

Abstract
Cardiovascular diseases are leading causes for death worldwide. Genetic disposition jointly with traditional risk factors precipitates their manifestation. Whereas the implications of a positive family history for individual risk have been known for a long time, only in the past few years have genome‐wide association studies (GWAS) shed light on the underlying genetic variations. Here, we review these studies designed to increase our understanding of the pathophysiology of cardiovascular diseases, particularly coronary artery disease and myocardial infarction. We focus on the newly established pathways to exemplify the translation from the identification of risk‐related genetic variants to new preventive and therapeutic strategies for cardiovascular disease.
Keywords: atherosclerosis, coronary artery disease, genome‐wide association studies, myocardial infarction
Subject Categories: Cardiovascular System; Chromatin, Epigenetics, Genomics & Functional Genomics
Glossary
- Coronary artery disease
Coronary artery disease is the manifestation of atherosclerosis in the coronary arteries, that is the vessels supplying the myocardium with oxygen and nutrients. Major complications of coronary artery disease include chest pain, myocardial infarction, arrhythmias and heart failure. It is one of the leading causes of death.
- Extracellular matrix
The extracellular matrix is a conglomerate of molecules and cells of often different types. The fibroblasts are the main cell type involved in the formation of the extracellular matrix. In addition to the effects on tissue/organ physical properties, for example elasticity and stiffness, the extracellular matrix also has effects on the behaviour of imbedded cells, that is migration but also gene expression and differentiation.
- Linkage disequilibrium
Linkage disequilibrium describes the occurrence that two alleles at different loci are not independently distributed in a population.
- Precision medicine
The response to a treatment modality may vary, even in patients with the same diagnosis. Precision medicine aims to tailor healthcare to individual patients. It can involve molecular diagnostics but also laboratory tests, imaging, environmental analyses and large‐scale data analysis, for example pattern recognition.
- Single Nucleotide Polymorphisms
Single nucleotide polymorphisms (SNPs) are variations in single nucleotides that occur in the genome with distinct frequencies. Association studies exploit the analysis of SNPs to investigate the association of these genetic variants with a particular disease.
- Statins
Statins are drugs used to lower lipid levels, especially low‐density lipid (LDL) cholesterol. The molecular target of statins is the enzyme HMG‐CoA reductase, a key player in the endogenous production of cholesterol. Statins have proven efficacy in the reduction in both LDL cholesterol and cardiovascular events.
- Systems medicine
Systems medicine is a novel discipline in biomedical research, closely related to systems biology. Systems medicine aims to study the individual in an integrated fashion, incorporating, for example, “‐omics” data, and biochemical and environmental interactions. As a further dimension, changes over time are included in the analyses. A major focus in systems medicine research is the development of computational models.
Introduction
Atherosclerosis is a systemic disorder with high prevalence in both industrialized and developing countries. Its manifestations in the form of cerebrovascular or coronary artery disease (CAD) account for 46% of all deaths in Europe (Nichols et al, 2014). Although progress in cardiovascular medicine has substantially reduced the death toll in the past decades, deeper insight into the pathophysiology of atherosclerosis is mandatory to further improve strategies for the prevention and treatment. Epidemiological studies, especially the Framingham Heart Study, spearheaded the efforts to track down the causes for myocardial infarction (MI) and led to the identification of several predictors for CAD, for example hypertension and hypercholesterolaemia (Kannel et al, 1961) which, together with obesity, smoking and diabetes, are condensed under the term “modifiable risk factors”, as they can be addressed by lifestyle changes and therapeutic interventions (Yusuf et al, 2004).
It is also known that CAD has a high heritability that is by far larger than the genetic influence on the modifiable risk factors (Marenberg et al, 1994). However, for many decades, the attempts to identify the genetic variants underlying this heritability were unsuccessful (Mayer et al, 2007). Over the last few years, methodological progress and collaborative efforts have changed this scenario. The invention of arrays for the low‐cost genotyping of hundreds of thousands of patient genomic markers, annotation of these markers in the human genome together with meta‐analyses of multiple case–control samples have enabled the identification of numerous genetic variants that are highly and reproducibly associated with CAD and MI risk.
Analysis of genotype frequencies with the aim to detect variants that occur at different frequency in cases and controls is now being carried out at the genome‐wide level. Variants found in such genome‐wide association studies (GWAS) to be associated with disease risk are not necessarily located in the coding regions. The fact that chromosomal loci tagged by respective variants do play a causal role in modulating risk is based on very strong statistical significance; however, the mechanisms involved remain often unclear. The hypothesis‐free method of the GWAS approach thereby differs from other emerging techniques such as exome sequencing (Table 1), where specific genes are being tested for association with disease. Moreover, the variant with the strongest association in a GWAS does not necessarily represent the causal one; it may rather be in linkage disequilibrium with a functionally relevant allele that is located in close proximity at this locus. Nevertheless, the GWAS approach has proven to be a powerful tool: as discussed in this review, more than 50 single nucleotide polymorphisms (SNPs) tagging distinct chromosomal regions have been annotated so far to be associated with CAD with high statistical significance. Here, we focus on variants that allow inference to the causal genes and molecular pathways and thus improve our understanding of the pathophysiology of CAD and MI, which may ultimately result in better treatment and prevention of these deadly diseases.
Table 1.
Association approaches in cardiovascular research (*) can be grouped into haplotypes, which may allow to derive more genetic information or imputation of other SNPs; #, can be further categorized if functional implications are known, for example effect of expression level = eSNP; +; may vary in different tissues or conditions
| GWAS | EWAS | Exome array | Exome sequencing | Genome sequencing | |
|---|---|---|---|---|---|
| Focus | Common SNPs*# | CpG methylation sites | Exonic variants | Coding sequences of all genes (high coverage) | Sequences of all genes (high coverage) |
| N of signals | ~8·106 | ~500·103 | ~220·103 | ~30·106 | 3·109 |
| Coverage | Whole genome | Whole genome+ | All genes | All genes | Whole genome |
| Costs per person [$] | ~100 | ~300 | ~100 | ~600 | > 1,000 |
bp, base pairs; EWAS, epigenome‐wide association study; GWAS, genome‐wide association study; N, number.
Genome‐wide association studies in coronary artery disease and myocardial infarction
In 2007, the first GWAS were published for CAD, all of which identified a locus on chromosome 9p21 to be genome‐wide significantly associated with the disease (Helgadottir et al, 2007; McPherson et al, 2007; Samani et al, 2007). This locus became the first claim of what has later been called a gold rush of CAD genetics. Interestingly, the chromosome 9p21 locus still has an outstanding position as it depicts the genetic variant with the highest population‐attributable risk (Table 2; Schunkert et al, 2008).
Table 2.
Loci identified to be associated with CAD/MI either by genome‐/exome‐wide association studies
| Chr. | Lead SNP | AF | OR | Gene at chr. locus | Bioinf. annot. | HTN | LIP | References |
|---|---|---|---|---|---|---|---|---|
| 1 | rs11206510 | T (0.82) | 1.08 | PCSK9 | No data | + | Myocardial Infarction Genetics Consortium (2009), Abifadel et al (2003), Cohen et al (2006), Teslovich et al (2010) | |
| rs17114036 | A (0.91) | 1.17 | PPAP2B | No data | Schunkert et al (2011) | |||
| rs17465637 | C (0.74) | 1.14 | MIA3 | MIA3, AIDA, C1orf58 | Samani et al (2007), Schunkert et al (2011) | |||
| rs599839 | A (0.78) | 1.11 | SORT1 | SORT1 | + | Schunkert et al (2011), Teslovich et al (2010), Samani et al (2007) | ||
| rs4845625 | T (0.47) | 1.06 | IL6R | IL6R, ATP8B2, CHTOP, UBAP2L | CARDIoGRAMplusC4D Consortium (2013) | |||
| 2 | rs6544713 | T (0.30) | 1.06 | ABCG5/ABCG8 | No data | + | Schunkert et al (2011), Teslovich et al (2010), IBC 50K CAD Consortium (2011) | |
| rs6725887 | C (0.15) | 1.14 | WDR12 | NA | Schunkert et al (2011), Myocardial Infarction Genetics Consortium (2009) | |||
| rs515135 | G (0.83) | 1.07 | APOB | No data | + | CARDIoGRAMplusC4D Consortium (2013), Teslovich et al (2010) | ||
| rs2252641 | G (0.46) | 1.06 | ZEB2 | No data | CARDIoGRAMplusC4D Consortium (2013) | |||
| rs1561198 | A (0.45) | 1.06 | VAMP5‐VAMP8‐GGCX | VAMP5/8 | CARDIoGRAMplusC4D Consortium (2013) | |||
| 3 | rs2306374 | C (0.18) | 1.12 | MRAS | MRAS, CEP70 | Erdmann et al (2009), Schunkert et al (2011) | ||
| 4 | rs7692387 | G (0.81) | 1.08 | GUCY1A3 | no data | + | CARDIoGRAMplusC4D Consortium (2013), Erdmann et al (2013), International Consortium for Blood Pressure Genome‐Wide Association Studies (2011) | |
| rs1878406 | T (0.15) | 1.10 | EDNRA | NA | CARDIoGRAMplusC4D Consortium (2013) | |||
| rs17087335 | T (0.21) | 1.06 | REST‐NOA1 | NA | Nikpay et al (2015) | |||
| 5 | rs2706399 | G (0.51) | 1.07 | IL5 | NA | IBC 50K CAD Consortium (2011) | ||
| rs273909 | C (0.14) | 1.07 | SLC22A4‐A5 | No data | CARDIoGRAMplusC4D Consortium (2013) | |||
| 6 | rs12526453 | C (0.67) | 1.10 | PHACTR1 | No data | Schunkert et al (2011), Myocardial Infarction Genetics Consortium (2009) | ||
| rs17609940 | G (0.75) | 1.07 | ANKS1A | NA | Schunkert et al (2011) | |||
| rs12190287 | C (0.62) | 1.08 | TCF21 | No data | Schunkert et al (2011) | |||
| rs3798220 | C (0.02) | 1.51 | LPA, SLC22A3, LPAL2 | LPA | + | Tregouet et al (2009), Schunkert et al (2011), Teslovich et al (2010) | ||
| rs10947789 | T (0.76) | 1.07 | KCNK5 | No data | CARDIoGRAMplusC4D Consortium (2013) | |||
| rs4252120 | T (0.73) | 1.07 | PLG | PLG, LPAL2 | CARDIoGRAMplusC4D Consortium (2013) | |||
| 7 | rs10953541 | C (0.80) | 1.08 | BCAP29 | No data | Coronary Artery Disease C4D Genetics Consortium (2011) | ||
| rs11556924 | C (0.62) | 1.09 | ZC3HC1 | ZC3HC1 | Schunkert et al (2011) | |||
| rs2023938 | G (0.10) | 1.08 | HDAC9 | No data | CARDIoGRAMplusC4D Consortium (2013) | |||
| rs3918226 | T (0.06) | 1.14 | NOS3 | NOS3 | Nikpay et al (2015) | |||
| 8 | rs2954029 | A (0.55) | 1.06 | TRIB1 | No data | + | IBC 50K CAD Consortium (2011), CARDIoGRAMplusC4D Consortium (2013), Teslovich et al (2010) | |
| rs264 | G (0.86) | 1.11 | LPL | LPL | + | CARDIoGRAMplusC4D Consortium (2013), Teslovich et al (2010), Stitziel et al (2016) | ||
| 9 | rs4977574 | G (0.46) | 1.29 | 9p21.3 | ANRIL | Samani et al (2007), McPherson et al (2007), Helgadottir et al (2007), Schunkert et al (2011), Holdt et al (2010) | ||
| rs579459 | C (0.21) | 1.10 | ABO | NA | + | Schunkert et al (2011), Teslovich et al (2010), Reilly et al (2011) | ||
| rs111245230 | C (0.04) | 1.14 | SVEP1 | NA | Stitziel et al (2016) | |||
| 10 | rs2505083 | C (0.38) | 1.07 | KIAA1462 | KIAA1462 | Erdmann et al (2009), Coronary Artery Disease C4D Genetics Consortium (2011) | ||
| rs1746048 | C (0.87) | 1.09 | CXCL12 | NA | Samani et al (2007), Schunkert et al (2011) | |||
| rs1412444 | T (0.42) | 1.09 | LIPA | NA | Coronary Artery Disease C4D Genetics Consortium (2011) | |||
| rs12413409 | G (0.89) | 1.12 | CYP17A1, NT5C2 | NA | + | Schunkert et al (2011), Newton‐Cheh et al (2009), Levy et al (2009) | ||
| 11 | rs974819 | T (0.32) | 1.07 | PDGFD | No data | Coronary Artery Disease C4D Genetics Consortium (2011) | ||
| rs964184 | G (0.13) | 1.13 | APOA1‐C3‐A4‐A5 | No data | + | Schunkert et al (2011), The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute (2014), Do et al (2015, 2013) | ||
| 12 | rs10840293 | A (0.55) | 1.06 | SWAP70 | NA | Nikpay et al (2015) | ||
| rs3184504 | T (0.44) | 1.07 | SH2B3, HNF1A | SH2B3, FLJ21127, ATXN2 | + | + | Schunkert et al (2011), Teslovich et al (2010), Newton‐Cheh et al (2009), Levy et al (2009), Gudbjartsson et al (2009) | |
| rs11830157 | G (0.36) | 1.12 | KSR2 | NA | Nikpay et al (2015) | |||
| 13 | rs4773144 | G (0.44) | 1.07 | COL4A1, COL4A2 | No data | Schunkert et al (2011) | ||
| rs9319428 | A (0.32) | 1.06 | FLT1 | No data | CARDIoGRAMplusC4D Consortium (2013) | |||
| 14 | rs2895811 | C (0.43) | 1.07 | HHIPL1 | YY1 | Schunkert et al (2011) | ||
| 15 | rs3825807 | A (0.57) | 1.08 | ADAMTS7 | ADAMTS7 | Schunkert et al (2011), Reilly et al (2011), Coronary Artery Disease C4D Genetics Consortium (2011) | ||
| rs17514846 | A (0.44) | 1.07 | FURIN‐FES | FURIN, MAN2A2 | + | CARDIoGRAMplusC4D Consortium (2013), International Consortium for Blood Pressure Genome‐Wide Association Studies (2011) | ||
| rs56062135 | C (0.79) | 1.07 | SMAD3 | NA | Nikpay et al (2015) | |||
| rs8042271 | G (0.9) | 1.10 | MFGE8‐ABHD2 | NA | Nikpay et al (2015) | |||
| 17 | rs216172 | C (0.37) | 1.07 | SMG6‐SRR | NA | Schunkert et al (2011) | ||
| rs12936587 | G (0.56) | 1.07 | PEMT, RASD1, SMCR3 | TOM1L2 | Schunkert et al (2011) | |||
| rs46522 | T (0.53) | 1.06 | UBE2Z, GIP, ATP5G1 | NA | Schunkert et al (2011) | |||
| rs7212798 | C (0.15) | 1.08 | BCAS3 | NA | Nikpay et al (2015) | |||
| 18 | rs663129 | A (0.26) | 1.06 | PMAIP1‐MC4R | NA | Nikpay et al (2015) | ||
| 19 | rs116843064 | G (0.98) | 1.14 | ANGTPL4 | NA | + | Teslovich et al (2010), Stitziel et al (2016) | |
| rs1122608 | G (0.77) | 1.14 | LDLR | SMARCA4 | + | Myocardial Infarction Genetics Consortium (2009), Schunkert et al (2011), Teslovich et al (2010), Do et al (2015) | ||
| rs2075650 | G (0.14) | 1.14 | APOE | TOMM40 | + | IBC 50K CAD Consortium (2011), Teslovich et al (2010) | ||
| rs12976411 | A (0.91) | 1.33 | ZNF507‐ LOC400684 | NA | Nikpay et al (2015) | |||
| 21 | rs9982601 | T (0.15) | 1.18 | MRPS6, SLC5A3, KCNE2 | No data | Myocardial Infarction Genetics Consortium (2009) | ||
| 22 | rs180803 | G (0.97) | 1.20 | POM121L9P‐ADORA2A | NA | Nikpay et al (2015) |
Chr., chromosome/chromosomal; SNP, single nucleotide polymorphism; AF, risk allele and its frequency; OR, odds ratio; HTN, associated with blood pressure; LIP, associated with LDL cholesterol/lipoprotein (a)/triglycerides; Bioinf. annot., bioinformatics annotation according to Braenne et al (2015) or others (Musunuru et al, 2010; Salvi et al, 2013); no data, no eQTL data or non‐coding variant; NA, not analysed.
In the first years of GWAS discoveries, identification of further loci came from individual genome‐wide association studies (Erdmann et al, 2009; Myocardial Infarction Genetics Consortium et al, 2009; Tregouet et al, 2009; IBC 50K CAD Consortium, 2011; Wang et al, 2011), whereas more recently the formation of large, international consortia has accumulated a sufficient statistical power for new discoveries. The joint forces of the CARDIoGRAM (Schunkert et al, 2011), C4D (Coronary Artery Disease C4D Genetics Consortium, 2011) and finally the CARDIoGRAMplusC4D (CARDIoGRAMplusC4D Consortium et al, 2013; Nikpay et al, 2015) consortium allowed the analysis of up to 180,000 individuals, about half of which had CAD. Interestingly, consequent studies revealed a strong relationship between the numbers of individuals investigated and the numbers of genome‐wide significant variants detected by the GWAS approach (Fig 1). This close correlation makes it likely that many more variants may eventually achieve genome‐wide significant association once even larger sample sets will be studied.
Figure 1. Numbers of individuals and SNPs investigated by GWAS influence the power for the detection of associated loci.
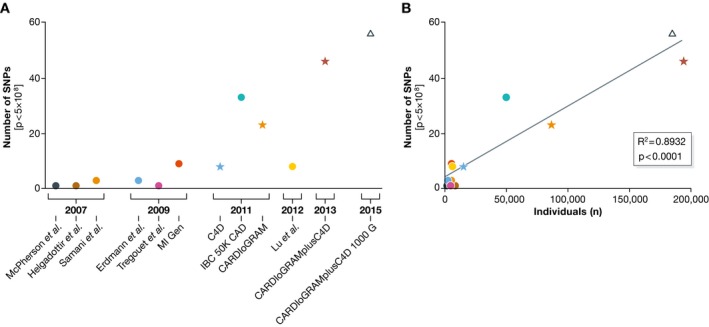
The number of investigated individuals in the discovery phases of the relative GWAS/meta‐analyses was plotted against the number of variants reaching genome‐wide level of significance in the overall analysis of the studies. Association between individuals in the discovery phase and the number of hits was evaluated by linear regression. P < 0.05 was considered as statistically significant. GraphPad Prism version 6.0c for Mac OS X (GraphPad Software, La Jolla, CA, USA) was used. (A) The number of SNPs detected at genome‐wide significant level for coronary artery disease in consecutive studies. (B) The number of SNPs detected with genome‐wide significance after replication correlates with the number of individuals included in the discovery studies. Symbols denote the numbers of genotyped SNPs [dots: ≤ 500,000 SNPs (Samani et al, 2007; McPherson et al, 2007; Helgadottir et al, 2007; Myocardial Infarction Genetics Consortium, 2009; Erdmann et al, 2009; Tregouet et al, 2009; IBC 50K CAD Consortium, 2011; Lu et al, 2012); asterisks: 2,500,000 SNPs (Coronary Artery Disease C4D Genetics Consortium, 2011; Schunkert et al, 2011; CARDIoGRAMplusC4D Consortium et al, 2013); arrow: 940,000 SNPs (Nikpay et al, 2015)].
Another means to increase the power of GWAS is by denser granularity of SNPs. This progress was initially driven by better arrays allowing the genotyping of more and more SNPs. More recently, improvement came from imputation of SNPs based on the 1,000 genomes sequencing data set (1000 Genomes Project Consortium et al, 2012). It is now possible to study more than 15 Mio. distinct SNPs in human DNA for association with disease. Denser genotyping and higher numbers of study subjects increased the number of loci with genome‐wide significant association for CAD to 56 (CARDIoGRAMplusC4D Nikpay et al, 2015). Table 2 gives an overview about the loci thus far identified to be genome‐wide significantly associated with CAD by GWAS.
Almost all risk variants identified by GWAS are commonly found in European populations. This led to the unexpected finding that an average European individual carries tens of these risk alleles. Moreover, almost all are located in non‐coding parts of the genome, which suggests that these variants are more likely to affect gene regulation rather than protein structure. Indeed, annotation of the risk alleles revealed that the majority are located in chromosomal regions relevant for gene regulation. The identification of the underlying pathophysiological mechanism(s) thus requires annotation of the affected gene(s), which may be challenging (Braenne et al, 2015). The chromosome 9p21 locus is a prominent example: whereas initial studies focused on the two genes situated at the locus, CDKN2A and CDKN2B, current studies rather point to radically new disease mechanisms. Indeed, it appears that the risk/non‐risk alleles at the 9p21 locus relate to different isoforms of ANRIL and subsequently to preferred synthesis of non‐circular/circular forms of this long non‐coding RNA, which affect via ribosomal function the expression of multiple genes leading ultimately to opposing effects on cell proliferation and apoptosis (Holdt et al, 2010, 2013).
Exome‐wide association study in coronary artery disease
The annotation to a gene is easier to achieve if the SNP leading a GWAS signal causes a mutation in the coding region presumably influencing protein function (Table 3). Important examples that will be discussed in more detail below include the PCSK9 and GUCY1A3 genes. Therefore, the idea arose to systematically investigate coding variants.
Table 3.
Genes associated with CAD/MI with lead SNPs, or proxy SNPs of the respective lead SNP, causing a deleterious variation, and genes identified by beneficial/deleterious mutations to be associated with CAD/MI
| Chr. | Gene | AA variation/type of mutation | Risk | References |
|---|---|---|---|---|
| 1 | CCDC181 | p.F238I | ↑ | Braenne et al (2015) |
| LMOD1 | p.T295M | ↑ | Braenne et al (2015) | |
| PCSK9 |
p.S127R/p.F216L p.Y142X/p.C679X |
↑ ↓ |
Abifadel et al (2003) Cohen et al (2006) |
|
| 2 | WDR12 | p.I75V | ↑ | Braenne et al (2015) |
| TNS1 | p.W1197R | ↑ | Braenne et al (2015) | |
| 3 | MAP4 | p.V628L/p.S427Y | ↑ | Braenne et al (2015) |
| 4 | GUCY1A3 | Nonsense/p.Gly537Arg | ↑ | Erdmann et al (2013) |
| 7 | ZC3HC1 | p.R363H | ↑ | Braenne et al (2015) |
| NPC1L1 | Nonsense/frameshift/splice site | ↓ | Myocardial Infarction Genetics Consortium Investigators et al (2014) | |
| 8 | LPL |
p.D36N nonsense |
↑ ↓ |
Stitziel et al (2016) |
| 9 | SVEP1 | p.D2702G | ↑ | Stitziel et al (2016) |
| 10 | BMPR1A | p.P2T | ↑ | Braenne et al (2015) |
| 11 | APOA5 | Non‐synonymous | ↑ | Do et al (2015) |
| APOC3 | Non‐synonymous/splice site/null | ↓ | The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute (2014) | |
| 12 | HNF1A | p.I27L | ↑ | Braenne et al (2015) |
| 17 | LRRC48 | p.R191W | ↑ | Braenne et al (2015) |
| 19 | LDLR | Non‐synonymous/null | ↑ | Do et al (2015) |
| ANGPTL4 | p.E40K | ↓ | Stitziel et al (2016) | |
| 20 | MYH7B | p.A25T | ↑ | Braenne et al (2015) |
| PROCR | p.S219G | ↑ | Braenne et al (2015) |
The results of the first exome‐wide association study on CAD have been just published (Stitziel et al, 2016). The authors included more than 72,000 cases and more than 120,000 controls, respectively. They confirmed signals in the LPA and PCSK9 genes but also identified novel variants in genes thus far not associated with CAD: a missense variant in the ANGPTL4 gene and a missense variant in the sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1 gene (SVEP1). SVEP1 was not associated with lipid levels, but with both systolic and diastolic blood pressure leading to an increase of 0.94 and 0.57 mmHg, respectively, in those carrying the risk allele (Stitziel et al, 2016). Moreover, SVEP1 has been investigated in septic shock and endotoxaemia (Nakada et al, 2015). Specifically, the inhibition of SVEP1 expression by RNAi was found to regulate the expression of adhesion molecules on the surface of endothelial cells (Schwanzer‐Pfeiffer et al, 2010). One paper also identified SVEP1 as the physiological ligand of integrin α9β1. Integrins are key molecules in the interaction of different cell types through the extracellular matrix (ECM) such that it may be feasible that SVEP1 influences cell adhesion mediated by integrin α9β1 (Sato‐Nishiuchi et al, 2012), which could be of pathophysiological relevance in CAD.
Pathophysiological insights
Bioinformatics analyses and experimental studies revealed the involvement of the genes affected by risk alleles in different pathophysiological pathways (Figs 2 and 3). In the following paragraphs, we highlight the effects on LDL cholesterol and triglyceride metabolism, blood pressure, NO/cGMP signalling, vascular remodelling and inflammation.
Figure 2. Genetic variation and pathophysiological pathways in atherosclerosis.
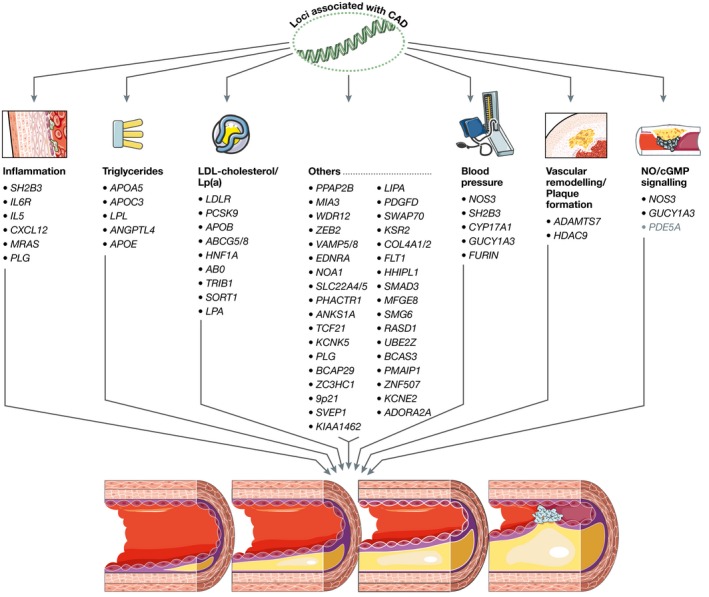
Figure 3. Genes involved in different pathophysiological pathways extracted from the 56 loci listed in Table 2 .
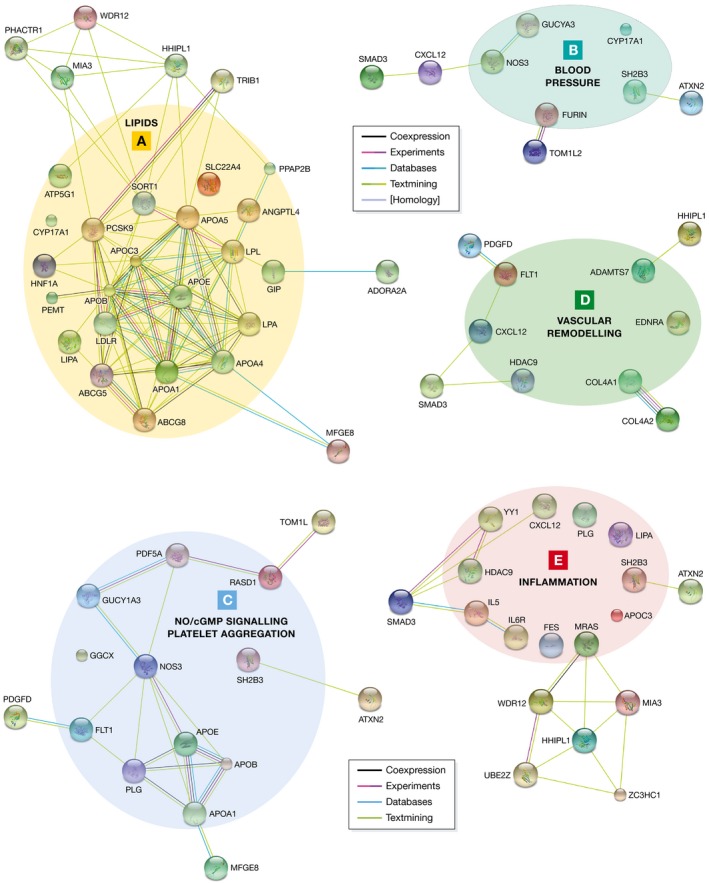
Functional annotations were collected from (i) the ConsensusPathDB database (http://consensuspathdb.org; Kamburov et al, 2013), (ii) the AmiGO 2 Gene Ontology (GO) browser (http://amigo.geneontology.org/amigo; Carbon et al, 2009), as well from (iii) the biomedical literature. Known and predicted associations among the genes within each functional category/pathway were retrieved from the STRING database (http://string-db.org; Franceschini et al, 2013) using default parameters. (A) Lipid metabolism. (B) Blood pressure. (C) NO‐cGMP signalling/platelet aggregation. (D) Vascular remodelling. (E) Inflammation.
LDL cholesterol metabolism
Of the 56 loci identified so far, a number affect lipid metabolism as an intermediary step, for example the LDL cholesterol receptor (LDLR) or LPA loci (Table 2). Interestingly, some of the genes found to associate with hypercholesterolaemia and CAD had never been implicated in these disorders before. Consequently, the novel loci suggest unexpected mechanisms affecting lipid metabolism. A prominent example is sortilin 1, encoded by the SORT1 locus on chromosome 1 (Samani et al, 2007; Musunuru et al, 2010). Another example is PCSK9, which is discussed in more detail below because of the striking therapeutic options (Fig 3A).
Triglyceride metabolism
Lipoprotein lipase (LPL), also genome‐wide significantly associated with CAD, seems to be a key player in CAD genetics, too. Under physiological conditions, LPL reduces triglyceride levels via the hydrolysis of lipoprotein‐bound triglycerides. LPL activity is increased by apolipoprotein A‐V (APOA5), but reduced by apolipoprotein C‐III (APOC3) and angiopoietin‐like 4 (ANGPTL4), all of which have been associated with CAD in a genome‐wide significant fashion (Table 3). It has been mentioned above that the ANGPTL4 gene has just recently been identified in an exome‐wide association study to be associated with CAD. Indeed, the protective allele p.E40K represents a loss‐of‐function variant. Our consortium was able to demonstrate that this and other loss‐of‐function variants led to significantly reduced triglyceride levels, whereas LDL and HDL cholesterol levels were not affected. In accordance, loss‐of‐function variants were associated with a lower risk for CAD. This becomes even more apparent since we also detected a missense variant in the LPL gene, which led to a 20% reduction in LPL activity, to be associated with increased risk for CAD (Stitziel et al, 2016). The findings for LPL and ANGPTL4 confirm recent results for two other regulators of LPL activity, APOA5 and APOC3, in that rare APOA5 mutations increase both plasma triglyceride levels and risk of CAD (Do et al, 2015), whereas rare loss‐of‐function mutations in the APOC3 gene have opposite effects (The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, 2014). Taken together, LPL activity seems to have a central role in triglyceride metabolism (Fig 3A) and consecutively CAD risk (Fig 4). Therefore, these proteins could represent possible targets for therapeutic intervention. Accordingly, attention should be paid to ongoing studies regarding APOC3 inhibitors, which are discussed in more detail below.
Figure 4. Novel insights into the genetic variation in LDL cholesterol metabolism and therapeutic modulation.
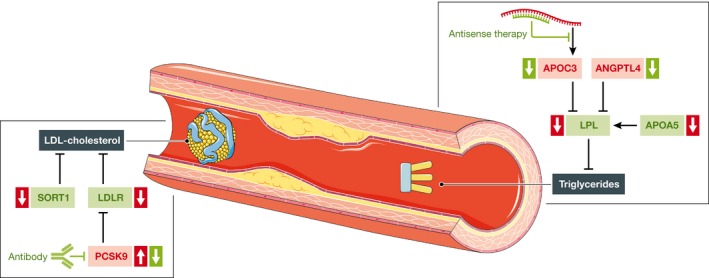
In low‐density lipoprotein (LDL) metabolism, sortilin 1 (SORT1), LDL cholesterol receptor (LDLR) and proprotein convertase subtilisin/kexin type 9 (PCSK9) are exemplarily shown (green, favourable effect regarding LDL cholesterol/triglycerides; red, unfavourable effect regarding LDL cholesterol/triglycerides; ↑, variants increase the risk of CAD; ↓, variants decrease the risk of CAD).
Blood pressure
Thanks to extensive genotyping efforts, several genomic loci have been identified to be associated with blood pressure. A score build on the cumulative effects of these loci displays a strong association with CAD (Levy et al, 2009; Newton‐Cheh et al, 2009; International Consortium for Blood Pressure Genome‐Wide Association Studies et al, 2011). Among the loci identified, there are several which are also genome‐wide significantly associated with CAD (Table 2, Fig 3B). It remains, however, unclear to which extent the effect on CAD is due to an increase in blood pressure given that the effect on blood pressure for each locus is very small (Lieb et al, 2013).
NO/cGMP signalling
Nitric oxide (NO) is an important signalling molecule (Fig 3C) in the cardiovascular system, and the expression of endothelial NO synthase (eNOS), which produces NO from arginine, is reduced in atherosclerosis (Kawashima, 2004). As NO inhibits proatherogenic mechanisms such as platelet aggregation, smooth muscle cell proliferation/migration and adhesion of inflammatory cells (Davignon, 2004), it is plausible that a lack of NO itself is deleterious in the context of atherosclerosis. Furthermore, uncoupling of eNOS, for example when levels of the NO precursor arginine are reduced, may produce reactive oxygen species instead of NO, further promoting atherosclerosis (Kawashima, 2004). Interestingly, the NOS3 gene has just recently been identified to be associated with CAD (Nikpay et al, 2015). Additionally, there is evidence that the particular variant (rs3918226), which is located in the promoter of the NOS3 gene, influences the expression of eNOS with a negative effect of the risk allele (Salvi et al, 2013). As briefly discussed above, the NOS3 gene has also been associated with blood pressure (Salvi et al, 2012, 2013). Bearing in mind that NO leads to vasodilatation, this is plausible. The effects on CAD risk are, however, larger than one would expect if just being mediated via increased blood pressure.
The major NO receptor, soluble guanylyl cyclase (sGC), also seems to play a role in the genetics of CAD. Indeed, the α1‐subunit (GUCY1A3) of sGC was shown by the meta‐analysis of the CARDIoGRAMplusC4D consortium to harbour a common variant associated with CAD (CARDIoGRAMplusC4D Consortium et al, 2013). However, even more evidence comes from the detection of a rare loss‐of‐function variant identified in an extended family with high prevalence of premature CAD and MI (Erdmann et al, 2013), in which exome sequencing identified a digenic mutation in GUCY1A3 and CCT7 to be responsible for the phenotype. Furthermore, mice lacking the α1‐subunit of the sGC have been shown to display accelerated thrombus formation (Erdmann et al, 2013). Platelets indeed play an important role not only in atherothrombosis but also in atherosclerosis (Von Hundelshausen & Weber, 2007). Studies investigating the effect of sGC on plaque formation in mouse models are eagerly awaited. A protective role of sGC is thereby strongly suggested by studies showing that cGMP formation is reduced in neointimal lesions (Melichar et al, 2004) and that cGMP leads to down‐regulation of the expression of adhesion molecules for the recruitment of leucocytes into atherosclerotic plaques (Ahluwalia et al, 2004). The common variant associated with CAD (rs7692387) is located in an intron however, and the exact mechanism from genotype to phenotype at this locus thus far remains elusive. Like NOS3, the GUCY1A3 gene is also associated with blood pressure (International Consortium for Blood Pressure Genome‐Wide Association Studies et al, 2011).
Vascular remodelling
Arterial remodelling is a crucial process in atherosclerosis (Fig 3D). Interestingly, constrictive remodelling and expansive remodelling of arteries exhibit different plaque morphologies: constrictive remodelling accompanied by lumen narrowing presents with more stable plaques, whereas expansive remodelling does not feature lumen narrowing but is associated with rather unstable plaques (Pasterkamp et al, 1998; Smits et al, 1999). A novel mechanism linking these observations might involve ADAMTS‐7, an extracellular matrix (ECM) protease. Variants in ADAMTS7 have been identified to be associated with CAD (Coronary Artery Disease C4D Genetics Consortium, 2011; Reilly et al, 2011; Schunkert et al, 2011). ADAMTS7 was more strongly associated with CAD rather than MI, suggesting a major role in plaque formation and not in plaque rupture (Reilly et al, 2011). At that time, little was known about ADAMTS7 in cardiovascular diseases. One study had shown that the expression of the rat counterpart Adamts‐7 is found to be increased in carotid arteries secondary to balloon injury (Wang et al, 2009). This effect was supposed to lead to the degradation of an abundantly expressed ECM protein, cartilage oligomeric matrix protein (COMP). COMP itself had been shown to affect vascular smooth muscle cells with intact COMP maintaining a differentiated, contractile phenotype (Wang et al, 2010). In the meantime, two studies investigating the role of ADAMTS7 in mouse models revealed evidence that mice lacking Adamts‐7 (Adamts7 −/−) display reduced atherosclerotic plaque formation compared to WT littermates when they are on a proatherogenic background and fed a western diet. This effect was most prominent in Adamts7 −/− Ldlr −/− mice with a strong reduction in plaque formation in the aortic root as well as in the en face whole aorta analysis (Bauer et al, 2015). The mechanism underlying this phenotype is still unclear. The second study focused on vascular remodelling. Adamts7 −/− mice displayed strongly reduced neointima formation after wire‐mediated vascular injury (Bauer et al, 2015; Kessler et al, 2015), a phenotype that is supposed to be mainly mediated by beneficial effects in endothelial cells (Kessler et al, 2015). Endothelial cells of Adamts7 −/− mice showed increased proliferation and migration in vitro. In accordance, reendothelialization of injured arteries was significantly accelerated in Adamts7 −/− compared to WT mice. This effect is thought to be due to the inhibition of the degradation of a novel ADAMTS‐7 target—thrombospondin‐1 (TSP‐1)—into bioactive fragments (Kessler et al, 2015). The concept of cell–matrix and ECM‐mediated cell–cell communication via finely regulated expression of proteases is attractive, especially regarding ECM proteins that contain several domains with differential functions such as TSP‐1 (Iruela‐Arispe et al, 1999). Interestingly, TSP1 has also been investigated in candidate gene studies in CAD (Stenina et al, 2007). As for many other candidates that have been investigated in the pre‐GWAS era, for example the ACE gene (Mayer et al, 2007), no signal has been detected for the TSP1 gene in GWAS so far.
Inflammation
Inflammation is a central feature of atherosclerosis (Fig 3E) (Swirski & Nahrendorf, 2013). This also holds true for its genetic component. Indeed, the genes encoding the cytokine CXCL12 and the interleukin 6 receptor (IL6) have been associated with CAD. In line, a pathway analysis of the CARDIoGRAMplusC4D meta‐analysis has also shown a major role for variants in inflammatory genes (CARDIoGRAMplusC4D Consortium, 2013). By contrast, C‐reactive protein (CRP) appears to be rather a marker than a causal factor, as its genetic variation does not translate to CAD risk (Schunkert & Samani, 2008; Zacho et al, 2008; Elliott et al, 2009; C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC) (2011)).
Novel insights might come from functional investigation of the SH2B3/LNK gene. The variant rs3184504 tagging the SH2B3 gene is associated with CAD (Gudbjartsson et al, 2009; CARDIoGRAMplusC4D Consortium, 2013). The interpretation of this association is complicated by the fact that this particular variant also represents an eSNP for a neighbour gene, ATXN2 (Braenne et al, 2015). Nevertheless, the SH2B3 gene, which encodes an adaptor protein expressed, for example, in leucocytes and platelets, revealed in a knock‐out mouse model (Lnk −/−) increased platelet counts, but reduced thrombus stability (Takizawa et al, 2010). Bearing in mind that the risk allele at the SH2B3 locus is also associated with increased platelet count (Soranzo et al, 2009), altered Sh2b3/Lnk function—as present in the mouse—seems to be a suitable model to analyse the functional impact of the variant in CAD. Besides the platelet phenotypes (Soranzo et al, 2009; Takizawa et al, 2010) and the also known association with blood pressure (Levy et al, 2009; Newton‐Cheh et al, 2009), SH2B3 seems to mainly influence inflammatory processes. From a genetic point of view, this is already noticeable from the associations of the locus with different inflammatory diseases, for example coeliac disease (Hunt et al, 2008), type 1 diabetes (Barrett et al, 2009; Concannon et al, 2009) and high eosinophil numbers (Gudbjartsson et al, 2009). An experimental study revealed that Sh2b3/Lnk influences the activation of dendritic cells, thereby modulating the immune response, that is dendritic cells from Lnk −/− mice are hyper‐responsive to IL‐15, resulting in an excessive production of IFN‐γ (Mori et al, 2014). So far, the results regarding atherosclerotic phenotypes are still lacking. In a rat myocardial infarction model, however, knockout of Sh2b3/Lnk led to significantly increased fibrosis and leucocyte infiltration, resulting in reduced cardiac function (Flister et al, 2015). Taken together, genetic and experimental results point to a role for SH2B3/LNK in inflammatory processes in CAD, making SH2B3/LNK a promising candidate for therapeutic interventions.
Therapeutic strategies
One major aim of the efforts to elucidate the genetic basis of CAD and MI is the identification of novel targets and the consequent development of novel strategies in prevention and therapy. Of interest, targets identified by genetic studies are estimated to be twice as successful in the development of new drugs compared to those identified by other means (Barrett et al, 2015; Nelson et al, 2015).
As discussed above, some genetic variants influence traditional risk factors, such as blood pressure and lipid metabolism, which may be targets for treatment themselves. In addition, based on genetic discoveries, targeted novel therapies may become available for focussed interference with lipid metabolism or currently unexploited mechanisms.
Novel therapeutics in lipid metabolism
LPA, PCSK9, LPL, ANGPTL4, APOA5 and APOC3 are several promising targets in lipid metabolism available for therapeutic interference (Fig 4). PCSK9 is the most established one, since it has been known for more than a decade that gain‐of‐function and loss‐of‐function variants in the PCSK9 gene increase (Abifadel et al, 2003) and decrease (Cohen et al, 2006) the risk of CAD and MI, respectively. GWAS also identified an association between common variants in the PCSK9 gene and both cholesterol levels (Teslovich et al, 2010) and CAD (Myocardial Infarction Genetics Consortium et al, 2009). PCSK9 functions in LDL cholesterol metabolism by tagging for the degradation of intracellular as well as extracellular LDL cholesterol receptors (Brautbar & Ballantyne, 2011). Thus, current therapeutic concepts exploit monoclonal antibodies to inhibit the effect of PCSK9 in the circulation but also RNAi‐ and small molecule‐based approaches are in the development and evaluation. Table 4 provides an overview over the compounds currently under investigation using PCSK9 as target.
Table 4.
Currently investigated PCSK9 inhibitors
| Name | Mechanism of action | Phase of development | Approved | LDL |
|---|---|---|---|---|
| Evolocumaba | Human mAb | Phase IV | Yese | ↓ 61% (Sabatine et al, 2015) |
| Alirocumabb | Human mAb | Phase IV | ↓ 62% (Robinson et al, 2015) | |
| Bococizumabc | Humanized mAb (Liang et al, 2012) | Phase II | No | ↓ 21–54% (Ballantyne et al, 2015) |
| ALN‐PCSd | Antisense oligo (Frank‐Kamenetsky et al, 2008) | Phase I | No | ↓ 40% (Fitzgerald et al, 2014) |
| NA | VLP‐based vaccine (Crossey et al, 2015) | Preclinical | No | NA |
mAb, monoclonal antibody; VLP, virus‐like particle; LDL., LDL cholesterol reduction; NA, not available.
Amgen.
Regeneron, Sanofi.
Pfizer.
Alnylam, The Medicines Company.
Approved for familial hypercholesterolaemia, statin intolerance and insufficient LDL cholesterol control with statins.
LPL and the proteins regulating its activity are also attractive targets for novel treatment strategies. As discussed above, APOC3 reduces LPL activity. Accordingly, inhibition of APOC3 by RNAi‐mediated knockdown is currently being investigated in clinical trials. In a dose‐ranging phase II study, ISIS 304801, an antisense APOC3 inhibitor, led to a dose‐dependent 31–71% reduction in triglycerides (Gaudet et al, 2015). Also ANGPTL4, a gene recently identified by an exome‐wide association study and also involved in the regulation of LPL activity, may be the target of novel drug approaches: mice deficient for Angptl4 display reduced triglyceride levels mainly due to increased clearance and reduced production of very low‐density lipoproteins (Desai et al, 2007).
Targeting NO/cGMP signalling
The NO/cGMP pathway appears to play an important role in atherosclerosis since two key enzymes, that is eNOS and sGC, are genome‐wide significantly associated with CAD (CARDIoGRAMplusC4D Consortium, 2013; Nikpay et al, 2015). Additionally, the importance of the pathway has been confirmed by the fact that loss‐of‐function mutations affecting the function of the sGC are responsible for premature CAD and MI in an extended family (Erdmann et al, 2013). Whereas the mechanism involving the common variants remains to be elucidated, several therapeutic options are already available. Although increasing the availability of NO might not be achieved the expected success due to adverse effects and pharmacokinetic obstacles, targeting the sGC directly could solve these problems at least in part. Riociguat is a sGC stimulator already in clinical use. In the PATENT trial, the drug has proven efficacy in patients with pulmonary hypertension (Ghofrani et al, 2013). Riociguat is also very interesting because it acts synergistically with NO (Stasch & Hobbs, 2009); that is, reduced sGC activity or expression could hypothetically be compensated by the presence of such modulators of sGC activity. Future experimental studies are needed to explore the effect of sGC stimulators on atherosclerosis phenotypes.
Precision medicine
One possibility of improving treatment is the identification and evaluation of novel drug targets. A second way to exploit GWAS findings is to develop individualized treatment strategies. The development of so‐called precision medicine is a major goal of current funding concepts (Jameson & Longo, 2015). Unfortunately, there is currently only limited evidence to suggest that genetic testing of individual common risk variants allows for stratification into different treatment modalities. However, individuals with a high genetic risk score appear to have a larger benefit from statin treatment than those with low genetic risk (Hughes et al, 2012; Mega et al, 2015; Schunkert & Samani, 2015). Further, stimulating input in this field might come from systems medicine approaches (Bjoerkegren et al, 2015). For example, an integrative analysis revealed an enrichment of CAD‐associated variants in differentially co‐expressed microRNA–mRNA pairs (Huan et al, 2013).
Outlook
GWAS have led to the identification of more than 50 genetic variants associated with CAD and MI so far, and likely more will follow. These variants reflect an important progress in defining the mechanisms leading to disease. However, the variants identified thus far only account for no more than 10% of the heritable risk. Future studies will aim to detect rare variants with strong impact on disease risk and might thus close this gap. Additionally, the field of epistasis, that is the interaction of different genes, is rapidly evolving. Finally, it has been shown for other complex disorders that the combined small effects of hundreds of thousands of SNPs may jointly explain the currently poorly understood inheritance patterns (Yang et al, 2015). The results achieved thus far do, however, provide valuable insight into the pathophysiology of CAD and MI and are starting points for individualized treatment strategies.
Conflict of interest
The authors declare that they have no conflict of interest.
Pending issues.
How can our understanding of the heritability of coronary artery disease be improved?
To which extent do the interactions of specific loci contribute to the heritability of the disease?
What are the molecular mechanisms by which non‐coding regions affect risk of coronary artery disease?
How can the acquired knowledge be translated into improved prevention and therapeutic strategies?
Acknowledgements
This work was supported by grants from the Fondation Leducq (CADgenomics: Understanding CAD Genes, 12CVD02), the German Federal Ministry of Education and Research (BMBF) within the framework of the e: Med research and funding concept (e:AtheroSysMed, grant 01ZX1313A‐2014) and the European Union Seventh Framework Programme FP7/2007‐2013 under grant agreement no. HEALTH‐F2‐2013‐601456 (CVgenes‐at‐target). Further grants were received from the DFG as part of the Sonderforschungsbereich CRC 1123 (B2). T.K. was supported by a DZHK Rotation Grant. Figures 2 and 4 contain modified image material available at Servier Medical Art under a Creative Commons Attribution 3.0 Unported License.
EMBO Mol Med (2016) 8: 688–701
See the Glossary for abbreviations used in this article.
References
- Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D et al (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34: 154–156 [DOI] [PubMed] [Google Scholar]
- Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perretti M, Moncada S, Hobbs AJ (2004) Antiinflammatory activity of soluble guanylate cyclase: cGMP‐dependent down‐regulation of P‐selectin expression and leukocyte recruitment. Proc Natl Acad Sci USA 101: 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne CM, Neutel J, Cropp A, Duggan W, Wang EQ, Plowchalk D, Sweeney K, Kaila N, Vincent J, Bays H (2015) Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo‐controlled, dose‐ranging study in statin‐treated subjects with hypercholesterolemia. Am J Cardiol 115: 1212–1221 [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C et al (2009) Genome‐wide association study and meta‐analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41: 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Dunham I, Birney E (2015) Using human genetics to make new medicines. Nat Rev Genet 16: 561–562 [DOI] [PubMed] [Google Scholar]
- Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, Ou K, Paschos GK, Zheng XL, Parmacek MS et al (2015) Knockout of adamts7, a novel CAD locus in humans, reduces atherosclerosis in mice. Circulation 131: 1202–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjoerkegren JL, Kovacic JC, Dudley JT, Schadt EE (2015) Genome‐wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol 65: 830–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braenne I, Civelek M, Vilne B, Di Narzo A, Johnson AD, Zhao Y, Reiz B, Codoni V, Webb TR, Foroughi Asl H et al (2015) Prediction of causal candidate genes in coronary artery disease loci. Arterioscler Thromb Vasc Biol 35: 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautbar A, Ballantyne CM (2011) Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol 8: 253–265 [DOI] [PubMed] [Google Scholar]
- C Reactive Protein Coronary Heart Disease Genetics Collaboration C (2011) Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ 342: d548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, AmiGO Hub , Web Presence Working Group (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25: 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDIoGRAMplusC4D Consortium (2013) Large‐scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45: 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354: 1264–1272 [DOI] [PubMed] [Google Scholar]
- Concannon P, Rich SS, Nepom GT (2009) Genetics of type 1A diabetes. N Engl J Med 360: 1646–1654 [DOI] [PubMed] [Google Scholar]
- Coronary Artery Disease C4D Genetics Consortium (2011) A genome‐wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 43: 339–344 [DOI] [PubMed] [Google Scholar]
- Crossey E, Amar MJA, Sampson M, Peabody J, Schiller JT, Chackerian B, Remaley AT (2015) A cholesterol‐lowering VLP vaccine that targets PCSK9. Vaccine 33: 5747–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon J (2004) Role of endothelial dysfunction in atherosclerosis. Circulation 109: III–27–III–32 [DOI] [PubMed] [Google Scholar]
- Desai U, Lee EC, Chung K, Gao C, Gay J, Key B, Hansen G, Machajewski D, Platt KA, Sands AT et al (2007) Lipid‐lowering effects of anti‐angiopoietin‐like 4 antibody recapitulate the lipid phenotype found in angiopoietin‐like 4 knockout mice. Proc Natl Acad Sci USA 104: 11766–11771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J et al (2013) Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 45: 1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O et al (2015) Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, Erdmann J, Braund P, Engert JC, Bennett D et al (2009) Genetic Loci associated with C‐reactive protein levels and risk of coronary heart disease. JAMA 302: 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann J, Grosshennig A, Braund PS, König IR, Hengstenberg C, Hall AS, Diemert P, Kathiresan S, Wright B, Tregouet DA et al (2009) New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 41: 280–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, De Wit C, Kaiser FJ, Braunholz D, Medack A, Fischer M et al (2013) Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 504: 432–436 [DOI] [PubMed] [Google Scholar]
- Fitzgerald K, Frank‐Kamenetsky M, Shulga‐Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J et al (2014) Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single‐blind, placebo‐controlled, phase 1 trial. Lancet 383: 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flister MJ, Hoffman MJ, Lemke A, Prisco SZ, Rudemiller N, O'Meara CC, Tsaih SW, Moreno C, Geurts AM, Lazar J et al (2015) SH2B3 is a genetic determinant of cardiac inflammation and fibrosis. Circ Cardiovasc Genet 8: 294–304 [DOI] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C et al (2013) STRING v9.1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41: D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank‐Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, Butler D, Charisse K, Dorkin R, Fan Y et al (2008) Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA 105: 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham MJ et al (2015) Antisense inhibition of apolipoprotein C‐III in patients with hypertriglyceridemia. N Engl J Med 373: 438–447 [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing Z‐C, Keogh AM, Langleben D, Kilama MO, Fritsch A et al (2013) Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369: 330–340 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, Thorleifsson G, Helgadottir H, Steinthorsdottir V, Stefansson H et al (2009) Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 41: 342–347 [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A et al (2007) A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316: 1491–1493 [DOI] [PubMed] [Google Scholar]
- Holdt LM, Beutner F, Scholz M, Gielen S, Gäbel G, Bergert H, Schuler G, Thiery J, Teupser D (2010) ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol 30: 620–627 [DOI] [PubMed] [Google Scholar]
- Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F et al (2013) Alu elements in ANRIL non‐coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans‐regulation of gene networks. PLoS Genet 9: e1003588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Zhang B, Wang Z, Joehanes R, Zhu J, Johnson AD, Ying S, Munson PJ, Raghavachari N, Wang R et al (2013) A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler Thromb Vasc Biol 33: 1427–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M, Saarela O, Stritzke J, Kee F, Silander K, Klopp N, Kontto J, Karvanen J, Willenborg C, Salomaa V et al (2012) Genetic markers enhance coronary risk prediction in men: the MORGAM prospective cohorts. PLoS ONE 7: e40922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt KA, Zhernakova A, Turner G, Heap GAR, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D et al (2008) Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 40: 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBC 50K CAD Consortium (2011) Large‐scale gene‐centric analysis identifies novel variants for coronary artery disease. PLoS Genet 7: e1002260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Consortium for Blood Pressure Genome‐Wide Association Studies (2011) Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iruela‐Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD (1999) Inhibition of angiogenesis by thrombospondin‐1 is mediated by 2 independent regions within the type 1 repeats. Circulation 100: 1423–1431 [DOI] [PubMed] [Google Scholar]
- Jameson JL, Longo DL (2015) Precision medicine–personalized, problematic, and promising. N Engl J Med 372: 2229–2234 [DOI] [PubMed] [Google Scholar]
- Kamburov A, Stelzl U, Lehrach H, Herwig R (2013) The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res 41: D793–D800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes J (1961) Factors of risk in the development of coronary heart disease–six year follow‐up experience. The Framingham Study. Ann Intern Med 55: 33–50 [DOI] [PubMed] [Google Scholar]
- Kawashima S (2004) Malfunction of vascular control in lifestyle‐related diseases: endothelial nitric oxide (NO) synthase/NO system in atherosclerosis. J Pharmacol Sci 96: 411–419 [DOI] [PubMed] [Google Scholar]
- Kessler T, Zhang L, Liu Z, Yin X, Huang Y, Wang Y, Fu Y, Mayr M, Ge Q, Xu Q et al (2015) ADAMTS‐7 inhibits re‐endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin‐1. Circulation 131: 1191–1201 [DOI] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T et al (2009) Genome‐wide association study of blood pressure and hypertension. Nat Genet 41: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Chaparro‐Riggers J, Strop P, Geng T, Sutton JE, Tsai D, Bai L, Abdiche Y, Dilley J, Yu J et al (2012) Proprotein convertase subtilisin/kexin type 9 antagonism reduces low‐density lipoprotein cholesterol in statin‐treated hypercholesterolemic nonhuman primates. J Pharmacol Exp Ther 340: 228–236 [DOI] [PubMed] [Google Scholar]
- Lieb W, Jansen H, Loley C, Pencina MJ, Nelson CP, Newton‐Cheh C, Reilly MP, Assimes TL, Boerwinkle E, Hall AS et al (2013) Genetic predisposition to higher blood pressure increases coronary artery disease risk. Hypertension 61: 995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wang L, Chen S, He L, Yang X, Shi Y, Cheng J, Zhang L, Gu CC, Huang J et al (2012) Genome‐wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet 44: 890–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330: 1041–1046 [DOI] [PubMed] [Google Scholar]
- Mayer B, Erdmann J, Schunkert H (2007) Genetics and heritability of coronary artery disease and myocardial infarction. Clin Res Cardiol 96: 1–7 [DOI] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg‐Hansen A, Folsom AR et al (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316: 1488–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM et al (2015) Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385: 2264–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melichar VO, Behr‐Roussel D, Zabel U, Uttenthal LO, Rodrigo J, Rupin A, Verbeuren TJ, Kumar HSA, Schmidt HHHW (2004) Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late‐stage atherosclerosis. Proc Natl Acad Sci USA 101: 16671–16676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Iwasaki Y, Seki Y, Iseki M, Katayama H, Yamamoto K, Takatsu K, Takaki S (2014) Lnk/Sh2b3 controls the production and function of dendritic cells and regulates the induction of IFN‐ ‐producing T cells. J Immunol 193: 1728–1736 [DOI] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank‐Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM et al (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466: 714–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium (2009) Genome‐wide association of early‐onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 41: 334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium Investigators (2014) Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med 371: 2072–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada T‐A, Russell JA, Boyd JH, Thair SA, Walley KR (2015) Identification of a nonsynonymous polymorphism in the SVEP1 gene associated with altered clinical outcomes in septic shock. Crit Care Med 43: 101–108 [DOI] [PubMed] [Google Scholar]
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J et al (2015) The support of human genetic evidence for approved drug indications. Nat Genet 47: 856–860 [DOI] [PubMed] [Google Scholar]
- Newton‐Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao J‐H, Heath SC, Eyheramendy S et al (2009) Genome‐wide association study identifies eight loci associated with blood pressure. Nat Genet 41: 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols M, Townsend N, Scarborough P, Rayner M (2014) Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J 35: 2950–2959 [DOI] [PubMed] [Google Scholar]
- Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC et al (2015) A comprehensive 1000 Genomes–based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet 47: 1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp G, Schoneveld AH, van der Wal AC, Haudenschild CC, Clarijs RJ, Becker AE, Hillen B, Borst C (1998) Relation of arterial geometry to luminal narrowing and histologic markers for plaque vulnerability: the remodeling paradox. J Am Coll Cardiol 32: 655–662 [DOI] [PubMed] [Google Scholar]
- Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett MS, Devaney JM, Knouff CW, Thompson JR et al (2011) Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome‐wide association studies. Lancet 377: 383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M et al (2015) Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 372: 1489–1499 [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM et al (2015) Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 372: 1500–1509 [DOI] [PubMed] [Google Scholar]
- Salvi E, Kutalik Z, Glorioso N, Benaglio P, Frau F, Kuznetsova T, Arima H, Hoggart C, Tichet J, Nikitin YP et al (2012) Genomewide association study using a high‐density single nucleotide polymorphism array and case‐control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension 59: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi E, Kuznetsova T, Thijs L, Lupoli S, Stolarz‐Skrzypek K, D'Avila F, Tikhonoff V, De Astis S, Barcella M, Seidlerova J et al (2013) Target sequencing, cell experiments, and a population study establish endothelial nitric oxide synthase (eNOS) gene as hypertension susceptibility gene. Hypertension 62: 844–852 [DOI] [PubMed] [Google Scholar]
- Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE et al (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357: 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato‐Nishiuchi R, Nakano I, Ozawa A, Sato Y, Takeichi M, Kiyozumi D, Yamazaki K, Yasunaga T, Futaki S, Sekiguchi K (2012) Polydom/SVEP1 is a ligand for integrin 91. J Biol Chem 287: 25615–25630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, Samani NJ (2008) Elevated C‐reactive protein in atherosclerosis–chicken or egg? N Engl J Med 359: 1953–1955 [DOI] [PubMed] [Google Scholar]
- Schunkert H, Götz A, Braund P, McGinnis R, Tregouet DA, Mangino M, Diemert P, Cambien F, Hengstenberg C, Stark K et al (2008) Repeated replication and a prospective meta‐analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation 117: 1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C et al (2011) Large‐scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43: 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, Samani NJ (2015) Statin treatment: can genetics sharpen the focus? Lancet 385: 2227–2229 [DOI] [PubMed] [Google Scholar]
- Schwanzer‐Pfeiffer D, Rossmanith E, Schildberger A, Falkenhagen D (2010) Characterization of SVEP1, KIAA, and SRPX2 in an in vitro cell culture model of endotoxemia. Cell Immunol 263: 65–70 [DOI] [PubMed] [Google Scholar]
- Smits PC, Pasterkamp G, van Ufford MAQ, Eefting FD, Stella PR, de Jaegere PPT, Borst C (1999) Coronary artery disease: arterial remodelling and clinical presentation. Heart 82: 461–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soranzo N, Spector TD, Mangino M, Kühnel B, Rendon A, Teumer A, Willenborg C, Wright B, Chen L, Li M et al (2009) A genome‐wide meta‐analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 41: 1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch J‐P, Hobbs AJ (2009) NO‐independent, haem‐dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol 191: 277–308 [DOI] [PubMed] [Google Scholar]
- Stenina OI, Topol EJ, Plow EF (2007) Thrombospondins, their polymorphisms, and cardiovascular disease. Arterioscler Thromb Vasc Biol 27: 1886–1894 [DOI] [PubMed] [Google Scholar]
- Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, König IR, Weeke PE, Webb TR, Auer PL, Schick UM et al (2016) Coding variation in ANGPTL4, LPL, and SVEP1 and risk of coronary disease. N Engl J Med 374: 1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M (2013) Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339: 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H, Nishimura S, Takayama N, Oda A, Nishikii H, Morita Y, Kakinuma S, Yamazaki S, Okamura S, Tamura N et al (2010) Lnk regulates integrin αIIbβ3 outside‐in signaling in mouse platelets, leading to stabilization of thrombus development in vivo . J Clin Invest 120: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466: 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute (2014) Loss‐of‐function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 371: 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregouet DA, König IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Diemert P, Perret C, Desuremain M et al (2009) Genome‐wide haplotype association study identifies the SLC22A3‐LPAL2‐LPA gene cluster as a risk locus for coronary artery disease. Nat Genet 41: 283–285 [DOI] [PubMed] [Google Scholar]
- Von Hundelshausen P, Weber C (2007) Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 100: 27–40 [DOI] [PubMed] [Google Scholar]
- Wang L, Zheng J, Bai X, Liu B, Liu C‐J, Xu Q, Zhu Y, Wang N, Kong W, Wang X (2009) ADAMTS‐7 mediates vascular smooth muscle cell migration and neointima formation in balloon‐injured rat arteries. Circ Res 104: 688–698 [DOI] [PubMed] [Google Scholar]
- Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu C‐J, Zhu Y, Gao Y, Xu Q et al (2010) Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res 106: 514–525 [DOI] [PubMed] [Google Scholar]
- Wang F, Xu CQ, He Q, Cai JP, Li XC, Wang D, Xiong X, Liao YH, Zeng QT, Yang YZ et al (2011) Genome‐wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet 43: 345–349 [DOI] [PubMed] [Google Scholar]
- Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, Robinson MR, Perry JR, Nolte IM, van Vliet‐Ostaptchouk JV et al (2015) Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet 47: 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J et al (2004) Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case‐control study. Lancet 364: 937–952 [DOI] [PubMed] [Google Scholar]
- Zacho J, Tybjaerg‐Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG (2008) Genetically elevated C‐reactive protein and ischemic vascular disease. N Engl J Med 359: 1897–1908 [DOI] [PubMed] [Google Scholar]