

Abstract
The rapid development of CRISPR technology greatly impacts the field of genetic engineering. The simplicity in design and generation of highly efficient CRISPR reagents allows more and more researchers to take on genome editing in different model systems in their own labs, even for those who found it daunting before. An active CRISPR complex contains a protein component (Cas9) and an RNA component (small guide RNA [sgRNA]), which can be delivered into cells in various formats. Cas9 can be introduced as a DNA expression plasmid, in vitro transcripts, or as a recombinant protein bound to the RNA portion in a ribonucleoprotein particle (RNP), whereas the sgRNA can be delivered either expressed as a DNA plasmid or as an in vitro transcript. Here we compared the different delivery methods in cultured cell lines as well as mouse and rat single-cell embryos and view the RNPs as the most convenient and efficient to use. We also report the detection of limited off-targeting in cells and embryos and discuss approaches to lower that chance. We hope that researchers new to CRISPR find our results helpful to their adaptation of the technology for optimal gene editing.
Introduction
CRISPR systems, short for clustered regularly interspaced short palindromic repeats, are a prevalent bacterial defense system against invading DNA by incorporating short foreign sequences into the bacterial genome and transcribing them into crRNA to recognize and degrade the same sequence upon reexposure. Since the 2012 report of using CRISPR/Cas9 complex to cleave a DNA target biochemically by the mere change of 20 nucleotides in the crRNA molecule1 and the first successful applications of CRISPR in human cells,2,3 the technology has been adapted in an astonishing speed and revolutionized the field of gene editing in almost any model systems,4 owing largely to the simplicity of target recognition via RNA/DNA base pairing. The history of discovery and development of CRISPRs is a remarkable story of turning decades of basic research into amazing applications.5
CRISPR/Cas is by no means the first nuclease technology applied to gene editing but no doubt the simplest to understand, easiest to generate, and cheapest to use, all contributing to its incredible development. Applications of earlier nucleases, on the other hand, have laid the foundation for and made significant breakthroughs in the field of gene editing.6
Meganucleases, also called homing endonucleases, identified in single-cell eukaryotic introns and involved in intron mobility, were the first reported endonucleases to be used in genome editing.7 The most used meganuclease is I-SceI from mitochondria of Saccharomyces cerevisiae.8 It has been used widely to introduce double-strand breaks in DNA repair studies.9 Homing nucleases are efficient and highly specific. However, the biggest limitation of meganucleases is the difficulty to reengineer the proteins to target a different sequence because the nuclease domain is also the DNA binding domain. Modifications to the protein to change binding specificity are highly likely to damage the nuclease activity in the meantime, making it problematic to target a new sequence. Zinc finger nucleases (ZFNs) are a fusion of zinc finger proteins and the DNase domain of FokI restriction endonuclease and were first reported to be able to cleave specific DNA sequences in 199610 and worked efficiently in mammalian cells.11–13 ZFNs contain two subunits that are designed to bind opposite strands of target site to allow FokI domain to dimerize. Mutations were introduced into FokI domain to form obligated heterodimers that significantly improved specificity.14 ZFNs were the first nuclease to be widely used in gene editing in cells and model systems6 and by far the furthest along in therapeutics.15 However, ZFNs require proprietary module libraries for design and assembly, at high costs and yet sometimes low success rates. Transcription activator-like effector nucleases (TALENs) are relatively easier to generate than ZFNs,16 and recent improvement on target site selection allows almost any sequence to be targeted.17 But commercially available reagents are still expensive and take weeks to obtain. Intrinsic to protein engineering, none of these technologies guarantees active nucleases from a given design. Other nucleases with constant protein portion and a variable nucleic acid portion, such as mobile group II introns, are more predictable with activity but difficult to adapt into systems other than prokaryotes.18
In contrast, CRISPR has the best of both worlds: with no need for protein engineering and active in almost all cell types. The most adopted CRISPR complex originated from Streptococcus pyogenes is composed of the Cas9 protein, harboring nuclease activities, and an RNA called small guide RNA (sgRNA) containing a constant backbone that binds Cas9 and a 20 bp variable region called spacer sequence that base pairs with the target DNA to provide specificity. Inside a cell, the CRISPR complex scans the genome for PAM (protospacer adjacent motif, usually with the sequence 5′-NGG) and unwinds double-stranded DNA at a PAM site, allowing sgRNA to base pair with now single-stranded DNA. The Cas9 protein then either cleaves close to PAM or the complex moves on to the next PAM, depending on the base pairing between sgRNA and DNA.19 The continuous effort on exploring other Cas systems20 and modification of Cas921 allows other PAM sites to be used and increase the possibility to target any sequence. In addition to nucleases, CRISPR can also be used in high-throughput screens22 and to regulate gene activation and inactivation by binding to the promoter region or fused to other functional domains.23
The active CRISPR complex is a ribonucleoprotein particle (RNP) containing a Cas9 molecule and an sgRNA. Both components of RNPs can be introduced into the cells in various ways, including plasmid, in vitro transcribed RNA, precomplexed RNP, or via viral vectors. There are pros and cons for each delivery method, primarily regarding efficiency and off-targeting. Plasmid DNA lasts longer inside the cells, potentially leading to more complete modifications as well as off-targeting. One also needs to consider the possibility of random integration into the genome and choice of promoters for different cell types. RNA reagents are easy to generate and can be cloning-free. RNP is the active nuclease entity delivered as is in vivo. Its relative short exposure to the genome might lower off-targeting.
We report here practical comparisons of nuclease activity from different formats of CRISPR reagents in both cells and embryos, and the RNP format is most convenient and functional in cells, embryos, as well as in biochemical assays. Occasional off-targeting was detected but likely tolerable in research systems.
Materials and Methods
Cas9 plasmid construction and mRNA in vitro transcription
A human codon-optimized Cas9 sequence2 was assembled from gBlocks (IDT) by Gibson method (NEB) and subcloned into a vector under the T7 promoter and a CMV promoter. Briefly, 9 gBlocks of roughly 500 bp each were designed with 15 nucleotides overlap. gBlocks were assembled 3 at a time using 2× Gibson mix, and incubated at 50°C for 1 hr. Final three fragments were also assembled, digested with EcoRI and XbaI, and subcloned into an expression vector. Linearized plasmid was used as a template for an in vitro transcription (IVT) of Cas9 mRNA using MessageMax T7 kit (CellScript) according to the manufacturer's protocol. Cas9 mRNA was purified by incubating with the equal volume of 5 M ammonium sulfate on ice for 15 min, followed by centrifugation at 4°C at top speed for 15 min. The RNA pellet was then washed with 70% ethanol and air-dried.
sgRNA template preparation
Two overlapping DNA oligos, one containing T7 promoter and 20 nucleotides of Cas9 target sequence (5′-oligoAAAATAATACGACTCACTATAGGGNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAG), and second containing sgRNA backbone (common reverse oligo, 5′-AAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC), were combined in a PCR, together with a T7 forward (5′-AAAATAATACGACTCACTATAGG) and a backbone reverse (5′-AAAAAAGCACCGACTCGGTGCCA) primer. PCR was performed using AccuPrime HiFi Taq polymerase (Invitrogen) under the following conditions: 95°C, 2 min, and then 35 cycles of 95°C, 30 sec; 60°C, 30 sec; and 68°C, 30 sec. PCR product was purified by QiaQuick PCR purification kit (Qiagen), and the DNA was used as a template for an in vitro sgRNA synthesis with HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB). The RNA was purified by precipitation with 2 volumes of ethanol plus 1/10 volume of 3 M sodium acetate, by precipitation with equal volume of 5 M ammonium acetate, or by MegaClear column purification. sgRNAs were quantified by using a Nanodrop or using the BR RNA assay for Qubit (Thermo Fisher).
Cas9 protein and ribonucleoprotein complex formation
We obtained the recombinant Cas9 protein from PNA Bio and New England Biolabs (NEB) as well as a custom prep by Aldevron. The protein from NEB does not contain a nuclear localization signal. RNP complexes were formed by incubating the recombinant Cas9 protein and sgRNA at 1:1 mass ratio (1: 4.6 molar ratio of the Cas9 protein to sgRNA) at 37°C for 5 min or on ice for at least 20 min before use or to be stored at −80°C.
Cell culture and transfections
Rat C6 glioma cells were maintained in F-12K media (ATTC) containing 15% horse serum, 2.5% FBS, and 1% penicillin/streptomycin at 37°C with 5% CO2. Mouse Neuro2a cells were maintained in EMEM (ATCC) supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. All cell transfections were performed with a Nucleofector (Lonza) according to the manufacturer's 96-well shuttle protocol for respective cell lines. After trypsinization, cells were counted, pelleted, and washed twice in Hanks balanced salt solution to minimize nuclease carryover from growth medium. Specifically, for the rat C6 cell line, SG solution and program FF-127 were used to transfect 200,000 cells. For the mouse Neuro-2a cell line, SF solution and program 96-DS-137 were used to transfect 200,000 cells per reaction. One microgram of a GFP plasmid was always transfected for each condition as a control. ZFN mRNAs were tranfected at 2 μg/reaction. Regardless of the delivery method of Cas9 (plasmid, mRNA or protein), sgRNAs were used at 2–12 μg/reaction. Cas9 expression plasmid was transfected at 2 μg/reaction, Cas9 mRNA at 2–4 μg/reaction, and recombinant Cas9 protein at 3–10 μg/reaction.
Cel-I assay for detection of nuclease-mediated NHEJ events
Transfected cells or tissue clips were added into 80 μl of QuickExtract (QE) DNA extraction solution (Epicentre, Madison, WI), and individual embryos into 5 μl QE each, incubated at 65°C for 15 min and 98°C for 3 min to release nucleic acids. Target regions or predicted off-target regions were PCR amplified (for PCR primer sequences see Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/hum) using the extracted genomic DNA samples as templates. Two Taq polymerase mixes and respective conditions were used interchangeably: AccuStart II PCR SuperMix (Quanta Biosciences) with the following program: 95°C, 2 min, and 35 cycles of 95°C, 15 sec, 60°C, 15 sec, and 72°C, 20 sec, and JumpStart ReadyMix (Sigma-Aldrich) with the following program: 95°C, 2 min, and 35 cycles of 95°C, 30 sec, 60°C, 30 sec, and 68°C, 45 sec, and final extension at 68°C for 5 min. Ten microliters of the above PCRs was incubated under the following program: 95°C, 10 min, 95°C to 85°C at −2°C/s, and 85°C to 25°C at −0.1°C/sec. One microliter each of nuclease S (Cel-I) and enhancer (Transgenomics) was added to digest the above reaction at 42°C for 20 min. The mixture was resolved on a 10% polyacrylamide TBE gel.
Stable cell line generation
Cells were nucleofected with linearized Cas9 plasmid, and single-cell clones were selected for hygromycin resistance. Each clone was transfected with an active sgRNA, and Cel-I assay was used to validate Cas9 expression. Cas9-positive clones were propagated and cryopreserved at early passages for future use.
In vitro cleavage assay
DNA targets for Cas9-sgRNA complexes were PCR amplified from rat genomic DNA, column purified, and quantified by using a Nanodrop. An in vitro cleavage reaction consisted specified amount of RNP mixture, 50 ng of PCR-amplified target region, and 1× NEB buffer 3 with BSA in 20 μl final volume. The reaction was incubated at 37°C, 40 min; 65°C, 10 min; and 10°C hold. The product was then resolved on a 10% acrylamide gel or a 2% agarose gel.
RT-qPCR
One-step RT-qPCR was performed from the above-mentioned QE samples by using qScript XLT One-Step RT-qPCR ToughMix (Quanta Biosciences). The custom TaqMan primers and probes were designed using IDT's PrimeTime qPCR software, and made by IDT. Cas9 mRNA was detected by using CasFor (5′-CGATTCTCCTACAGTCGCTTAC) and CasRev (5′-AGCTTGATCGCTCCATGA TT) primers as well as a TaqMan probe: FAM-AGACTTCCCTTTCTCCACTTTGGCC-BKFQ. sgRNAs were detected using the following primer and probe set: sgFor (5′-GTTTTAGAGCTAGAAATAGCAAGTTA), sgRev (5′-AAAAAAGCACCGACTCGGTGCCA), and a TaqMan probe: FAM-AGGCTAGTCCGTTATCAACTTGAAA-BKFQ. Twenty-microliter reactions contained 0.5 μl of QE reaction, 500 nM primers, 100 nM TaqMan probe, and 1× of PCR mix. PCR was run on Bio-Rad CFX-96 instrument as follows: 50°C, 10 min; 95°C, 5 min; then 40 cycles of 95°C, 15 sec; and 60°C, 1 min. The Cas9 and sgRNA expression data were normalized to a housekeeping gene (rat GAPDH, VIC, Rn01775763_g1; Life Technologies; mouse GAPDH, FAM, Mm.PT.39a.1, IDT), and the expression data were analyzed by ΔΔCt method. A no-RT reaction was run on each sample with the same template input and the resulting Ct value was subtracted from the Ct value of respective RT-PCR.
Ethics statement
All animal work was performed in accordance with the approved animal protocols overseen by SAGE Labs's (now Horizon Discovery Group Company) Institutional Animal Care and Use Committee (IACUC). Both mice and rats were housed in standard cages and maintained on a 12 hr light/dark cycle with ad libitum access to food and water. Routine health monitoring of the colony was performed at IDEXX (Columbia, MO) and revealed no evidence of infection with serious known pathogens.
Microinjection
Four- to five-week-old female donors were injected with 5 units (mice) or 20 units (rats) of PMS followed by 5 units (mice) or 50 units of hCG injection after 48 hr of the PMS injection and then immediately mated with stud males after the hCG injection. Fertilized eggs were harvested a day later for microinjection. Injected eggs were either transferred into pseudopregnant females for live births or allowed to develop in vitro to reach blastocysts for analyzing nonhomologous end joining (NHEJ) activity.
Off-target activity analysis
sgRNA target sequences were run against rat genome using the Optimized CRISPR Design algorithm from the lab of Feng Zhang at MIT. Top eight off-target regions for each target were selected according to the score. Flanking PCR primers were designed to amplify 300–600 bp fragments. Amplified fragments were analyzed using Cel-I assay, and the results were screened for the predicted cutting pattern.
Results and Discussion
Local concentrations of the Cas9 protein and sgRNA are critical to nuclease activity
One advantage of CRISPR technology is that the Cas9 protein is constant, and only up to 20 bases of sequence in sgRNA has to be changed to recognize and cleave a new target. sgRNAs for a given target can be synthesized via PCR amplification of oligo DNA template and in vitro transcription, completely bypassing cloning and allowing generation and validation of a CRISPR reagent in a matter of days. RNA is also friendly to transfection and microinjection, as we have shown working with ZFN mRNA in both cultured cells and embryo manipulation.24,25 We used in vitro-transcribed sgRNAs throughout this study.
Unlike our experience with ZFN mRNAs, co-transfection of in vitro-transcribed Cas9 mRNA or Cas9-expressing plasmid DNA with in vitro-transcribed sgRNAs rarely led to efficient cleavage at the target sites in the rat C6 cell line by nucleofection. As shown in Fig. 1A as an example, two sgRNAs, ApoE #7 targeting the 3′ end of the coding sequence of the rat ApoE gene and rRosa26 targeting intron 1 of the rat Rosa26 locus, were co-transfected with Cas9 mRNA into C6 cells. sgRNA target site and primer sequences are listed in Supplementary Table S1. Transfected cells were collected at various time points posttransfection and analyzed using Cel-I assay, in which CRISPR cleavage-mediated modifications via NHEJ at a target site result in cleavage of the PCR amplicon of the target region into fragments of predicted sizes.11 No cleavage was detected in cells up to 48 hr posttransfection with either target. The mouse Neuro-2a cell line usually worked slightly better. In Fig. 1B, sgRNAs targeting the mouse Ptsg1 gene (mPtsg1) and the Rosa26 locus (mRosa26), respectively, were transfected with Cas9 mRNA into Neuro-2a cells, and each target site was analyzed using Cel-I assay. Cleavage activity was detected at 48 hr posttransfection with sgRNA mPtsg1 but not with mRosa26. In the meantime, respective ZFN mRNAs against the Rosa26 locus were transfected into C6 and Neuro-2a cells as controls, and expected cleavage pattern of the target amplicons was readily detected at as early as 8 hr after transfection. Increasing Cas9 mRNA and sgRNA to tens of micrograms in each nucleofection reaction occasionally improved cleavage rates, but the results were still not consistent (not shown).
Figure 1.

Cotransfection of Cas9 mRNA and sgRNA does not lead to consistent cleavage activity in cells. (A) Two sgRNAs, one targeting the rat ApoE gene (ApoE #7, lanes 1–6) and the other, the Rosa26 locus (rRosa26, lanes 8–16), were nucleofected along with Cas9 mRNA (R) or Cas9 expression plasmid (D) into the rat C6 cell line, and at 8, 24, and 48 hr postnucleofection, cells were collected, and target sites were PCR amplified and analyzed by using Cel-I assay (see Materials and Methods). Transfection of ZFN mRNA (ZR) targeting the Rosa26 locus introduced in/dels and generated cleaved bands of expected 234 and 161 bp in Cel-I assay (lanes 17–19). Lanes 7 and 20 are transfection controls. The positions of expected but absent cleaved bands are marked with arrowheads. (B) The same test in the mouse Neuro-2a cell line: lanes 1–7, transfected with sgRNA mPtsg1 (against Ptsg1 gene); lanes 8–16, sgRNA mRosa26, targeting the mouse Rosa26 locus; lanes 17–19, ZFN mRNA (ZR) against Rosa26 locus.
However, when the Cas9 expression plasmid was first transfected and followed by transfection of in vitro-transcribed sgRNA 24 hr later, we observed consistent and efficient cleavage at target sites in both C6 and Neuro-2a cells. Figure 2A shows an example of C6 cells sequentially transfected with Cas9 plasmid and sgRNA ApoE #1, targeting the 5′ end of the rat ApoE coding sequence. In the time course, CRISPR activity was detected starting at 24 hr posttransfection.
Figure 2.

Alternative formats of CRISPR delivery led to efficient modification of the target sites. (A) The rat C6 cells were first transfected with the Cas9 expression plasmid and followed by transfection of sgRNA ApoE #1 24 hr later. Cells were then collected at different time points after sgRNA transfection for analysis. Expected bands are marked with asterisks, and sizes in number of base pairs. The bands below the 231 and 224 bp doublet are nonspecific amplifications (also see Fig. 3A). (B) sgRNAs ApoE #7 and rRosa26 were transfected into a rat C6 stable line expressing the Cas9 protein, and Cel-I assay was done at different time posttransfection. (C) sgRNAs mPtsg1 and mRosa26 were tested in a mouse Neuro-2a cell line stably expressing Cas9. (D) A more detailed time course of nuclease activity detected in sgRNA mRosa26 transfected into a Neuro-2a Cas9 stable cell line.
The results from sequential transfections seem to imply that the presence of Cas9 protein at the time of introducing sgRNAs into the cells helps CRISPR activity. We thus created stable Cas9-expressing cell lines. As shown in Fig. 2B, sgRNAs ApoE #7 and rRosa26 were nucleofected into a C6 stable line, and as shown in Fig. 2C, the same two sgRNAs from Fig. 1B were nucleofected into a Neuro-2a stable line. In both cases, CRISPR-mediated NHEJ events were detected at 8 hr posttransfection, the earliest time point taken. We took a closer look at mRosa26 sgRNA in the Neuro-2a stable line. NHEJ events were detected as early as 5 hr posttransfection (Fig. 2D). We observed similar results with sequential transfections in human cell lines, such as HEK293 and K562, and in stable cell lines created in HEK293 cells (not shown). With the Cas9 protein expressed constitutively and consistently, transfecting sgRNAs into Cas9 stable cell lines eliminates the need for the extra transfection step using Cas9 plasmid, reduces experimental variability, and provides consistent results, again suggesting that the presence of the Cas9 protein at the time of sgRNA introduction helps complex formation. We believe that using Cas9 stable cell lines is the most efficient and reliable means to validate sgRNA activity.
Ribonucleoprotein particles
It may not be desirable or feasible to have a Cas9 transgene in each cell line of interest for gene editing. The successful use of ribonucleoprotein particle (RNP) formed by incubating the Cas9 protein and sgRNA has been demonstrated to mediate both knockouts and knockins in various systems, including Caenorhabditis elegans,26 zebrafish,27,28 mice,28,29 rats,30 and cell lines,31,32 plants,33 as well as primary human T-cells.34 We tested the recombinant Cas9 protein from several different sources, with or without an NLS, for its ability to cleave the genomic targets. We chose 1:1 mass ratio (about 1:5 molar ratio) of the Cas9 protein to sgRNA(s) in this work to maximize inclusion of the Cas9 protein into RNPs for two reasons: one, RNA is known to be of little toxicity for cells and embryos, even at high concentrations; two, compared with the commercial Cas9 protein, sgRNAs are easy and inexpensive to produce. Even though each protein preparation had a different concentration and specific activity, reflecting active portion in the prep, overall, ribonucleoprotein complex worked very efficiently in a variety of cells. Figure 3A shows a direct comparison of RNP transfection to co-transfection of mRNA and plasmid with the same amount of sgRNAs. When compared with a stable line in parallel, RNPs cleaved the target site at least as efficiently, if not better, implied by earlier detection of NHEJ events (Fig. 3B).
Figure 3.

Comparison of delivery of Cas9 as protein, RNA, and DNA plasmid as well as stable expression in cultured cells. (A) sgRNA ApoE #1 against was transfected after forming a complex with recombinant Cas9 protein (P), along with Cas9 mRNA (R) or with the Cas9 expression plasmid (D) into the rat C6 cells. Most samples were transfected with 12 μg of sgRNA (R). R* indicates the samples transfected with 2 μg sgRNA. Cells were collected at 2, 6, 24, or 48 hr posttransfection and analyzed by using Cel-I assay. Expected bands are marked with asterisks, and sizes in number of base pairs. The bands below the 231 and 224 bp doublet are nonspecific amplifications. (B) sgRNA ApoE #7 RNP transfection into C6 cells was compared with sgRNA transfected into the C6 Cas9 stable cell line in a time course and analyzed by using Cel-I assay. Band sizes are labeled on the right.
We did not observe a difference between Cas9 proteins with or without nuclear localization signal (NLS). As a side-by-side comparison, we complexed two Cas9 protein preparations, one without NLS (NEB) and one with NLS present (Aldevron), with various amounts of sgRNAs ApoE #3 and #7 separately or combined and nucleofected into the rat C6 cells. No significant difference was observed between the two protein preparations (Supplementary Fig. S1). It is possible that nucleofection delivers RNPs directly to the nucleus. It is also possible that the positively charged Cas9 protein naturally localizes to the nucleus. If so, a Cas9 expression plasmid or mRNA without an NLS should work equally well as those with an NLS.
Transfected sgRNA is not rapidly degraded
To find out whether sgRNAs were degraded before the Cas9 protein was produced in sufficient amount, we measured Cas9 mRNA and sgRNA levels by using quantitative RT-PCRs in the transfected cells from Figs. 1 and 2, comparing co-transfection of rRosa26 and mRosa26 sgRNAs with Cas9 plasmid or Cas9 mRNA, and sgRNA alone into C6 or Neuro-2a cells and into Cas9 stable cell lines in a time course (Supplementary Fig. S2A–D). Surprisingly, we detected relatively stable levels of sgRNA at all time points tested. We went further to test sgRNA ApoE #1 with mRNA co-transfection and sgRNA alone into the rat C6 cells up to 96 hr posttransfection. Not surprisingly, there was no activity detected in either transfection sample sets (Supplementary Fig. S2E). Cas9 mRNA (Supplementary Fig. S2F) and sgRNA (Supplementary Fig. S2G) levels had similar trend as in Supplementary Fig. S2A–D. The gradual reduction with time seems to be a simple correlation to cell division given that the expression was normalized to GAPDH. In the sgRNA ApoE #1-transfected C6 stable cell line, where CRISPR activity was detected from 3 hr posttransfection (Supplementary Fig. S2H), the sgRNA level over time maintained the same trend and was still detected 96 hr after transfection (Supplementary Fig. S2I). Whereas the assay is by no means precisely measuring the RNA levels, given that raw extractions were used as input to avoid yield loss of small sgRNAs, the results from transfection of different sgRNAs demonstrated that unbound sgRNAs were not rapidly degraded. It is possible that in the absence of the Cas9 protein, transfected sgRNA is sequestered inside the cell, thus becoming unavailable to the Cas9 protein translated later, or sgRNA simply diffused in the cell and its concentration goes below the threshold needed for effective complex formation by the time the Cas9 protein is made.
sgRNA concentration determination
There are different methods to clean up in vitro transcription reactions and quantify sgRNA. For purification, we compared precipitation with equal volume of 5 M ammonium sulfate, ethanol precipitation, and column purification. For quantification, we used both a Nanodrop and a Qubit, which measure fluorescence emitted from a dye upon specific binding to RNA but not free NTPs. sgRNAs prepared using either of the precipitation methods in the past were remeasured using Nanodrop and Qubit side by side. There was a dramatic overestimation by Nanodrop because of the co-precipitation of NTPs from the in vitro transcription reaction, ranging 2–10-fold (Supplementary Table S2), whereas samples purified by using the MegaClear columns were measured only slightly higher on Nanodrop (Supplementary Table S3). We found that length and temperature of incubation with the precipitation methods are both contributing factors to the amount of NTP co-precipitated, rendering unpredictable overestimation by Nanodrop. At the same time, larger Cas9 mRNA preparations (Supplementary Table S4), also purified by ammonium precipitation, and column-purified PCR products or plasmid preparations (Supplementary Table S5) were measured comparably by Nanodrop and Qubit. To obtain accurate sgRNA concentration, one needs to either use column purification or measure with a method that free NTPs will not interfere.
In vitro analysis
To assay CRISPR activity in vitro, ribonucleoprotein complexes were formed by combining the Cas9 protein and each of the following four sgRNAs at 1:1 mass ratio: rRosa26, ApoE #3, targeting the 5′ end of the rat ApoE coding sequence, and ApoE #7 and ApoE C2, both targeting at the 3′ end of the ApoE coding sequence. DNA target sequences and primers are listed in Supplementary Table S1. Fifty nanograms of PCR-amplified DNA targets was incubated with 125 ng to 2 μg of respective RNP complexes, and cleavage of targets was visualized on 2% agarose gels. Given that at 1:1 mass ratio of the Cas9 protein and sgRNA the Cas9 protein is limiting, the molar ratio to target DNA was calculated based on the amount of the Cas9 protein, and marked as Cas9/target ratio in Fig. 4. However, it is not clear how much RNPs in fact form in each mixture. The Rosa26 target was cleaved close to completion at all molar ratios (Fig. 4A). The DNA targets for sgRNAs #3 and #7 were not completely cleaved even at a 37- and 32-fold of excess Cas9, respectively, and the percentage of uncut target increased with decreasing amount of the RNP mixture. The least active was the C2 RNPs, with dramatic reduction of cleavage from Cas9/target ratio of 32:1 to 16:1.
Figure 4.

In vitro cleavage assay. RNP complexes were formed by incubating the Cas9 protein and each of the four sgRNAs against rat targets: rRosa26 (A), ApoE #3 (B), #7 (C), and C2 (D). Fifty nanograms of the PCR amplicon of each target region was incubated with 2, 1, 0.5, 0.25, and 0.125 μg of respective RNP complex (see Materials and Methods), and the resulting reactions were resolved on 10% acrylamide/1× TBE gels. Without knowing what percentage of Cas9 and sgRNA in the mixture actually form RNPs and given that Cas9 is limiting, we calculated the molar ratios of Cas9 to target, which are marked above the lanes. Expected sizes of amplicons and cleaved products are marked to the right of each panel.
The relatively high ratio of RNP to target could also be a result of inefficient complex formation and/or low protein specific activity. However, the fact that different sgRNAs reached maximum cleavage at different Cas9/target ratio argues against that the requirement of high RNP-to-target ratio is simply caused by the inactive protein. The binding affinity between an sgRNA and Cas9 protein and base pairing between sgRNA and the target site likely both contribute to the cleavage activity.
We always form the complex at high concentrations and observed that, afterward, dilution and freezing RNPs at −80°C did not detectably affect activity. The in vitro assay is also a convenient method to monitor and/or compare activity of the recombinant Cas9 protein from different batches as well as reconfirming activity of RNPs stored at ready-to-inject concentrations.
Microinjection of RNA mixture and RNPs
Interestingly, unlike co-transfection, co-injection of Cas9 mRNA and sgRNA into mouse or rat fertilized eggs efficiently modifies the target sites, as reported by many other labs as the main format for microinjection.35,36 We injected varying concentrations of Cas9 mRNA (60, 20, and 10 ng/μl) while keeping sgRNA at 10 ng/μl into single-cell mouse embryos. The embryos were then allowed to develop in vitro to blastocysts, and Cel-1 assay was performed on each embryo separately (Supplementary Fig. S3A). The results showed that Cas9 mRNA was not critical in the range tested, and NHEJ level was above 50% in all combinations. In a separate experiment, when we kept Cas9 mRNA stable (5 ng/μl) and compared varied concentration of sgRNA at 15 and 2.5 ng/μl, we observed a reduction of NHEJ rate at lower sgRNA concentration from ∼45% to 28% (Table 1 and Supplementary Fig. S3B), which is still significantly more active than what we observed in cells.
Table 1.
Titration of Cas9 mRNA and sgRNA concentrations for microinjection
| Cas9 mRNA (ng/μl) | sgRNA (ng/μl) | Total embryos | NHEJ-positive embryos | Cleavage rate (%) |
|---|---|---|---|---|
| 60 | 10 | 12 | 9 | 75 |
| 20 | 10 | 13 | 9 | 69 |
| 10 | 10 | 12 | 10 | 83 |
| 5 | 15 | 22 | 9 | 41 |
| 5 | 2.5 | 25 | 7 | 28 |
NHEJ, nonhomologous end joining; sgRNA, small guide RNA.
The difference in CRISPR activity between transfecting and microinjecting Cas9 mRNA might lie in the fact that much larger quantity can be delivered via microinjection than transfection, and upon translation of the Cas9 mRNA, even with diffusion, the local sgRNA concentration is still sufficient for RNP complex formation in embryos. Similarly, the observation of higher CRISPR activity detected in some cell lines than others by co-transfection of Cas9 mRNA and sgRNA may reflect simply the difference in transfection efficiency.
For microinjection into single-cell embryos, RNPs were formed by combining the Cas9 protein and sgRNA at 1:1 mass ratio (see Methods) and then diluted to various concentrations just before injection or stored at −80°C. We observed comparable efficiencies of target site modification with each component at between 100 and 25 ng/μl, either freshly diluted or stored frozen.
Like with transfection, we did not observe a difference between Cas9 proteins with or without nuclear localization signal in microinjection. On the other hand, we found out that the reducing agent DTT in the protein buffer was deleterious to the development of single-cell embryos at as low as 0.1 mM final concentration and should be avoided in the buffer to be used to dilute the Cas9 protein for injection purpose. Whereas DTT is critical to maintain the stability of a protein preparation, we observed that once Cas9 forms a complex with sgRNA, DTT was not required to keep RNP active (not shown).
Competition
The low toxicity of CRISPR in cells and embryos allows multiplexing in targeting. One natural concern is whether there is competition between sgRNAs for binding the Cas9 protein and form the complex. We formed RNPs by combining the Cas9 protein and a target sgRNA with a competing sgRNA in one tube, and the Cas9 protein to total sgRNA at 1:1 mass ratio to maximize potential competition. Two of the four sgRNAs against rat targets, Rosa26, ApoE #3, #7, and C2, were co-complexed with the Cas9 protein. These RNP complexes were assayed at a molar ratio to PCR target of 37–32:1 (variation because of sizes of target DNA; Fig. 5, top panels) and 9.3–8:1 (Supplementary Fig. S4). The three more active sgRNAs, Rosa26, #3, and #7, were not detectably affected by the presence of other sgRNAs in the reaction, and all their target sites were cleaved as efficiently as by single RNPs.
Figure 5.

Competition assay in vitro and in cells. RNPs were formed by combining the Cas9 protein with one or two sgRNAs, as indicated above the panels. Target sites are marked on top of the panels. Each RNP complex was tested for cleaving each target site in vitro (top panels, marked as “Biochemistry”) and by transfection into the rat C6 cells (bottom panels, marked “Transfection”). Amplicon sizes and sizes for cleaved bands are marked to the left of each panel for target sites Rosa26, ApoE #3, and #7, to the right of the panels for C2. The dotted vertical lines in the bottom left panel indicate lanes 8 through 10 were assembled from different gels.
sgRNAs ApoE #7 and C2 cleave 46 bp apart on opposite strands. The larger bands from both cleavages can be recut by the other RNP (Supplementary Fig. S4B). The fact that the 234 bp band from C2 is absent from both ratios indicates that every product from C2 cleavage was cleaved again by #7, and the presence of 203 bp product from the #7 cleavage indicates that C2 did not always cut. And at a lower RNP ratio, the low intensity of 154 bp and high intensity of 203 bp demonstrated that C2 is much less efficient than #7 and itself without #7 competing, indicating a competition as well. The in vitro assay is more informative than Cel-I assay because it detects true cleavage of the target site rather than relying on heteroduplex formation that can be difficult to resolve when two sgRNAs target the same region. There is also slight competition of #3 to C2 at a lower RNP ratio. Interestingly, the most active sgRNA Rosa26 did not affect C2 activity. We conclude that there is some competition against the relatively weak sgRNA at low concentrations, but the competition is not predictable or universal, even at protein limiting conditions.
Similarly, we went on to test the same complexes in rat C6 cells and analyzed modifications at each target site. sgRNAs rRosa 26, ApoE #3, and #7 efficiently induced in/dels at their respective target sites, without interfering each other's activity (Fig. 5). sgRNA ApoE C2, the least active in vitro, was not active in cells (lanes 24, 26, and 28 in bottom right panel), nor its presence interfered with other sgRNA activity. Interestingly, ApoE #3, more active in vitro than ApoE #7, generated less NHEJ-mediated in/dels in C6 cells (Fig. 5). The difference in activity was more dramatic in embryos. When the Cas9 protein was precomplexed with sgRNAs ApoE #3 and #7 at a 3:1 ratio, target site #7 was more than 3 times more frequently modified than that of #3 (Fig. 6). These results demonstrate that biochemistry assays in vitro are not always in agreement with observations in vivo, and that validation of sgRNA activity in transfected cells is necessary, especially when downstream experiments are time-consuming and costly.
Figure 6.
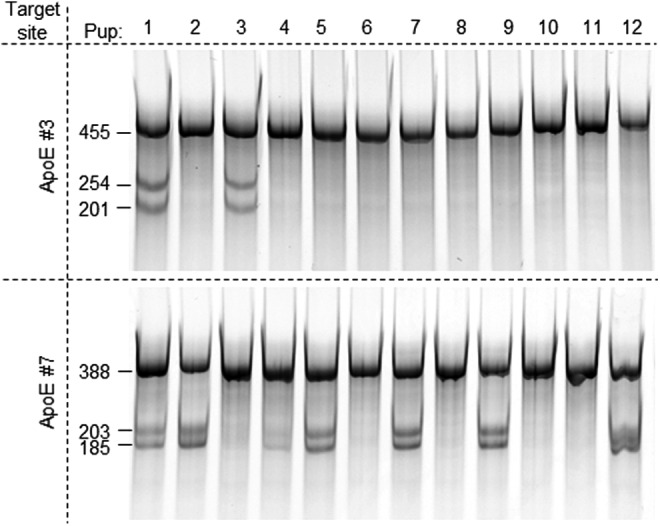
In vivo activity comparison between sgRNAs ApoE #3 and #7. sgRNAs #3 and #7 were mixed at a 3:1 ratio and incubated with an equal amount of the Cas9 protein. The mixture was then diluted in an injection buffer to the final concentration of 100 ng/μl of the Cas9 protein, 150 ng/μl of #3, and 50 ng/μl of #7 and injected into rat embryos and transferred. DNA was prepared from clipped tissues of 12 live births and analyzed with Cel-I assay to detect NHEJ events at both #3 (top panel) and #7 (bottom panel) target sites in each pup.
Under the conditions we tested and with these specific target sites, we did not observe conclusive, prominent competition between sgRNAs even when the Cas9 protein is limiting. Overall, transfection of RNPs into cultured cells and microinjection of embryos result in comparable trends of relative activity. Empirically, when we coinject two sgRNAs, we found it helpful to adjust the ratio of the sgRNAs according to their relative activity to achieve more balanced cleavage at both sites, especially after the analysis of first litter (not shown).
Off-targeting is rare
To test off-targeting events in both cells and embryos, we chose the rat ApoE targets. Among the four ApoE sgRNAs we used in this study, sgRNAs #1 and #3 target the 5′ end of the coding sequence, and sgRNAs #7 and C2, the 3′ end of the coding sequence. sgRNA C2 is not active in transfected cells, whether as part of RNPs or in stable lines. We transfected sgRNAs #1, #3, and #7 into a C6 Cas9 stable line and as RNPs into C6 cells and tested for modification at their top predicted off-target sites, respectively, at various time points using Cel-I assay. Only one of the seven tested off-target sites of sgRNA #1 (OT1-3) was modified weakly in both stable cell line and by RNP transfection (Supplementary Fig. S5A). None of the 8 off-target sites of either #3 (not shown) or #7 (Supplementary Fig. S5B) were cleaved by CRISPR in either format. When we co-injected sgRNAs #1 and #7 with Cas9 mRNA and obtained 3 pups with both sites targeted, OT1-3 again was the only predicted off-target site that was modified in one of the three founders (Fig. 7). Interestingly, sgRNA#1 was the least specific predicted by the sgRNA design algorithm (see Methods) and could be avoided when more specific designs, such as ApoE #3, were available. We have since analyzed numerous founders (both mice and rats) and observed occasional off-targeting events at 1 or 2 out of 8–10 sites per target, but off-targeting was never in all founder animals for a given project (not shown), indicating off-targeting is less efficient than on-targeting and a founder animal without off-targeting was always obtained. Off-targeting can be further lowered by choosing more specific sgRNA designs when possible. Additionally, delivery Cas9 as mRNA or recombinant protein drastically shortens length of exposure of genome to CRISPR complexes. By adjusting the amount of CRISPR reagents delivered to the cells/embryos should also further reduce off-targeting, as shown recently in cells.37
Figure 7.

Off-target events detected in live births were rare. sgRNAs ApoE #1 and #7 were coinjected with Cas9 mRNA into single-cell rat embryos and transferred to pseudopregnant females. Three pups were modified at both target sites. Top 7 predicted off-target sites of sgRNA ApoE #1 and top 8 predicted off-target sites of sgRNA ApoE #7 were analyzed for modifications in all 3 founders. In agreement with the results from transfected cells (Supplementary Fig. S4), only off-target site 3 for sgRNA #1 was modified in one of the three animals. Shown here as an example, Cel-I assay on target site and off-target sites 1–3 (OT 1-1, 1-2, and 1-3) of sgRNA ApoE #1. The cleaved bands in OT 1-1 samples were caused by SNPs. Positions of expected bands are marked by arrows on the right of the gel.
Obviously, we only sampled a small fraction of all possible off-target sites. Several genome-wide approaches38–41 have been developed for much thorough analysis of misfiring of nucleases. For therapeutic purposes, even off-target sites that are modified several magnitude less frequently than respective on-target site can be detrimental. On the research front, however, low level of off-targeting is more tolerable. Our data demonstrated here that the simple sgRNA design tool can be used to choose more specific sgRNAs. Given the cost associated with genome-wide methods and high efficiency of CRISPR targeting, when off-targeting is really a concern, it is likely cheaper and more useful to create the same cell line or animal model using two different sgRNAs, hence different off-targeting sites, and help to resolve true phenotypes from off-targeting effects.
Summary
In this study, we reported our experience of using CRISPR in different formats in both cells and embryos. The highest nuclease activity was obtained when Cas9 was either in preformed RNPs or stably expressed in a cell line, implying that local concentrations of sgRNA and the Cas9 protein are critical for effective CRISPR complex formation. RNP is the only format that is reliably active in cells, embryos, as well as in vitro, and the in vitro assay can be used as a convenient method for reconfirming RNP activity after storage. However, the differences we observed between CRISPR activities in vitro and in vivo (cells or embryos) also demonstrate the importance of sgRNA validation in cells, and Cas9 stable cell lines are the most convenient and reliable for using in sgRNA validation. We detected minimal off-targeting events in founder animals, and off-targeting can be further reduced by avoiding less-specific sgRNA designs and by using RNPs at controlled concentrations. We did not observe strong competition between sgRNAs in multiplex targeting; however, empirically we found it helpful to adjust the amount of each sgRNA according to their relative activity in vivo.
Supplementary Material
Acknowledgments
We thank Bret Robb at NEB for a sample of the Cas9 protein, and Andre Chambers for technical assistance.
Author Disclosure
All authors are full-time employees of Horizon Discovery, which sells CRISPR reagents and provides CRISPR-based genetic engineering services in cell lines and rodents.
References
- 1.Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science 2013;339:823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sontheimer EJ, Barrangou R. The bacterial origins of the CRISPR genome editing revolution. Hum Gene Ther 2015;26:412–424 [DOI] [PubMed] [Google Scholar]
- 5.Mojica FJM, Garrett RA. Discovery and seminal developments in the CRISPR field. In: CRISPR-Cas Systems: RNA-Mediated Adaptive Immunity in Bacteria and Archaea. Barrangou R. and van der Oost J, eds. (Springer, New York, NY: ). 2013; pp. 1–31 [Google Scholar]
- 6.Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol 2015;428:963–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacquier A, Dujon B. An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell 1985;41:383–394 [DOI] [PubMed] [Google Scholar]
- 8.Plessis A, Perrin A, Haber JE, et al. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics 1992;130:451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kass EM, Helgadottir HR, Chen CC, et al. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc Natl Acad Sci U S A 2013;110:5564–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 1996;93:1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005;435:646–651 [DOI] [PubMed] [Google Scholar]
- 12.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003;300:763. [DOI] [PubMed] [Google Scholar]
- 13.Durai S, Mani M, Kandavelou K, et al. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 2005;33:5978–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller JC, Holmes MC, Wang J, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 2007;25:778–785 [DOI] [PubMed] [Google Scholar]
- 15.Jo YI, Kim H, Ramakrishna S. Recent developments and clinical studies utilizing engineered zinc finger nuclease technology. Cell Mol Life Sci 2016;72:3819–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 2012;14:49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb BM, Mercer AC, Barbas CF., 3rd Directed evolution of the TALE N-terminal domain for recognition of all 5' bases. Nucleic Acids Res 2013;41:9779–9785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Troung DM, Hewitt FC, Hanson JH, et al. Retrohoming of a mobile group II intron in human cells suggests how eukaryotes limit group II intron proliferation. PLoS Genet 2015;11:e1005422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sternberg SH, Redding S, Jinek M, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014;507:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015;520:186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015;523:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 2015;6:299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sternberg SH, Doudna JA. Expanding the biologist's toolkit with CRISPR-Cas9. Mol Cell 2015;58:568–574 [DOI] [PubMed] [Google Scholar]
- 24.Carbery ID, Ji D, Harrington A, et al. Targeted genome modification in mice using zinc-finger nucleases. Genetics 2010;186:451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui X, Ji D, Fisher DA, et al. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol 2011;29:64–67 [DOI] [PubMed] [Google Scholar]
- 26.Cho SW, Lee J, Carroll D, et al. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 2013;195:1177–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gagnon JA, Valen E, Thyme SB, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 2014;9:e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sung YH, Kim JM, Kim HT, et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 2014;24:125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Shao Y, Guan Y, et al. Large genomic fragment deletion and functional gene cassette knock-in via Cas9 protein mediated genome editing in one-cell rodent embryos. Sci Rep 2015;5:17517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ménoret S, De Cian A, Tesson L, et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Sci Rep 2015;5:14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S, Kim D, Cho SW, et al. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 2014;24:1012–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang X, Potter J, Kumar S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 2015;208:44–53 [DOI] [PubMed] [Google Scholar]
- 33.Woo JW, Kim J, Kwon SI, et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotech 2015;33:1162–1164 [DOI] [PubMed] [Google Scholar]
- 34.Schumann K, Lin S, Boyer E, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A 2015;112:10437–10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu X, Chang N, Wang X, et al. Heritable gene-targeting with gRNA/Cas9 in rats. Cell Res 2013;23:1322–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li D, Qiu Z, Shao Y, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotech 2013;31:681–683 [DOI] [PubMed] [Google Scholar]
- 37.Davis KM, Pattanayak V, Thompson DB, et al. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol 2015;11:316–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai SQ, Zheng Z, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotech 2015;33:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 2015;12:237–243 [DOI] [PubMed] [Google Scholar]
- 40.Crosetto N, Mitra A, Silva MJ, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 2013;10:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frock RL, Hu J, Meyers RM, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotech 2015;33:179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.