

Abstract
Background
Adaptive evolution is not possible without the generation of phenotypic variants. The origin of these variations has been a central topic in evolutionary biology. Up to now, it was commonly accepted that standing genetic variation is the only cause of phenotypic variants. However, epigenetic information is emerging as a complementary source of heritable phenotypic variation that contributes to evolution. The relative importance of genetics and epigenetics in generating heritable phenotypic variation is nevertheless a matter of debate.
Results
We used a host–parasite system to address this question. The human blood fluke Schistosoma mansoni can adapt rapidly to new intermediate snail hosts. The interaction between parasite and mollusk is characterized by a compatibility polymorphism illustrating the evolutionary dynamics in this system. The principal molecular marker for compatibility (infection success) is the expression pattern of a group of polymorphic mucins (SmPoMuc) in the parasite. We show here that chromatin structure changes as the SmPoMuc promoters are the cause for SmPoMuc transcription polymorphism leading to phenotypic novelty and increase in infection success, i.e., fitness.
Conclusion
We establish that epigenetic changes can be the major if not only cause of adaptive phenotypic variants in Schistosoma mansoni, suggesting that epimutations can provide material for adaptive evolution in the absence of genetic variation in other systems. In addition, our results indicate that epidrugs can be used to control parasite development but also parasite evolution.
Electronic supplementary material
The online version of this article (doi:10.1186/s13072-016-0076-2) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Adaptive evolution, Compatibility polymorphism, Schistosoma mansoni
Background
Adaptive evolution relies on the generation of heritable phenotypic variants on which selection can act. The origin of variation has puzzled researchers since Darwinian times [1]. There is now a relative broad consensus that genotype environment interactions represent the major, if not exclusive, source of phenotypic novelty. However, this view has been challenged by theoretical considerations [2–5], and it has been suggested that a substantial part of variability is the result of variations in the epigenetic component of the genome [6, 7]. A concept emerges in which upon environmental changes, the epigenetic (“low-fidelity”) system permits a population to generate new phenotypes while keeping genetic information invariant. This allows for exploring the fitness landscape after an environmental change and will “buy time” for this population. If the new environment persists, genetically encoded phenotypic variants could emerge and the new phenotype could be genetically assimilated. Heritability of epialleles has been clearly established for numerous examples (e.g., [8, 9]), and several epigenetically encoded phenotypic characters were described (e.g., [10, 11]). However, none of them is adaptive in the sense that the phenotype provides a fitness gain and is favored by selection under ecologically realistic conditions. It remains therefore an open question whether epigenetic inheritance contributes significantly to adaptive evolution. We addressed this question choosing a metazoan host/parasite system. Selective pressures are strong in these systems, evolution is fast and effective population sizes are small. We provide here, to the best of our knowledge, for the first time experimental evidence for an epigenetically encoded adaptive phenotype.
The parasite/host system we used is the interaction between Schistosoma mansoni and its intermediate host Biomphalaria glabrata. S. mansoni is a parasite of humans and causes intestinal schistosomiasis, the second most important human parasitic disease after malaria [12]. The life cycle requires passage through two obligatory hosts, the freshwater snail Biomphalaria spp. where the parasite multiplies asexually, and human or rodents as definitive hosts for sexual reproduction [13]. The snail/schistosome interaction is characterized by a phenomenon called compatibility polymorphism [14], i.e., specific strains of S. mansoni can only infect specific strains of B. glabrata, while others cannot be infected [15]. These incompatible snail strains are, however, not resistant to S. mansoni since they can be infected by other strains of the parasite. We had earlier identified S. mansoni polymorphic mucins (SmPoMucs) as key molecular markers for compatibility [16, 17]. For several reasons, these proteins appear to be essential for the parasites to penetrate into the snail: (1) SmPoMucs are expressed only in the snail infecting miracidia where they are excreted from the apical gland of the larvae. (2) SmPoMucs present the strongest qualitative and quantitative differences between compatible and incompatible strains on the level of the proteome. Their exact function remains unknown but their amino acid sequence and glycosylation level [18] suggests that and (3) they form mucus that facilitates penetration through the snail epidermis. (4) They are recognized by the immune receptors of the invertebrate host [19] which triggers probably elimination of sporocysts of incompatible S. mansoni strains. SmPoMucs are encoded by a multigene family that comprises at least 10 genes, organized in four clusters on the genome. These genes belong to a class of genes that are probably specific to Platyhelminthes and are called micro-exon genes (MEGs) [20]. The unusual structure of MEGs allows parasites to generate a “controlled chaos” of a polymorphic SmPoMuc protein repertoire from a small number of genes using transcriptional, post-transcriptional and post-translational mechanisms [17]. We recently demonstrated that transcriptional control is based on epigenetic mechanisms [21]. We reasoned therefore that the compatibility phenotype would be a suitable model to disentangle the effects of genetically and epigenetically heritable components on adaptive traits. We applied a classical pedigree study and used two pure lines of S. mansoni: the Brazilian SmBRE strain and the Guadeloupian SmGH2 strain with their sympatric Brazilian snail strain BgBRE and the Guadeloupian strain BgGUA, respectively. Both strains show strong differences in compatibility (measured by prevalence and intensities) toward the same reference B. glabrata strain [15]. As mentioned above, SmPoMucs are the key elements in the compatibility polymorphism [16], and expression of this gene family is regulated by epigenetic mechanisms [21, 22]. We therefore investigated expression of these genes and studied chromatin structure and DNA sequence in the promoter regions. We show that the compatibility phenotype and SmPoMuc expression follow non-Mendelian segregation, that epialleles and alleles of SmPoMuc do not cosegregate, and that treatment with an epimutagen modifies SmPoMuc expression and increases fitness of the treated parasite larvae. Taken together our results indicate that not only SmPoMuc expression but also infection success are at least partially under epigenetic control, thus providing an example for an epigenetically encoded adaptive trait.
Results
Inbred S. mansoni strains used in the study are pure lines
Our laboratory maintains the life cycle of currently five inbred strains of Schistosoma mansoni and their corresponding sympatric Biomphalaria glabrata host strains. These strains show a specific heritable reaction norm in their compatibility with B. glabrata strains [15]. Every strain possesses also a specific heritable pattern of SmPoMuc so that we can “fingerprint” them by Western blots. To investigate whether the capacity to infect allopatric hosts can be selected for, we performed infections in allopatric combinations SmBRE/BgGUA and SmBRE/BgVEN and measured compatibility. The SmBRE strain was originally sampled in Recife, Brazil, in the 1960s. It was provided to our laboratory in 1975 by Pr. Y. Golvan (Faculté de Médecine de Paris—Saint Antoine) and since then has been maintained in its sympatric intermediate host strain BgBRE. BgBRE is also from Recife and was also acquired in 1975. The allopatric mollusk strain BgGUA originates from the town of Dans Fond and arrived at our laboratory in 2005. Snail strains BgVEN were originally sampled in Venezuela. Prevalence (percentage of infected snails) is roughly 80 % in the allopatric combinations (SmBRE on BgGUA or BgVEN) compared to the sympatric combination (SmBRE on BgBRE) where it is 100 % and this without significant variations for the six generations (Fig. 1a, b). Whatever the generation, offspring of both Schistosoma lines passed through allopatric snail hosts showed the same compatibility phenotype toward the original sympatric BgBRE snail strain (100 %, Fig. 1a, b). Similar results were obtained for another S. mansoni strain, SmGH2, hence fewer generations were followed. SmGH2 was collected in 1983 from eggs shed by a patient in the hospital of Pointe-à-Pitre. Since then it was maintained in the laboratory in its sympatric snail host BgGUA. About 60 % of snails are infected in the sympatric (SmGH2 on BgGUA) combination and less than 5 % in the allopatric combination (SmGH2 on BgBRE). Neither a gain of compatibility with the allopatric host nor a loss of compatibility with the sympatric host was detected. This means that the compatibility character cannot be selected for, i.e., offspring from allopatric combinations shows the same reaction norm as the original population. Therefore, even though the used strains are not clonal [23], they are pure lines in the sense of classical genetics.
Fig. 1.
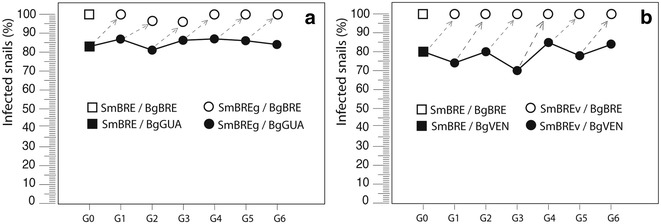
Compatibility of the SmBRE schistosome strain with its homopatric snail host BgBRE and two heteropatric snail strains, BgGUA and BgVEN: a SmBREg corresponds to the SmBRE strain passed for 6 generations (G1–G6) on BgGUA snails; b SmBREv corresponds to the SmBRE strain passed for 6 generations (G1–G6) on the BgVEN snails. At each generation, compatibility of the SmBREg (a) and SmBREv (b) was tested on the original BgBRE snail strain
SmPoMucs are involved in an early stage of host/parasite interaction
In order to infect its host, a parasite must find the host, adhere to the surface, penetrate and finally evade immune response inside the host. SmPoMucs could be involved in any of these processes, but their constitutive expression in the apical glands of miracidia suggested involvement in the early steps, i.e., adherence and penetration. We reasoned that if this would be true, then bypassing penetration by transfer of sporocysts into snails would lead to no difference in prevalence between allopatric and sympatric combinations. Our results show that there is indeed no difference in the development of S. mansoni after transfer into the allopatric (SmBRE in BgGUA) vs the sympatric combination (SmBRE in BgBRE). In other words, if SmBRE arrives inside the BgGUA snail it behaves there as in a sympatric host (Additional file 1). This shows that when infecting allopatric BgGUA snails, the limiting step is penetration and not sporocyst SpI development, and lends further support to the hypothesis that SmPoMucs are involved in penetration or very early transformation steps.
Strain hybrids express more SmPoMuc variants than each parent
We decided to introduce diversity by generating crosses between SmBRE and SmGH2. Thirty five to fifty snails from each strain (BgBRE and BgGUA) were exposed individually to 20 miracidia. Prevalence and intensity (number of mother sporocysts spI per snail) were measured. At each step, miracidia were set aside to perform molecular biology analyses. The experiments were carried out with miracidia obtained from SmBRE × SmBRE and SmGH2 × SmGH2 crosses, F1 miracidia obtained from reciprocal crosses of SmBRE × SmGH2, F2 miracidia were obtained from F1 × F1 and F3 miracidia obtained from crosses of two different clonal populations of F2 cercariae. The experimental scheme is summarized in Fig. 2. Quantitative RT-PCR shows that SmPoMuc 3.1(r1r2) is on average 18.3-fold more transcribed in SmGH2 F0 than in SmBRE F0 strains (n = 4), and ChIP indicates a 12.05-fold higher acetylation of H3K9 in SmGH2 F0 compared to SmBRE F0 (n = 7) (Fig. 3). This confirms our earlier results that major control of expression of these genes occurs at the transcriptional level [21]. The hybrid lines F1 and F2 show higher transcription levels compared to SmBRE F0 parent (data not shown). In the F3 generation, SmPoMuc 3.1(r1r2) transcription is statistically not anymore different from SmBRE F0 (Fig. 3). We wondered if such differences in transcription levels concerned also other genes and tested 14 arbitrarily chosen protein coding genes and transcribed repeats by qRT-PCR (genes and primers in Additional file 4). In none of the cases, we detected significant differences in transcription between the 4 generations (data not shown). We then used Western blots to test if SmPoMuc transcripts are translated. SmPoMucs proteins were detected using an antibody that recognizes the C-terminal conserved sequence by Western blot [19]. Proteins were extracted from 1000 miracidia for each condition. We knew already that although there is very little nucleotide differences in the promoter regions of SmPoMucs family genes [21], the expression profiles between the parental strains SmBRE F0 and SmGH2 F0 were different [19], and we confirmed this here by Western blot (Fig. 4a). F1 hybrid miracidia express all bands of F0 parents, leading to a combined profile that corresponds almost perfectly to a superposition of SmGH2 and SmBRE parental profiles (Fig. 4a). In F2 and F3, we still find all bands of F0 parents even if their proportions change (Fig. 4b). Western blots were repeated with three F3 clones, and regardless of their genotype we always found the combined pattern of SmPoMuc expression. Since SmPoMucs are key markers for host–parasite compatibility, we expected the hybrids with a combined SmPoMuc profile to show changes in compatibility. We therefore investigated their capacity to infest different snail strains.
Fig. 2.
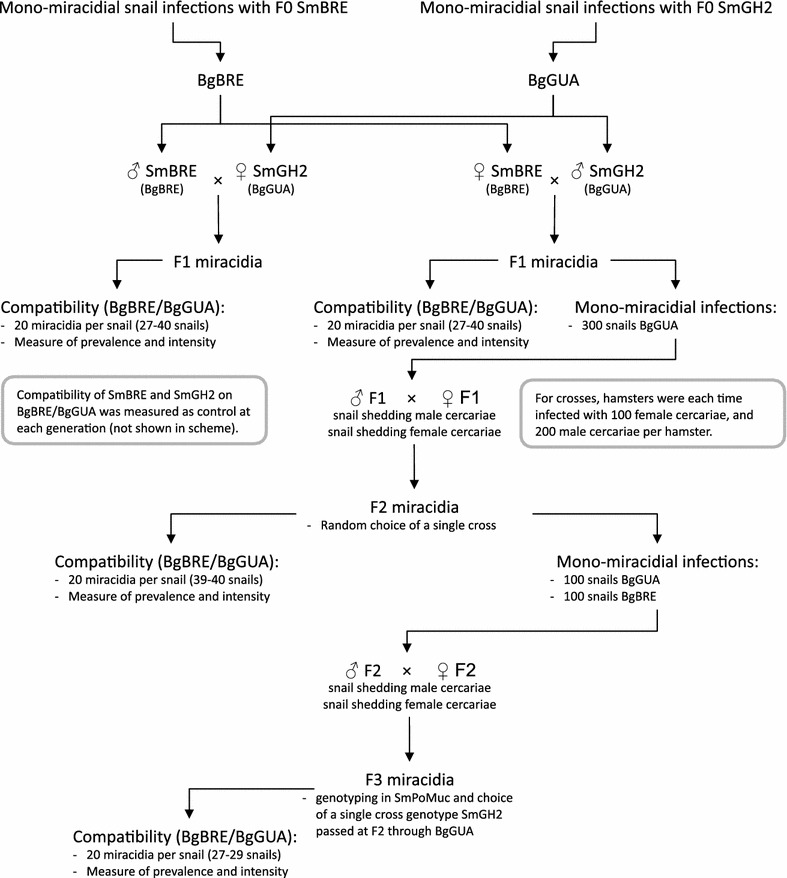
Crossing scheme for pedigree study. After monomiracidial infection of their sympatric hosts in F0, crosses within each generation were produced in each generation until F3
Fig. 3.
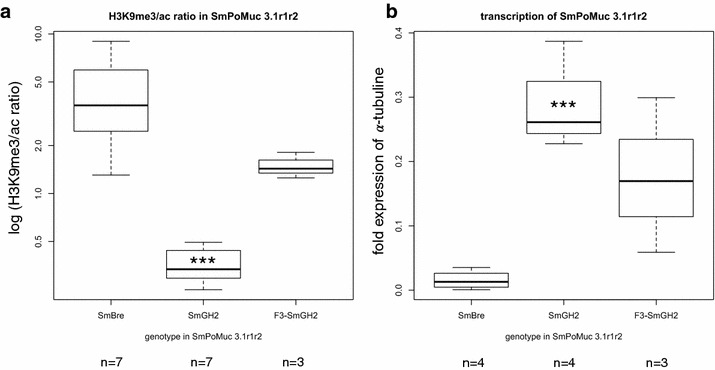
Boxplot of chromatin structure and transcription level in the promoter region of SmPoMuc3.1 (r1r2). Two technical replicates for each biological replicate (n). a ChIP H3K9me3 to H3K9ac ratio at the promoter of SmPoMuc 3.1(r1r2). b Relative transcription of SmPoMuc 3.1(r1r2) compared to alpha-tubulin measured by RT-qPCR. ***indicate statistical significant differences according to ANOVA followed by Dunnett’s post hoc test to F0 SmBRE that was defined as reference (ANOVA F 2,8 = 13.128; p = 0.003 for transcription and F 2,14=7.26; p = 0.007 for H3K9me3/ac ChIP ratio). Only SmGH2 is significantly different in chromatin structure and transcription level from the reference. Genotype SmGH2 of the F3 is similar (not different) to reference genotype F0 SmBre in terms of chromatin structure and transcription. For additional pairwise comparison we used Students T-Test. We found for H3K9me3/ac ratio (a) t = 10.045, 8 df, p < 0.0001, i.e. there is statistically significant difference between SmGH2 and F3-SmGH2. For transcription (b) the difference between SmGH2 and F3-SmGH2 is statistically not significant (t = 1.514; 5 df; p = 0.1904) since variance is strong. Nevertheless, there is a clear shift of transcription in F3-SmGH2 towards the lower level of transcription observed in SmBRE
Fig. 4.
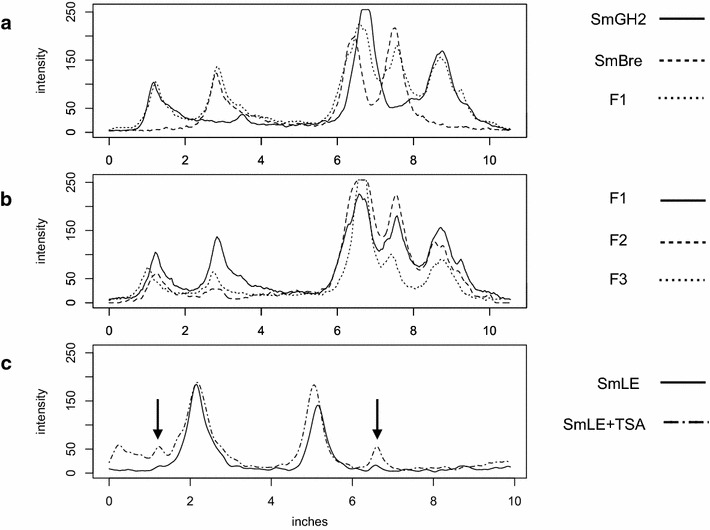
Expression profiles of SmPoMucs. Western blot experiments were performed using a standardized method on proteins extracts from 1000 miracidia. Image was analyzed and grayscale (intensity on y axis) indicates protein abundance. X-axis migration distance in inches. a SmPoMucs expression in F0 (full and dashed lines) and F1 (dotted line). F1 profile corresponds almost perfectly to the combination of both F0. b Comparison of SmPoMucs expression in F1, F2 and F3. Global profile remains constant but 3 major bands decrease in intensity. c SmPoMucs expression in SmLE with (dot-and-dash line) and without TSA (full line) treatment. After TSA treatment, latent variants are now expressed (indicated by arrowheads)
F3 Hybrids show higher fitness than the parents
Prevalences in BgBRE are 96–100 % for SmBRE F0 and 4–6 % for SmGH2 F0. Prevalences increase significantly (Fisher exact test, p < 0.0001) compared to SmGH2 in F1, F2 and F3 to reach maximal values in F2 and F3 (Fig. 5***). Prevalences are statistically not different from SmBRE F0 in all hybrid generations, thus we consider them to be similar. Intensity in BgBRE is 7.1 ± 0.25 (mean ± SE) for SmBRE (n = 34) and 1 ± 0 for SmGH2 (n = 50). Similar to prevalence, intensity values increase significantly compared to SmGH2 in F1, F2 and F3 (Fig. 5).
Fig. 5.
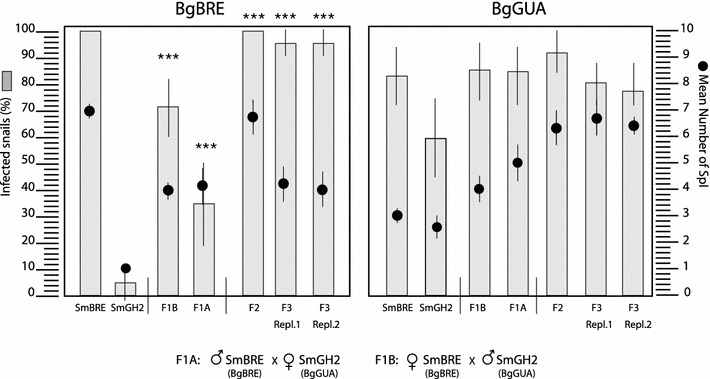
Prevalence and intensity of S. mansoni infection in B. glabrata snail strains. Prevalence is expressed as % of infected snails, intensity as mean number of SpI sporocysts per snail. ***indicate were prevalences and intensities increase significantly (Fisher’s exact test, p < 0.0001) compared to SmGH2. They are not different from SmBRE F0 (p < 0.05). Each individual snail was exposed to 20 miracidia
In BgGUA snails, prevalences are 80–83 and 55–60 % for parental miracidia SmBRE F0 and SmGH2 F0, respectively. F1–F3 have infectivities that are statistically not different from SmBRE F0 (p < 0.05, Fisher exact test) but higher than those of the SmGH2 F0 parents (Fig. 5). For infection intensities of F0–F3 on BgGUA, there is no significant difference between SmBRE and SmGH2 F0 parents, but we observe an increase in intensity values across generations F1–F3 (Fig. 5). The measure of the superior performance, i.e., heterosis on intensity parameter was calculated following the formula ((Crossbred average − Parental average)/Parental average) × 100. On BgBRE, the crossbred F1 intensity is roughly 40 % greater than the average parental intensity. Consequently, all crosses of the two different strains of the parasite S. mansoni show increasing infection success of hybrids over three generations, i.e., an increase in global fitness. Interestingly, while the SmPoMuc expression (molecular) phenotype is additive, compatibility phenotypes in F2 and F3 hybrids show uniparental phenotypic dominance of SmBRE. Segregation of SmPoMuc patterns and infection success is uncoupled, which is inconsistent with Mendelian genetic inheritance of a single locus or few loci. Consequently, we explored other mechanisms responsible for fitness increase. Since we had earlier shown that chromatin structure controls SmPoMuc expression [21], we decided to study segregation of chromatin marks at the SmPoMuc locus that shows the strongest chromatin structure differences and expression differences between SmBRE and SmGH2: SmPoMuc 3.1 (r1r2).
Chromatin structure in SmPoMuc 3.1(r1r2) shows non-Mendelian segregation
SmPoMuc promoter sequences do not have methylated cytosines [21]. Therefore, we focused our analysis on histone modifications in the promoter regions by native chromatin immunoprecipitation (N-ChIP) using two different antibodies that recognize histone H3 acetylated on lysine 9 (H3K9ac) and histone H3 tri-methylated on lysine 9 (H3K9me3). ChIP was followed by qPCR analysis to quantify the immunoprecipitated DNA. Since acetylation and methylation at H3K9 are mutually exclusive, we express chromatin status here as ratio of H3K9me3/H3K9ac. SmPoMuc 3.1(r1r2) promoter shows a very different chromatin structure between the two F0 parents SmBRE and SmGH2 (Fig. 3). This is correlated with differential expression of this gene between the two parents. In F3, chromatin structure is closer to the SmBRE F0 parent (Fig. 3). The promoter regions of SmPoMuc 3.1 (r1r2) contain 3 diagnostic SNPs between SmBRE F0 and SmGH2 F0. To identify the genotypes at SmPoMuc 3.1 (r1r2), we PCR-amplified and sequenced the SmPoMuc 3.1 (r1r2) region using DNA of miracidia from all generations SmBRE F0, SmGH2 F0, F1, F2 and F3. As expected, F1 and F2 hybrid populations are heterozygous with two different parental alleles. We then studied in more detail F3 clones homozygous for the SmGH2 genotype. We found that the SmPoMuc 3.1 (r1r2) SmGH2 genotype had aquired in F3 an epigenotype that is statistically not different from SmBRE F0. One might argue that this is part of a genome-wide change in chromatin structure following the hybridization event. We therefore investigated another locus for which we had earlier identified differences in H3K9ac enrichment between SmBRE and SmGH2 [24]: Smp_171100.
Smp_171100 codes for a putative M13 metallo-endopeptidases and is located on Schisto_mansoni.Chr_2 22,414,795–22,470,759. The function of the gene is not of particular importance here. The gene is not anymore listed in the latest version of the genome annotation, but we have confirmed the presence of a transcript by RT-PCR. We had shown earlier that chromatin differences exist in the gene body between SmBRE and SmGH2 [24], and we used them here as epigenetic markers for both strains. We confirmed our findings that SmBRE F0 and SmGH2 F0 show different H3K9me3/H3K9ac ratios. These differences in chromatin structure are correlated with transcription differences (Fig. 6). It should be noted that the difference is located in the body of the gene and not in the TSS, which explains the inverse relation of acetylation and expression. In the F3, the H3K9me3/H3K9ac ratio is not different from SmBRE F0 (Fig. 6). We sequenced a region of the Smp_171100 gene sequence containing 6 SNPs between SmBRE and SmGH2. As expected and as in SmPoMuc 3.1 (r1r2), F1 and F2 miracidial populations are heterozygous. The F3 clone that has the SmGH2 alleles for SmPoMuc 3.1 (r1r2)) is homozygous for SmBRE in Smp_171100.
Fig. 6.
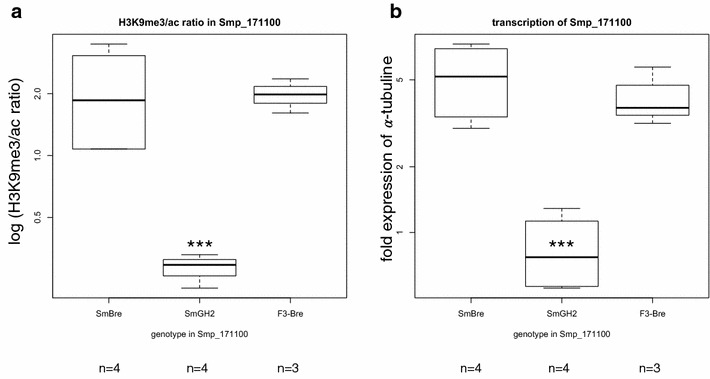
Boxplot of transcription level and chromatin structure in the gene body of Smp_171100. Two technical replicates for each biological replicate (n). a ChIP H3K9me3 to H3K9ac ratio. b Relative transcription compared to alpha-tubulin measured by RT-qPCR. ***indicate statistically significant differences as in Fig. 3 (ANOVA F 2,8 = 7.56; p = 0.014 for transcription and F 2,8 = 7.020; p = 0.017 for H3K9me3/ac ChIP ratio). Transcription level and chromatin structure co-segregate with the genotype
In conclusion, while in the SmPoMuc 3.1(r1r2) locus, the SmGH2 genotype had changed the epigenotype in the F3 (Fig. 3), in Smp_171100 we see a co-segregation of epigenotype and genotype (both are SmBRE in F3). In other words, for the two loci in which to our knowledge there is a clear epigenetic difference between SmBRE F0 and SmGH2 F0, we distinguish two modes of chromatin heritability, one case of non-Mendelian inheritance (SmPoMuc 3.1(r1r2)), and one case of Mendelian inheritance and cosegregation of genotype and epigenotype (Smp_171100).
Pharmacological induction of chromatin structure changes leads to transcription of new SmPoMuc variants and renders incompatible (avirulent) strain compatible (virulent)
All lines of evidence indicated that it was the H3K9me3/ac ratio that controlled specifically SmPoMuc 3.1(r1r2) expression and that the change in chromatin structure resulted in new phenotypic variants based on new combinations of SmPoMuc. This in turn led probably to the increase in infection success of the hybrids. If this was true, then perturbation of H3K9me3/ac ratio by other means should also lead to new SmPoMuc variants and should have an impact on parasite–host compatibility. Locus-specific epigenetic engineering of SmPoMuc is not yet feasible. We therefore opted for pharmacological treatment of S. mansoni eggs with the histone deacetylase inhibitor Trichostatin A (TSA). We had shown earlier that treatment with 20 µM TSA increased the number of alternative transcripts (number of polymorphic bands detected by RT-PCR) in SmPoMuc but did not alter transcription of the abovementioned control genes [22]. Also in other systems, it was shown that TSA modulates transcription of around 5 % of genes and that genes can be up-regulated but also down-regulated by TSA [25–28]. In other words, TSA does not induce a global increase in transcription but will activate or inactivate certain loci only. We treated the S. mansoni eggs three times with 20 µM TSA before hatching. This dose have negligible cytotoxic effect [22]. For this part of the study, we used again two pure lines of S. mansoni: the Brazilian SmLE strain (originally from Belo Horizonte) and SmGH2 strain with the Brazilian snail BgBAR2 (sympatric with SmLE and also coming initially from Belo Horizonte). The SmLE/SmGH2—BgBAR combinations were chosen because they have a very pronounced phenotype both in terms of compatibility (SmGH2 has almost no capacity to infect of BgBAR, but SmLE is highly compatible) and in SmPoMuc expression (only 2 strong bands in Western blots in SmLE). Analysis of Western blots shows that TSA treatment leads to increased expression of SmPoMuc protein variants in the miracidia that were latent in the untreated control (arrowheads in Fig. 4c), i.e., the already known transcription variation translates also into changes of the protein level. We subsequently investigated the compatibility phenotype using prevalences and intensities with mock treatment and with TSA treatment. BgBAR snails were exposed individually to 20 miracidia. Using only the solvent ethanol as treatment, mean prevalences in BgBAR are 95.65 % (number of exposed snails n = 23) for SmLE, and 21.5 % for SmGH2 (n = 97). Mean prevalences increased after treatment with TSA by 4–9 %: with TSA they reach 31.1 % (n = 103) for SmGH2, and 100 % in SmLE (n = 22). This is, however, not yet significant (Fisher exact test, p < 0.05). For intensity values, we detected an increase for SmGH2 from 2.9 to 4.0 after TSA treatment, and for SmLE from 2 to 9.55. This increase in individual infection success is significant (Fisher exact test, p < 0.05). Experiments were repeated four times by different experimenters (raw data provided as Additional file 2).
In conclusion, treatment with an epimutagenic agent does not only lead to changes in the SmPoMuc expression patterns but also allows a parasite with low compatibility to increase its compatibility with a new snail host.
Discussion
In parasite–host interactions, infection success can be used as an estimation for parasite fitness. It can be measured as number of infected host individuals (prevalence) and number of successful infection events per host individual (intensity). Using these parameters, we show here that after a hybridization event between two different S. mansoni strains, the offspring shows increased fitness. Increased fitness of hybrids is not surprising [29, 30], but in general such hybrid vigor or heterosis is attributed to increased genetic diversity in the offspring. This is probably also the case in S. mansoni, and host compatibility has necessarily a genetic basis. However, here we demonstrate that genetic diversity is not the only mechanism by which phenotypic diversity increases in hybrids. At least for the SmPoMuc loci, epigenetic plays a more important role. This makes sense in light of the fact that nucleotide diversity between the two parental strains at these loci is extremely low [21]. One of the caveats of our study is that we did not address the question by which mechanism chromatin structure is modified. It could be that the SmBRE locus is paramutagenic for SmGH2. Paramutation is a heritable change in the expression of a paramutable allele, initiated by interaction in heterozygotes with a paramutagenic allele. Paramutations are meiotically stable and inherited in the absence of the inducing allele (e.g., [10]). Recent work places DNMT2, an enzyme that is able to methylate cytosine residues in tRNA, in the center of the paramutation pathway in animals [31] and a DNMT2 homologue (Smp_198180) exists in S. mansoni [32]. Another limitation in our approach comes from the non-specificity of the pharmacological induction of epimutations. We observed new SmPoMuc combinations without changing the genetic background, and we tested a large number of reference genes that did not change transcription after TSA treatment, but we cannot exclude that transcription in other loci is modified. Nevertheless, we confirmed our earlier findings that epigenetic mechanisms control transcription of the SmPoMuc genes and we show now that epigenetic information is indeed the origin of phenotypic novelty in these loci. The exact function of SmPoMucs is still elusive; nevertheless, our earlier studies had firmly established SmPoMuc variability as a key marker for compatibility between S. mansoni and its intermediate host [17, 19], and here, we show that SmPoMucs are involved in the very early steps of infection (adherence, penetration and/or pre-Sp1 development). We conclude that strain hybridization and TSA treatment lead to changes in the epigenetic information that establish novel developmental trajectories leading to new and more phenotypic variants in the population. This allows the parasite population to explore the fitness landscape. If new matching phenotypes between parasites and mollusk host are produced, the latter can be infected, i.e., the phenotype is adaptive. This strategy might have evolved as a result of the combination of characteristics of the two hosts. Snail populations in endemic areas show very low prevalence (e.g., [33]), and it is often impossible to catch infected snail. This high selective pressure has led to local adaptation and compatibility with allopatric snail strains. Sexual reproduction of the parasite takes place within the vertebrate host, and when miraciadia are released they must immediately find a suitable snail host: at 25 °C, infection success declines rapidly 4 h after hatching and is zero after 12 h [34]. Non-migrating human hosts (e.g, school children) will shed eggs good for infection of sympatric snails. However, vertebrate hosts might also disperse (e.g., herdsmen) and parasite offspring might encounter allopatric snail hosts in this case. If the parasite cannot sense migration, a mixed stable/bet-hatching strategy would be best. However, if the parasite could sense migration of the host through dietary changes (e.g., different food, beverages, and starvation), modification of biorhythm (e.g., migration at night) or hormonal changes (e.g., stress), a switch from stable transmission to bet-hatching would be even more successful. The need of the parasite to receive triggers from the vertebrate host to complete its maturation is well documented and several hormones have been identified that alter parasite development (reviewed in [35]). In this sense, our TSA treatment might have mimicked events that trigger the switch between low and high phenotypic diversity. A schematic representation of this scenario is given in Additional file 3. Further work is needed to identify the biological trigger that leads to increased phenotypic variation in S. mansoni miracidia. Nevertheless, our findings fit already perfectly into theoretical models that predict such a function for the epigenetic information [2, 4] and are to the best of our knowledge the first experimental evidence for an epigenetic basis of adaptive evolution.
Conclusion
We show here that histone modifications, i.e., changes in the epigenetic information can be a source of phenotypic variants in the parasite S. mansoni. These phenotypic variants are adaptive since they confer a higher fitness to the parasite by increasing its infection success in the intermediate mollusk host. It is conceivable that environmental clues trigger epigenetic variation and thus contribute to exploring the adaptive landscape. The genetic and molecular bases for the generation of epigenetic variants have not been investigated in this study.
Methods
Ethics statement
Our laboratory has permission A 66040 from both French Ministère de l’agriculture et de la pêche and French Ministère de l’Education Nationale de la Recherche et de la Technologie for experiments on animals and certificate for animal experimentation (authorization 007083, decree 87-848 and 2012201-0008) for the experimenters. Housing, breeding and animal care followed the national ethical requirements.
Culture of Schistosoma mansoni parents and hybrids strains
We used in this part of the study different strains of S. mansoni: SmBRE, SmGH2 and SmLE. Each strain was maintained in its sympatric mollusk Biomphalaria glabrata strain (BgBRE, BgGUA and BgBAR2, respectively) as intermediate host and in hamsters (Mesocricetus auratus) as definitive host as described previously [36]. Three generations of hybrids of SmBRE and SmGH2 were produced as follows. Monomiracidial infections were performed within each strain of mollusk BgBRE and BgGUA. We obtained clonal populations of cercariae after 4 weeks. Sex was determined by PCR as described before [37, 38]. Strain hybrids were produced by infection of hamsters with 300 cercariae: 200 males from a clonal cercarial population and 100 females from another clonal population for each generation. Different combinations of parental cercariae were used for each generation in order to generate biological replicates. Three months later, eggs were collected from hamster livers and hatched in spring water. Miracidia were divided into three parts: (1) one part was used for life trait studies; (2) a second part was used for pedigree; and (3) a third part was concentrated by sedimentation on ice for 30 min and stored for molecular analysis at −20 °C.
SmBRE sporocysts transfer into BgBRE and BgGUA mollusks
SmBRE miracida freshly hatched from a mouse liver were used to perform single miracidium infection of a dozen BgBRE, as detailed in [39]. Thirty days later, mollusks were screened for presence of parasites. Two mollusks with a large number of sporocysts were selected as donors for sporocyst grafts.
Grafts were performed as described in [40, 41]. This technique allows to transplant secondary sporocysts from one mollusk to another. Grafted secondary sporocysts revert to primary sporocysts and then underdo normal development until cercariae emission. Large (10–12 mm) BgBRE and BgGUA mollusks were selected as receivers and anesthetized by incubating 4 h in spring water mixed with sodium pentobarbital at a final concentration of 1.2 mg/mL. Shells of mollusk donors were carefully removed with tweezers, and their digestive gland (where secondary sporocysts are located) was recovered. Explants of 1 mm3 containing 1–3 sporocysts were prepared and grafted (within an hour) in the cephalopedal sinus of receiver snails with a custom-made glass microneedle attached to a 1-mL syringe. A small incision of the tegument above the genital pore was made to reach the cephalopedal sinus. Grafted snails were maintained in normal growing conditions. Sporocysts from the first mollusk donor were grafted in 16 BgGUA and 14 BgBRE, and sporocysts from the second mollusk donor were transplanted in 17 BgGUA and 15 BgBRE.
Sixty days after transplantation, mollusks were screened for presence of parasites by searching for secondary sporocysts under a stereomicroscope and by looking for cercariae emission. Grafts were considered successful if both conditions were met. Fisher’s exact test for count data was used to compare between sympatric and allopatric sporocyst transfer success. The experiment was done in duplicates.
Sex identification of cercariae
Four weeks after the monomiracidial infestation of mollusk for each generation SmBRE F0, SmGH2 F0, F1 and F2, four clonal cercariae were selected from each mollusk in order to identify the sex by PCR [37, 38]. After DNA extraction from single cercariae, PCR was performed using two pairs of primers, two control primers that amplify Rhodopsin [Smp_scaff001984 (49840–50016)] on male and female, and two female-specific primers that amplify SmWSPP2 [Smp_scaff002739 (2682–3046)] only on female (Additional file 4). If all four reactions delivered the same result, we considered sex as identified. We then chose male and female clones to infect hamsters and produce the next generation.
Pharmacological induction by Trichostatin A treatment
After dissection of 4 infested hamsters (2 by strain), livers were collected and divided into two equal parts to compensate for a potential host bias. Two half livers for each strain SmLE and SmGH2 were incubated in 20 mL 150 mM NaCl and 0.1–0.3 % ethanol as control. For the treatment, two half livers for each strain SmLE and SmGH2 were incubated in 20 mL NaCl, and 20 µl of 20 mM Trichostatin A (TSA) (InvivoGen met-tsa-5) dissolved in ethanol was added two times at an interval of 12 h. Livers were incubated overnight at 4 °C. The next day, directly after grinding livers, another 20 µl of 20 mM TSA was added to the eggs from treated livers for SmLE and SmGH2. This strategy was chosen since TSA is known to be instable [42]. The final concentration of TSA was 20 µM in 150 mM NaCl and 0.1–0.3 % ethanol (solvent), and total treatment time was 16 h. Miracidia (non-treated SmGH2, treated SmGH2, non-treated SmLE and treated SmLE) were divided into two parts (1) for prevalence and intensity analysis and (2) for Western blot analysis.
Compatibility of S. mansoni with mollusk hosts
Single mollusks (n = 30–50) were exposed to 20 miracidia each in 5 ml of spring water overnight at 24–25 °C. Prevalence was measured 15 days post-exposure, by determining the rate of infected mollusks over the entire mollusks that were exposed to parasites. The intensities were evaluated by counting the number of mother sporocysts that developed within infected mollusk as previously described [43]. For selection experiments, the SmBRE strain was used for 6 generations on the BgGUA and BgVEN snail strains, respectively. Prevalences were determined at each passage. We named SmBREg and SmBREv these schistosome strains maintained on heteropatric snail hosts. At each generation, infectivity of SmBREg and SmBREv was tested vis-à-vis the original sympatric BgBRE snail strain.
Western blot
One thousand miracidia were individually collected, counted and incubated in 30 µl Lämmli buffer, 5 min at 99 °C. Fifteen µl were separated by electrophoresis through a 10 % SDS-PAGE gel and blotted on a nitrocellulose membrane (Trans-Blot turbo, Bio-Rad). The membrane was blocked with 5 % skimmed dry milk in TBST (TBS buffer containing 0.05 % Tween 20) for 1 h at room temperature and then incubated with the primary antibody “anti-SmPoMuc” [19] diluted 1/500 in TBST for 90 min at room temperature. The membrane was then incubated with secondary antibody (peroxidase conjugated, purified anti-rabbit IgG) diluted 1/5000 in TBST for 1 h and washed three times with TBST. Finally, proteins were detected with a ChemiDoc MP Imaging system (Bio-Rad) using ECL reagents. Images were converted into 256 grayscales, and analysis was done with ImageJ [44].
RNA extraction, reverse transcription and quantitative PCR
Messenger RNAs were isolated from 500 to 5000 miracidia from each strain using the Dynabeads® mRNA DIRECT Micro Kit (Invitrogen). The samples were put directly into 100 µl lysis buffer at −80 °C and processed according to the manufacturer’s instructions. After washing, the samples were resuspended directly in 20 µl of DNase treatment mix (Ambion RNA by Life Technologies DNA-free). cDNA were synthesized from 13 µl of the total mRNA preparation, in a final volume of 20 µl using the RevertAid Premium First Strand cDNA Synthesis Kit by Thermo Scientific. Quantitative PCR analyses were carried out using 2.5 µl of cDNA diluted 1/10 in a final volume of 10 µl (1.5 µl H2O, 0.5 µM of each primer, 5 µl of master mix), using a LightCycler® 480 Real-Time Instrument, and 2.5 µl of mRNA diluted 1/20 as negative control for the specific exon junctions amplifications. Alpha-tubulin (α-tub) was used as reference. Primer sequences are listed in Additional file 4. The following protocol was used: denaturation, 95 °C 10 min, amplification and quantification (45 cycles), 95 °C for 10 s, 60 °C for 5 s, 72 °C for 20 s; melting curve, 65–97 °C with a heating rate of 0.11 C/s and continuous fluorescence measurement, and cooling step to 40 °C. For each reaction, the crossing point cycle threshold (Ct) was determined using the “second derivate” method of the LightCycler® 480 Software release 1.5.0. Reactions were carried out in duplicate then the mean Ct was calculated. The amplification of a unique band of each locus was verified by size separation on a LabChip GX capillary electrophoretic system.
Chromatin immunoprecipitation (ChIP) followed by qPCR
Native chromatin immunoprecipitation was performed as described before [45]. The following antibodies against histone isoforms were used to precipitate chromatin in miracidia: Abcam anti-H3K9me3 (ab8898, Lot 733951) and Millipore anti-H3K9ac (07-352, Lot DAM1576933). Immunoprecipitated DNA was extracted by phenol/chloroform protocol and analyzed by quantitative PCR using specific primers for SmPoMuc 3.1(r1–r2) promoter (primer sequences in Additional file 4) and for the Smp_171100. The amount of target DNA recovered in the immunoprecipitated fraction was quantified by calculating the percent of input recovery (% IR) normalized with the percent input recovery obtained with α-tubulin gene. The percent input recovery of the bound immunoprecipitated fraction for each amplicon was calculated by the following formula: % input recovery = 100 × E(Ct (input) − Ct (IPBound)). The percent background was calculated by the following formula: % background = 100 × E(Ct (input) − Ct (C-Bound)), where E is the primer efficiency designed to amplify the amplicon, Ct (IPBound) is the Ct of the bound fraction obtained in the immunoprecipitated sample, Ct (C-Bound) is the Ct of the bound fraction obtained in the negative control (fraction without antibody) and Ct (input) is the Ct of the unbound fraction obtained in the negative control. It represents the quantity of chromatin that was used for the study minus the fraction that bound non-specifically to the protein A Sepharose beads. Finally, the ratios of the H3K9me3 to H3K3Ac for each gene was calculated using formulas (2ct (α-tub)/2ct (SmPoMuc 3.1(r1r2)) and 2ct (α-tub)/2ct (Smp_171100)).
DNA extraction, PCR amplification and Sanger sequencing
Genomic DNA for SmBRE F0, SmGH2 F0, F1, F2 and F3 miracidial population, was prepared by the incubation during 3 h at 55 °C with 500 µl of lysis buffer (20 mM Tris/Cl Ph 8; 1 Mm EDTA; 100 mM NaCl; 0.5 % SDS) and 0.15 mg of proteinase K. The samples were extracted twice with equal volumes of phenol/chloroform, followed by two extractions of equal volumes of chloroform. DNA was precipitated with equal volumes of isopropoanol/NaOAc (3 M, pH 5.2) at room temperature. After centrifugation and washing with 1 ml of 70 % ethanol, the pellet was dissolved in 50 µl of 1 mM Tris/Cl pH 8.
In order to determine whether the chromatin structure follows the Mendelian transmission or not (co-segregate with the allele or not), we focus specifically on sequences where we determined the chromatin structure by ChIP-qPCR for SmBRE F0, GH2 F0, F1, F2 and F3 miracidial population. We identified 3 SNPs between SmBRE F0 and GH2 F0 for SmPoMuc 3.1(r1r2) promoter sequence and 6 SNPs for Smp_171100 sequence using Sequencer software. Primers including SNPs for the two loci were designed, and the regions were amplified by qPCR. For the SmPoMuc 3.1(r1r2) promoter, qPCR was applied on a 9 kb PCR product containing the 2-kb region upstream of the transcription start site (TSS) [21], and for Smp_171100 sequence, qPCR was applied directly on genomic DNA for SmBRE F0, SmGH2 F0, F1, F2 and F3 miracidial population. qPCR products were sequenced using Eurogentec facilities (http://www.eurogentec.com/life-science.html). We checked and aligned nucleotide sequences manually using Sequencer and BioEdit softwares.
Statistical analysis
Fisher’s exact test was used to compare prevalences and T test to compare intensities. For the statistical differences in the amount of transcripts, we used ANOVA with Dunnett’s post hoc test using the parental strain SmBRE as control group.
Authors’ contributions
SF, AT, AR, BA, DD, JBu and CG designed and performed the experiments. SF, CC, GM and CG wrote the manuscript. AR and DR performed sporocyst transfer. NA raised the animals. JBo performed the statistical analysis. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Benjamin Gourbal, Claudine Junien and Richard Galinier for fruitful discussions. We thank also Anne Rognon and late Bernard Dejean for providing valuable technical support. Special thanks go to G. Olivieira (Instituto Nacional de Ciência e Tecnologia em Doenças Tropicais, Fundacao Oswaldo Cruz, Brazil) for the generous gift BgBAR / SmLE strains (SIBIO license 42195). This work is dedicated to our friend and colleague Jérôme Buard who passed away during the writing of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the French National Agency for Research (ANR-2010-BLAN-1720-01; EPIGEVOL).
Abbreviations
- SmPoMuc
Schistosoma mansoni polymorphic mucins
- SmBRE
Schistosoma mansoni strain Brazil BRE
- SmLE
Schistosoma mansoni strain Brazil LE
- SmGH2
Schistosoma mansoni strain Guadeloupe GH2
- BgBRE
Biomphalaria glabrata strain Brazil BRE
- BgGUA
Biomphalaria glabrata strain Guadeloupe GUA
- BgBAR
Biomphalaria glabrata strain Brazil BAR
- ChIP
chromatin Immunoprecipitation
- H3K9ac
histone H3 acetylated on lysine 9
- H3K9me3
histone H3 tri-methylated on lysine 9
- TSA
trichostatin A
Additional files
10.1186/s13072-016-0076-2 Infection success after sporocyst transfer. SmBRE sporocysts succeed to infect the two strains of mollusks BgBRE and BgGUA and the vertebrate host.
10.1186/s13072-016-0076-2 TSA treatment results. This table contains results of all TSA experiments.
10.1186/s13072-016-0076-2 Schematic representation of compatibility polymorphism and influence of changes in the epigenotype. On the left a high compatibility situation: the hypothetical inbred S. mansoni strain “white” can infect successfully the B. glabrata strain “MOSTLY WHITE” because most snails are compatible “white.” However, SmWHITE cannot infect B. glabrata “dark” snails. On the right, the incompatible situation in which the same SmWHITE strain is exposed to a BgDARK strain. None of the BgDARK can be infected by SmWHITE. If SmWHITE is treated with TSA, new phenotypic variants develop. Some of them (e.g., SmBLACK) are also incompatible with the snail hosts; others can infect (e.g., SmGRAY). By increasing the phenotypic variants in the inbred strain through epimutations, the reaction norm of the strain becomes larger and previously incompatible hosts can be infected.
10.1186/s13072-016-0076-2 Primers used in this study.
Contributor Information
Sara Fneich, Email: sfneich@jouy.inra.fr.
André Théron, Email: theron@univ-perp.fr.
Céline Cosseau, Email: celine.cosseau@univ-perp.fr.
Anne Rognon, Email: rognon@univ-perp.fr.
Benoit Aliaga, Email: benoit.aliaga@univ-perp.fr.
Jérôme Buard, Email: christoph.grunau@univ-perp.fr.
David Duval, Email: david.duval@univ-perp.fr.
Nathalie Arancibia, Email: nathalie.arancibia@univ-perp.fr.
Jérôme Boissier, Email: boissier@univ-perp.fr.
David Roquis, Email: david.roquis@yahoo.com.
Guillaume Mitta, Email: mitta@univ-perp.fr.
Christoph Grunau, Phone: +33(0)468662180, Email: christoph.grunau@univ-perp.fr.
References
- 1.Darwin C. The variation of animals and plants under domestication. London: John Murray publisher;1868.
- 2.Klironomos FD, Berg J, Collins S. How epigenetic mutations can affect genetic evolution: model and mechanism. BioEssays. 2013;35(6):571–578. doi: 10.1002/bies.201200169. [DOI] [PubMed] [Google Scholar]
- 3.Maynard Smith J. Models of a dual inheritance system. J Theor Biol. 1990;143:41–53. [DOI] [PubMed]
- 4.Pal C, Miklos I. Epigenetic inheritance, genetic assimilation and speciation. J Theor Biol. 1999;200:19–37. [DOI] [PubMed]
- 5.Waddington CH. The epigenotype. Int J Epidemiol. 1942;41(1):10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 6.Tal O, Kisdi E, Jablonka E. Epigenetic contribution to covariance between relatives. Genetics. 2010;184(4):1037–1050. doi: 10.1534/genetics.109.112466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frésard L, Morisson M, Brun JM, Collin A, Pain B, Minvielle F, Pitel F. Epigenetics and phenotypic variability: some interesting insights from birds. Genet Sel Evol. 2013;45:16. doi: 10.1186/1297-9686-45-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23(3):314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 9.Cortijo S, Wardenaar R, Colomé-Tatché M, Gilly A, Etcheverry M, Labadie K, Caillieux E, Hospital F, Aury JM, Wincker P. Mapping the epigenetic basis of complex traits. Science. 2014;343(6175):1145–1148. doi: 10.1126/science.1248127. [DOI] [PubMed] [Google Scholar]
- 10.Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-Mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441(7092):469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- 11.Manning K, Tör M, Poole M, Hong Y, Thompson AJ, King GJ, Giovannoni JJ, Seymour GB. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38(8):948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 12.Hotez PJ, Kamath A. Neglected tropical diseases in sub-Saharan Africa: review of their prevalence, distribution, and disease burden. PLoS Negl Trop Dis. 2009;3(8):e412. doi: 10.1371/journal.pntd.0000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan JA, Dejong RJ, Adeoye GO, et al. Origin and diversification of the human parasite Schistosoma mansoni. Mol Ecol. 2005;14:3889–902. [DOI] [PubMed]
- 14.Mitta G, Adema CM, Gourbal B, Loker ES, Theron A. Compatibility polymorphism in snail/schistosome interactions: from field to theory to molecular mechanisms. Dev Comp Immunol. 2012;37(1):1–8. doi: 10.1016/j.dci.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theron A, Rognon A, Gourbal B, Mitta G. Multi-parasite host susceptibility and multi-host parasite infectivity: A new approach of the Biomphalaria glabrata/Schistosoma mansoni compatibility polymorphism. Infect Genet Evol. 2014;26:80–88. doi: 10.1016/j.meegid.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Roger E, Mitta G, Moné Y, Bouchut A, Rognon A, Grunau C, Boissier J, Théron A, Gourbal B. Molecular determinants of compatibility polymorphism in the Biomphalaria glabrata/Schistosoma mansoni model: New candidates identified by a global comparative proteomics approach. Mol Biochem Parasitol. 2008;157(2):205–216. doi: 10.1016/j.molbiopara.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Roger E, Grunau C, Pierce RJ, et al. Controlled chaos of polymorphic mucins in a metazoan parasite (Schistosoma mansoni) interacting with its invertebrate host (Biomphalaria glabrata). PLoS Negl Trop Dis. 2008;2:e330. [DOI] [PMC free article] [PubMed]
- 18.Roger E, Gourbal B, Grunau C, Pierce RJ, Galinier R, Mitta G. Expression analysis of highly polymorphic mucin proteins (SmPoMuc) from the parasite Schistosoma mansoni. Mol Biochem Parasitol. 2008;157(2):217–227. doi: 10.1016/j.molbiopara.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 19.Moné Y, Gourbal B, Duval D, Du Pasquier L, Kieffer-Jaquinod S, Mitta G. A large repertoire of parasite epitopes matched by a large repertoire of host immune receptors in an invertebrate host/parasite model. PLoS Negl Trop Dis. 2010;4:e813. [DOI] [PMC free article] [PubMed]
- 20.DeMarco R, Mathieson W, Manuel SJ, Dillon GP, Curwen RS, Ashton PD, Ivens AC, Berriman M, Verjovski-Almeida S, Wilson RA. Protein variation in blood-dwelling schistosome worms generated by differential splicing of micro-exon gene transcripts. Genome Res. 2010;20(8):1112–1121. doi: 10.1101/gr.100099.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrin C, Lepesant JM, Roger E, et al. Schistosoma mansoni mucin gene (SmPoMuc) expression: epigenetic control to shape adaptation to a new host. PLoS Pathog. 2013;9:e1003571. [DOI] [PMC free article] [PubMed]
- 22.Cosseau C, Azzi A, Rognon A, Boissier B, Gourbière S, Roger E, Mitta G, Grunau C. Epigenetic and phenotypic variability in populations of Schistosoma mansoni—a possible kick-off for adaptive host/parasite evolution. Oikos. 2010;119(4):669–678. doi: 10.1111/j.1600-0706.2009.18040.x. [DOI] [Google Scholar]
- 23.Clément JA, Toulza E, Gautier M, Parrinello H, Roquis D, Boissier J, Rognon A, Moné H, Mouahid G, Buard J, et al. Private selective sweeps identified from next-generation pool-sequencing reveal convergent pathways under selection in two inbred Schistosoma mansoni strains. PLoS Negl Trop Dis. 2013 doi: 10.1371/journal.pntd.0002591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lepesant JM, Grunau C, Cosseau C. Towards an understanding of the epigenetics of schistosomes: a comparative epigenomic study. Mem Inst Oswaldo Cruz. 2011;106:823–30. [DOI] [PubMed]
- 25.Lopez-Atalaya JP, Ito S, Valor LM, Benito E, Barco A. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 2013;41:8072–84. [DOI] [PMC free article] [PubMed]
- 26.Halsall J, Gupta V, O’Neill LP, Turner BM, Nightingale KP. Genes are often sheltered from the global histone hyperacetylation induced by HDAC inhibitors. PLoS One. 2012;7(3):e33453. doi: 10.1371/journal.pone.0033453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews KT, Gupta AP, Tran TN, Fairlie DP, Gobert GN, Bozdech Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PLoS One. 2012;7(2):e31847. doi: 10.1371/journal.pone.0031847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrenkaufer GM, Eichinger DJ, Singh U. Trichostatin A effects on gene expression in the protozoan parasite Entamoeba histolytica. BMC Genom. 2007;8(1):216. doi: 10.1186/1471-2164-8-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hochholdinger F, Hoecker N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007;12:427–32. [DOI] [PubMed]
- 30.Baranwal VK, Mikkilineni V, Zehr UB, Tyagi AK, Kapoor S. Heterosis: emerging ideas about hybrid vigour. J Exp Bot (Soc Experiment Biol). 2012;63:6309–14. [DOI] [PubMed]
- 31.Kiani J, Grandjean V, Liebers R, Tuorto F, Ghanbarian H, Lyko F, Cuzin F, Rassoulzadegan M. RNA–mediated epigenetic heredity requires the cytosine methyltransferase Dnmt2. PLoS Genet. 2013;9(5):e1003498. doi: 10.1371/journal.pgen.1003498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geyer KK, Chalmers IW, Mackintosh N, Hirst JE, Geoghegan R, Badets M, Brophy PM, Brehm K, Hoffmann KF. Cytosine methylation is a conserved epigenetic feature found throughout the phylum Platyhelminthes. BMC Genom. 2013;14(1):462. doi: 10.1186/1471-2164-14-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sire C, Durand P, Pointier JP, Théron A. Genetic diversity and recruitment pattern of Schistosoma mansoni in a Biomphalaria glabrata snail population: a field study using random-amplified polymorphic DNA markers. J Parasitol. 1999;85:436–41. [PubMed]
- 34.Anderson R, Mercer J, Wilson R, Carter N. Transmission of Schistosoma mansoni from man to snail: experimental studies of miracidial survival and infectivity in relation to larval age, water temperature, host size and host age. Parasitology. 1982;85(02):339–360. doi: 10.1017/S0031182000055323. [DOI] [PubMed] [Google Scholar]
- 35.De Mendonça RL, Escrivá H, Bouton D, Laudet V, Pierce RJ. Hormones and nuclear receptors in schistosome development. Parasitol Today. 2000;16(6):233–240. doi: 10.1016/S0169-4758(00)01641-0. [DOI] [PubMed] [Google Scholar]
- 36.Théron A, Pages JR, Rognon A. Schistosoma mansoni: distribution patterns of miracidia among biomphalaria glabrata snail as related to host susceptibility and sporocyst regulatory processes. Exp Parasitol. 1997;1–9. [DOI] [PubMed]
- 37.Portela J, Grunau C, Cosseau C, Beltran S, Dantec C, Parrinello H, Boissier J. Whole-genome in-silico subtractive hybridization (WISH) - using massive sequencing for the identification of unique and repetitive sex-specific sequences: the example of Schistosoma mansoni. BMC Genom. 2010;11:387. doi: 10.1186/1471-2164-11-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepesant JMJ, Boissier J, Climent D, Cosseau C, Grunau C. Female biased sex-ratio in Schistosoma mansoni after exposure to an allopatric intermediate host strain of Biomphalaria glabrata. Exp Parasitol. 2013;135:350–356. doi: 10.1016/j.exppara.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Boissier J, Mone H. Experimental observations on the sex ratio of adult Schistosoma mansoni, with comments on the natural male bias. Parasitology. 2000;121(04):379–383. doi: 10.1017/S0031182099006393. [DOI] [PubMed] [Google Scholar]
- 40.Jourdane J, Theron A. Schistosoma mansoni: cloning by microsurgical transplantation of sporocysts. Exp Parasitol. 1980;50(3):349–357. doi: 10.1016/0014-4894(80)90038-7. [DOI] [PubMed] [Google Scholar]
- 41.Jourdane J. Maintenance of a male and a female clone of Schistosoma mansoni by microsurgical transplantation of sporocysts. Reliability of the method. Annales de parasitologie humaine et comparee. 1983;59(4):361–367. [PubMed] [Google Scholar]
- 42.Grunau C, Buard J, Brun ME, De Sario A. Mapping of the juxtacentromeric heterochromatin-euchromatin frontier of human chromosome 21. Genome Res. 2006;16(10):1198–1207. doi: 10.1101/gr.5440306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Théron A, Gerard C. Development of accessory sexual organs in Biomphalaria glabrata (Planorbidae) in relation to timing of infection by Schistosoma mansoni: consequences for energy utilization patterns by the parasite. J Molluscan Stud (Malacol Soc Lond). 1994;60:25–31.
- 44.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with imagej. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 45.Cosseau C, Azzi A, Smith K, Freitag M, Mitta G, Grunau C. Native chromatin immunoprecipitation (N-ChIP) and ChIP-Seq of Schistosoma mansoni: Critical experimental parameters. Mol Biochem Parasitol. 2009;166(1):70–76. doi: 10.1016/j.molbiopara.2009.02.015. [DOI] [PubMed] [Google Scholar]