead to a block in maturation of T-helper 1 cells and reduced production of proinflammatory cytokines (Bergman et al. 2004).
The first evidence that a single strain of H. pylori alters its LPS antigenic phenotype during the course of infection was demonstrated by investigating the expression of Lewis antigens in 127 isolates recovered from serial biopsies of 26 asymptomatic subjects (Rasko et al. 2000). This alteration of LPS biosynthesis in H. pylori occurs during host colonization in response to several stimuli (Salaün, Ayraud and Saunders 2005; Nilsson et al. 2008) such as interaction with T-helper cells Bergman, 2004 #5473} and gastric pH (Skoglund et al. 2009). More recently, several studies have focused on the genomic changes occurring in H. pylori isolates that have been recovered several years apart from patients with chronic infection (Falush et al. 2001; Israel et al. 2001; Kraft et al. 2006; Alvi et al. 2007; Morelli et al. 2010; Kennemann et al. 2011).
A whole-genome analysis of 10 H. pylori sequential isolates recovered from four patients over 16 years of chronic gastritis revealed five SNPs affecting LPS genes, including genes involved in the biosynthesis of lipid A (phosphoethanolamine transferase), core (kdsA and waaF) and O-specific oligosaccharide (wecA) synthesis, as well as in a putative LPS biosynthetic protein (Kennemann et al. 2011). A cluster of nucleotide polymorphisms in the fucT (fucosyltransferase) gene, presumably facilitating its expression, was identified in whole-genome analyses of two H. pylori strains isolated from spouses (Linz et al. 2013). Hyperexpression of fucT promotes posttranslational fucosylation of the O-specific oligosaccharide, generating Lewis antigens (Ge et al. 1997; Martin et al. 1997; Moran 2008; Linz et al. 2013). The alteration of H. pylori LPS during chronic gastric infection, either by altering LPS biosynthesis or by adding fucosyl residues to O-specific oligosaccharides, generates Lewis structures that mimic host antigens and contribute to immune system evasion.
MOLECULAR MECHANISMS OF LPS VARIATION
Antigenic variation of surface structures is a powerful mechanism for pathogen evasion of adaptive immune responses (Lerouge and Vanderleyden 2002; van der Woude and Bäumler 2004; Lukácová, Barák and Kazár 2008). One of these adaptions involves phase variation, which is a reversible, yet heritable form, of gene regulation that results in heterogeneous clonal populations and can be mediated by various molecular mechanisms (van der Woude and Bäumler 2004). LPS phase variation can occur by addition of carbohydrates through the activity of glycosyltransferases or sialyltransferases, or addition of phosphorylcholine (ChoP) resulting in changes that affect antigenicity, serum sensitivity and adhesion (van der Woude and Bäumler 2004). Phase variation has been described for human pathogens such as S. enterica serovar Typhimurium, C. jejuni, Neisseria spp. and H. pylori but because variable LPS modification is not easily identified, it is possible that phase variation is more widespread than currently known. Genetic and epigenetic mechanisms behind LPS variation are discussed below.
Adaptive mutagenesis and altered gene expression
Acquisition of adaptive mutations is a common theme in microbial persistence. In patients with CF with chronic lung infection, P. aeruginosa strains accumulate a large proportion of mutator strains (Oliver et al. 2000) that contribute to selection of mucoid variants (Oliver et al. 2000; Mathee et al. 2008; Ciofu et al. 2010; Hogardt and Heesemann 2010). The proinflammatory microenvironment in the airways including polymorphonuclear cells, hydrogen peroxide production and antibiotics (Blázquez et al. 2006) has been associated with mutagenesis and mucoid conversion in vitro (Mathee et al. 1999; Sanders et al. 2006; Moyano et al. 2007). Cationic antimicrobial peptides can also exert a mutagenic inducing effect, as recently demonstrated for human cathelicidin LL-37 (Limoli et al. 2014). Mutagenesis depended on LL-37 entering the bacterial cytosol and binding to DNA, which in turns promotes abnormal DNA synthesis by the error-prone polymerase DinB (Sanders et al. 2006; Limoli et al. 2014).
Environmental cues, such as ionic concentration, can lead to O antigen structural variations resulting from altered gene expression regulated by two-component signal transduction systems. One of the best examples of this type of regulation is the PhoP/PhoQ system in Salmonella (Prost and Miller 2008; Needham and Trent 2013). PhoQ is a membrane sensor histidine kinase and PhoP is its cognate response regulator. Activation of the PhoP/PhoQ system by acidic pH, specific antimicrobial peptides, and depletion of Mg2+ and Ca2+ stimulates transcription of pagP and pagL (among other genes) and subsequent upregulation of the encoded proteins, which acylate and deacylate lipid A, respectively (Prost and Miller 2008; Needham and Trent 2013). Further, CF clinical isolates of P. aeruginosa obtained from patients treated with inhaled colistin (polymyxin E) can develop resistance by loss-of-function mutations in the phoQ gene (Miller et al. 2011). Disruption of phoQ in the presence of an intact phoP stimulated Arap4N addition to lipid A by upregulated expression of the Arap4N synthesis operon. Therefore, this adaptive mutagenesis strategy results in high-level polymyxin resistance clinical strains of P. aeruginosa.
Slipped-strand mispairing
One of the molecular mechanisms of phase variation involves slipping of one of the DNA strands, which causes mispairing between daughter and parent strands during DNA replication (slipped-strand mispairing) (Lukácová, Barák and Kazár 2008). Short DNA repeats, microsatellites and tandem repeats are particularly prone to slipped-strand mispairing (van Belkum et al. 1997; Torres-Cruz and van der Woude 2003). In H. pylori, phase variation is related to an increase in the number of poly-C tract repeats in the β-(1,3)-galactosyl transferase (GalT), which leads to a switching on Leb expression (Pohl et al. 2009). Also, repetitive poly-A and poly-C sequences in the fucosyltransferase fucT mediate slipped-strand mispairing, which in turn results in production of Lewis antigens with different fucosylated oligosaccharides (Wang et al. 2000; Nilsson et al. 2008). Further, the α-(1,2)-fucosyltransferase gene futC contains an heptameric sequence (AAAAAAG) next to the ribosome-binding site, which may cause a phase shift in the reading frame during translation (Wang et al. 2000).
Lateral gene transfer, recombination and genetic rearrangements
The heterogeneity of O antigens is mostly due to variation within the O antigen gene cluster, but it is unclear how such variation was generated (Reeves et al. 2013). Genes involved in O antigen biosynthesis are generally arranged in large operons with low G + C content relative to the average G + C characteristic of each species, which suggests that these clusters were acquired by horizontal gene transfer from a species with low G + C content (Lerouge and Vanderleyden 2002). The G + C content within the O antigen clusters also greatly differs from gene to gene, indicating that the gene clusters might have been assembled from multiple horizontal transmission events and from several sources over a much longer time (Lerouge and Vanderleyden 2002). The role of lateral gene transfer in the evolution of O antigen clusters and O antigen diversification has been well described in Salmonella (Perepelov et al. 2011; Reeves et al. 2013), Escherichia (D'Souza, Samuel and Reeves 2005; Hu et al. 2010; Azmuda et al. 2012), Vibrio (González-Fraga et al. 2008; Wildschutte et al. 2010), Yersinia (Cunneen and Reeves 2007) and Brucella (Wattam et al. 2014). Another mechanism of variation involves large chromosomal rearrangements. For example, more than half of the P. aeruginosa clone C isolates from CF lung infection exhibit large chromosomal inversions mediated an IS6100-induced coupled insertion-inversion mechanism. This creates also a selective advantage by insertion of IS6100 into wbpM, pilB and mutS, which leads to common CF phenotypes such as O-antigen and type IV pili deficiency and hypermutability (Kresse et al. 2003).
CONCLUDING REMARKS
The LPS is an abundant molecule of the outer membrane of most Gram-negative bacteria and plays a key role during host–pathogen interaction and the establishment of chronic infection. LPS-mediated virulence resides both in the endotoxic activity of lipid A and in the ability of the core and O antigen to provide the bacterium with resistance to host defense mechanisms. O antigen modification in general contributes to enhance the bacteria's ability to establish infection. For example, P. aeruginosa O antigen modification directed by the D3 prophage promotes adhesion to epithelial cells (Vaca-Pacheco et al. 1999), while in H. pylori, expression of the Lewis antigen LeX promotes bacterial adhesion to the gastric epithelia by interacting with host lectins. Further, O antigen modification can contribute to host immune evasion either by mimicry of host molecules (e.g. Lewis antigens in H. pylori) or by inhibiting activation of the host complement system (Raetz and Whitfield 2002). It is also well established that during chronic infection there is an increase of mutator phenotypes (Oliver et al. 2000), which leads to a higher mutation rate and will consequently contribute to the accumulation of modifications in LPS structure during colonization.
Several studies have shown alterations in the LPS molecule during chronic infection, which are thought to contribute to adhesion, host colonization, immune defenses evasion and adaptation to the infection niche. Different mechanisms both at the genetic and epigenetic levels have been implied in LPS variation, creating LPS diversity and thus contributing to the success of the infection.
Future progress in LPS research will require interdisciplinary experimental approaches, combining the application of genome-wide approaches (such as genomics, transcriptomics, proteomics and metabolomics), structural biology, animal knockout models, enzymology, carbohydrate chemistry and membrane biochemistry. LPS phase variation has been described for some human pathogens (S. enterica serovar Typhimurium, C. jejuni, Neisseria spp. and H. pylori) and future research should address the investigation of these mechanisms in other species as well. An in-depth understanding of LPS variation and its effects on pathogenicity and virulence is of paramount importance in the understanding of infection establishment and progression.
FUNDING
Research in the authors' laboratories has been supported the grants from the Canadian Institutes of Health Research, Cystic Fibrosis Canada, and the UK Cystic Fibrosis Trust (to M.A.V.) and from the Portuguese Foundation for Science and Technology (UID/BIO/04565/2013) and Programa Operacional Regional de Lisboa 2020 (Project N. 007317) to the Institute for Bioengineering and Biosciences (to I.S-C.). R.F.M. was supported by the PhD fellowship SFRH/BD/84233/2012. I.S-C. and M.A.V. were members of the EU COST Action BM1003: Microbial cell surface determinants of virulence as targets for new therapeutics in cystic fibrosis (http://www.cost-bm1003.info/).
Conflict of interest. None declared.
REFERENCES
- Aaron SD, Vandemheen KL, Ramotar K, et al. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA. 2010;304:2145–53. doi: 10.1001/jama.2010.1665. [DOI] [PubMed] [Google Scholar]
- Abeyrathne PD, Daniels C, Poon KK, et al. Functional characterization of WaaL, a ligase associated with linking O-antigen polysaccharide to the core of Pseudomonas aeruginosa lipopolysaccharide. J Bacteriol. 2005;187:3002–12. doi: 10.1128/JB.187.9.3002-3012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvi A, Devi SM, Ahmed I, et al. Microevolution of Helicobacter pylori type IV secretion systems in an ulcer disease patient over a ten-year period. J Clin Microbiol. 2007;45:4039–43. doi: 10.1128/JCM.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspinall GO, Monteiro MA, Pang H, et al. Lipopolysaccharide of the Helicobacter pylori type strain NCTC 11637 (ATCC 43504): structure of the O antigen chain and core oligosaccharide regions. Biochemistry. 1996;35:2489–97. doi: 10.1021/bi951852s. [DOI] [PubMed] [Google Scholar]
- Azmuda N, Rahman MZ, Sultana M, et al. Evidence of interspecies O antigen gene cluster transfer between Shigella boydii 15 and Escherichia fergusonii. APMIS. 2012;120:959–66. doi: 10.1111/j.1600-0463.2012.02926.x. [DOI] [PubMed] [Google Scholar]
- Babu M, Diaz-Mejia JJ, Vlasblom J, et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011;7:e1002377. doi: 10.1371/journal.pgen.1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PJ, Hraba T, Taylor CE, et al. Molecular structures that influence the immunomodulatory properties of the lipid A and inner core region oligosaccharides of bacterial lipopolysaccharides. Infect Immun. 1994;62:2257–69. doi: 10.1128/iai.62.6.2257-2269.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DE, Hoffman PS, Appelmelk BJ, et al. The Helicobacter pylori genome sequence: genetic factors for long life in the gastric mucosa. Trends Microbiol. 1997;5:468–74. doi: 10.1016/s0966-842x(97)01164-5. [DOI] [PubMed] [Google Scholar]
- Bergman MP, Engering A, Smits HH, et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med. 2004;200:979–90. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MC, McGhee GC, Zhao Y, et al. Effect of a waaL mutation on lipopolysaccharide composition, oxidative stress survival, and virulence in Erwinia amylovora. FEMS Microbiol Lett. 2009;291:80–7. doi: 10.1111/j.1574-6968.2008.01438.x. [DOI] [PubMed] [Google Scholar]
- Blázquez J, Gómez-Gómez JM, Oliver A, et al. PBP3 inhibition elicits adaptive responses in Pseudomonas aeruginosa. Mol Microbiol. 2006;62:84–99. doi: 10.1111/j.1365-2958.2006.05366.x. [DOI] [PubMed] [Google Scholar]
- Bowden SD, Hale N, Chung JC, et al. Surface swarming motility by Pectobacterium atrosepticum is a latent phenotype that requires O antigen and is regulated by quorum sensing. Microbiology. 2013;159:2375–85. doi: 10.1099/mic.0.070748-0. [DOI] [PubMed] [Google Scholar]
- Bragonzi A, Wiehlmann L, Klockgether J, et al. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology. 2006;152:3261–9. doi: 10.1099/mic.0.29175-0. [DOI] [PubMed] [Google Scholar]
- Brown LM. Helicobacter pylori: epidemiology and routes of transmission. Epidemiol Rev. 2000;22:283–97. doi: 10.1093/oxfordjournals.epirev.a018040. [DOI] [PubMed] [Google Scholar]
- Bystrova OV, Knirel YA, Lindner B, et al. Structures of the core oligosaccharide and O-units in the R- and SR-type lipopolysaccharides of reference strains of Pseudomonas aeruginosa O-serogroups. FEMS Immunol Med Mic. 2006;46:85–99. doi: 10.1111/j.1574-695X.2005.00004.x. [DOI] [PubMed] [Google Scholar]
- Chmiela M, Miszczyk E, Rudnicka K. Structural modifications of Helicobacter pylori lipopolysaccharide: an idea for how to live in peace. World J Gastroentero. 2014;20:9882–97. doi: 10.3748/wjg.v20.i29.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HS, Raetz CR. Dioxygenases in Burkholderia ambifaria and Yersinia pestis that hydroxylate the outer Kdo unit of lipopolysaccharide. P Natl Acad Sci USA. 2011;108:510–5. doi: 10.1073/pnas.1016462108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigana C, Curcurù L, Leone MR, et al. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS One. 2009;4:e8439. doi: 10.1371/journal.pone.0008439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofu O, Mandsberg LF, Bjarnsholt T, et al. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology. 2010;156:1108–19. doi: 10.1099/mic.0.033993-0. [DOI] [PubMed] [Google Scholar]
- Ciofu O, Tolker-Nielsen T, Jensen PO, et al. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliver Rev. 2015;85:7–23. doi: 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Coutinho CP, de Carvalho CC, Madeira A, et al. Burkholderia cenocepacia phenotypic clonal variation during a 3.5-year colonization in the lungs of a cystic fibrosis patient. Infect Immun. 2011a;79:2950–60. doi: 10.1128/IAI.01366-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho CP, Dos Santos SC, Madeira A, et al. Long-term colonization of the cystic fibrosis lung by Burkholderia cepacia complex bacteria: epidemiology, clonal variation, and genome-wide expression alterations. Front Cell Infect Microbiol. 2011b;1:12. doi: 10.3389/fcimb.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer N, Klockgether J, Wrasman K, et al. Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol. 2011;13:1690–704. doi: 10.1111/j.1462-2920.2011.02483.x. [DOI] [PubMed] [Google Scholar]
- Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens. 2015;4:66–89. doi: 10.3390/pathogens4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen TW, Giles DK, Wolf LN, et al. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011;7:e1002454. doi: 10.1371/journal.ppat.1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunneen MM, Reeves PR. The Yersinia kristensenii O11 O-antigen gene cluster was acquired by lateral gene transfer and incorporated at a novel chromosomal locus. Mol Biol Evol. 2007;24:1355–65. doi: 10.1093/molbev/msm058. [DOI] [PubMed] [Google Scholar]
- D'Argenio DA, Wu M, Hoffman LR, et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol. 2007;64:512–33. doi: 10.1111/j.1365-2958.2007.05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP, Cunningham MD, Bailey T, et al. Ability of bacteria associated with chronic inflammatory disease to stimulate E-selectin expression and promote neutrophil adhesion. Infect Immun. 1995;63:1311–7. doi: 10.1128/iai.63.4.1311-1317.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bernard M, Josenhans C. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2014;19:11–8. doi: 10.1111/hel.12160. [DOI] [PubMed] [Google Scholar]
- Delucia AM, Six DA, Caughlan RE, et al. Lipopolysaccharide (LPS) inner-core phosphates are required for complete LPS synthesis and transport to the outer membrane in Pseudomonas aeruginosa PAO1. MBio. 2011;2 doi: 10.1128/mBio.00142-11. e00142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman JR, Rodrigue N, Aaron SD, et al. Evolutionary genomics of epidemic and nonepidemic strains of Pseudomonas aeruginosa. P Natl Acad Sci USA. 2013;110:21065–70. doi: 10.1073/pnas.1307862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo F, Kubik L, Oblak A, et al. Activation of human toll-like receptor 4 (TLR4). Myeloid differentiation factor 2 (MD-2) by hypoacylated lipopolysaccharide from a clinical isolate of Burkholderia cenocepacia. J Biol Chem. 2015a;290:21305–19. doi: 10.1074/jbc.M115.649087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo F, Silipo A, Bianconi I, et al. Persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73 exhibits an under-acylated LPS structure responsible of its low inflammatory activity. Mol Immunol. 2015b;63:166–75. doi: 10.1016/j.molimm.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect. 2010;16:821–30. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- D'Souza JM, Samuel GN, Reeves PR. Evolutionary origins and sequence of the Escherichia coli O4 O-antigen gene cluster. FEMS Microbiol Lett. 2005;244:27–32. doi: 10.1016/j.femsle.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Duerr CU, Zenk SF, Chassin C, et al. O-antigen delays lipopolysaccharide recognition and impairs antibacterial host defense in murine intestinal epithelial cells. PLoS Pathog. 2009;5:e1000567. doi: 10.1371/journal.ppat.1000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst RK, Adams KN, Moskowitz SM, et al. The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J Bacteriol. 2006;188:191–201. doi: 10.1128/JB.188.1.191-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst RK, Hajjar AM, Tsai JH, et al. Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. J Endotoxin Res. 2003;9:395–400. doi: 10.1179/096805103225002764. [DOI] [PubMed] [Google Scholar]
- Ernst RK, Moskowitz SM, Emerson JC, et al. Unique lipid a modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J Infect Dis. 2007;196:1088–92. doi: 10.1086/521367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller G, Steininger H, Kränzlein J, et al. Antigastric autoantibodies in Helicobacter pylori infection: implications of histological and clinical parameters of gastritis. Gut. 1997;41:619–23. doi: 10.1136/gut.41.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Kraft C, Taylor NS, et al. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. P Natl Acad Sci USA. 2001;98:15056–61. doi: 10.1073/pnas.251396098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler M, Thomas RJ, Atherton J, et al. Galectin-3 binds to Helicobacter pylori O-antigen: it is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell Microbiol. 2006;8:44–54. doi: 10.1111/j.1462-5822.2005.00599.x. [DOI] [PubMed] [Google Scholar]
- Ge Z, Chan NW, Palcic MM, et al. Cloning and heterologous expression of an α1,3-fucosyltransferase gene from the gastric pathogen Helicobacter pylori. J Biol Chem. 1997;272:21357–63. doi: 10.1074/jbc.272.34.21357. [DOI] [PubMed] [Google Scholar]
- Geurtsen J, Steeghs L, Hove JT, et al. Dissemination of lipid A deacylases (pagL) among Gram-negative bacteria: identification of active-site histidine and serine residues. J Biol Chem. 2005;280:8248–59. doi: 10.1074/jbc.M414235200. [DOI] [PubMed] [Google Scholar]
- González-Fraga S, Pichel M, Binsztein N, et al. Lateral gene transfer of O1 serogroup encoding genes of Vibrio cholerae. FEMS Microbiol Lett. 2008;286:32–8. doi: 10.1111/j.1574-6968.2008.01251.x. [DOI] [PubMed] [Google Scholar]
- Govan JRW, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–74. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield LK, Whitfield C. Synthesis of lipopolysaccharide O-antigens by ABC transporter-dependent pathways. Carbohydr Res. 2012;356:12–24. doi: 10.1016/j.carres.2012.02.027. [DOI] [PubMed] [Google Scholar]
- Hancock RE, Mutharia LM, Chan L, et al. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: a class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect Immun. 1983;42:170–7. doi: 10.1128/iai.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, King JD, Huszczynski S, et al. Five new genes are important for common polysaccharide antigen biosynthesis in Pseudomonas aeruginosa. MBio. 2013;4:e00631–12. doi: 10.1128/mBio.00631-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15:306–16. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Hauser AR, Jain M, Bar-Meir M, et al. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev. 2011;24:29–70. doi: 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LR, Kulasekara HD, Emerson JC, et al. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros. 2009;8:66–70. doi: 10.1016/j.jcf.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogardt M, Heesemann J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol. 2010;300:557–62. doi: 10.1016/j.ijmm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Hu B, Perepelov AV, Liu B, et al. Structural and genetic evidence for the close relationship between Escherichia coli O71 and Salmonella enterica O28 O-antigens. FEMS Immunol Med Mic. 2010;59:161–9. doi: 10.1111/j.1574-695X.2010.00676.x. [DOI] [PubMed] [Google Scholar]
- Hug I, Couturier MR, Rooker MM, et al. Helicobacter pylori lipopolysaccharide is synthesized via a novel pathway with an evolutionary connection to protein N-glycosylation. PLoS Pathog. 2010;6:e1000819. doi: 10.1371/journal.ppat.1000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel DA, Salama N, Krishna U, et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. P Natl Acad Sci USA. 2001;98:14625–30. doi: 10.1073/pnas.251551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Ramirez D, Seshadri R, et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J Clin Microbiol. 2004;42:5229–37. doi: 10.1128/JCM.42.11.5229-5237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadurugamuwa JL, Lam JS, Beveridge TJ. Interaction of gentamicin with the A band and B band lipopolysaccharides of Pseudomonas aeruginosa and its possible lethal effect. Antimicrob Agents Ch. 1993;37:715–21. doi: 10.1128/aac.37.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Blaser MJ. Bacterial populations as perfect gases: genomic integrity and diversification tensions in Helicobacter pylori. Nat Rev Microbiol. 2006;4:826–36. doi: 10.1038/nrmicro1528. [DOI] [PubMed] [Google Scholar]
- Keenleyside WJ, Whitfield C. A novel pathway for O-polysaccharide biosynthesis in Salmonella enterica serovar Borreze. J Biol Chem. 1996;271:28581–92. doi: 10.1074/jbc.271.45.28581. [DOI] [PubMed] [Google Scholar]
- Kennemann L, Didelot X, Aebischer T, et al. Helicobacter pylori genome evolution during human infection. P Natl Acad Sci USA. 2011;108:5033–8. doi: 10.1073/pnas.1018444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JD, Kocincova D, Westman EL, et al. Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009;15:261–312. doi: 10.1177/1753425909106436. [DOI] [PubMed] [Google Scholar]
- Knirel YA, Bystrova OV, Kocharova NA, et al. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res. 2006;12:324–36. doi: 10.1179/096805106X118906. [DOI] [PubMed] [Google Scholar]
- Kraft C, Stack A, Josenhans C, et al. Genomic changes during chronic Helicobacter pylori infection. J Bacteriol. 2006;188:249–54. doi: 10.1128/JB.188.1.249-254.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresse AU, Dinesh SD, Larbig K, et al. Impact of large chromosomal inversions on the adaptation and evolution of Pseudomonas aeruginosa chronically colonizing cystic fibrosis lungs. Mol Microbiol. 2003;47:145–58. doi: 10.1046/j.1365-2958.2003.03261.x. [DOI] [PubMed] [Google Scholar]
- Lam JS, Taylor VL, Islam ST, et al. Genetic and Functional Diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front Microbiol. 2011;2:118. doi: 10.3389/fmicb.2011.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Haagensen JAJ, Ciofu O, et al. Heterogeneity of biofilms formed by nonmucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. J Clin Microbiol. 2005;43:5247–55. doi: 10.1128/JCM.43.10.5247-5255.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev. 2002;26:17–47. doi: 10.1111/j.1574-6976.2002.tb00597.x. [DOI] [PubMed] [Google Scholar]
- Lieberman TD, Michel J-B, Aingaran M, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–80. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli DH, Rockel AB, Host KM, et al. Cationic antimicrobial peptides promote microbial mutagenesis and pathoadaptation in chronic infections. PLoS Pathog. 2014;10:e1004083. doi: 10.1371/journal.ppat.1004083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz B, Windsor HM, Gajewski JP, et al. Helicobacter pylori genomic microevolution during naturally occurring transmission between adults. PLoS One. 2013;8:e82187. doi: 10.1371/journal.pone.0082187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungh Å, Moran AP, Wadström T. Interactions of bacterial adhesins with extracellular matrix and plasma proteins: pathogenic implications and therapeutic possibilities. FEMS Immunol Med Mic. 1996;16:117–26. doi: 10.1111/j.1574-695X.1996.tb00128.x. [DOI] [PubMed] [Google Scholar]
- Logan SM, Conlan JW, MA Monteiro, et al. Functional genomics of Helicobacter pylori: identification of a β-1,4 galactosyltransferase and generation of mutants with altered lipopolysaccharide. Mol Microbiol. 2000;35:1156–67. doi: 10.1046/j.1365-2958.2000.01784.x. [DOI] [PubMed] [Google Scholar]
- Lorè NI, Cigana C, De Fino I, et al. Cystic fibrosis-niche adaptation of Pseudomonas aeruginosa reduces virulence in multiple infection hosts. PLoS One. 2012;7:e35648. doi: 10.1371/journal.pone.0035648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutet SA, Valvano MA. A decade of Burkholderia cenocepacia virulence determinant research. Infect Immun. 2010;78:4088–100. doi: 10.1128/IAI.00212-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukácová M, Barák I, Kazár J. Role of structural variations of polysaccharide antigens in the pathogenicity of Gram-negative bacteria. Clin Microbiol Infect. 2008;14:200–6. doi: 10.1111/j.1469-0691.2007.01876.x. [DOI] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira A, dos Santos SC, Santos PM, et al. Proteomic profiling of Burkholderia cenocepacia clonal isolates with different virulence potential retrieved from a cystic fibrosis patient during chronic lung infection. PLoS One. 2013;8:e83065. doi: 10.1371/journal.pone.0083065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira A, Santos PM, Coutinho CP, et al. Quantitative proteomics (2-D DIGE) reveals molecular strategies employed by Burkholderia cenocepacia to adapt to the airways of cystic fibrosis patients under antimicrobial therapy. Proteomics. 2011;11:1313–28. doi: 10.1002/pmic.201000457. [DOI] [PubMed] [Google Scholar]
- Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62:596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3:144–56. doi: 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- Malnick SD, Melzer E, Attali M, et al. Helicobacter pylori: friend or foe? World J Gastroenterol. 2014;20:8979–85. doi: 10.3748/wjg.v20.i27.8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamat U, Skurnik M, Bengoechea JA. LPS core oligosaccharide biosynthesis and assembly. In: Knirel YA, Valvano MA, editors. Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Internaction with Host Cells. Wien: Springer; 2011. pp. 237–73. [Google Scholar]
- Martin SL, Edbrooke MR, Hodgman TC, et al. Lewis X biosynthesis in Helicobacter pylori. Molecular cloning of an α(1,3)-fucosyltransferase gene. J Biol Chem. 1997;272:21349–56. doi: 10.1074/jbc.272.34.21349. [DOI] [PubMed] [Google Scholar]
- Mathee K, Ciofu O, Sternberg C, et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology. 1999;145:1349–57. doi: 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- Mathee K, Narasimhan G, Valdes C, et al. Dynamics of Pseudomonas aeruginosa genome evolution. P Natl Acad Sci USA. 2008;105:3100–5. doi: 10.1073/pnas.0711982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsby-Baltzer I, Mielniczuk Z, Larsson L, et al. Lipid A in Helicobacter pylori. Infect Immun. 1992;60:4383–7. doi: 10.1128/iai.60.10.4383-4387.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, Sherman DJ, Simpson BW, et al. Lipopolysaccharide transport to the cell surface: periplasmic transport and assembly into the outer membrane. Philos Trans R Soc B. 2015;370 doi: 10.1098/rstb.2015.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mégraud F, Bessède E, Varon C. Helicobacter pylori infection and gastric carcinoma. Clin Microbiol Infect. 2015;9:222–3. doi: 10.1016/j.cmi.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Miller AK, Brannon MK, Stevens L, et al. PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrob Agents Ch. 2011;55:5761–9. doi: 10.1128/AAC.05391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira NP, Madeira A, Moreira AS, et al. Genomic expression analysis reveals strategies of Burkholderia cenocepacia to adapt to cystic fibrosis patients' airways and antimicrobial therapy. PLoS One. 2011;6:e28831. doi: 10.1371/journal.pone.0028831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro MA, Chan KH, Rasko DA, et al. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. Molecular mimicry between H. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. J Biol Chem. 1998;273:11533–43. doi: 10.1074/jbc.273.19.11533. [DOI] [PubMed] [Google Scholar]
- Moran AP. Relevance of fucosylation and Lewis antigen expression in the bacterial gastroduodenal pathogen Helicobacter pylori. Carbohydr Res. 2008;343:1952–65. doi: 10.1016/j.carres.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Moran AP, Aspinall GO. Unique structural and biological features of Helicobacter pylori lipopolysaccharides. Prog Clin Biol Res. 1998;397:37–49. [PubMed] [Google Scholar]
- Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol. 1997;179:6453–63. doi: 10.1128/jb.179.20.6453-6463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran AP, Prendergast MM, Appelmelk BJ. Molecular mimicry of host structures by bacterial lipopolysaccharides and its contribution to disease. FEMS Immunol Med Mic. 1996;16:105–15. doi: 10.1111/j.1574-695X.1996.tb00127.x. [DOI] [PubMed] [Google Scholar]
- Moran AP, Sturegård E, Sjunnesson H, et al. The relationship between O-chain expression and colonisation ability of Helicobacter pylori in a mouse model. FEMS Immunol Med Mic. 2000;29:263–70. doi: 10.1111/j.1574-695X.2000.tb01532.x. [DOI] [PubMed] [Google Scholar]
- Moreira AS, Coutinho CP, Azevedo P, et al. Burkholderia dolosa phenotypic variation during the decline in lung function of a cystic fibrosis patient during 5.5 years of chronic colonization. J Med Microbiol. 2014;63:594–601. doi: 10.1099/jmm.0.069849-0. [DOI] [PubMed] [Google Scholar]
- Morelli G, Didelot X, Kusecek B, et al. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6:e1001036. doi: 10.1371/journal.pgen.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz SM, Ernst RK. The role of Pseudomonas lipopolysaccharide in cystic fibrosis airway infection. Subcell Biochem. 2010;53:241–53. doi: 10.1007/978-90-481-9078-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz SM, Ernst RK, Miller SI. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol. 2004;186:575–9. doi: 10.1128/JB.186.2.575-579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat E, Paterson S, Fothergill JL, et al. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Resp Crit Care. 2011;183:1674–9. doi: 10.1164/rccm.201009-1430OC. [DOI] [PubMed] [Google Scholar]
- Moyano AJ, Luján AM, Argaraña CE, et al. MutS deficiency and activity of the error-prone DNA polymerase IV are crucial for determining mucA as the main target for mucoid conversion in Pseudomonas aeruginosa. Mol Microbiol. 2007;64:547–59. doi: 10.1111/j.1365-2958.2007.05675.x. [DOI] [PubMed] [Google Scholar]
- Muotiala A, Helander IM, Pyhala L, et al. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun. 1992;60:1714–6. doi: 10.1128/iai.60.4.1714-1716.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray GL, Attridge SR, Morona R. Altering the length of the lipopolysaccharide O antigen has an impact on the interaction of Salmonella enterica serovar Typhimurium with macrophages and complement. J Bacteriol. 2006;188:2735–9. doi: 10.1128/JB.188.7.2735-2739.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol. 2013;11:467–81. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini R, Savio A, Poiesi C, et al. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology. 1996;111:655–65. doi: 10.1053/gast.1996.v111.pm8780570. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol R. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson C, Skoglund A, Moran AP, et al. Lipopolysaccharide diversity evolving in Helicobacter pylori communities through genetic modifications in fucosyltransferases. PLoS One. 2008;3:e3811. doi: 10.1371/journal.pone.0003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki EM, O'Brien JP, Brodbelt JS, et al. Extracellular zinc induces phosphoethanolamine addition to Pseudomonas aeruginosa lipid A via the ColRS two-component system. Mol Microbiol. 2015;97:166–78. doi: 10.1111/mmi.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt MA, Goldberg JB, Hogardt M, et al. Metabolic network analysis of Pseudomonas aeruginosa during chronic cystic fibrosis lung infection. J Bacteriol. 2010;192:5534–48. doi: 10.1128/JB.00900-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, et al. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–4. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Otero LL, Ruiz VE, Perez Perez GI. Helicobacter pylori: the balance between a role as colonizer and pathogen. Best Pract Res Cl Ga. 2014;28:1017–29. doi: 10.1016/j.bpg.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Paixão TA, Roux CM, den Hartigh AB, et al. Establishment of systemic Brucella melitensis infection through the digestive tract requires urease, the type IV secretion system, and lipopolysaccharide O antigen. Infect Immun. 2009;77:4197–208. doi: 10.1128/IAI.00417-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Parkins MD, Floto RA. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J Cyst Fibros. 2015;14:293–304. doi: 10.1016/j.jcf.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Perepelov AV, Liu B, Guo D, et al. Structure elucidation of the O-Antigen of Salmonella enterica O51 and its structural and genetic relation to the O-Antigen of Escherichia coli O23. Biochemistry. 2011;76:774–9. doi: 10.1134/S0006297911070078. [DOI] [PubMed] [Google Scholar]
- Pérez-Pérez GI, Shepherd VL, Morrow JD, et al. Activation of human THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect Immun. 1995;63:1183–7. doi: 10.1128/iai.63.4.1183-1187.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl MA, Romero-Gallo J, Guruge JL, et al. Host-dependent Lewis (Le) antigen expression in Helicobacter pylori cells recovered from Leb-transgenic mice. J Exp Med. 2009;206:3061–72. doi: 10.1084/jem.20090683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. Efflux-mediated antimicrobial resistance. J Antimicrob Chemoth. 2005;56:20–51. doi: 10.1093/jac/dki171. [DOI] [PubMed] [Google Scholar]
- Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11:77. doi: 10.1186/1478-811X-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prost LR, Miller SI. The Salmonellae PhoQ sensor: mechanisms of detection of phagosome signals. Cell Microbiol. 2008;10:576–82. doi: 10.1111/j.1462-5822.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- Raetz CR, Reynolds CM, Trent MS, et al. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Wilson TJ, Zopf D, et al. Lewis antigen expression and stability in Helicobacter pylori isolated from serial gastric biopsies. J Infect Dis. 2000;181:1089–95. doi: 10.1086/315354. [DOI] [PubMed] [Google Scholar]
- Reeves PR, Cunneen MM, Liu B, et al. Genetics and evolution of the Salmonella galactose-initiated set of O antigens. PLoS One. 2013;8:e69306. doi: 10.1371/journal.pone.0069306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PR, Hobbs M, Valvano MA, et al. Bacterial polysaccharide synthesis and gene nomenclature. Trends Microbiol. 1996;4:495–503. doi: 10.1016/s0966-842x(97)82912-5. [DOI] [PubMed] [Google Scholar]
- Ruan X, Pérez JM, Marolda CL, et al. The WaaL O-antigen lipopolysaccharide ligase has features in common with metal ion-independent inverting glycosyltransferases. Glycobiology. 2012;22:288–99. doi: 10.1093/glycob/cwr150. [DOI] [PubMed] [Google Scholar]
- Rubin EJ, Trent MS. Colonize, evade, flourish: how glyco-conjugates promote virulence of Helicobacter pylori. Gut Microbes. 2013;4:439–53. doi: 10.4161/gmic.25721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadovskaya I, Brisson JR, Thibault P, et al. Structural characterization of the outer core and the O-chain linkage region of lipopolysaccharide from Pseudomonas aeruginosa serotype O5. Eur J Biochem. 2000;267:1640–50. doi: 10.1046/j.1432-1327.2000.01156.x. [DOI] [PubMed] [Google Scholar]
- Salaün L, Ayraud S, Saunders NJ. Phase variation mediated niche adaptation during prolonged experimental murine infection with Helicobacter pylori. Microbiology. 2005;151:917–23. doi: 10.1099/mic.0.27379-0. [DOI] [PubMed] [Google Scholar]
- Saldías MS, Ortega X, Valvano MA. Burkholderia cenocepacia O antigen lipopolysaccharide prevents phagocytosis by macrophages and adhesion to epithelial cells. J Med Microbiol. 2009;58:1542–8. doi: 10.1099/jmm.0.013235-0. [DOI] [PubMed] [Google Scholar]
- Sanders LH, Rockel A, Lu H, et al. Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J Bacteriol. 2006;188:8573–85. doi: 10.1128/JB.01481-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder TH, Lee MM, Yacono PW, et al. CFTR is a pattern recognition molecule that extracts Pseudomonas aeruginosa LPS from the outer membrane into epithelial cells and activates NF-kappa B translocation. P Natl Acad Sci USA. 2002;99:6907–12. doi: 10.1073/pnas.092160899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab U, Abdullah LH, Perlmutt OS, et al. Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect Immun. 2014;82:4729–45. doi: 10.1128/IAI.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semeraro N, Montemurro P, Piccoli C, et al. Effect of Helicobacter pylori lipopolysaccharide (LPS) and LPS derivatives on the production of tissue factor and plasminogen activator inhibitor type 2 by human blood mononuclear cells. J Infect Dis. 1996;174:1255–60. doi: 10.1093/infdis/174.6.1255. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, Gu RF, Gao N. Dimerization of isolated Pseudomonas aeruginosa lipopolysaccharide transporter component LptA. Biochem Bioph Res Co. 2014;450:1327–32. doi: 10.1016/j.bbrc.2014.06.138. [DOI] [PubMed] [Google Scholar]
- Silipo A, Molinaro A, Cescutti P, et al. Complete structural characterization of the lipid A fraction of a clinical strain of B. cepacia genomovar I lipopolysaccharide. Glycobiology. 2005;15:561–70. doi: 10.1093/glycob/cwi029. [DOI] [PubMed] [Google Scholar]
- Silipo A, Molinaro A, Ierano T, et al. The complete structure and pro-inflammatory activity of the lipooligosaccharide of the highly epidemic and virulent Gram-negative bacterium Burkholderia cenocepacia ET-12 (strain J2315) Chemistry. 2007;13:3501–11. doi: 10.1002/chem.200601406. [DOI] [PubMed] [Google Scholar]
- Simpson BW, May JM, Sherman DJ, et al. Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos Trans R Soc B. 2015;370 doi: 10.1098/rstb.2015.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund A, Bäckhed HK, Nilsson C, et al. A changing gastric environment leads to adaptation of lipopolysaccharide variants in Helicobacter pylori populations during colonization. PLoS One. 2009;4:e5885. doi: 10.1371/journal.pone.0005885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Buckley DG, Wu Z, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. P Natl Acad Sci USA. 2006;103:8487–92. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandeo P, Dehò G, Polissi A. Lipopolysacchairde export to the outer membrane. In: Knirel Y, Valvano MA, editors. Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Interaction With Host Cells. Wien: Springer; 2011. pp. 311–37. [Google Scholar]
- Sperandeo P, Villa R, Martorana AM, et al. New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J Bacteriol. 2011;193:1042–53. doi: 10.1128/JB.01037-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramulu DD, Lunsdorf H, Lam JS, et al. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol. 2005;54:667–76. doi: 10.1099/jmm.0.45969-0. [DOI] [PubMed] [Google Scholar]
- Stead CM, Beasley A, Cotter RJ, et al. Deciphering the unusual acylation pattern of Helicobacter pylori lipid A. J Bacteriol. 2008;190:7012–21. doi: 10.1128/JB.00667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead CM, Zhao J, Raetz CR, et al. Removal of the outer Kdo from Helicobacter pylori lipopolysaccharide and its impact on the bacterial surface. Mol Microbiol. 2010;78:837–52. doi: 10.1111/j.1365-2958.2010.07304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaipisuttikul I, Hittle LE, Chandra R, et al. A divergent Pseudomonas aeruginosa palmitoyltransferase essential for cystic fibrosis-specific lipid A. Mol Microbiol. 2014;91:158–74. doi: 10.1111/mmi.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Cruz J, van der Woude MW. Slipped-strand mispairing can function as a phase variation mechanism in Escherichia coli. J Bacteriol. 2003;185:6990–4. doi: 10.1128/JB.185.23.6990-6994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AX, Karbarz MJ, Wang X, et al. Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A. J Biol Chem. 2004;279:55780–91. doi: 10.1074/jbc.M406480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AX, Whittimore JD, Wyrick PB, et al. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol. 2006;188:4531–41. doi: 10.1128/JB.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trancassini M, Iebba V, Citerà N, et al. Outbreak of Achromobacter xylosoxidans in an italian cystic fibrosis center: genome variability, biofilm production, antibiotic resistance, and motility in isolated strains. Front Microbiol. 2014;5:138. doi: 10.3389/fmicb.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse CC, Mayo-Smith LM, Poltak SR, et al. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. P Natl Acad Sci USA. 2013;110:E250–9. doi: 10.1073/pnas.1207025110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent MS, Pabich W, Raetz CRH, et al. A PhoP/PhoQ-induced lipase (PagL) that catalyzes 3-O-deacylation of lipid A precursors in membranes of Salmonella typhimurium. J Biol Chem. 2001;276:9083–92. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- Uehlinger S, Schwager S, Bernier SP, et al. Identification of specific and universal virulence factors in Burkholderia cenocepacia strains by using multiple infection hosts. Infect Immun. 2009;77:4102–10. doi: 10.1128/IAI.00398-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca-Pacheco S, Paniagua-Contreras GL, Garcia-Gonzalez O, et al. The clinically isolated FIZ15 bacteriophage causes lysogenic conversion in Pseudomonas aeruginosa PAO1. Curr Microbiol. 1999;38:239–43. doi: 10.1007/pl00006794. [DOI] [PubMed] [Google Scholar]
- Valvano MA. Common themes in glycoconjugate assembly using the biogenesis of O-antigen lipopolysaccharide as a model system. Biochemistry. 2011;76:729–35. doi: 10.1134/S0006297911070029. [DOI] [PubMed] [Google Scholar]
- Valvano MA. Genetics and biosynthesis of lipopolysaccharide. In: Tang Y-W, Sussman M, Liu D, et al., editors. Molecular Medical Microbiology. Vol. 1. Amsterdam: Academic Press; 2015. pp. 55–89. [Google Scholar]
- Valvano MA, Messner P, Kosma P. Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology. 2002;148:1979–89. doi: 10.1099/00221287-148-7-1979. [DOI] [PubMed] [Google Scholar]
- Valvano MA, Patel KB, Furlong SE. Genetics, biosynthesis and assembly of O antigen. In: Knirel Y, Valvano MA, editors. Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Interaction With Host Cells. Wein: Springer; 2011. pp. 275–310. [Google Scholar]
- van Belkum A, Scherer S, van Leeuwen W, et al. Variable number of tandem repeats in clinical strains of Haemophilus influenzae. Infect Immun. 1997;65:5017–27. doi: 10.1128/iai.65.12.5017-5027.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude MW, Bäumler AJ. Phase and antigenic variation in bacteria. Clin Microbiol Rev. 2004;17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidigal PG, Dittmer S, Steinmann E, et al. Adaptation of Stenotrophomonas maltophilia in cystic fibrosis: molecular diversity, mutation frequency and antibiotic resistance. Int J Med Microbiol. 2014;304:613–9. doi: 10.1016/j.ijmm.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Walsh AG, Matewish MJ, Burrows LL, et al. Lipopolysaccharide core phosphates are required for viability and intrinsic drug resistance in Pseudomonas aeruginosa. Mol Microbiol. 2000;35:718–27. doi: 10.1046/j.1365-2958.2000.01741.x. [DOI] [PubMed] [Google Scholar]
- Wang G, Ge Z, Rasko DA, et al. Lewis antigens in Helicobacter pylori: biosynthesis and phase variation. Mol Microbiol. 2000;36:1187–96. doi: 10.1046/j.1365-2958.2000.01934.x. [DOI] [PubMed] [Google Scholar]
- Warren AE, Boulianne-Larsen CM, Chandler CB, et al. Genotypic and phenotypic variation in Pseudomonas aeruginosa reveals signatures of secondary infection and mutator activity in certain cystic fibrosis patients with chronic lung infections. Infect Immun. 2011;79:4802–18. doi: 10.1128/IAI.05282-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ME, Burns JL, Smith AL. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology. 2004;150:2947–58. doi: 10.1099/mic.0.27230-0. [DOI] [PubMed] [Google Scholar]
- Wattam AR, Foster JT, Mane SP, et al. Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J Bacteriol. 2014;196:920–30. doi: 10.1128/JB.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West NP, Sansonetti P, Mounier J, et al. Optimization of virulence functions through glucosylation of Shigella LPS. Science. 2005;307:1313–7. doi: 10.1126/science.1108472. [DOI] [PubMed] [Google Scholar]
- Whitfield C, Trent MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem. 2014;83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- WHO. Schistosomes, liver flukes and Helicobacter pylori. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. World Health Organization, International Agency for Research on Cancer. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- Wildschutte H, Preheim SP, Hernandez Y, et al. O-antigen diversity and lateral transfer of the wbe region among Vibrio splendidus isolates. Environ Microbiol. 2010;12:2977–87. doi: 10.1111/j.1462-2920.2010.02274.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Jelsbak L, Marvig RL, et al. Evolutionary dynamics of bacteria in a human host environment. P Natl Acad Sci USA. 2011;108:7481–6. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlosnik JE, Costa PS, Brant R, et al. Mucoid and nonmucoid Burkholderia cepacia complex bacteria in cystic fibrosis infections. Am J Resp Crit Care. 2011;183:67–72. doi: 10.1164/rccm.201002-0203OC. [DOI] [PubMed] [Google Scholar]
- Zlosnik JE, Mori PY, To D, et al. Swimming motility in a longitudinal collection of clinical isolates of Burkholderia cepacia complex bacteria from people with cystic fibrosis. PLoS One. 2014;9:e106428. doi: 10.1371/journal.pone.0106428. [DOI] [PMC free article] [PubMed] [Google Scholar]