

Abstract
Mitochondria are separated from the remainder of the eukaryotic cell by the mitochondrial outer membrane (MOM). The MOM plays an important role in different transport processes like lipid trafficking and protein import. In yeast, the ER–mitochondria encounter structure (ERMES) has a central, but poorly defined role in both activities. To understand the functions of the ERMES, we searched for suppressors of the deficiency of one of its components, Mdm10, and identified a novel mitochondrial protein that we named Mdm10 complementing protein 3 (Mcp3). Mcp3 partially rescues a variety of ERMES‐related phenotypes. We further demonstrate that Mcp3 is an integral protein of the MOM that follows a unique import pathway. It is recognized initially by the import receptor Tom70 and then crosses the MOM via the translocase of the outer membrane. Mcp3 is next relayed to the TIM23 translocase at the inner membrane, gets processed by the inner membrane peptidase (IMP) and finally integrates into the MOM. Hence, Mcp3 follows a novel biogenesis route where a MOM protein is processed by a peptidase of the inner membrane.
Keywords: ERMES complex, IMP, Mdm10, mitochondria, outer membrane
Subject Categories: Membrane & Intracellular Transport
Introduction
Mitochondria are involved in many metabolic pathways and serve as interface for processes like cell differentiation, growth, ageing and death. Therefore, they are of eminent importance to the eukaryotic cell. Their optimal function relies on the composition of two distinct membranes, the mitochondrial inner membrane and mitochondrial outer membrane (MIM and MOM, respectively).
Most mitochondrial and all integral MOM proteins are encoded in the nucleus, imported into mitochondria and sorted inside the organelle to their correct location. Proper positioning of proteins is achieved by the MOM complexes TOM (translocase of the outer membrane), TOB (topogenesis of outer membrane β‐barrel proteins, synonymously named SAM for sorting and assembly machinery) and MIM (mitochondrial import) as well as the MIM and intermembrane space (IMS) complexes TIM23, TIM22 (translocase of the inner membrane), MIA (mitochondrial intermembrane space assembly) and OXA (derived from its subunit Oxa1) complexes (for review see 1, 2, 3, 4). In addition, most of the imported proteins are cleaved by processing peptidases inside mitochondria (for review, see 5).
Several proteins destined to the MIM or IMS are first recognized by the TOM complex in the outer membrane and are then handed over to the TIM23 complex. This latter complex allows the integration of precursor proteins into the inner membrane. A subgroup of proteins is processed by the heterotrimeric protease complex inner membrane peptidase (IMP) 6. In yeast, the IMP complex contains two catalytically active subunits Imp1 and Imp2. The cleavage by IMP results in either a soluble IMS protein like Mcr1 or a MIM anchored protein like Mgr2 7, 8. So far only MIM or IMS proteins were found to be substrates of IMP.
Over the last decade, it became obvious that the lipid composition of mitochondria depends on contact sites with other cellular compartments. The functional importance of these contacts is still not completely understood, and the molecular mechanisms by which lipid exchange between the organelles occurs are yet to be unravelled. Several contacts of mitochondria have been described in yeast. Among them are tethers between mitochondria and the vacuole 9, 10 or the plasma membrane (reviewed in 11).
Mitochondria–ER contacts that recently attracted substantial attention are mediated by the ERMES (ER–mitochondria encounter structure) complex that is composed of the mitochondrial membrane proteins Mdm10, Mdm34, the soluble protein Mdm12 and the ER membrane protein Mmm1 12. Loss of one ERMES component leads to several defects in yeast cells, such as reduced insertion of β‐barrel proteins (reviewed in 13), structurally altered mitochondria, loss of the mitochondrial genome and inability to grow on non‐fermentable carbon sources (for an overview, see 14). ERMES complex is furthermore involved in mitochondrial division 15, and lack of the ERMES complex leads to reduced mitophagy 16. Moreover, deletion of ERMES subunits causes alterations in the lipid composition of mitochondria, especially reduction in the levels of phosphatidylethanolamine (PE) and cardiolipin (CL) 12, 17, 18, 19, 20, 21, as well as elevation of the yeast sterol ergosterol 20. Some ERMES components were predicted to harbour SMP (synaptotagmin‐like, mitochondrial and lipid‐binding proteins) domains that could be involved in lipid binding and transport between membranes 22. Indeed, a recent study reported that SMP domains of Mdm12 and Mmm1 bind phospholipids 23. Of note, several studies indicate that the ERMES‐mediated contact is not the only one between ER and mitochondria 24, 25. Yet an additional player, Lct1/Lam6, coordinates the extent of ER–mitochondria and vacuole–mitochondria contacts 9, 26.
Despite this progress in understanding the function of the ERMES complex, the actual molecular mechanisms that lead to all the defects described above are still not clearly defined. Recently, we identified two high‐copy suppressors of the growth defect of mdm10Δ cells 20. We showed that the two novel mitochondrial proteins (Mcp1/2) can complement the loss of Mdm10 by partial restoration of the mitochondrial lipid composition 20. Here, we report a third suppressor that we named Mdm10 complementing protein 3 (Mcp3). We demonstrate that Mcp3 is a MOM protein exposed to the cytosol. Remarkably, the correct biogenesis of Mcp3 depends on the mitochondrial membrane potential and the protein is processed by the inner membrane peptidase (IMP). Thus, Mcp3 is the first MOM substrate of this peptidase and follows a unique and novel import pathway.
Results
Mcp3 is a high‐copy suppressor of cells lacking Mdm10
Recently, we identified two novel mitochondrial proteins that partially rescue the loss of ERMES components, Mcp (Mdm10 complementing protein) 1 and Mcp2 20. Re‐evaluation of all clones led to the identification of a third gDNA segment that rescued the growth defect of mdm10Δ cells at elevated temperatures on non‐fermentable carbon sources. The segment contained a C‐terminally truncated part of the open reading frame (ORF) of Mdm10 and the genes FUN14, SPO7 and ERP2. SPO7 encodes for a putative nuclear phosphatase involved in control of nuclear growth 27. Erp2 is a member of the p24 family involved in ER to Golgi transport 28. FUN14 encodes a protein of so far unknown function (Function unknown now, 29) that was found in two high‐throughput studies to be a mitochondrial protein 30, 31. We cloned the FUN14 coding sequence in an ARS/CEN plasmid containing a rather strong triose phosphate isomerase (TPI) promoter. The resulting expression of Fun14 in mdm10Δ cells led to a partial rescue of the growth phenotype (Fig 1A). We renamed the protein Mcp3 for Mdm10 complementing protein 3.
Figure 1. Mcp3 is a multicopy suppressor of mdm10Δ.
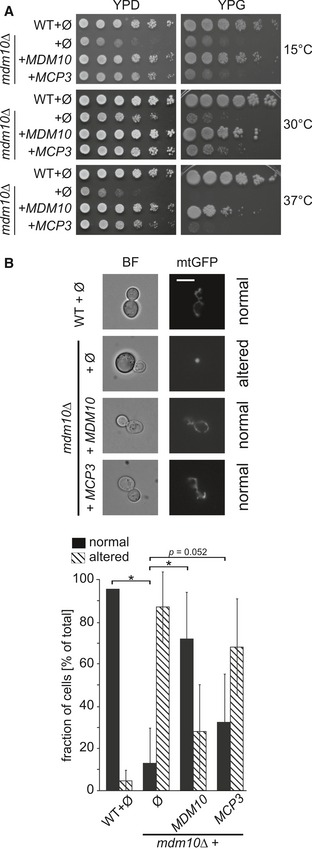
- Rescue of growth phenotype. Wild‐type (WT) or mdm10Δ cells were transformed with the empty plasmid pYX142 (∅). In addition, mdm10Δ cells were transformed with pYX142 encoding MDM10 or MCP3. Cells were grown to an OD600 of 1.0 and spotted on YPD or YPG plates in a 1:5 dilution series. Plates were incubated for growth at the indicated temperatures.
- Over‐expression of Mcp3 partially rescues the mitochondrial morphology defect in mdm10Δ cells. Cells described in (A) expressing mitochondrially targeted GFP (mtGFP) were analysed by fluorescence microscopy. Typical images (fluorescence and bright field, BF) of the four different strains are shown (scale bar = 5 μm). The quantification shows the average percentage with standard deviation bars of three independent experiments (n = 3; SD; *P < 0.05 and as indicated; two‐tailed Student's t‐test) with at least 100 cells per experiment.
Over‐expression of Mcp3 partially restores mitochondrial defects of cells lacking Mdm10
Yeast cells lacking any of the ERMES subunits do not show the typical mitochondrial network but rather contain spherical mitochondria 32, 33, 34, 35. To test whether over‐expression of Mcp3 rescues the altered mitochondrial morphology, cells were additionally transformed with a plasmid encoding a mitochondria‐targeted GFP and analysed by fluorescence microscopy. Over‐expression of Mcp3 in these cells resulted in a recovery in about 30% of the cells (Fig 1B). Thus, the partial rescue of the growth phenotype of mdm10Δ cells upon over‐expression of Mcp3 is in line with the partial restoration of mitochondrial morphology.
The lipid composition of mitochondrial membranes influences the stability of different membrane protein complexes. In this context, we reported before that the loss of Mdm10 leads to alterations in the formation of respiratory chain super‐complexes 20. Another consequence of loss of Mdm10 is a change in the assembly of TOM and TOB complexes as shown by blue native PAGE (BN‐PAGE) analysis. We first wanted to investigate whether the over‐expression of Mcp3 in mdm10Δ cells has an effect on respiratory chain super‐complexes assembly. To this end, we isolated mitochondria from wild‐type and mdm10Δ cells with or without over‐expression of Mcp3. As expected, mitochondria isolated from mdm10Δ cells showed severely reduced amounts of respiratory chain super‐complexes in comparison with wild‐type cells (Fig 2A). In line with the partial growth rescue, over‐expression of Mcp3 led only to a minor increase of the different complex species.
Figure 2. Mcp3 rescues assembly defects of protein complexes and lipid alterations of mdm10Δ cells, and restores growth of other ERMES deletion mutants.
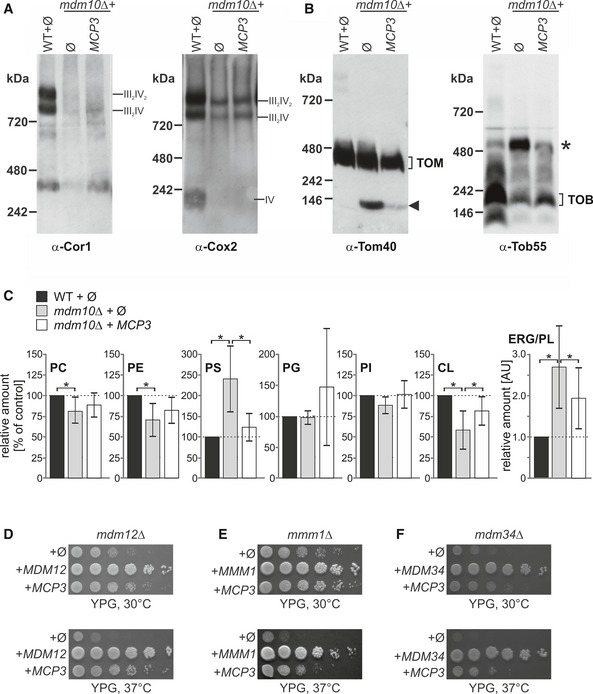
- Respiratory chain super‐complex levels are slightly restored by over‐expression of Mcp3. Mitochondria isolated from the indicated strains were lysed in 1% digitonin and subjected to a 4–8% BN‐PAGE. Proteins were analysed by immunodecoration with antibodies against either Cor1 of complex III or Cox2 of complex IV.
- Assembly defects in TOM and TOB complexes in mdm10Δ cells are partially restored by Mcp3 over‐expression. Mitochondria of the indicated cells were lysed in 1% digitonin and subjected to a 6–13% BN‐PAGE and immunoblotting with antibodies against Tom40 (left) or Tob55 (right). The assembled TOM complex and an unassembled Tom40 species at c. 100 kDa (arrowhead) are indicated as well as the TOB complex and an additional higher molecular weight species of the TOB complex (asterisk).
- Over‐expression of Mcp3 partially rescues the alterations in lipid composition of cells lacking Mdm10. Lipids were extracted from highly pure mitochondria isolated from the indicated cells and then analysed by mass spectrometric analysis. The level of each phospholipid species in wild‐type mitochondria was set to 100% and the ERG/PL ratio to 1. Relative changes in mitochondria from other cells are presented. The mean with standard deviations of three biological repeats with two technical repeats for each (n = 6; SD; *, P < 0.05; two‐tailed Student's t‐test) is given.
- Mcp3 rescues loss of Mdm12. Wild‐type (WT) or mdm12Δ cells transformed with the empty plasmid pYX142 (∅) or mdm12Δ cells over‐expressing either Mcp3 or Mdm12 (as a control) were analysed at 30°C or 37°C by drop dilution assay on rich medium containing glycerol (YPG).
- Mcp3 rescues loss of Mmm1. WT and mmm1Δ cells were transformed and analysed as described in (D).
- Mcp3 rescues loss of Mdm34. WT and mdm34Δ cells were transformed and analysed as described in (D).
Next, we asked whether the assembly of TOM and TOB complexes is influenced by over‐expression of Mcp3 in mdm10Δ cells. The same mitochondria as above were analysed by BN‐PAGE and immunodecoration with antibodies against the subunits of TOM and TOB complex, Tom40 and Tob55, respectively. As reported before, an unassembled Tom40 species emerges in organelles lacking Mdm10 (20, 36 and Fig 2B, left panel, arrowhead). This Tom40 species is nearly absent in mitochondria from mdm10Δ cells over‐expressing Mcp3. Similar observations were obtained for the TOB complex. In control mitochondria, the main assembly form is TOB core complex with a molecular mass of around 250 kDa consisting of Tob55, Tob38 and Mas37. An additional higher molecular weight species emerges in the absence of Mdm10 (Fig 2B, right panel, asterisk). This species resembles the reported TOB‐Tom5/Tom40 form 37. Although the over‐expression of Mcp3 does not fully rescue the amount of TOB core complex, the TOB‐Tom5/Tom40 species is reduced to control levels (Fig 2B, right panel, third lane). We conclude that Mcp3 over‐expression has partial restoring effects on the TOM and TOB complexes.
Next, we analysed the influence of Mcp3 on the mitochondrial lipid profile in cells lacking Mdm10. To this end, we extracted lipids from highly pure mitochondria and subjected them to mass spectrometric analysis. As described before, cells lacking Mdm10 show reduced amounts of the phospholipids PE and CL, an increase in PS levels and twofold increase in the ratio of ergosterol (ERG) to phospholipids (PL) (Fig 2C and 20). Over‐expression of Mcp3 leads to a partial rescue of the alteration in the levels of the non‐bilayer forming phospholipids PE and CL. Of note, in contrast to the earlier reported suppressors Mcp1 and Mcp2, the effect is slightly more pronounced for CL (compare Fig 2C to 20). The precursor of CL, PG, seems also to be affected although due to its overall very low abundance in mitochondria, the changes in PG vary strongly among the different preparations. Furthermore, the increase in both the amounts of PS and the ERG/PL ratio observed in mdm10Δ mitochondria is partially reduced by over‐expression of Mcp3.
Taken together, the partial rescue of growth defect of mdm10Δ cells by over‐expression of Mcp3 is in line with a partial restoration of mitochondrial morphology, protein complex assembly and lipid profile.
Over‐expression of Mcp3 rescues the loss of other ERMES components
Next, we asked whether Mcp3 can rescue the deletion of any ERMES component. To address this question, we over‐expressed Mcp3 in cells lacking either Mdm12, Mdm34 or Mmm1 and analysed the growth behaviour of the transformed cells. Over‐expression of Mcp3 led to a similar partial rescue of the growth phenotype of cells lacking Mdm12 (Fig 2D), the ER located Mmm1 (Fig 2E) or the MOM protein Mdm34 (Fig 2F). We conclude that Mcp3 can compensate the absence of functional ERMES complex rather than the missing function of an individual subunit.
Loss of Mcp3 leads to minor effects on yeast growth and mitochondrial morphology
We wondered whether deletion of MCP3 has an influence on viability. Loss of Mcp3 in different yeast wild‐type backgrounds had no severe impact on growth of yeast cells except a moderate growth defect that was observed in the BY4741 background on non‐fermentable carbon sources (Fig 3A, YPG). In addition, we investigated the mitochondrial morphology of mcp3Δ cells expressing a mitochondrial targeted GFP. In agreement with the hampered growth observed, loss of Mcp3 leads to measurable, but statistically insignificant alterations in the branched tubular network of the organelle to shortened, thickened and less branched tubules (Fig 3B). Accordingly, the ultrastructure of mitochondria, that is morphology and number of cristae, was as assessed by electron microscopy (EM) not altered in mcp3Δ cells (Fig EV1A). To investigate whether deletion of Mcp3 has a direct effect on ERMES assembly, we performed in mcp3Δ cells fluorescence microscopic analysis of a labelled form of the ERMES subunit Mmm1.
Figure 3. Elevated levels of Mcp3 influence mitochondrial morphology of yeast cells.
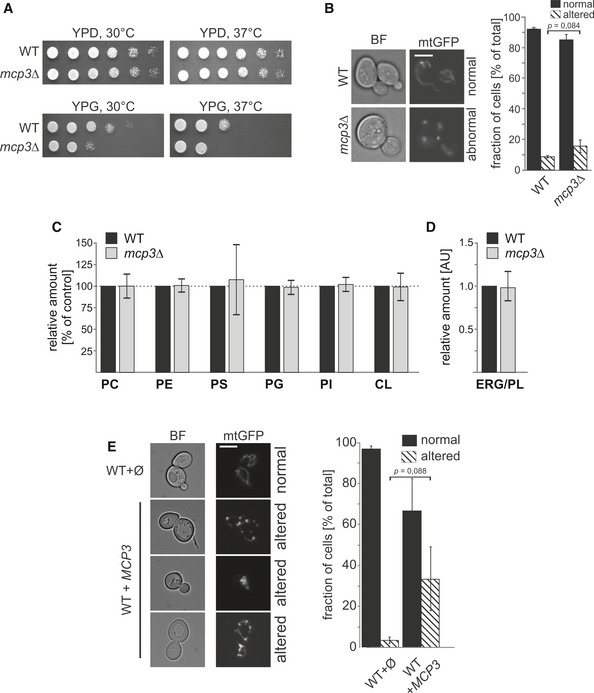
-
AGrowth phenotype of mcp3Δ cells. WT or mcp3Δ cells were grown to an OD600 of 1.0 in YPD medium and spotted on YPD or YPG plates in a 1:5 dilution series. Plates were incubated for growth at the indicated temperatures.
-
Bmcp3Δ cells have normal mitochondrial morphology. WT and mcp3Δ cells expressing mitochondrially targeted GFP (mtGFP) were grown to mid‐logarithmic phase then analysed by fluorescence microscopy. Typical images of the two different strains are shown (scale bar = 5 μm). The quantification of the depicted strains shows the average percentage with standard deviation bars of three independent experiments with at least 100 cells per experiment (n = 3; SD; P as indicated; two‐tailed Student's t‐test).
-
C, DLoss of Mcp3 has no effect on the phospholipid composition of mitochondria. Highly pure mitochondria were isolated from the indicated yeast cells. Lipids were extracted and then analysed by mass spectrometric analysis. The level of each phospholipid species in wild‐type mitochondria was set to 100% and the ERG/PL ratio to 1. Relative changes in mitochondria from mcp3Δ cells are presented. The mean with standard deviations of three biological repeats with two technical repeats for each (n = 6; SD) is given.
-
EOver‐expression of Mcp3 leads to alterations in the tubular mitochondrial network. Wild‐type cells expressing mitochondrially targeted GFP (mtGFP) were transformed with a plasmid over‐expressing Mcp3 (MCP3) or an empty plasmid as control (∅). Cells were grown to mid‐logarithmic phase and then analysed by fluorescence microscopy. Typical images of the two different strains are shown (scale bar = 5 μm). The quantification shows the average percentage with standard deviation bars of three independent experiments with at least 100 cells per experiment (n = 3; SD; P as indicated; two‐tailed Student's t‐test).
Figure EV1. Altered levels of Mcp3 have no effect on mitochondrial ultrastructure.
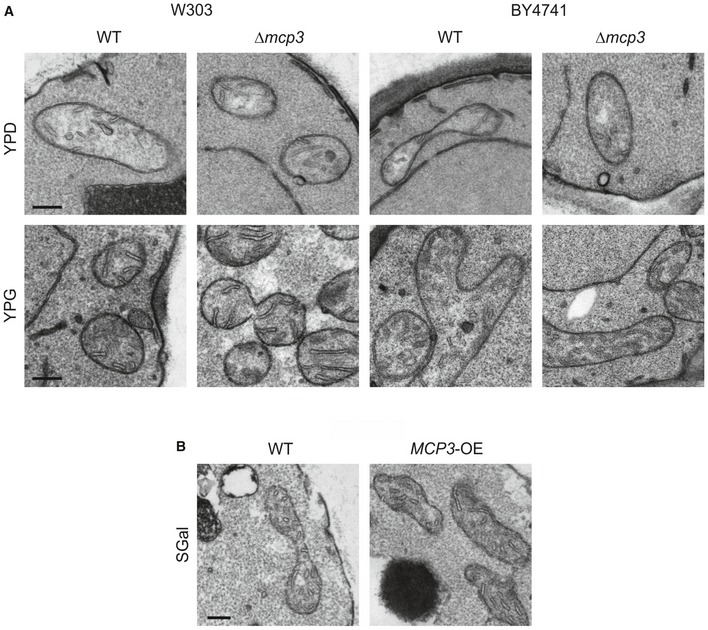
- WT and mcp3Δ cells in two different genetic backgrounds (W303 and BY4741) were grown in glucose (YPD) or glycerol (YPG) containing medium and analysed by thin‐section transmission electron microscopy. Representative images of mitochondria are shown. Scale bars, 200 nm.
- WT cells carrying an empty plasmid (WT) and cells overexpressing MCP3 from a plasmid under control of the TPI promoter (MCP3‐OE) were grown in galactose‐containing minimal medium (SGal) and analysed by thin‐section transmission electron microscopy. Representative images of mitochondria are shown. Scale bar, 200 nm.
Employing two different wild‐type backgrounds, we observed that the ERMES complex assembled similarly in both control and mcp3Δ cells and co‐localized with mitochondria in punctate structures (Fig EV2A and B). Hence, Mcp3 is required neither for the formation of cristae structures nor for proper assembly of the ERMES complex.
Figure EV2. Altered levels of Mcp3 have no effect on ERMES foci formation and colocalization with mitochondria.
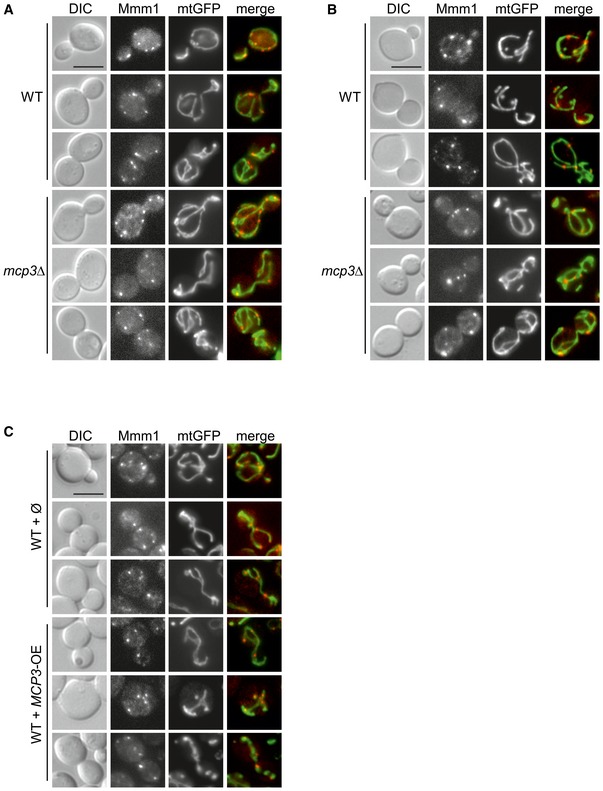
-
A, BWT and mcp3Δ cells (from BY4741 (A) or W303 (B) background) expressing mitochondrially targeted GFP (mtGFP) and RFP‐tagged Mmm1 were grown to mid‐logarithmic phase and then analysed by fluorescence microscopy. Representative images are shown. Scale Bars, 5 μm.
-
CWT cells harbouring an empty plasmid or a plasmid overexpressing Mcp3 (BY4741 background) were transformed with plasmids encoding mitochondrially targeted GFP (mtGFP) and RFP‐tagged Mmm1 and were analysed as in (A). Scale Bar, 5 μm.
Next, we asked whether the loss of Mcp3 leads to a change in the mitochondrial lipid profile. Lipids were extracted from highly pure mitochondria and analysed by mass spectrometry. Neither the phospholipid nor ergosterol levels were altered in mitochondria from mcp3Δ cells as compared to control organelles (Fig 3C and D, respectively).
Although over‐expression of Mcp3 did not result in any growth phenotype (Appendix Fig S1), we asked whether it can cause alterations in mitochondrial morphology. Interestingly, elevated levels of Mcp3 led in some of the cells to partially fragmented mitochondria (Fig 3E), suggesting an influence of Mcp3 over‐expression on mitochondrial morphology, although this trend is statistically not significant. In addition, mitochondrial ultrastructure as monitored by EM was not affected upon over‐expression of Mcp3 (Fig EV1B). These findings might suggest a yet undefined role of Mcp3 in mitochondrial morphogenesis. To test whether over‐expression of Mcp3 has a direct effect on ERMES complex formation, we analysed ERMES foci in cells with elevated Mcp3 levels. Over‐expression of Mcp3 did not alter ERMES punctae formation or co‐localization with mitochondria (Fig EV2C).
Collectively, loss of Mcp3 has no severe effect on yeast growth, cristae formation, ERMES assembly, morphology and lipid composition of mitochondria. Yet over‐expression of the protein leads to slight mitochondrial morphology changes, with no obvious effects on the other tested characteristics.
Mcp3 resides in the mitochondrial outer membrane
In silico analysis of the sequence of Mcp3 predicts a canonical mitochondrial targeting sequence, followed by a hydrophobic segment and two putative transmembrane domains (TMDs, Fig 4A). A first attempt to C‐terminally label Mcp3 with an HA‐tag in order to analyse the protein by immunodetection resulted in a non‐functional protein (see also below). A previous report predicted Mcp3 to be a putative substrate of the Imp1 subunit of the IMP peptidase and suggested the cleavage site to be between Asn69 and Asp70 38. In agreement with our observation that C‐terminally tagged Mcp3 is non‐functional, Esser et al were unable to test this prediction since C‐terminally tagged Mcp3 was not detectable. An independent support for processing of Mcp3 by IMP is provided by high‐throughput analysis of the N‐terminal sequences of mitochondrial proteins 39. This study found the N‐terminus of endogenous Mcp3 to start with Asp70 (DSLG…) in agreement with the predicted processing site of Imp1. To further analyse Mcp3, we constructed an internal tagged version of Mcp3. The HA‐tag was inserted four amino acids C‐terminally to the processed N‐terminus identified by mass spectrometry (DSLG‐HA Tag‐…). Functionality of the internally tagged Mcp3 (HA‐Mcp3) was shown by growth rescue of mdm10Δ cells (Fig 4B). To test whether this construct can be detected, we over‐expressed HA‐Mcp3 in cells lacking endogenous Mcp3. Using mitochondria isolated from these cells, we observed that the mature HA‐Mcp3 migrates as a 17‐kDa protein in line with processing of the N‐terminal region (Fig 4C and see below).
Figure 4. Subcellular and submitochondrial localization of Mcp3.
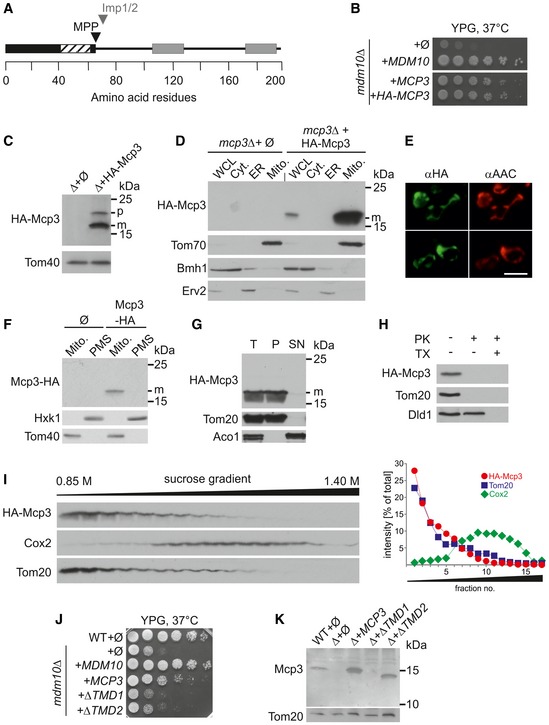
- Predicted protein domains for Mcp3. Mcp3 is predicted to contain a canonical N‐terminal mitochondrial targeting signal (black bar) that contains a hydrophobic stretch (hatched part). The predicted MPP and the Imp1/2 cleavage sites are indicated by a black or grey arrow head, respectively. The two predicted transmembrane domains are highlighted as grey bars.
- Internally tagged HA‐Mcp3 is functional. mdm10Δ cells were transformed with the empty plasmid pYX142 (∅) or pYX142 encoding Mdm10, Mcp3, or an internally HA‐tagged Mcp3 (MDM10, MCP3, HA‐MCP3). Cells were grown to an OD600 of 1.0 and spotted on YPG plates in a 1:5 dilution series. Plates were incubated for growth at 37°C for 4 days.
- HA‐Mcp3 is processed in vivo. Mitochondria isolated from mdm10Δ (Δ) cells containing the empty plasmid or expressing HA‐Mcp3 were analysed by SDS–PAGE and immunodecoration with antibodies against the indicated proteins. Two different bands are detectable in case of HA‐Mcp3 (p, precursor; m, mature protein). The MOM protein Tom40 serves as loading control.
- Mcp3 is a mitochondrial protein. Whole‐cell lysate (WCL) and fractions corresponding to cytosol (Cyt.), light microsomal fraction (ER) and mitochondria (Mito.) of mcp3Δ cells either containing the empty plasmid (∅) or expressing HA‐Mcp3 were analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐tag, the mitochondrial protein Tom70, a marker protein for the cytosol (Bmh1) and an ER marker protein (Erv2). m, mature protein.
- Mcp3 co‐localizes with the mitochondrial marker protein AAC. Cells over‐expressing HA‐Mcp3 were fixed and analysed by fluorescence microscopy using antibodies against the HA‐epitope (αHA) and the mitochondrial ADP/ATP carrier (αAAC) (scale bar = 5 μm).
- C‐terminally tagged Mcp3‐HA is located to mitochondria. A mitochondria‐enriched fraction (Mito.) and the post‐mitochondrial supernatant (PMS) of wild‐type cells carrying the empty plasmid (∅) or over‐expressing the C‐terminally HA‐tagged Mcp3 (Mcp3‐HA) were analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐Tag, the mitochondrial protein Tom40 and a marker protein for the cytosol (Hxk1). m, mature protein.
- Mcp3 is membrane‐embedded. Mitochondria isolated from mcp3Δ cells expressing HA‐Mcp3 were left untreated (I, input) or subjected to alkaline extraction. The supernatant (SN) and pellet (P) fractions were analysed by SDS–PAGE and immunodecoration with antibodies against the indicated proteins. Tom20, an integral MOM proteins; Aco1, soluble matrix protein. m, mature protein.
- Mcp3 is a MOM protein. Mitochondria isolated from cells expressing HA‐Mcp3 were treated with proteinase K (PK, 100 μg/ml) in the absence or presence of the detergent Triton X‐100 (TX). Samples were precipitated with TCA and analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐tag or the indicated mitochondrial proteins. Tom20, a MOM protein exposed to the cytosol; Dld1, a protein located in the IMS.
- Mcp3 behaves like an outer membrane protein in density gradient centrifugation. Mitochondrial vesicles isolated from cells over‐expressing HA‐Mcp3 were subjected to sucrose density gradient centrifugation. Fractions of the gradient were collected and analysed by SDS–PAGE and immunodecoration with antibodies against the indicated proteins. The processed form of HA‐Mcp3 is shown. Right panel: The intensities of the various bands were quantified and depicted. The sum of all intensities for each protein was set to 100%.
- Mcp3 lacking either transmembrane domain fails to rescue loss of Mdm10. Wild‐type cells carrying the empty plasmid (wt + ∅) or mdm10Δ cells transformed with the empty plasmid (∅) or plasmid encoding Mdm10, Mcp3, or Mcp3 lacking TMD1 or TMD2 (MDM10, MCP3, ΔTMD1, ΔTMD2) were grown to an OD600 of 1.0 and spotted on YPG plates in a 1:5 dilution series. Plates were incubated for growth at 37°C for 4 days.
- Mcp3 lacking either of the two transmembrane domains is unstable. Crude mitochondria were obtained from wild‐type cells containing an empty plasmid (Ø) or mcp3Δ cells (Δ) transformed with plasmid encoding Mcp3 or Mcp3 lacking TMD1 or TMD2 (MCP3, ΔTMD1, ΔTMD2). Samples were analysed by SDS–PAGE and immunodecoration with antibodies against Mcp3 and Tom20 as loading control.
Next, we wanted to confirm the predicted mitochondrial localization of Mcp3. To this end, we fractionated the aforementioned cells into various subcellular compartments. HA‐Mcp3 and the mitochondrial marker Tom70 were solely detected in the mitochondrial fraction (Fig 4D). The mitochondrial localization could be confirmed by immunofluorescence microscopy. The fluorescence signal for HA‐Mcp3 closely resembled the mitochondrial staining of AAC (Fig 4E). We wondered whether also non‐functional C‐terminally HA‐tagged Mcp3 was localized to mitochondria. As shown before with the C‐terminally GFP‐tagged Mcp3 30, processed Mcp3‐HA was found in enriched mitochondrial fractions (Fig 4F). This suggests that an alteration of the C‐terminus leads to loss of function rather than unstable or mis‐targeted protein.
Several in silico prediction programmes suggest two TMDs for Mcp3 (Fig 4A). To investigate whether Mcp3 is indeed an integral membrane protein, mitochondria containing HA‐Mcp3 were treated with an alkaline carbonate solution to extract soluble proteins and those that are peripheral attached to the membrane. Similarly to the integral membrane protein Tom20, HA‐Mcp3 was found in the pellet fraction (Fig 4G). Next, we wanted to know in which of the mitochondrial membranes Mcp3 is embedded. First, we treated isolated mitochondria containing HA‐Mcp3 with proteinase K (PK). As shown in Fig 4H, HA‐Mcp3 is digested in intact mitochondria similarly to the MOM protein Tom20, whereas the IMS protein Dld1 is degraded only upon lysis of the membranes with detergent. This suggests that Mcp3 is a MOM protein exposed to the cytosol. To substantiate this conclusion, we separated the two mitochondrial membranes by density gradient centrifugation. Membrane vesicles from mitochondria expressing HA‐Mcp3 were obtained by hypo‐osmotic swelling and sonication. These vesicles were separated by a sucrose gradient, and the different fractions were analysed for the presence of HA‐Mcp3 and marker proteins for the MIM and MOM (Fig 4I). HA‐Mcp3 and the MOM marker protein Tom20 are predominantly detectable in the lighter fractions corresponding to the MOM. In contrast, the MIM protein Cox2 is found, as expected, mainly in the heavier fractions (Fig 4I). Next, we wondered whether the predicted TMDs are important for the function of Mcp3. We constructed two additional versions of Mcp3 that lack either TMD1 (amino acid 106–128) or TMD2 (amino acids 172–198) and tested their rescue capacity in cells lacking Mdm10. Both constructs failed to rescue the growth defect of mdm10Δ cells (Fig 4J). We then asked whether the absence of the TMDs results in changes in protein stability. Using cells expressing these Mcp3 variants, we observed that the absence of TMD1 resulted in a non‐detectable species, whereas loss of TMD2 caused lower detected amount of the protein in comparison with native Mcp3 (Fig 4K). We conclude that both TMDs are important for the stability and function of the protein.
Taken together, our results show that Mcp3 is an integral MOM protein and are in line with a high‐throughput study where Mcp3 was found in the MOM proteome 40.
Mcp3 is processed in vitro and imported by the TOM complex
Our findings raise the unique possibility that Mcp3 is a MOM protein and also a substrate of the IMP peptidase. This led us to study the biogenesis of Mcp3. First, we asked whether Mcp3 is processed in an in vitro import assay. To address this question, we incubated radiolabelled Mcp3 with isolated mitochondria. Mcp3 was indeed processed into a mature form of about 15 kDa in a time‐dependent manner (Fig 5A). We also noted an additional band at around 10 kDa that we assume to be the cleaved N‐terminal segment of the precursor protein (Fig 5A, arrowhead). Thus, the processing of Mcp3 under both in vivo and in vitro conditions allows us to use the formation of the mature band as a read‐out for correct import.
Figure 5. Mcp3 is imported by the TOM complex.
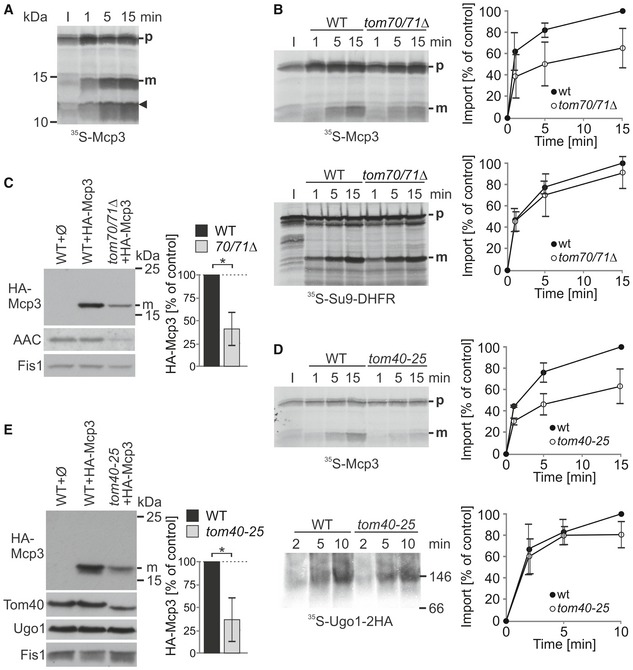
- Isolated mitochondria were incubated with radiolabelled Mcp3 precursor protein for the indicated time periods. After import, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography. The arrowhead marks an additional band of the size of the cleaved N‐terminus of Mcp3. p and m, precursor and mature forms, respectively.
- Import of Mcp3 is dependent on the receptor Tom70. Mitochondria isolated from wild‐type or tom70/71Δ cells were incubated with radiolabelled Mcp3 or pSu9‐DHFR (as control) for the indicated time periods. After import, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography. p and m, precursor and mature forms, respectively. I, 20% of radiolabelled precursor protein used in each import reaction. Bands corresponding to the mature (m) form were quantified. Import into wild‐type mitochondria after 15 min was set to 100%. The mean with standard deviations is depicted (n = 3; SD).
- Reduced steady‐state levels of HA‐Mcp3 in cells lacking Tom70. Crude mitochondria isolated from the indicated cells were analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐tag, AAC as control substrate of Tom70, and Fis1 as a loading control. HA‐Mcp3 levels were quantified in relation to Fis1 levels, and the levels in WT cells were set to 100%. The bar diagram shows the mean with standard deviation (n = 3; SD; *, P < 0.05; two‐tailed Student's t‐test).
- Import of Mcp3 is dependent on Tom40. Mitochondria isolated from wild‐type and tom40‐25 cells were incubated with radiolabelled Mcp3 for the indicated time periods. Next, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography as described in (B). As a control, Ugo1 was imported into the same mitochondria. Organelles were reisolated and analysed by BN‐PAGE and autoradiography. The band corresponding to the Ugo1 dimer at around 150 kDa was quantified. The mean with standard deviation is shown (n = 3; SD).
- Steady‐state levels of HA‐Mcp3 are lower in cells harbouring a mutant TOM40 allele. Crude mitochondria from wild‐type and tom40‐25 cells were obtained and analysed as described in (C). Immunodecoration was performed with antibodies against the HA‐tag, Tom40, and Fis1 as loading control.
We first aimed to find out which import receptor recognizes the precursor protein on the surface of the organelles. We isolated mitochondria from strains deleted in either TOM20 or TOM70/71. In vitro import of Mcp3 was not altered when Tom20 was absent (Appendix Fig S2A). On the other hand, the in vitro import of Mcp3 was strongly diminished in organelles lacking Tom70/71, whereas the bona fide Tom20 substrate pSu9‐DHFR was not affected (Fig 5B). Accordingly, in vivo steady‐state levels of HA‐Mcp3 were strongly reduced in the absence of TOM70/71 (Fig 5C). Taken together, Mcp3 is imported via recognition by Tom70.
Recently, an import pathway for MOM multispan proteins was described that requires Tom70 but is independent of other components of the TOM complex 41, 42. To investigate whether this is also the case for Mcp3, we tested whether its import relies on functional Tom40. To this end, we used mitochondria from cells harbouring the temperature‐sensitive allele tom40‐25 43. Mcp3 was imported into the altered organelles with highly reduced efficiency as compared to control mitochondria. In contrast, as expected, organelles harbouring the tom40‐25 mutant showed only a slightly reduced capacity to assemble the TOM pore‐independent import substrate Ugo1 (Fig 5D). In line with the in vitro results, the detected steady‐state levels of HA‐Mcp3 are strongly reduced in the tom40‐25 mutant in comparison with control cells even at the permissive temperature (Fig 5E). Of note, the non‐processed precursor was degraded by addition of external protease, suggesting that Mcp3 does not reach the intermembrane space in these mitochondria (Fig EV3A). Thus, in contrast to other multispan MOM proteins, the import of Mcp3 requires functional Tom40.
Figure EV3. Mcp3 precursor is degraded by PK in vitro if it does not reach the inter membrane space.
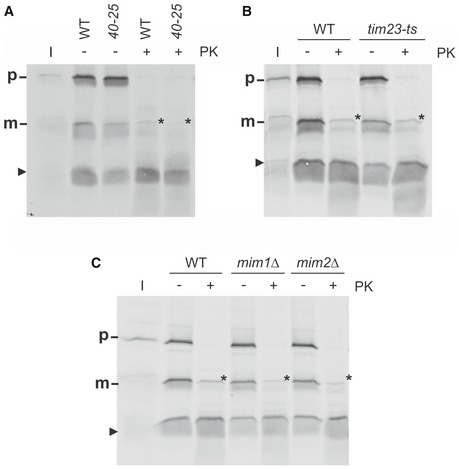
- Mitochondria from wild‐type and tom40‐25 cells were isolated and incubated with radiolabelled Mcp3 for 15 min. After import, mitochondria were reisolated, left untreated or incubated with proteinase K (PK) and analysed by SDS–PAGE and autoradiography.
- Mitochondria from wild‐type and tim23ts cells were isolated and incubated with radiolabelled Mcp3 for 15 min. Analysis was performed as in (A).
- Mitochondria from wild‐type and mim1Δ or mim2Δ cells were isolated and incubated with radiolabelled Mcp3 for 15 min. Analysis was performed as in (A).
We also aimed at studying physical interactions of the precursor protein with the different TOM components. First, we performed a pull‐down assay with 35S‐Mcp3 and purified GST fusion proteins of the soluble cytosolic domains of Tom70, Tom20 and Tom22 44. In line with our previous results and as shown in Fig 6A, the highest amount of Mcp3 binds to GST‐Tom70. Furthermore, also Tom22 binds in vitro Mcp3 precursor (Fig 6A). To further confirm this direct interaction in organello, we used mitochondria that contain His‐tagged Tom22 45. After in vitro import and lysis of the organelles, proteins pulled down by Ni‐NTA beads were analysed. We observed specific binding of Mcp3 to the resin only if mitochondria contained the His‐tagged Tom22 (Fig 6B). Finally, we analysed the in vitro import reaction into the His‐Tom22 mitochondria by BN‐PAGE and addition of an antibody against the His‐tag. Indeed, the import intermediate of radiolabelled Mcp3 associated with the TOM complex (at about 480 kDa) was shifted to higher molecular weight (Fig 6C, arrowhead). Taken together, these results strongly favour a passage of Mcp3 through the TOM complex, where Tom22 mediates the relay to Tom40 after initial binding by Tom70.
Figure 6. Mcp3 precursor physically interacts with components of the TOM complex.
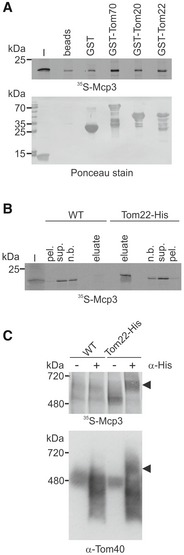
- Mcp3 precursor binds in vitro to cytosolic domains of TOM receptors. Radiolabelled Mcp3 was incubated with recombinant GST alone or GST fused to the cytosolic domains of Tom70, Tom20 or Tom22 prebound to glutathione beads or with unloaded beads. After washing, bound material was eluted with sample buffer. Proteins were analysed by SDS–PAGE, blotting onto a membrane and autoradiography. To demonstrate equal amounts of GST fusion proteins, the membrane was stained with Ponceau S. I, input 5% of precursor used.
- Mcp3 precursor binds to Tom22 in organello. Radiolabelled Mcp3 was incubated with mitochondria isolated from WT His‐Tom22 containing cells. Mitochondria were pre‐incubated with CCCP (40 μM) to halt import. After lysis with β‐dodecyl maltoside, samples were incubated with Ni‐NTA beads. After washing, bound proteins were eluted with sample buffer and analysed by SDS–PAGE and autoradiography. I, 5% of 35S‐Mcp3 used; pel., 2% insoluble material after clarifying spin; sup., 2% of material incubated with Ni‐NTA; n.b. 2% of non‐bound material after binding to Ni‐NTA; eluate, 100% of bound material.
- Radiolabelled Mcp3 was incubated with organelles described in (B). After import, mitochondria were solubilized in digitonin, and the lysate was cleared by centrifugation and incubated with or without an antibody against the His‐tag (α‐His). Samples were analysed by BN‐PAGE, autoradiography and afterwards immunodecoration with an antibody against Tom40. Bands shifted by the anti‐His antibody are indicated by arrowhead.
Biogenesis of Mcp3 is dependent on the TIM23 complex and the inner membrane peptidase
So far specific peptidases at the MOM were not identified. Thus, the processing of Mcp3 raised the possibility that the import pathway of the protein includes contact with a peptidase anchored to the inner membrane. Furthermore, presequence‐containing proteins that are imported via the general import pathway are transferred from TOM complex to the TIM23 complex in the MIM. To test whether the Mcp3 precursor follows a similar pathway, we used a yeast strain bearing a temperature‐sensitive allele of TIM23, tim23ts 46. In vitro import of Mcp3 into mitochondria harbouring the altered Tim23 was strongly impaired already at permissive temperature, whereas the import of the Tim23‐independent MOM protein Ugo1 was not influenced (Fig 7A). Accordingly, the steady‐state levels of HA‐Mcp3 were reduced in the organelles of mutant cells, whereas the levels of the MOM proteins Ugo1 and Fis1 were unaffected (Fig 7B). Hence, the presequence translocase TIM23 complex is involved in the biogenesis of Mcp3.
Figure 7. Biogenesis of Mcp3 depends on the TIM23 complex and processing by IMP.
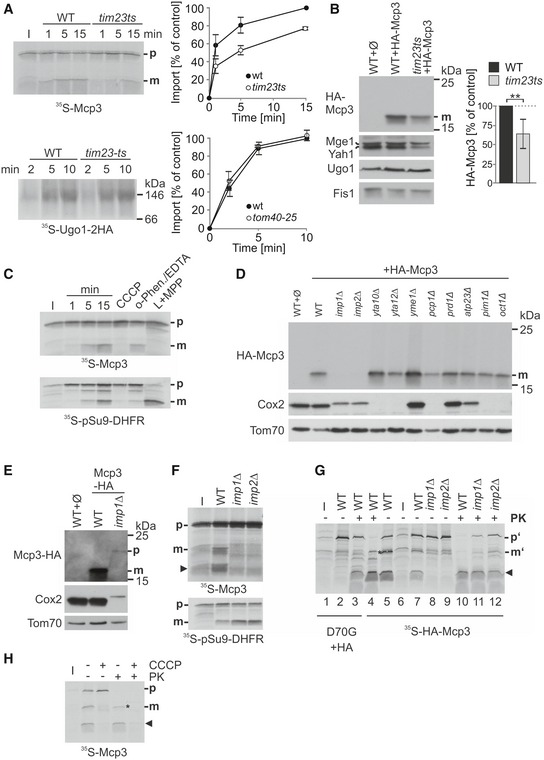
- Import of Mcp3 is dependent on the TIM23 complex. Mitochondria isolated from wild‐type and tim23ts cells were incubated with radiolabelled Mcp3 for the indicated time periods. Further treatment and analysis (n = 3; SD) was as described for Fig 5D.
- Steady‐state levels of HA‐Mcp3 are lower in cells harbouring a temperature‐sensitive TIM23 allele. Crude mitochondria were obtained at the non‐permissive temperature from WT or tim23ts cells containing a plasmid expressing HA‐Mcp3. Samples were analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐tag, the matrix proteins Mge1 and Yah1 as typical TIM23 substrates, Ugo1 and Fis1 as TIM23‐independent substrates. HA‐Mcp3 levels were quantified in relation to Fis1 levels. Levels in WT cells were set to 100%. The bar diagram shows the mean with standard deviation of six independent experiments (n = 6; SD; **, P < 0.01; two‐tailed Student's t‐test).
- Import and processing of Mcp3 is dependent on the mitochondrial membrane potential. Isolated mitochondria were incubated with radiolabelled Mcp3 or pSu9‐DHFR as control for the indicated time periods or for 15 min in the presence of either CCCP, or o‐phenanthroline and EDTA (o‐Phen./EDTA). After import, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography. L + MPP: radiolabelled proteins were incubated with recombinant MPP.
- Mcp3 is processed by Imp1/2. Single‐deletion strains of known mitochondrial proteases and peptidases were transformed with a plasmid expressing HA‐Mcp3. Crude mitochondria were isolated and analysed by SDS–PAGE and immunodecoration with antibodies against the indicated proteins and the HA‐epitope. Cox2, a known substrate of IMP; Tom70, a mitochondrial outer membrane protein.
- C‐terminally tagged Mcp3‐HA is processed by Imp1. WT and imp1Δ cells were transformed with a plasmid expressing Mcp3‐HA, and further analysis was performed as described in (D).
- Mcp3 is processed by Imp1/2 after in vitro import into mitochondria. Mitochondria isolated from WT, imp1Δ or imp2Δ strains were incubated with radiolabelled precursor of Mcp3 or pSu9‐DHFR as control. Further treatment and analysis was as described in Fig 5A.
- Full‐length Mcp3 precursor accumulates during import if IMP processing is abolished. Radiolabelled internally HA‐tagged Mcp3 without (HA‐Mcp3) or with mutation in the Imp1 cleavage site (D70G + HA) were incubated with mitochondria as in (F). After import, samples were incubated with or without PK (20 μg/ml). Samples were analysed by SDS–PAGE and autoradiography.
- Mitochondrial membrane potential is required for efficient import of Mcp3 precursor into mitochondria. Isolated mitochondria were incubated with radiolabelled Mcp3 in the presence or absence of CCCP. After import, mitochondria were incubated with PK as in (G) and analysed by SDS–PAGE and autoradiography. I, 20% of radiolabelled precursor protein used in each import reaction; the arrowhead marks an additional band of the size of the cleaved N‐terminus of Mcp3.
Establishing a requirement for the TIM23 machinery at the inner membrane, we next asked whether Mcp3 import requires the membrane potential. Hence, we tested the processing efficiency in the presence of the uncoupler CCCP and valinomycin (Fig 7C for CCCP and Fig EV4). In the presence of both substances that depolarize mitochondria, we could hardly detect a mature form. We conclude that processing of Mcp3 is dependent on the mitochondrial membrane potential. Since Mcp3 has in its N‐terminal domain a predicted MTS, we wondered whether the observed processing can be mediated by the matrix processing peptidase (MPP). To that end, we added to the in vitro import reaction two chelators for bivalent cations, ortho‐phenanthroline and EDTA, which are known to inhibit MPP. This addition had no influence on processing of Mcp3 (Fig 7C, sixth lane). Additionally, incubation of 35S‐Mcp3 precursor with recombinant MPP did not result in cleavage of the precursor molecules while the known MPP substrate, pSu9‐DHFR, was almost completely processed (Fig 7C, L + MPP). Next, we isolated mitochondria from a temperature‐sensitive mutant strain of the MPP subunit Mas1 and performed in vitro import assays under various conditions. Whereas the mutation had a clear effect on cleavage of the model substrate pSu9‐DHFR, processing of Mcp3 was not influenced (Appendix Fig S3). Taken together, the observed mature form of Mcp3 is not a result of MPP cleavage.
Figure EV4. Import of Mcp3 is dependent on the mitochondrial membrane potential.
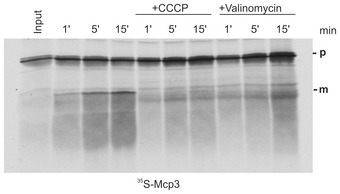
Mitochondria isolated from wild‐type yeast cells were incubated with radiolabelled precursor protein Mcp3 for the indicated time periods (1, 5 or 15 min) in the absence or presence of CCCP or valinomycin. After import, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography. Input, 20% of radiolabeled precursor applied in the reaction.
To identify the peptidase that cleaves Mcp3, we over‐expressed HA‐Mcp3 in different yeast deletion strains each lacking one of the known enzymes involved in processing and/or degradation of mitochondrial proteins and analysed the steady‐state levels of HA‐Mcp3 in these cells. Only in the absence of the two subunits of the inner membrane peptidase, Imp1 and Imp2, the mature form could not be detected (Fig 7D). Interestingly, also the C‐terminally tagged Mcp3 (Mcp3‐HA) is processed by Imp1 (Fig 7E). This observation confirms that the C‐terminal tag results in an inactive mitochondrial protein rather than mislocalization to another organelle or protein instability. To show that Mcp3 is a substrate of the IMP complex, we isolated mitochondria from imp1Δ or imp2Δ cells and performed an in vitro import assay. The mature form of Mcp3 was not formed in mitochondria lacking either Imp1 or Imp2 (Fig 7F). To finally confirm the predicted Imp1 cleavage site N‐terminally to Asp70 38, we performed site‐directed mutagenesis of the internally HA‐tagged Mcp3 variant to obtain the D70G substitution mutant. Such an Asp to Gly exchange is known to prevent cleavage of substrate proteins by IMP 6, 8. HA‐tagged 35S‐Mcp3 was incubated in vitro with isolated mitochondria and was as expected processed, in an IMP‐dependent manner, upon its import into a mature form (Fig 7G, m', lanes 5 and 7–9). In contrast, the D70G mutant lacking the Imp1 consensus sequence was not processed (Fig 7G, lane 3).
Next, we analysed the D70G variant. We incubated mitochondria after import with proteinase K (PK). Interestingly, the D70G precursor was resistant to PK treatment (Fig 7G, lane 3, p'), whereas the non‐processed HA‐tagged native Mcp3 precursor is fully degraded. Thus, it seems that the import intermediate of Mcp3‐D70G, which cannot be cleaved by IMP, accumulates in the intermembrane space. This implicates that the N‐terminus of Mcp3 reaches the intermembrane space before the C‐terminal TMD is inserted into the outer membrane. Indeed, in the absence of the IMP peptidases also the precursor of native HA‐Mcp3 accumulates in the intermembrane space (Fig 7G, lanes 10–12).
Of note, we observed a PK protected band of lower molecular weight of HA‐Mcp3 in wild‐type mitochondria (Fig 7G, lane 4, asterisk). This band could correspond to a processed Mcp3 intermediate in the IMS. We wanted to know whether, similarly to the overall import of Mcp3, the formation of this species is dependent on the membrane potential. Fig 7H shows that indeed, the corresponding band for Mcp3 disappears in the presence of the uncoupler CCCP. We detected this processed PK‐resistant variant in many experiments, but we did not find appropriate conditions for chasing this form to a fully membrane‐inserted species. Hence, we assume that it is a non‐productive import intermediate of Mcp3.
Our observations confirm that the IMP complex cleaves the precursor form of Mcp3. Since Imp1 and Imp2 are only functional in a multimeric Imp1/Imp2 complex, we cannot determine which subunit is actually responsible for the processing of Mcp3. However, all results are in agreement with the in silico prediction of Mcp3 as substrate of Imp1 38. Taken together, our results demonstrate that after crossing the MOM Mcp3 is taken over by TIM23 complex and processed by IMP in the IMS.
MIM complex is involved in the biogenesis of Mcp3
Next, we asked how Mcp3 is integrated into the MOM. Two major complexes are known to mediate insertion of proteins into the MOM, namely the TOB/SAM and MIM complexes. To test whether the β‐barrel insertion machinery is involved in the membrane integration of Mcp3, we employed a yeast strain deleted for the TOB subunit Mas37/Sam37. No obvious difference in Mcp3 import was observed when mitochondria from mas37Δ cells were compared to wild‐type organelles (Appendix Fig S2B). In addition, we did not observe any influence of the small TIM chaperones that are involved in β‐barrel and carrier‐type TIM22 complex substrate handling on the import of Mcp3 (Appendix Fig S2C and D). Hence, it appears that Mcp3 follows a different membrane integration pathway than β‐barrel proteins.
The MIM complex, which has two known subunits Mim1 and Mim2, mediates membrane integration of various MOM helical proteins 41, 42. To investigate its role in the import of Mcp3, mitochondria from mim1Δ or mim2Δ cells were analysed. Import of Mcp3 was strongly reduced in both cases (Fig 8A). Similarly, the steady‐state levels of HA‐Mcp3 were highly diminished in these cells (Fig 8B). As expected, the levels of Tom20, a known substrate of MIM complex, are reduced in mim1Δ or mim2Δ cells. Since we could show that complete loss of Tom20 has no effect on import of Mcp3 (Appendix Fig S2A), this reduction should not influence Mcp3 import. Yet it is known that also the assembly of the TOM complex is affected by the absence of functional MIM complex 47, 48, 49, 50. Therefore, the import of most proteins is indirectly hampered in these cells as exemplified by the matrix destined model substrate pSu9‐DHFR (Appendix Fig S4). To investigate whether MIM components can directly bind Mcp3 precursor, we performed an in vitro pull‐down assay. The fusion protein MBP‐Mim1 and MBP alone were expressed in E. coli, bound to amylose beads and further incubated with 35S‐Mcp3. Fig 8C shows that the amount of Mcp3 precursor pulled down with MBP‐Mim1 is strongly increased in comparison with MBP alone. This result suggests a direct interaction of Mim1 and Mcp3. Taken together, we suggest that the MIM complex might play a role in the import of Mcp3 into the MOM.
Figure 8. Insertion of Mcp3 into the MOM involves the MIM complex.
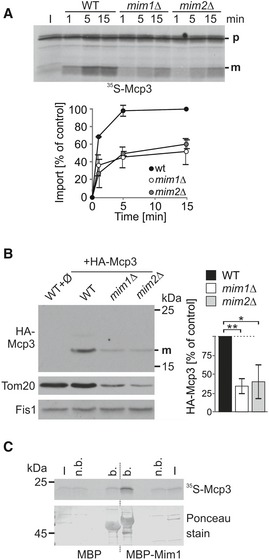
- Import of Mcp3 is mediated by the MIM complex. Mitochondria isolated from WT, mim1Δ, or mim2Δ cells were incubated with radiolabelled Mcp3 for the indicated time periods. After import, mitochondria were reisolated and analysed by SDS–PAGE and autoradiography. Bands corresponding to the mature (m) form were quantified. Import after 15 min into wild‐type mitochondria was set to 100%. The mean with standard deviations is depicted (n = 3; SD). I, 20% of radiolabelled precursor protein used in each import reaction.
- Steady‐state levels of over‐expressed HA‐Mcp3 are reduced in cells lacking MIM complex subunits. Crude mitochondria were obtained from WT cells containing an empty plasmid (Ø) and from wild‐type, mim1Δ or mim2Δ cells containing a plasmid encoded HA‐Mcp3. Samples were analysed by SDS–PAGE and immunodecoration with antibodies against the HA‐tag, Tom20 as MIM substrate and Fis1 as a loading control. HA‐Mcp3 levels were quantified in relation to Fis1 levels. Levels in wild‐type cells were set to 100%. The bar diagram shows the mean with standard deviation (n = 3; SD; *, P < 0.05; **, P < 0.01; two‐tailed Student's t‐test).
- Mcp3 precursor binds in vitro to Mim1. Radiolabelled Mcp3 was incubated with amylose beads harbouring recombinant MBP or MBP fused to Mim1. After washing, proteins were eluted with sample buffer. Analysis was performed by SDS–PAGE, blotting and autoradiography. To demonstrate equal amounts of MBP fusion proteins, the membrane was stained with Ponceau S. I, input 5% of precursor used; n.b., 5% of unbound material; b, 100% of bound material.
Discussion
Contacts of ER and mitochondria and the presence of the ERMES tether are of great relevance for yeast cells regarding mitochondrial function. Evidence arises that ERMES does not “only” serve as a mechanical tether between the ER and mitochondria but has additional functions (for a review, see 14). For example, several ERMES components were predicted and shown to have lipid binding capacity 22, 23, and Mdm10 was reported to interact with the TOB complex in β‐barrel protein import (reviewed in 13). The whole picture is still blurry since all these processes are interdependent, and the observed phenotypes can be interpreted as an indirect outcome of a different defect.
ER–mitochondria encounter structure itself is crucial for mitochondrial function, and cells lacking this complex suffer from a severe phenotype. Its importance becomes more obvious by the fact that cells lacking one of its components easily acquire endogenous suppressors, which was already reported in the early years of the discovery of the individual components 32, 51. Furthermore, S. cerevisiae seems to have backup systems that can partially complement the ERMES function. We recently reported that over‐expression of two mitochondrial proteins Mcp1/2 can partially rescue the defects caused by loss of ERMES function 20. Furthermore, a close interaction of mitochondria with the vacuole was shown to contribute to mitochondrial lipid exchange and can serve as a potential backup system for ERMES‐mediated interactions 9, 10. In addition, a second ER–mitochondria contact mediating complex has been described 25. The overall picture becomes even more complex since ER–vacuole and ER–mitochondria contact sites are coordinated by the ER protein Ltc1/Lam6, which is suggested to be involved in sterol exchange between membranes 26, 52.
Since contacts between mitochondria and other organelles are mediated by the MOM, it is not surprising that two of the suppressors of the ERMES identified by us (Mcp1 in 20 and Mcp3 in this work) are located in the outer membrane. Although the molecular mechanism by which these proteins ameliorate the lack of ERMES is not known, better understanding of their mode of function requires comprehensive insights into their membrane topology and biogenesis pathways.
In this respect, Mcp3 provides an example for a protein that follows a unique import pathway which was not described so far. Although it shares certain import steps with other MOM proteins, the overall import and assembly process is very unusual (Fig 9). At the surface of the organelle, Mcp3 is recognized by Tom70 (Step 1). Of note, also other presequence‐containing proteins share this first recognition step by Tom70 53. These are proteins with a mature part that tends to aggregate and hence are protected by the chaperone‐like function of Tom70. Considering the two predicted transmembrane domains and the hydrophobic stretch close to the IMP cleavage site, Tom70 might function as a receptor with a chaperone function in the import of Mcp3. This dependence on Tom70 is shared by other multispan MOM proteins but not by the single‐span protein Om45 41, 42, 43, 54. However, whereas multispan proteins like Ugo1 and Scm4 are relayed directly from Tom70 to the MIM complex, Mcp3 is translocated across the outer membrane via the Tom40 pore (Fig 9, Step 2).
Figure 9. Biogenesis pathway of Mcp3.
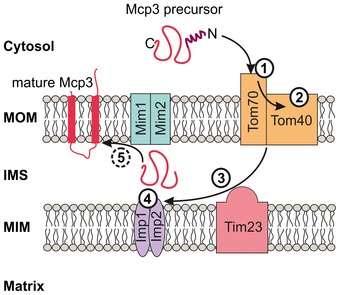
Working model for the import route of Mcp3. The different steps (1–5) are described in the text.
Mcp3 then interacts with the TIM23 complex, and the hydrophobic patch close to its N‐terminus suggests a stop‐transfer mechanism followed by a lateral release (Fig 9, Step 3). An interaction of a MOM protein with the TIM23 complex was reported recently for OM45 43, 54. Yet, while Om45 is directed from the TIM23 to the MIM complex without any processing events 43, 54, Mcp3 is processed by IMP. Of note, although Mcp3 import depends on the mitochondrial membrane potential, in contrast to classical MTS bearing proteins it is not processed by the MPP but rather by IMP. Hence, although Mcp3 shares some import steps with the pathway taken by Om45, its recognition by Tom70 and cleavage by IMP are obvious divergences from this pathway. To our knowledge, this is the first MOM protein that is found to be processed by the peptidase of the inner membrane.
A tempting hypothesis is that after its processing, Mcp3 is relayed to the MIM machinery (Fig 9, steps 4 and 5). Indeed, Mim1 can bind Mcp3 precursor in vitro and Mim1 dependency was also suggested for the membrane integration of the MOM protein Om45 43. However, it cannot be excluded that the reduction in both in vitro import and steady‐state levels of Mcp3 in organelles lacking Mim1 and Mim2 is due to the overall import defects of such mitochondria. Thus, to finally prove that this complex is able to mediate the membrane integration of single‐ and multispan MOM proteins from both sides of the membrane, further experimental evidence is needed. Such a remarkable ability would be unique and was not reported so far for other cellular membrane insertases like the YidC/Oxa/Alb3, Sec or the Get1/2 machineries.
A prominent example of a protein functioning in the mitochondrial outer membrane where the mitochondrial membrane potential plays an important role in its biogenesis is the protein kinase PINK1. Mutations in this protein, which is found in higher eukaryotes, can cause familiar Parkinson's disease. The membrane potential is crucial for the fate and the activity of PINK1. It is suggested that PINK1, as a sensor for mitochondrial dysfunction, is targeted to mitochondria similarly to Mcp3 by its canonical MTS 55. In intact mitochondria, PINK1 is imported, reaches the MIM and is processed by the MIM protease PARL. Next, PINK1 is completely degraded and PINK/parkin signalling is suppressed 55. The PARL processing event reminds remotely on the processing of Mcp3 by IMP. In case of mitochondrial defects, PINK1 is not completely imported into mitochondria since the driving force by the membrane potential is missing. As a result, PINK1 is inserted into the MOM in a TOM‐dependent process where it initiates PINK1/parkin signalling. Although the exact function of Mcp3 is unclear, the example of PINK1 demonstrates that the physiological status of the organelle might affect the function of Mcp3 as well.
Taken together, Mcp3 follows a novel import route into mitochondria. Mcp3 is first recognized by the receptor Tom70, next channelled through the TOM complex and further imported by the TIM23 machinery. It is the first MOM protein shown to be processed by IMP.
Materials and Methods
Yeast strains and growth conditions
Yeast strains (all isogenic to W303a/α, or BY4741/2) were grown in and on standard rich or synthetic liquid and solid media, respectively 56. For drop dilution assays, cells were cultured to an OD600 of 1.0 and diluted in fivefold increments followed by spotting 5 μl of each dilution on the corresponding medium. To delete complete ORFs by homologous recombination, gene‐specific primers were used to amplify either the HIS3MX6 cassette from the plasmid pFA6a‐HIS3MX6 57 or the KanMX4 cassette from the plasmid pFA6a‐KanMX4 58. All deletion strains were confirmed by PCR with gene‐specific primers. Transformation of yeast cells was performed by the lithium acetate method. Yeast strains used in this study are summarized in Appendix Table 1, and primer sequences are listed in Appendix Table 2.
Recombinant DNA techniques
The ORF of the MCP3 gene was amplified by PCR from yeast genomic DNA with specific primers and cloned into the plasmid pYX142. For expression under the control of the endogenous promoter and terminator, 500 bp upstream and downstream, respectively, of the Mcp3 ORF were amplified by PCR and together with the ORF were subcloned into the vector pRS316. Mcp3 with an internal HA‐tag (HA‐Mcp3) was constructed by sequential amplification of the N‐terminal and C‐terminal part of MCP3 from genomic DNA and insertion into the plasmid pYX142. Primer sequences were designed in a way that the HA‐tag was inserted four amino acids downstream to the anticipated Imp1 cleavage site 39. The C‐terminally HA‐tagged version of Mcp3 was obtained by cloning MCP3 without its stop codon into the plasmid pYX142. Primers for the transmembrane domain deletion constructs were designed to omit amino acids 106–128 (ΔTMD1) or 172–198 (ΔTMD2). All specific primer sequences and restriction sites used for cloning are listed in Appendix Table 2. All constructs were confirmed by DNA sequencing. Plasmids used for expression of ERMES components were described previously 20. For synthesis of radiolabelled precursor of Mcp3, the different variants were subcloned via EcoRI and HindIII restriction sites into the vector pGEM4. Site‐directed mutagenesis to obtain the D70G amino acid exchange mutant was performed initially with the pGEM4 construct.
Biochemical methods
Subcellular fractionation was performed according to published procedures 59. Mitochondria were isolated by differential centrifugation as described previously 60. Further purification of mitochondria was achieved via a self‐generated Percoll gradient or a sucrose step gradient 61. For swelling experiments, mitochondria were incubated in hypotonic buffer (20 mM Hepes, pH 7.2) for 30 min on ice. Soluble proteins were precipitated by trichloroacetic acid (TCA). Protein samples were analysed by SDS–PAGE and immunoblotting using the ECL system. Carbonate extraction to precipitate integral membrane proteins and sucrose gradient centrifugation to separate OM and IM vesicles were performed as described previously 20. Pull‐down experiments with radiolabelled precursor and GST fusion proteins or MBP fusion proteins were performed as described before 42, 44. Proteinase K accessibility of imported proteins was analysed by incubation of reisolated mitochondria with PK (20–100 μg/ml) for 30 min on ice, inhibiting the protease by 1 mM PMSF, and finally TCA precipitation of the samples.
Blue native PAGE
Mitochondria were lysed in 40 μl buffer containing digitonin (1% digitonin, 20 mM Tris–HCl, 0.1 mM EDTA, 50 mM NaCl, 10% (v/v) glycerol, 1 mM PMSF, pH 7.2). After incubation on ice for 15 min and clarifying spin (30,000 g, 15 min, 2°C), 5 μl of sample buffer (5% (w/v) Coomassie blue G, 500 mM 6‐amino‐N‐caproic acid, 100 mM Bis‐Tris, pH 7.0) was added, and the mixture was analysed by electrophoresis in a 6–13% or 4–8% gradient BN‐PAGE 62. Gels were blotted on polyvinylidene fluoride membranes, and proteins were further analysed by autoradiography or immunodecoration.
Fluorescence microscopy
For visualization of mitochondria, yeast cells were transformed with a vector harbouring the mitochondrial presequence of subunit 9 of F o‐ATPase of N. crassa fused to GFP 63. Microscopy images were acquired with an Axioskop 20 fluorescence microscope equipped with an Axiocam MRm camera using the 43 Cy3 filter set and the AxioVision software (Carl Zeiss). For studies of ERMES foci formation and their colocalization with mitochondria, yeast cells were transformed with a plasmid expressing an ERFP tagged Mmm1 16 and with either plasmids pYX142‐mtGFP or pYX122‐mtGFP expressing mitochondrially targeted GFP (mtGFP) 63. Microscopy images were acquired with an Axioplan 2 epifluorescence microscope (Carl Zeiss) equipped with an Evolution VF Mono Cooled monochrome camera (Intas) using a Plan Neofluar 100×/1.30 Ph3 oil objective (Carl Zeiss) with QCapture Pro 6.0 software (Media Cybernetics).
Electron microscopy
Preparation of cells for electron microscopy was according to published procedures 64. Ultrathin sections (70 nm) were cut with a diamond knife on a Leica Ultracut UCT microtome and mounted on pioloform‐coated copper grids. Samples were analysed in a Zeiss CEM 902 transmission electron microscope operated at 80 kV. Micrographs were taken using a 1,350 × 1,350 pixel Erlangshen ES500W CCD camera (Gatan) and Digital Micrograph software (version 1.70.16; Gatan).
Analysis of phospholipids and ergosterol
Cells at the log phase (OD600 1.0–2.0) grown on selective medium containing galactose were harvested, and mitochondria were isolated first by differential centrifugation, and then, crude organelles were purified further by a self‐generated Percoll or sucrose density gradient (see above). Mass spectrometric analysis was performed as described 65. In brief, ion mode on a triple quadrupole–linear ion trap hybrid mass spectrometer (QTRAP 5500, AB Sciex) was applied, except for analysis of cardiolipin and ergosterol, which was done in negative ion mode on a quadrupole time‐of‐flight mass spectrometer (QStar Elite, AB Sciex) 18. Ergosterol quantification was performed as described before 66.
In vitro import assays and monitoring of steady‐state levels of proteins
Mitochondria for import assays were isolated from yeast cells as described above. Cells were grown at 30°C in lactate containing medium to mid‐logarithmic phase with the following exceptions: tom40‐25, mas37Δ, tim10‐1ts and tim23ts and the corresponding wild type were grown at 24°C. Radiolabelled precursor proteins were synthesized in the presence of [35S]‐methionine in rabbit reticulocyte lysate. The import reaction was performed by incubating the radiolabelled protein with isolated mitochondria in import buffer (250 mM sucrose, 0.25 mg/ml BSA, 80 mM KCl, 10 mM MOPS‐KOH, 5 mM MgCl2, 2 mM ATP and 2 mM NADH, pH 7.2). Mitochondria from tom40‐25 and tim10‐1ts cells and from their corresponding wild‐type cells were shifted in vitro for 15 min to the non‐permissive temperature 37°C prior to the import reaction. For depletion of the membrane potential, mitochondria were preincubated for 5 min with either 40 μM CCCP or 10 μg/ml valinomycin prior to the import reaction. MPP was inhibited by the addition of 5 mM EDTA and 100 μM o‐phenanthroline. The import reaction was terminated by diluting the reaction in a buffer (250 mM sucrose, 80 mM KCl, 10 mM MOPS, 2 mM PMSF, 1 mM EDTA, pH 7.2) and placing the samples on ice. In some cases, the import reactions were treated with PK as described above.
For preparation of crude mitochondria, cells were grown in minimal medium containing sucrose to mid‐logarithmic phase at 30°C except cells from tom40‐25 and its corresponding wild type that were grown at 24°C. Cells from strain tim23ts and its corresponding wild type were grown at 24°C and shifted for 5–6 h to 37°C prior to lysis. Cells were disrupted by repeated cycles of vortexing with glass beads at 4°C. Mitochondria were enriched by differential centrifugation.
Statistical analysis
Data are presented as the mean ± sample‐based standard deviation (SD). Two‐tailed Student's t‐tests were used to evaluate statistical significance of quantifications (*, P < 0.05; **, P < 0.01). Analyses were performed using Excel (Microsoft).
Author contributions
MS, TT, PW, HK, CÖ, XC, BW and BB performed experiments and analysed data together with DR and KSD; DR and KSD designed the study and wrote the manuscript with advice from MS, BB, BW and HK.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank E. Kracker and Rita Grotjahn for technical assistance, and Drs. C. Koehler, K. Okamoto, T. Langer, D. Mokranjac, C. Meisinger, N. Pfanner, T. Becker and B. Guiard for yeast strains. This work was supported by the Deutsche Forschungsgemeinschaft (DI 1386/2‐1 to K.S.D) and (DR 1028/7‐1 to D.R.)
EMBO Reports (2016) 17: 965–981
References
- 1. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Endo T, Yamano K (2009) Multiple pathways for mitochondrial protein traffic. Biol Chem 390: 723–730 [DOI] [PubMed] [Google Scholar]
- 3. Mokranjac D, Neupert W (2009) Thirty years of protein translocation into mitochondria: unexpectedly complex and still puzzling. Biochim Biophys Acta 1793: 33–41 [DOI] [PubMed] [Google Scholar]
- 4. Neupert W, Herrmann JM (2007) Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723–749 [DOI] [PubMed] [Google Scholar]
- 5. Teixeira PF, Glaser E (2013) Processing peptidases in mitochondria and chloroplasts. Biochim Biophys Acta 1833: 360–370 [DOI] [PubMed] [Google Scholar]
- 6. Luo W, Fang H, Green N (2006) Substrate specificity of inner membrane peptidase in yeast mitochondria. Mole Genet Genomics 275: 431–436 [DOI] [PubMed] [Google Scholar]
- 7. Hahne K, Haucke V, Ramage L, Schatz G (1994) Incomplete arrest in the outer membrane sorts NADH‐cytochrome b5 reductase to two different submitochondrial compartments. Cell 79: 829–839 [DOI] [PubMed] [Google Scholar]
- 8. Ieva R, Heisswolf AK, Gebert M, Vogtle FN, Wollweber F, Mehnert CS, Oeljeklaus S, Warscheid B, Meisinger C, van der Laan M et al (2013) Mitochondrial inner membrane protease promotes assembly of presequence translocase by removing a carboxy‐terminal targeting sequence. Nat Commun 4: 2853 [DOI] [PubMed] [Google Scholar]
- 9. Elbaz‐Alon Y, Rosenfeld‐Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M (2014) A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 30: 95–102 [DOI] [PubMed] [Google Scholar]
- 10. Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30: 86–94 [DOI] [PubMed] [Google Scholar]
- 11. Westermann B (2015) The mitochondria‐plasma membrane contact site. Curr Opin Cell Biol 35: 1–6 [DOI] [PubMed] [Google Scholar]
- 12. Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science 325: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hohr AI, Straub SP, Warscheid B, Becker T, Wiedemann N (2014) Assembly of beta‐barrel proteins in the mitochondrial outer membrane. Biochim Biophys Acta 1853: 74–88 [DOI] [PubMed] [Google Scholar]
- 14. Klecker T, Bockler S, Westermann B (2014) Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol 24: 537–545 [DOI] [PubMed] [Google Scholar]
- 15. Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J (2013) ER‐associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2: e00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bockler S, Westermann B (2014) Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 28: 450–458 [DOI] [PubMed] [Google Scholar]
- 17. Nguyen TT, Lewandowska A, Choi JY, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA, Shaw JM (2012) Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13: 880–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T (2009) The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol 184: 583–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tamura Y, Onguka O, Aiken Hobbs AE, Jensen RE, Iijima M, Claypool SM, Sesaki H (2012) Role for two conserved intermembrane space proteins, Ups1p and Up2p, in intra‐mitochondrial phospholipid trafficking. J Biol Chem 287: 15205–15218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan T, Ozbalci C, Brugger B, Rapaport D, Dimmer KS (2013) Mcp1 and Mcp2, two novel proteins involved in mitochondrial lipid homeostasis. J Cell Sci 126: 3563–3574 [DOI] [PubMed] [Google Scholar]
- 21. Yamano K, Tanaka‐Yamano S, Endo T (2010) Mdm10 as a dynamic constituent of the TOB/SAM complex directs coordinated assembly of Tom40. EMBO Rep 11: 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kopec KO, Alva V, Lupas AN (2010) Homology of SMP domains to the TULIP superfamily of lipid‐binding proteins provides a structural basis for lipid exchange between ER and mitochondria. Bioinformatics 26: 1927–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ah Young AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF (2015) Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci USA 112: E3179–E3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS et al (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323: 1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lahiri S, Chao JT, Tavassoli S, Wong AK, Choudhary V, Young BP, Loewen CJ, Prinz WA (2014) A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol 12: e1001969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J (2015) Ltc1 is an ER‐localized sterol transporter and a component of ER‐mitochondria and ER‐vacuole contacts. J Cell Biol 209: 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siniossoglou S, Santos‐Rosa H, Rappsilber J, Mann M, Hurt E (1998) A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J 17: 6449–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marzioch M, Henthorn DC, Herrmann JM, Wilson R, Thomas DY, Bergeron JJ, Solari RC, Rowley A (1999) Erp1p and Erp2p, partners for Emp24p and Erv25p in a yeast p24 complex. Mol Biol Cell 10: 1923–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark MW, Keng T, Storms RK, Zhong W, Fortin N, Zeng B, Delaney S, Ouellette BF, Barton AB, Kaback DB et al (1994) Sequencing of chromosome I of Saccharomyces cerevisiae: analysis of the 42 kbp SPO7‐CENI‐CDC15 region. Yeast 10: 535–541 [DOI] [PubMed] [Google Scholar]
- 30. Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- 31. Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B et al (2003) The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci USA 100: 13207–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berger KH, Sogo LF, Yaffe MP (1997) Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J Cell Biol 136: 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burgess SM, Delannoy M, Jensen RE (1994) MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J Cell Biol 126: 1375–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B (2002) Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell 13: 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sogo LF, Yaffe MP (1994) Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol 126: 1361–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meisinger C, Rissler M, Chacinska A, Szklarz LK, Milenkovic D, Kozjak V, Schonfisch B, Lohaus C, Meyer HE, Yaffe MP et al (2004) The mitochondrial morphology protein Mdm10 functions in assembly of the preprotein translocase of the outer membrane. Dev Cell 7: 61–71 [DOI] [PubMed] [Google Scholar]
- 37. Thornton N, Stroud DA, Milenkovic D, Guiard B, Pfanner N, Becker T (2010) Two modular forms of the mitochondrial sorting and assembly machinery are involved in biogenesis of alpha‐helical outer membrane proteins. J Mol Biol 396: 540–549 [DOI] [PubMed] [Google Scholar]
- 38. Esser K, Jan PS, Pratje E, Michaelis G (2004) The mitochondrial IMP peptidase of yeast: functional analysis of domains and identification of Gut2 as a new natural substrate. Mole Genet Genomics 271: 616–626 [DOI] [PubMed] [Google Scholar]
- 39. Vogtle FN, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, Gevaert K, Kellermann J, Voos W, Sickmann A, Pfanner N et al (2009) Global analysis of the mitochondrial N‐proteome identifies a processing peptidase critical for protein stability. Cell 139: 428–439 [DOI] [PubMed] [Google Scholar]
- 40. Zahedi RP, Sickmann A, Boehm AM, Winkler C, Zufall N, Schonfisch B, Guiard B, Pfanner N, Meisinger C (2006) Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell 17: 1436–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Becker T, Wenz LS, Kruger V, Lehmann W, Muller JM, Goroncy L, Zufall N, Lithgow T, Guiard B, Chacinska A et al (2011) The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J Cell Biol 194: 387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papic D, Krumpe K, Dukanovic J, Dimmer KS, Rapaport D (2011) Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1‐dependent import pathway. J Cell Biol 194: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wenz LS, Opalinski L, Schuler MH, Ellenrieder L, Ieva R, Bottinger L, Qiu J, van der Laan M, Wiedemann N, Guiard B et al (2014) The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. EMBO Rep 15: 678–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Papic D, Elbaz‐Alon Y, Koerdt SN, Leopold K, Worm D, Jung M, Schuldiner M, Rapaport D (2013) The role of Djp1 in import of the mitochondrial protein Mim1 demonstrates specificity between a cochaperone and its substrate protein. Mol Cell Biol 33: 4083–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meisinger C, Ryan MT, Hill K, Model K, Lim JH, Sickmann A, Muller H, Meyer HE, Wagner R, Pfanner N (2001) Protein import channel of the outer mitochondrial membrane: a highly stable Tom40‐Tom22 core structure differentially interacts with preproteins, small tom proteins, and import receptors. Mol Cell Biol 21: 2337–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gevorkyan‐Airapetov L, Zohary K, Popov‐Celeketic D, Mapa K, Hell K, Neupert W, Azem A, Mokranjac D (2009) Interaction of Tim23 with Tim50 Is essential for protein translocation by the mitochondrial TIM23 complex. J Biol Chem 284: 4865–4872 [DOI] [PubMed] [Google Scholar]
- 47. Waizenegger T, Schmitt S, Zivkovic J, Neupert W, Rapaport D (2005) Mim1, a protein required for the assembly of the TOM complex of mitochondria. EMBO Rep 6: 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ishikawa D, Yamamoto H, Tamura Y, Moritoh K, Endo T (2004) Two novel proteins in the mitochondrial outer membrane mediate beta‐barrel protein assembly. J Cell Biol 166: 621–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dimmer KS, Papic D, Schumann B, Sperl D, Krumpe K, Walther DM, Rapaport D (2012) A crucial role for Mim2 in the biogenesis of mitochondrial outer membrane proteins. J Cell Sci 125: 3464–3473 [DOI] [PubMed] [Google Scholar]
- 50. Hulett JM, Lueder F, Chan NC, Perry AJ, Wolynec P, Likic VA, Gooley PR, Lithgow T (2008) The transmembrane segment of Tom20 is recognized by Mim1 for docking to the mitochondrial TOM complex. J Mol Biol 376: 694–704 [DOI] [PubMed] [Google Scholar]
- 51. Hanekamp T, Thorsness MK, Rebbapragada I, Fisher EM, Seebart C, Darland MR, Coxbill JA, Updike DL, Thorsness PE (2002) Maintenance of mitochondrial morphology is linked to maintenance of the mitochondrial genome in Saccharomyces cerevisiae. Genetics 162: 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elbaz‐Alon Y, Eisenberg‐Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T, Schuldiner M (2015) Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep 12: 7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamamoto H, Fukui K, Takahashi H, Kitamura S, Shiota T, Terao K, Uchida M, Esaki M, Nishikawa S, Yoshihisa T et al (2009) Roles of Tom70 in import of presequence‐containing mitochondrial proteins. J Biol Chem 284: 31635–31646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song J, Tamura Y, Yoshihisa T, Endo T (2014) A novel import route for an N‐anchor mitochondrial outer membrane protein aided by the TIM23 complex. EMBO Rep 15: 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Okatsu K, Kimura M, Oka T, Tanaka K, Matsuda N (2015) Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci 128: 964–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Green SR, Moehle CM (2001) Media and culture of yeast. Curr Protoc Cell Biol Chapter 1: Unit 1.6 [DOI] [PubMed] [Google Scholar]
- 57. Wach A, Brachat A, Alberti‐Segui C, Rebischung C, Philippsen P (1997) Heterologous HIS3 marker and GFP reporter modules for PCR‐targeting in Saccharomyces cerevisiae. Yeast 13: 1065–1075 [DOI] [PubMed] [Google Scholar]
- 58. Wach A, Brachat A, Pohlmann R, Philippsen P (1994) New heterologous modules for classical or PCR‐based gene disruptions in Saccharomyces cerevisiae. Yeast 10: 1793–1808 [DOI] [PubMed] [Google Scholar]
- 59. Walther DM, Papic D, Bos MP, Tommassen J, Rapaport D (2009) Signals in bacterial beta‐barrel proteins are functional in eukaryotic cells for targeting to and assembly in mitochondria. Proc Natl Acad Sci USA 106: 2531–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Daum G, Bohni PC, Schatz G (1982) Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem 257: 13028–13033 [PubMed] [Google Scholar]
- 61. Graham JM (2001) Purification of a crude mitochondrial fraction by density‐gradient centrifugation. Curr Protoc Cell Biol Chapter 3: Unit 3.4 [DOI] [PubMed] [Google Scholar]
- 62. Schagger H (2002) Respiratory chain supercomplexes of mitochondria and bacteria. Biochim Biophys Acta 1555: 154–159 [DOI] [PubMed] [Google Scholar]
- 63. Westermann B, Neupert W (2000) Mitochondria‐targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 16: 1421–1427 [DOI] [PubMed] [Google Scholar]
- 64. Bauer C, Herzog V, Bauer MF (2001) Improved Technique for Electron Microscope Visualization of Yeast Membrane Structure. Microsc Microanal 7: 530–534 [DOI] [PubMed] [Google Scholar]
- 65. Ozbalci C, Sachsenheimer T, Brugger B (2013) Quantitative analysis of cellular lipids by nano‐electrospray ionization mass spectrometry. Methods Mol Biol 1033: 3–20 [DOI] [PubMed] [Google Scholar]
- 66. Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A (2009) Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci USA 106: 2136–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File