

Abstract
Objective
Rolandic epilepsy is a common genetic focal epilepsy of childhood characterized by centrotemporal sharp waves on electroencephalogram. In previous genome‐wide analysis, we had reported linkage of centrotemporal sharp waves to chromosome 11p13, and fine mapping with 44 SNPs identified the ELP4‐PAX6 locus in two independent US and Canadian case–control samples. Here, we aimed to find a causative variant for centrotemporal sharp waves using a larger sample and higher resolution genotyping array.
Methods
We fine‐mapped the ELP4‐PAX6 locus in 186 individuals from rolandic epilepsy families and 1000 population controls of European origin using the Illumina HumanCoreExome‐12 v1.0 BeadChip. Controls were matched to cases on ethnicity using principal component analysis. We used generalized estimating equations to assess association, followed up with a bioinformatics survey and literature search to evaluate functional significance.
Results
Homozygosity at the T allele of SNP rs662702 in the 3′ untranslated region of PAX6 conferred increased risk of CTS: Odds ratio = 12.29 (95% CI: 3.20–47.22), P = 2.6 × 10−4 and is seen in 3.9% of cases but only 0.3% of controls.
Interpretation
The minor T allele of SNP rs662702 disrupts regulation by microRNA‐328, which is known to result in increased PAX6 expression in vitro. This study provides, for the first time, evidence of a noncoding genomic variant contributing to the etiology of a common human epilepsy via a posttranscriptional regulatory mechanism.
Introduction
Rolandic epilepsy (RE), or benign epilepsy of childhood with centrotemporal spikes (BECTS) (OMIM #117100), is the most common childhood epilepsy syndrome with a prevalence of ~1 in 2500 children, showing onset of seizures in a narrow age range of 4–12 years, and invariable remission by 14 years.1, 2 Another specific feature of focal seizures in RE is the selective involvement of the vocal tract, with both sensory and motor disturbances including paresthesia and clonic movements of the lower face, dysarthria, and hypersalivation, and often a subsequent period of speech arrest. Seizures occur almost exclusively in sleep at the transition between rapid eye movement (REM) and non‐REM cycles, and may secondarily generalize. Approximately 30% of RE patients have an antecedent history of speech sound disorder (SSD, developmentally inappropriate errors in speech production that limit intelligibility, usually caused by a mild speech dyspraxia3), and 42% meet ICD‐10 criteria for reading disorder.4 RE clinically overlaps with more severe epilepsy syndromes in the “epilepsy‐aphasia spectrum” such as atypical benign partial epilepsy (ABPE, OMIM #604827), continuous spikes in slow wave sleep (CSWSS), and Landau–Kleffner syndromes (LKS, OMIM #245570)5 as well as atypical forms6 that all share the common electroencephalographic (EEG) signature of centrotemporal spikes (CTS). CTS is also seen in 2–4% of the school‐aged population7 and is over‐represented in neurodevelopmental disorders such as autism8 and attention‐deficit hyperactivity disorder.9
While some question whether RE is genetically influenced,10, 11 rare Mendelian variants exist.12, 13, 14 Following the success of exome sequencing in discovering mostly de novo mutations15 for severe infantile epileptic encephalopathies, several studies have identified GRIN2A mutations in epilepsies of the epilepsy‐aphasia spectrum, ranging in frequency from 2.1% in RE to 20% in CSWSS.16 Other very rare sequence mutations have been found in KCNQ2, KCNQ3, RBFOX1, GABRG2, and DEPDC5.17, 18, 19, 20 Recurrent structural genomic variation has been found at 16p11.2 in 1.3% of RE, but incomplete penetrance suggests the presence of additional genetic and/or environmental factors.21 Alternative approaches to identifying genetic contributors to RE have focused on the genetic model of CTS, the EEG signature necessary for diagnosis, which serves as an electrophysiological endophenotype of RE. The inheritance of CTS in RE is consistent with an autosomal dominant pattern.22 Early candidate gene studies reported linkage of CTS to chromosome 15q13.33,23 but we subsequently reported strong genome‐wide linkage evidence for CTS in RE families at 11p13,24 and found the locus to be pleiotropic for speech dyspraxia in RE.3 Two small case–control samples with limited SNP coverage allowed localization of allelic association at the 11p13 locus to SNPs at the ELP4‐PAX6 24 locus which was not independently replicated.25 Deep sequencing failed to reveal rare causative coding mutations at this locus,26 although the prevalence of CTS (2–4%) in the general population7 renders a rare variant genetic model unlikely. Here we report localization evidence of CTS at the ELP4‐PAX6 locus in an expanded sample, refining the association to a noncoding SNP previously reported to regulate PAX6 expression, and with suggested evidence of a novel mechanism of epilepsy susceptibility via reduced microRNA binding affinity.
Subjects and Methods
Study design
Probands with RE (who have CTS by definition) and their parents and siblings were recruited as described previously from the US, Canada, Argentina, France and the UK24 with ethics approval by local institutional review boards. Written informed consent was obtained from all of the patients’ legal guardians to share clinical, neuroimaging, and electroencephalographic data and provide blood and saliva when available. Briefly, cases with RE, as defined in accordance with the International League Against Epilepsy,28 were enrolled and their families recruited (2005–2014); ascertainment was through the proband, with no other family member required to be affected with RE. Patients were excluded if the cause of the seizures was determined to be due to alternative structural, inflammatory, or metabolic cause.29 In addition, cases with unwitnessed episodes or with only secondary generalized seizures were excluded. Siblings aged 4–16 years underwent EEG to detect the presence of CTS, which has age‐dependent penetrance and is detectable between 4 and 16 years;22 the EEGs were assessed blind to identity by two independent experts. This study analyzed two groups of case participants of European descent (1) with only CTS (152 subjects from 126 families), and (2) with either CTS or SSD (186 subjects from 128 families) to test for pleiotropy. Phenotyping for CTS and SSD was conducted as previously reported.24, 30 One‐thousand population controls of European descent, determined by principal component analysis,31 were selected at random from a pool of 4491 unrelated individuals who took part in a population‐based study of children visiting the Ontario Science Centre,32 frequency matched by sex, and self‐reported to be unaffected with epilepsy. Genotypes of 646 RE probands and their family members, and the 4491 unrelated population controls ascertained at the Ontario Science Centre were obtained from the Illumina HumanCoreExome‐12 v1.0 BeadChip (538,448 SNPs), and five additional cases with CTS on the HumanOmniExpress‐12 v1.1 BeadChip (730,525 SNPs) (four from Argentina, one from USA). Genotypes for 50 of these RE cases were previously obtained at 44 SNPs at the 11p13 locus and included in our previously published CTS case–control association study.24
Quality control of genotype data
PLINK v1.0733 and R statistical software34 were used for quality control of genotype data. Individuals with a genotype missing rate of 10% or greater and SNPs with a call rate <90% were removed. Duplicated SNPs for each platform were also identified and the SNP with the highest call rate was kept. Sex was assessed against reported gender using heterozygosity from the X chromosome. In addition, samples that were outliers for heterozygosity on autosomal chromosomes were removed. Heterozygous haploid SNPs on the sex chromosomes were removed.
Pairwise relatedness was assessed using PLINK's calculation of the kinship coefficient (–genome option). The individual with the lowest missing genotype rate was kept from identified monozygotic twins and duplicated samples. Family relationships were recorded, and information from PLINK's kinship coefficient calculations were used in conjunction to build the pedigrees de novo using in‐house scripts. The pedigrees were then checked for errors using the kinship2 package in R.35
KING31 was used in cases and controls for principal component analysis with subjects from the International Hapmap Project (Phase 3)36 from various ethnic backgrounds, while correcting for family relationships. Subjects who clustered close to Hapmap‐defined populations other than CEPH (Utah residents with ancestry from northern and western Europe) (CEU) or Toscani in Italy (TSI) were excluded from the analysis. Principal component analysis was then re‐run with the remaining sample individuals and Hapmap CEU and TSI subjects; sample individuals who were more than six standard deviations from the mean for any of the first three principal components were also excluded from the analysis. Furthermore, cases and controls were compared in a principal component analysis against a broader European reference population from the 1000 Genomes Project37 and Human Genome Diversity Project38 to ensure homogeneity between cases and controls. The Tracy–Widom test was used to determine the number of principal components needed to correct for population stratification in the association model.
Statistical analysis and bioinformatics
We restricted statistical analysis to the genotyped markers at the chromosome 11p13 CTS locus (chr11:30,862,638–31,815,896; hg19) defined by a 1‐LOD score interval from the previously reported linkage study.24 We estimated the number of independent tests, and calculated a Bonferroni‐corrected critical value for declaring regional statistical significance using the Genetic Type I Error Calculator (GEC).39 The P‐value required for statistical significance was 3.09 × 10−3 in the ELP4‐PAX6 region. We used generalized estimating equations to account for the relatedness with an independence correlation structure. All association tests were conducted using the geeglm function in the geepack package in R.40 In the primary analysis, we coded SNPs as additive, and adjusted for sex and principal components estimated using KING.31 The independent association test per continental region for CTS was conducted as follows: for the top SNP associated with CTS, 69 CTS cases from USA and 13 from Canada were pooled into the “North American” group site and checked for association with 539 independent population controls; and 59 CTS cases from UK and seven from France were pooled into the “European” group site and checked for association with 434 nonoverlapping population controls. Forest plots were generated using the forestplot package in R.41
We visualized the association P‐values using LocusZoom's web interface.42 For finer resolution in post hoc analysis, BEAGLE 4.0 version r139943 was used to impute the genotypes of all samples in the region of interest (chr11:30,862,638–31,815,896; NCBI build 37). Whenever available, parent‐child relationships were used for imputation. All 2504 individuals in the 1000 Genomes Project (phase 3 version 5) were used as the reference during the phasing and imputation steps. For strand alignment of genotyped SNPs, however, the conform‐gt program (version r1174) was used with only the European‐identified individuals in the 1000 Genomes Project37 as reference. Only SNPs with an allelic r 2 > 0.8 were retained for further analysis.
We evaluated results in the context of annotations in the Database of Genomic Variants (DGV),44 the NIH Roadmap Epigenomics Mapping Consortium (REMC),45 the Encyclopedia of DNA Elements (ENCODE),46 and an integrative analysis of public ChIP‐seq experiments (ReMap).47 Haploreg (v4)48 was queried for transcription factor affinity binding predictions and the presence of suggested transcription factor motifs were verified using JASPAR.49 The Probability of Interaction by Target Accessibility (PITA)50 tool was used to verify microRNA target accessibility and recognition at the PAX6 3′UTR region.
Results
Association of CTS in the ELP4‐PAX6 region
In the regional analysis of 11p13, 152 individuals with CTS from Canada, the United States, Argentina, and Europe (Tables 1 and 2) were compared to 1000 ethnically matched controls of European origin. One genotyped SNP, rs662702, in the 3′‐untranslated region (UTR) of PAX6 reached regional significance (P = 1.53 × 10−3) under an additive model with an estimated odds ratio (OR) of 1.97 (95% CI: 1.29–2.99; Fig. 1A and Table S1). The T allele was present in 14% of the individuals with CTS and only 7.6% of controls. Follow‐up fine mapping with imputation, and conditional analysis on rs662702, did not reveal other variants with greater evidence of association (Fig. 1A and Table S1). The evidence at rs662702 was consistent across continents, with ORs of 1.92 (95% CI: 1.08–3.43) for North America (69 cases from the US and 13 from Canada), and 2.20 (95% CI: 1.17–4.13) for the independent sample from Europe (59 from the UK and 7 from France) (Fig. 1B). The rs662702 variant was not genotyped as part of the original linkage and association study implicating the 11p13 locus,24 but ELP4 associated variants from that study are in LD with rs662702 (D′ = 0.65). Because the 11p13 locus is pleiotropic for CTS and SSD,3 we tested the hypothesis that SNP rs662702 also contributes to SSD. We reanalyzed using the expanded sample of 186 individuals with CTS or SSD; the results argued against a pleiotropic effect, with the OR decreasing from 1.97 to 1.84 and the P‐value increasing (P = 4.17 × 10−3) despite the larger sample size. The homozygosity frequency of the rs662702 T risk allele among Ontario Science Centre population controls is 0.30% (3/1000). To confirm the population estimate of this genotype frequency we consulted the 1000 Genomes Project's European sample37 and an unrelated European subset of the Human Genome Diversity Project,38 which report comparable frequencies of 0.60% (3/503) and 0.64% (1/157), respectively. Since the minor T allele frequency differs across ethnic backgrounds, we further verified by principal component analysis considering up to 10 components, that T allele carriers in CTS cases overlap with T allele carriers from OSC controls, and overlap with a broader panel of European individuals from the 1000 Genomes Project37 and from the Human Genome Diversity Project38 (Fig. S1). Among those with CTS, the rs662702 homozygosity frequency is almost 4% (6/152,with all six TT homozygotes unrelated to one another), resulting in a 12.29 (95% CI: 3.20–47.22) times greater odds of CTS (after adjusting for sex and population structure) among individuals homozygous for the rs662702 T allele (P = 2.58 × 10−4). Five of the six TT homozygotes who had CTS also had RE. Seven additional individuals from RE families were homozygous for the rs662702 T allele, but were removed from the study prior to association analysis because they were either of non‐European origin (N = 4) or their CTS status was unknown (N = 3). Of the 13 individuals from RE families that were homozygous for the TT allele, eight had RE (62%) and all either showed CTS on EEG or did not undergo EEG because they were beyond the age range for CTS detection.
Table 1.
Distribution of sex, CTS, SSD, and ethnicity of 651 RE cases and their RE‐unaffected family members ascertained for this study. Six hundred and forty‐six individuals were genotyped on the Illumina HumanCoreExome platform and five cases with CTS on the HumanOmniExpress platform
| Sex | CTS | SSD | Ethnicity | |||||
|---|---|---|---|---|---|---|---|---|
| Males | Females | Present | Unknown/Absent | Present | Unknown/Absent | European | Other | |
| RE cases | 106 (62.7%) | 63 (37.3%) | 167a (98.8%) | 2 (1.2%) | 42 (24.9%) | 127 (75.1%) | 132b (78.1%) | 37 (21.9%) |
| RE‐unaffected family members; unaffected siblings | 206; 56 (42.7%; 39.4%) | 276; 86 (57.3%; 60.6%) | 28; 19 (5.8%; 13.4%) | 454; 123c (94.2%; 86.6%) | 51; 29 (10.6%; 20.4%) | 431; 113 (89.4%; 79.6%) | 365; 103 (75.7%; 72.5%) | 117; 39 (24.3%; 27.5%) |
CTS association analysis was restricted to the subset of individuals in the table who were of European origin (determined by principal component analysis), with established CTS on EEG. CTS, centrotemporal spikes; SSD, speech sound disorder, RE, Rolandic epilepsy, EEG, electroencephalographic.
One hundred and fifty‐two of the 167 individuals with CTS were of European origin.
One hundred and thirty‐two of 152 individuals with CTS also had RE.
Thirty‐three of these 123 were unaffected with CTS; the remainder had an unknown CTS status.
Table 2.
Distribution of CTS cases and their family members used in the analysis per geographic location
| USA | UK | Canada | France | Argentina | |
|---|---|---|---|---|---|
| CTS cases (Europeana) | 76 (69) | 66 (59) | 15 (13) | 8 (7) | 4 (4) |
| Family members without/unknown CTS | 285 | 188 | 0 | 9 | 0 |
Genotypes from the four individuals from Argentina and one from USA were obtained from the HumanOmniExpress BeadChip. Genotypes from the remaining 646 individuals were obtained from the HumanExomeCore BeadChip. RE, Rolandic epilepsy; CTS, centrotemporal spikes.
Only those RE cases with CTS of European origin were used in the association analysis.
Figure 1.
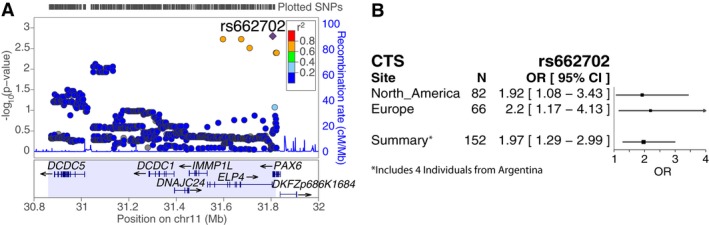
Association of centrotemporal spikes (CTS) in the 11p13 linkage locus. (A) LocusZoom42 plot for the association of 152 CTS cases with 1000 population controls using an additive model under the CTS 1‐LOD linkage interval with rs662702 (purple diamond) and two imputed SNPs annotated to ELP4 in linkage disequilibrium with rs662702, providing a region‐wide significant association with CTS. (B) Association evidence for rs662702 with 95% confidence intervals for independent North American and European samples. The summary row reflects the association analysis at rs662702 presented in Figure 1A, which includes the North American CTS cases, the European CTS cases, four additional CTS cases from Argentina who are not included in the North American‐only or European‐only analyses, and all 1000 population controls, with genotype distribution of 6/31/115 and 3/145/852 (TT/TC/CC) for CTS cases and controls, respectively.
Bioinformatic assessment
rs662702 resides in the 3′UTR of PAX6. The C allele is conserved in mammals and overlaps a region bound by several transcription factors46, 47 (Fig. 2), some of which play an established role in brain gene expression (e.g., FOXP227 and TFAP2C51). DNase hypersensitivity, epigenetic modifications and chromatin state52 annotations strongly support that rs662702 is an enhancer utilized in several brain tissues and developmental stages. HaploReg48 predicts that the rs662702 T allele slightly alters the transcription factor binding sites of two TALE homeobox transcription factors, MEIS2 and TGIF1 (Fig. 2C). MEIS2 and TGIF1 have been shown to bind overlapping motifs and compete for the same binding site leading to differential regulation of brain genes with TGIF1 acting as a repressor.53
Figure 2.
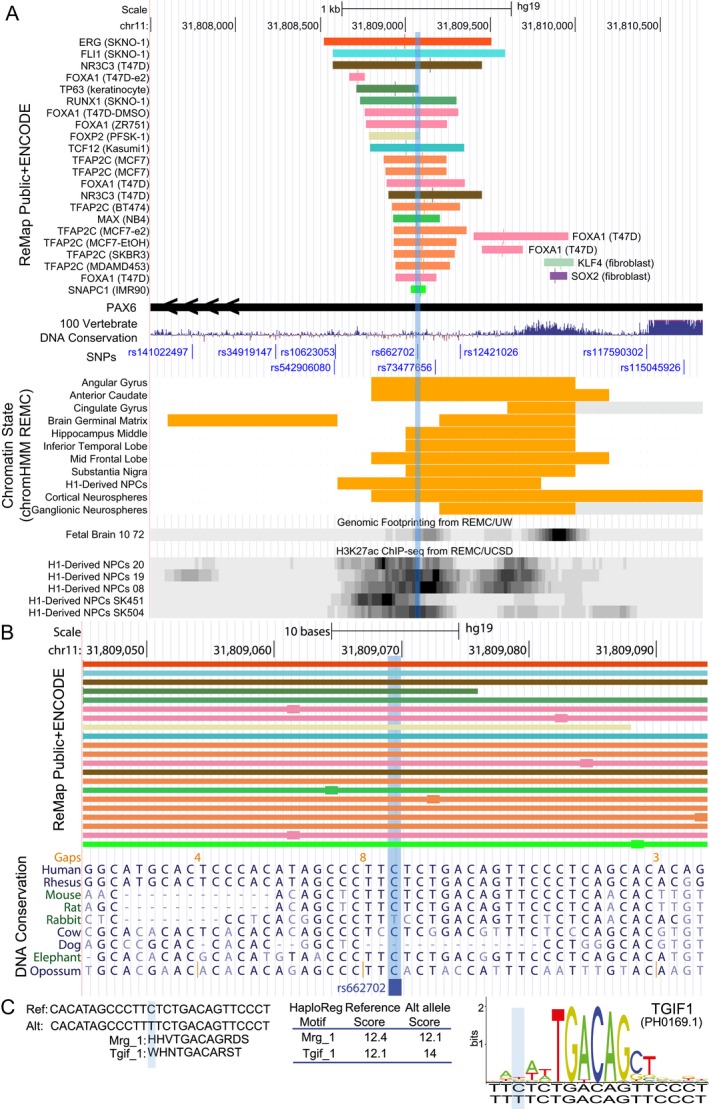
Overview of epigenetic interactions and evolutionary conservation at the rs662702 locus. (A) UCSC genome browser view of a curated set of transcription factor binding sites47 obtained by chromatin immunoprecipitation followed by DNA sequencing (ChIP‐seq) from both ENCODE46 and public datasets47 shows that the region surrounding rs662702 (light blue shading) overlaps several transcription factor binding sites. Most of the transcription factor binding sites within 250 bp of rs662702 come from breast cancer (13/21: NR3C3, FOXA1, and TFAP2C) and leukemia (5/21: ERG, FLI1, RUNX1, TCF12, MAX) cell lines. The remainder of the binding events come from a neuroectodermal cell line (FOXP2), differentiated keratinocytes (TP63), and an immortalized fibroblast (SNAPC1). When considering the summit of ChIP‐seq signal (larger box on colored line) of these bound transcription factors, TFAP2C summit is the closest to rs662702. This region is also annotated as an enhancer chromatin state (orange bars) in several regions of the brain and is a DNase I hypersensitive site in fetal brain (data from REMC).45 (B) Zoomed in view of rs662702 shows the close proximity of transcription factor binding summits for TFAP2C and FOXA1 as well as a strong conservation of the major C allele in mammals (Multiz69 alignment shown). (C) Summary of HaploReg (v4)48 predictions of the transcription factor binding site affinity changes due to rs662702. The reference and alternative allele and the affinity scores are shown. Both matches are to homeobox proteins belonging to the TALE family of homeodomain‐containing proteins. Mrg–1 motif is related to MEIS2 and Tgif–1 is related to TGIF1 (transforming factor growth beta (TGFβ)‐induced factor 1). The TGIF1 motif logo shown was obtained from the JASPAR database, which showed a score threshold >80% for the murine‐derived motif for the 60 bp sequence surrounding the SNP; the scoring is made against 200 random matrix models permuted for the motif sequence).49 According to Haploreg,48 the T allele increases the predicted binding affinity to TGIF1. NPC, neuronal progenitor cell; REMC, Roadmap Epigenomics Mapping Consortium.
However, the strongest experimental evidence as to how rs662702 impacts PAX6 expression comes from published experiments showing that the T allele of rs662702 disrupts the seed region of microRNA‐328 (miR‐328).50, 54 Using reporter assays it has been shown that the T allele prevents the downregulation of PAX6 by miR‐328,54 which gives rise to higher PAX6 expression as shown in retinal pigment epithelial cells.55
Discussion
The results of this association study suggest rs662702 in the 3′UTR of PAX6 may contribute to centrotemporal spikes in rolandic epilepsy. Each T allele at rs662702 doubles the odds of CTS, whereas T allele homozygosity displays a 12‐fold increase in risk. One possible explanation for this observation is that other undiscovered variants at the ELP4‐PAX6 locus contribute to CTS as compound heterozygotes. This 3′ UTR association with CTS invites one to question the role of rs662702 variants in related epilepsies and neurodevelopmental disorders that feature CTS. Although the 11p13 locus is pleiotropic for speech dyspraxia and CTS in RE, this study does not support the hypothesis that pleiotropy is mediated through rs662702. The functional effects of rs662702 variation have been investigated in other studies54, 55 and suggest increased expression of PAX6 via disrupted binding of microRNA‐328, a novel mechanism for epilepsy susceptibility.
The association of rs662702 is consistent across North American and European samples providing similar estimates of effect size for CTS (OR 1.92 vs. 2.20). Although homozygosity of the T allele at rs662702 is extremely rare among the Ontario Science Centre controls (0.30%), 1000 Genomes Project Europeans (0.60%) and Human Genome Diversity Project Europeans (0.64%), ~4% of individuals with CTS are homozygous for the T allele at rs662702; a 12‐fold increase in odds over controls after correction for sex and population stratification. All individuals from the RE families that were homozygous for the T allele and underwent EEG analysis during the critical age range during which CTS is detectable displayed CTS, suggesting that TT homozygosity may be a highly penetrant genotype. Five of the six rs662702 TT homozygotes had RE, suggesting therefore that TT homozygosity may also contribute to seizure susceptibility. However, since CTS is necessary but not sufficient for RE, it is likely that other interacting genetic and/or environmental factors contribute to the seizure expression or modify the neurodevelopmental phenotype in RE. Such factors may include reported rare sequence variants in other genes such as GRIN2A 16, 17, 18, 19, 20, 56, 57 and other recurrent or private structural variations, for example, 16p11.2, or other undiscovered variants in cis‐regulatory modules in the ELP4‐PAX6 locus. The investigation of such hypothetical gene–gene interactions (statistical or physical) will require larger scale studies.
The appearance of CTS in related epilepsies of the epilepsy‐aphasia spectrum as well as in autism and attention‐deficit hyperactivity disorder8, 9 raises the intriguing hypothesis that rs662702 might also be a marker for a broader range of neurodevelopmental disorders. This hypothesis should be evaluated in disease‐specific cohorts to determine the neurodevelopmental phenotype associated with rs662702 T allele homozygosity.
Although we did not find evidence for a pleiotropic effect of rs662702 on CTS and speech dyspraxia, this relationship may be indirect through regulatory effects on FOXP2, disruption of which causes severe speech dyspraxia.58 pax6 has been reported as a major regulator of foxp2 expression in zebrafish through direct binding to a highly conserved enhancer (ECR1).59 As such, increased PAX6 expression through miR‐328 regulation could affect the gene expression of FOXP2, and we speculate that alterations in the functional interaction between PAX6 and FOXP2 might contribute to the vocal tract symptoms in the phenotype of RE.
The transcription factor PAX6 is a highly conserved “master regulator” crucial for development of the eye, brain, olfactory system and endocrine pancreas. It is a major determinant of patterning and regionalization in the developing nervous system, as well as regulating cell fate and proliferation,60 and is expressed in the developing mouse telencephalon only dorsally. PAX6 displays complex spatiotemporal and quantitative expression patterns determined by a large array of posttranscriptional and cis‐regulatory control elements, some of which are known to be located upstream, within introns, and some of which are known to be sited in the highly conserved downstream regulatory region residing within ELP4 introns.61 The functional effects of rs662702 TT in CTS are more consistent with a relative spatio‐temporal alteration in gene function rather than the total loss of function usually associated with hemizygous mutations and classic PAX6 ocular or brain malformations.62
The rs662702 T allele increases PAX6 expression in vitro55 and the possible relevance of this for RE is suggested by experimental overexpression in vivo. Manuel and colleagues63 used a human multicopy transgene to show that overexpression causes cell‐autonomous defects of late cortical progenitor proliferation in the fetal mouse brain.63 Specifically, overexpression resulted in abnormalities of cortical thickness and layering in rostral and central regions,63 in striking similarity to recent findings obtained through longitudinal magnetic resonance imaging (MRI) structural studies in RE,64 where RE patients show areas of reduced frontal, temporal, and occipital cortical thickness. In silico analysis of gene ontology and phenologs across human, mouse, chicken, zebrafish, worm, yeast, and plant ranks the probability that PAX6 is associated with epilepsy as one of its top predictions.65
We propose that the principal mechanism by which the rs662702 T allele leads to pathogenicity is through increased PAX6 expression by reduction in the binding affinity of miR‐328,54 disrupting PAX6 autoregulation. MicroRNAs are noncoding single‐stranded RNA molecules that generally lead to mRNA degradation or reduce translation by binding to complementary mRNA at the 3′ UTR. The anatomical (vocal tract) and temporal (mid childhood) specificity of the seizures in RE tantalisingly supports an etiologic role for microRNAs, which are known to be key influences in the timing and tissue specificity of late developmental transitions.66 This does not exclude a role for the transcription factors MEIS2 and TGIF1, which bind in the same region and may be involved in the coregulation of PAX6 during brain development.67 MicroRNA binding has not been reported as a pathological mechanism in epilepsy previously, although miR‐134 was found to be upregulated in refractory temporal lobe epilepsy tissue and in a mouse model.68 This novel finding raises the possibilities: (1) that other regulatory mechanisms in noncoding regions of ELP4‐PAX6 may explain remaining risk for CTS in individuals lacking the TT genotype; that (2) miR‐328 is implicated in epilepsies and neurodevelopmental disorders related through CTS; and (3) that microRNAs may play a role in other common epilepsies of complex genetic inheritance.
Author Contributions
NP, DKP, and LJS researched data; contributed to discussion; and wrote, reviewed, and edited the manuscript. MDW researched data; reviewed and edited the manuscript. LA, JC, EW, SA, RHC, MK, DM, CO, JTa, JTr, TC, CIA, SLK, DEM, PM, SMW, PA, and RS made substantial contribution to acquisition of the data; and reviewed, and edited the manuscript. LJS and DKP are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of Interest
R.S. is a consultant to Highland Therapeutics, BNAS, Lilly Corporation, and Purdue Pharma. D.K.P. is a scientific advisor to Amplexa Genetics. L.A. is a contractor for Eli Lilly and Company. The remaining authors declare no conflicts of interest.
Supporting information
Figure S1. Principal component analysis (PCA) using KING31 of the first 10 principal components. One hundred and fifty‐two individuals with CTS, 1000 OSC controls and Europeans from the 1000 Genomes Project and the Human Genome Diversity Project reference panels were included in the PCA while adjusting for relatedness. CTS and OSC individuals who are carriers of the T allele for SNP rs662702 are marked with a blue triangle and those who are homozygous are marked with a red cross.
Table S1. Genotyped SNPs in the ELP4 region (chr11: 30,862,638–31,815,896) tested for association in 152 CTS cases and 1000 Ontario Science Centre population controls. Only one genotyped SNP, rs662702, was regionally significant (P < 3.06 × 10−3). This SNP falls in the 3′‐UTR region of the PAX6 gene. CHR, chromosome; BP, base‐pair position; SNP, single‐nucleotide polymorphism; OR, odds ratio; LogP, −log10(P‐value); MAF, minor allele frequency; 1K, 1000 Genomes Project (phase 3 version 5); MAF 1K EUR, MAF in Europeans of the 1K project.
Acknowledgments
We acknowledge the contributions of the Ontario Science Centre, Toronto, Ontario. We thank members of the IRELAND Study consortium including those who referred patients to the study: Huntley Hardison, Linda Leary, Edward Novotny, Frances Rhoads, Maria Younes, Kum Gomez, Elaine Hughes, John Jackman, David Scott, Jan Stanek, and Rajesh Gupta. We acknowledge Sushma Goyal, Zaloa Agirre‐Arrizubieta and the late Lewis Kull for reporting EEGs. We thank Veronica van Heyningen for reviewing and providing comments on early versions of this manuscript. Finally, thanks to the many families across the world who contributed to the study. The Canadian Institutes of Health Research (201503MOP‐342469 to LJS and DKP; and MOP‐106573 and MOP‐93696 to JC, RS and PA); Ontario Ministry of Research and Innovation Early Researcher's Award (LJS); Waterloo Foundation (DKP); European Union Marie Curie International Reintegration Award of the Seventh Framework Programme (DKP); European Union Grant agreement 602531: “Strategies for Innovative Research to Improve Diagnosis, Prevention and Treatment in Children with Difficult to Treat Epilepsy (DESIRE)” of the Seventh Framework Programme (DKP); Charles Sykes Epilepsy Research Trust (DKP); Epilepsy Research UK (DKP); NIHR Specialist Biomedical Research Centre for Mental Health of South London and Maudsley NHS Foundation Trust (DKP); American Epilepsy Society, the Epilepsy Foundation, Anna and Jim Fantaci, Fight Against Childhood Epilepsy and Seizures (faces), Neurotherapy Ventures Charitable Research Fund (DKP); Charles L Shor Foundation for Epilepsy Research Inc. (DKP); People Against Childhood Epilepsy (PACE) (DKP); Ali Paris Fund (DKP); National Institutes of Health grants NS047530 (DKP); SickKids Foundation, Canada Research Chair and NSERC (436194‐2013) (MDW); and Eli Lilly LIFA Fellowship (LA).
References
- 1. Astradsson A, Olafsson E, Ludvigsson P, et al. Rolandic epilepsy: an incidence study in Iceland. Epilepsia 1998;39:884–886. [DOI] [PubMed] [Google Scholar]
- 2. Sidenvall R, Forsgren L, Heijbel J. Prevalence and characteristics of epilepsy in children in northern Sweden. Seizure 1996;5:139–146. [DOI] [PubMed] [Google Scholar]
- 3. Pal DK, Li W, Clarke T, et al. Pleiotropic effects of the 11p13 locus on developmental verbal dyspraxia and EEG centrotemporal sharp waves. Genes Brain Behav 2010;9:1004–1012. [DOI] [PubMed] [Google Scholar]
- 4. Vega YH, Smith A, Cockerill H, et al. Risk factors for reading disability in families with rolandic epilepsy. Epilepsy Behav 2015;53:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guerrini R, Pellacani S. Benign childhood focal epilepsies. Epilepsia 2012;53(Suppl 4):9–18. [DOI] [PubMed] [Google Scholar]
- 6. Fejerman N, Caraballo R, Tenembaum SN. Atypical evolutions of benign localization‐related epilepsies in children: are they predictable? Epilepsia 2000;41:380–390. [DOI] [PubMed] [Google Scholar]
- 7. Eeg‐Olofsson O, Petersen I, Sellden U. The development of the electroencephalogram in normal children from the age of 1 through 15 years. Paroxysmal activity. Neuropadiatrie 1971;2:375–404. [DOI] [PubMed] [Google Scholar]
- 8. Ballaban‐Gil K, Tuchman R. Epilepsy and epileptiform EEG: association with autism and language disorders. Ment Retard Dev Disabil Res Rev 2000;6:300–308. [DOI] [PubMed] [Google Scholar]
- 9. Holtmann M, Becker K, Kentner‐Figura B, Schmidt MH. Increased frequency of rolandic spikes in ADHD children. Epilepsia 2003;44:1241–1244. [DOI] [PubMed] [Google Scholar]
- 10. Vears DF, Tsai MH, Sadleir LG, et al. Clinical genetic studies in benign childhood epilepsy with centrotemporal spikes. Epilepsia 2012;53:319–324. [DOI] [PubMed] [Google Scholar]
- 11. Vadlamudi L, Milne RL, Lawrence K, et al. Genetics of epilepsy: the testimony of twins in the molecular era. Neurology 2014;83:1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kugler SL, Bali B, Lieberman P, et al. An autosomal dominant genetically heterogeneous variant of rolandic epilepsy and speech disorder. Epilepsia 2008;49:1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheffer IE, Jones L, Pozzebon M, et al. Autosomal dominant rolandic epilepsy and speech dyspraxia: a new syndrome with anticipation. Ann Neurol 1995;38:633–642. [DOI] [PubMed] [Google Scholar]
- 14. Guerrini R, Bonanni P, Nardocci N, et al. Autosomal recessive rolandic epilepsy with paroxysmal exercise‐induced dystonia and writer's cramp: delineation of the syndrome and gene mapping to chromosome 16p12‐11.2. Ann Neurol 1999;45:344–352. [DOI] [PubMed] [Google Scholar]
- 15. Epilepsy Precision Medicine Consortium . A roadmap for precision medicine in the epilepsies. Lancet Neurol 2015;14:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lemke JR, Lal D, Reinthaler EM, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 2013;45:1067–1072. [DOI] [PubMed] [Google Scholar]
- 17. Lal D, Reinthaler EM, Altmuller J, et al. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS ONE 2013;8:e73323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lal D, Reinthaler EM, Schubert J, et al. DEPDC5 mutations in genetic focal epilepsies of childhood. Ann Neurol 2014;75:788–792. [DOI] [PubMed] [Google Scholar]
- 19. Neubauer BA, Waldegger S, Heinzinger J, et al. KCNQ2 and KCNQ3 mutations contribute to different idiopathic epilepsy syndromes. Neurology 2008;71:177–183. [DOI] [PubMed] [Google Scholar]
- 20. Reinthaler EM, Dejanovic B, Lal D, et al. Rare variants in gamma‐aminobutyric acid type A receptor genes in rolandic epilepsy and related syndromes. Ann Neurol 2015;77:972–986. [DOI] [PubMed] [Google Scholar]
- 21. Reinthaler EM, Lal D, Lebon S, et al. 16p11.2 600 kb Duplications confer risk for typical and atypical Rolandic epilepsy. Hum Mol Genet 2014;23:6069–6080. [DOI] [PubMed] [Google Scholar]
- 22. Bali B, Kull LL, Strug LJ, et al. Autosomal dominant inheritance of centrotemporal sharp waves in rolandic epilepsy families. Epilepsia 2007;48:2266–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neubauer BA, Fiedler B, Himmelein B, et al. Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology 1998;51:1608–1612. [DOI] [PubMed] [Google Scholar]
- 24. Strug LJ, Clarke T, Chiang T, et al. Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur J Hum Genet 2009;17:1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reinthaler EM, Lal D, Jurkowski W, et al. Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 2014;55:e89–e93. [DOI] [PubMed] [Google Scholar]
- 26. Derkach A, Chiang T, Gong J, et al. Association analysis using next‐generation sequence data from publicly available control groups: the robust variance score statistic. Bioinformatics 2014;30:2179–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pal DK. Epilepsy and neurodevelopmental disorders of language. Curr Opin Neurol 2011;24:126–131. [DOI] [PubMed] [Google Scholar]
- 28. Commission on Classification and Terminology of the International League Against Epilepsy . Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389–399. [DOI] [PubMed] [Google Scholar]
- 29. Boxerman JL, Hawash K, Bali B, et al. Is Rolandic epilepsy associated with abnormal findings on cranial MRI? Epilepsy Res 2007;75:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clarke T, Strug LJ, Murphy PL, et al. High risk of reading disability and speech sound disorder in rolandic epilepsy families: case‐control study. Epilepsia 2007;48:2258–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu X, Li S, Cooper RS, Elston RC. A unified association analysis approach for family and unrelated samples correcting for stratification. Am J Hum Genet 2008;82:352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crosbie J, Arnold P, Paterson A, et al. Response inhibition and ADHD traits: correlates and heritability in a community sample. J Abnorm Child Psychol 2013;41:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 35. Sinnwell JP, Therneau TM, Schaid DJ. The kinship2 R package for pedigree data. Hum Hered 2014;78:91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. International HapMap Consortium ; Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Genomes Project Consortium ; Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cann HM, de Toma C, Cazes L, et al. A human genome diversity cell line panel. Science 2002;296:261–262. [DOI] [PubMed] [Google Scholar]
- 39. Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet 2012;131:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hojsgaard S. The R Package geepack for Generalized Estimating Equations. J Stat Softw 2006;15:1–11. [Google Scholar]
- 41. Gordon M, Lumley T. forestplot: advanced forest plot using ‘grid’ graphics. Vienna, Austria: R Foundation for Statistical Computing, 2015. [Google Scholar]
- 42. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing‐data inference for whole‐genome association studies by use of localized haplotype clustering. Am J Hum Genet 2007;81:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacDonald JR, Ziman R, Yuen RK, et al. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42(Database issue): D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roadmap Epigenomics Consortium ; Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Encode Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Griffon A, Barbier Q, Dalino J, et al. Integrative analysis of public ChIP‐seq experiments reveals a complex multi‐cell regulatory landscape. Nucleic Acids Res 2015;43:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40(Database issue):D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mathelier A, Fornes O, Arenillas DJ, et al. JASPAR 2016: a major expansion and update of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 2015;44:D110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kertesz M, Iovino N, Unnerstall U, et al. The role of site accessibility in microRNA target recognition. Nat Genet 2007;39:1278–1284. [DOI] [PubMed] [Google Scholar]
- 51. Coelho DJ, Sims DJ, Ruegg PJ, et al. Cell type‐specific and sexually dimorphic expression of transcription factor AP‐2 in the adult mouse brain. Neuroscience 2005;134:907–919. [DOI] [PubMed] [Google Scholar]
- 52. Ernst J, Kellis M. Large‐scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 2015;33:364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang Y, Hwang CK, D'Souza UM, et al. Three‐amino acid extension loop homeodomain proteins Meis2 and TGIF differentially regulate transcription. J Biol Chem 2000;275:20734–20741. [DOI] [PubMed] [Google Scholar]
- 54. Chen KC, Hsi E, Hu CY, et al. MicroRNA‐328 may influence myopia development by mediating the PAX6 gene. Invest Ophthalmol Vis Sci 2012;53:2732–2739. [DOI] [PubMed] [Google Scholar]
- 55. Liang CL, Hsi E, Chen KC, et al. A functional polymorphism at 3′UTR of the PAX6 gene may confer risk for extreme myopia in the Chinese. Invest Ophthalmol Vis Sci 2011;52:3500–3505. [DOI] [PubMed] [Google Scholar]
- 56. Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy‐aphasia spectrum disorders. Nat Genet 2013;45:1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 2013;45:1061–1066. [DOI] [PubMed] [Google Scholar]
- 58. Lai CS, Fisher SE, Hurst JA, et al. A forkhead‐domain gene is mutated in a severe speech and language disorder. Nature 2001;413:519–523. [DOI] [PubMed] [Google Scholar]
- 59. Coutinho P, Pavlou S, Bhatia S, et al. Discovery and assessment of conserved Pax6 target genes and enhancers. Genome Res 2011;21:1349–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stoykova A, Gotz M, Gruss P, Price J. Pax6‐dependent regulation of adhesive patterning, R‐cadherin expression and boundary formation in developing forebrain. Development 1997;124:3765–3777. [DOI] [PubMed] [Google Scholar]
- 61. McBride DJ, Buckle A, van Heyningen V, Kleinjan DA. DNaseI hypersensitivity and ultraconservation reveal novel, interdependent long‐range enhancers at the complex Pax6 cis‐regulatory region. PLoS ONE 2011;6:e28616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sisodiya SM, Free SL, Williamson KA, et al. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet 2001;28:214–216. [DOI] [PubMed] [Google Scholar]
- 63. Manuel M, Georgala PA, Carr CB, et al. Controlled overexpression of Pax6 in vivo negatively autoregulates the Pax6 locus, causing cell‐autonomous defects of late cortical progenitor proliferation with little effect on cortical arealization. Development 2007;134:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garcia‐Ramos C, Jackson DC, Lin JJ, et al. Cognition and brain development in children with benign epilepsy with centrotemporal spikes. Epilepsia 2015;56:1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Woods JO, Singh‐Blom UM, Laurent JM, et al. Prediction of gene‐phenotype associations in humans, mice, and plants using phenologs. BMC Bioinformatics 2013;14:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let‐7 heterochronic regulatory RNA. Nature 2000;408:86–89. [DOI] [PubMed] [Google Scholar]
- 67. Agoston Z, Heine P, Brill MS, et al. Meis2 is a Pax6 co‐factor in neurogenesis and dopaminergic periglomerular fate specification in the adult olfactory bulb. Development 2014;141:28–38. [DOI] [PubMed] [Google Scholar]
- 68. Jimenez‐Mateos EM, Engel T, Merino‐Serrais P, et al. Silencing microRNA‐134 produces neuroprotective and prolonged seizure‐suppressive effects. Nat Med 2012;18:1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Blanchette M, Kent WJ, Riemer C, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 2004;14:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Principal component analysis (PCA) using KING31 of the first 10 principal components. One hundred and fifty‐two individuals with CTS, 1000 OSC controls and Europeans from the 1000 Genomes Project and the Human Genome Diversity Project reference panels were included in the PCA while adjusting for relatedness. CTS and OSC individuals who are carriers of the T allele for SNP rs662702 are marked with a blue triangle and those who are homozygous are marked with a red cross.
Table S1. Genotyped SNPs in the ELP4 region (chr11: 30,862,638–31,815,896) tested for association in 152 CTS cases and 1000 Ontario Science Centre population controls. Only one genotyped SNP, rs662702, was regionally significant (P < 3.06 × 10−3). This SNP falls in the 3′‐UTR region of the PAX6 gene. CHR, chromosome; BP, base‐pair position; SNP, single‐nucleotide polymorphism; OR, odds ratio; LogP, −log10(P‐value); MAF, minor allele frequency; 1K, 1000 Genomes Project (phase 3 version 5); MAF 1K EUR, MAF in Europeans of the 1K project.