

Abstract
Treatment of Guillain‐Barré syndrome with a standard course of high‐dose intravenous immunoglobulin (IVIg) results in a variable clinical recovery which is associated with changes in serum IgG levels after treatment. The neonatal Fc‐receptor protects IgG from degradation, and a genetic polymorphism in its promoter region that influences the expression of Fc‐receptor, may in part explain the variation in IgG levels and outcome. This polymorphism was determined by polymerase chain reaction in a cohort of 257 patients with Guillain‐Barré syndrome treated with IVIg. We could not demonstrate a relation between this polymorphism, the pharmacokinetics of IVIg, or the clinical course and outcome.
Introduction
Intravenous immunoglobulins (IVIg) is the first choice treatment for the Guillain‐Barré syndrome (GBS).1 Patients with GBS are usually treated with a 5‐day standard regimen of in total 2 g/kg bodyweight of IVIg, independent of disease severity.1 A previous study showed that the serum IgG levels reached after 2 weeks vary largely between patients, and that a relatively low IgG increase is related to poor recovery.2 At present it is unknown which mechanism determines this variation in the pharmacokinetics of IVIg. Identification of an underlying genetic factor(s) influencing IgG's kinetics would be a crucial step toward individualized dosing.
The neonatal Fc receptor (FcRn) protects IgG from degradation by binding and recycling IgG back into the circulation.3, 4, 5 When saturated by supraphysiological levels of IgG, the excess IgG will be degraded instead of recycled.3 The expression level of this receptor is influenced by a polymorphism in the promoter region of the gene‐encoding FcRn.6 A variable number of tandem repeats (VNTR) in the promoter of the FcRn alpha‐chain gene, ranging from VNTR1‐5, results in different expression levels of FcRn mRNA and protein.7 Previous studies in immunodeficient patients showed that this polymorphism influences the pharmacokinetics of IVIg.8 Patients homozygous for the VNTR3 allele (VNTR 3/3, relatively high FcRn expression) had higher IgG trough levels and higher IVIg efficiency than VNTR 3/2 heterozygous patients (relatively low FcRn expression).8
We hypothesized that the VNTR in the FcRn promoter region influences the pharmacokinetics of IVIg in patients with GBS, thereby explaining the variation in IgG levels found previously and possibly the poor clinical outcome associated with a low IgG increase. This hypothesis was tested in a large cohort of patients for whom standardized serial serum samples and detailed clinical follow‐up data were available.
Materials and Methods
Patients
DNA and clinical data were available from 257 patients with GBS, previously included in clinical trials coordinated by our center that investigated the therapeutic effect of IVIg.9, 10, 11 All gave informed consent after prior approval by the appropriate Institutional Review Board, additional informed consent was obtained for genetic studies.12 All patients met the diagnostic criteria for GBS,13 were included within 2 weeks of onset of weakness, and were treated with a standard regimen of IVIg of 2 g per kg bodyweight administered over 5 days. Additional serum samples obtained pretreatment, at 2 and 4 weeks, and at 3 and 6 months after start of IVIg were available from 98 (38%) patients to determine IgG concentrations. These 98 patients did not differ from the remaining patients with respect to demographic characteristics, treatment, disease severity at entry, or clinical course and outcome. The delta‐IgG level (∆IgG) was defined as the increase in IgG level after 2 weeks compared to the pretreatment sample.2 Predefined measures for clinical course and outcome were used, including the GBS disability score (ranging 0–6, healthy death), the need for mechanical ventilation, and the Medical Research Council (MRC) sum score (ranging 0–60 [total paralysis – normal strength]) before treatment, at nadir and at 6 months.13, 14 All samples were stored at −80°C until use.
Data collection
Genomic DNA was isolated from EDTA anticoagulated peripheral blood samples, using invicorb® MaxiBlood kit (Invitek, Berlin, Germany) according to the instructions of the manufacturer. The VNTR genotype was determined using the polymerase chain reaction (PCR) on GeneAmp PCR System 2700 according to Sachs et al., with minor modifications.7 Briefly, 80 ng of DNA was added to a mastermix of 2 μL MgCl (25 mmol/L), 1.0 μL (12.5 pmol/μL) of both forward (5′‐GGCTGGGGGTCTCGACACT‐ 3′) and reverse (5′‐AGTCTGGACCGAGCCCGC‐3′) primer (flanking the VNTR sequence), 0.5 μL deoxynucleotide triphosphate (dNTP) (20 nmol/L), and 0.2 μL Taq polymerase, in a total volume of 20 μL. Analysis of PCR products was performed using 2.0% agarose gels. We performed fluorescent fragment length analysis (GeneScan®) according to the manufacturer protocols using a 6FAM labeled reverse primer (5′‐(6FAM)GCGGGCTCGGTCCAGACT‐3′), in a select number of patients (n = 25) to validate the PCR and gel electrophoresis technique.
Statistical analysis
Statistical analysis was performed using SPSS (IBM Corp., Version 21.0. Armonk, NY, USA) and Graphpad Prism software (GraphPad Software, Version 5.0, La Jolla, CA, USA). The effect of VNTR genotype on IgG serum levels was tested with an independent samples t‐test. Clinical outcome scores between VNTR genotypes were compared using χ2‐test for categorical data and independent samples t‐test for numerical data. Mann–Whitney U test was used for nonnormal distributed numerical data. When necessary we corrected for confounding factors using ANCOVA, otherwise ANOVA was used. Survival analysis was performed by Kaplan–Meier analysis (log rank). Statistical significance was defined as a two‐sided P < 0.05.
Results
PCR and gel electrophoresis analysis of DNA from 257 GBS patients identified five different FcRn VNTR alleles, resulting in 7 distinctive FcRn VNTR genotypes (Fig. 1). The VNTR allele frequencies were 0.2% (VNTR1), 9.1% (VNTR2), 89.5% (VNTR3), 0.8% (VNTR4), and 0.4% (VNTR5). The distributions of allele frequencies followed the Hardy–Weinberg equilibrium. The two predominant genotypes were VNTR 3/3 found in 207 patients (80.5%) and VNTR 3/2 in 40 patients (15.6%). Other rarer genotypes were found in the remaining 10 patients (3.8%), for example. VNTR 5/3, 4/3, 4/2, 3/1, and 2/2 (Fig. 1). The VNTR genotypes found by PCR were confirmed by fluorescent fragment length analysis (Fig. S1).
Figure 1.
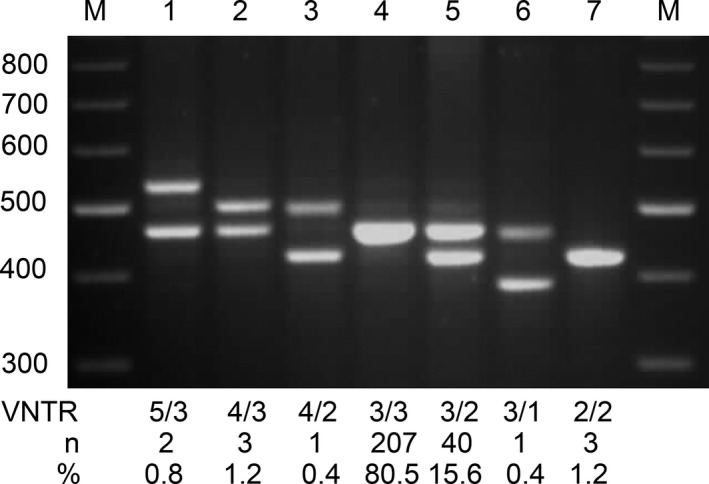
Representative examples of the different variable number of tandem repeats (VNTR) genotypes identified using polymerase chain reaction (PCR) and gel electrophoresis. PCR revealed 5 distinctive VNTR alleles, based on a repetitive genetic motif of 37 base pairs in size, and 7 different genotypes (lane 1–7). The derived VNTR sizes were as follows: VNTR 1 (389 bp); VNTR 2 (426 bp); VNTR 3 (463 bp); VNTR 4 (500 bp); and VNTR 5 (537 bp). The two flanking lanes are PCR molecular weight ladders (M) with 100 bp difference in size. For each genotype, the number and percentage of patients was indicated.
Patients with the VNTR 3/3 genotype did not differ from patients with the VNTR 3/2 genotype regarding demographics or signs of preceding infection, with the exception of a higher percentage of males in the VNTR 3/2 group (Table 1). In the subgroup where IgG levels were available (n = 98), 82 patients (84%) had VNTR 3/3, 15 patients (16%) had VNTR 3/2, and 1 patient (1%) had VNTR 4/3. The mean dose of administered IVIg did not differ between the genotype groups: 135 (SD = 57 g) in the VNTR 3/3 subgroup and 146 (SD = 51 g) in the VNTR 3/2 subgroup. Serum IgG level before and at 2 and 4 weeks after start of IVIg and the ∆IgG did not differ between the VNTR 3/3 and VNTR 3/2 subgroups (Fig 2). However, serum IgG levels at 3 and 6 months after IVIg treatment were higher in patients with VNTR 3/2 than in the VNTR 3/3 homozygotes (P = 0.004, Fig 2).
Table 1.
Baseline characteristics, clinical severity, and clinical outcome of 247 patients based on variable number of tandem repeats (VNTR) in the promoter of the FcRn alpha‐chain gene
| Baseline characteristics | Genotype | P‐value | |
|---|---|---|---|
| VNTR 3/3 (n = 207) | VNTR 3/2 (n = 40) | ||
| Age (years) | 49.5 (16.1) | 52.0 (17.0) | ns |
| Males | 108 (52.2%) | 28 (70.0%) | 0.038 |
| Body weight | 74.4 (14.5) (n = 112) | 78.4 (12.0) (n = 22) | ns |
| Preceding diarrhea | 57 (28%) | 12 (31%) | ns |
| Preceding upper respiratory tract infection | 78 (39%) (n = 201) | 13 (33%) (n = 39) | ns |
| Clinical severity at entry | |||
| Mean GBS disability score at entry | 3.6 (0.8) (n = 206) | 3.4 (0.9) (n = 39) | ns |
| Mean MRC sum score at entry | 44.4 (9.9) (n = 202) | 43.5 (12.5) (n = 38) | ns |
| Clinical severity during follow‐up | |||
| Mean GBS disability score at nadir | 3.9 (0.9) | 3.8 (1.0) | ns |
| Mean MRC sum score at nadir | 38.0 (15.4) (n = 206) | 37.6 (17.4) (n = 40) | ns |
| Mechanical ventilation | 41 (20%) | 11 (28%) | ns |
| GBS disability score at 6 months | 1.4 (1.1) (n = 199) | 1.5 (1.1) (n = 40) | ns |
Data are presented as mean (sd) or as number (percentage).
Ns, not significant; IVIg, intravenous immunoglobulin
Figure 2.
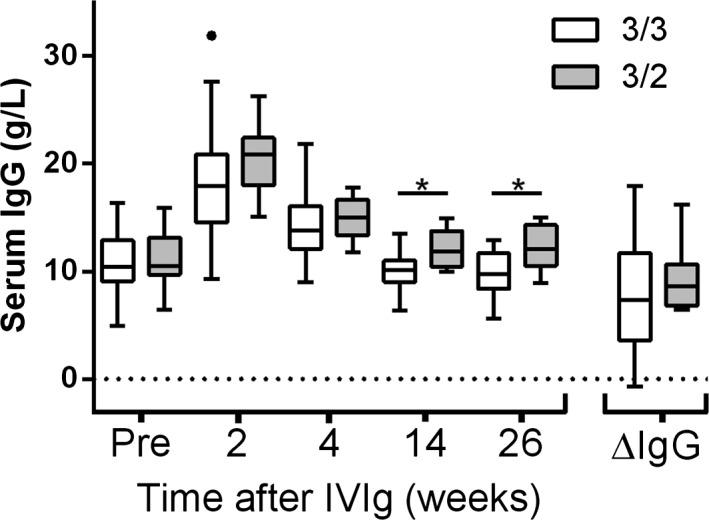
Serum Immunoglobulin G (IgG) levels before treatment, after treatment at standardized time points (2, 4, 14, and 26 weeks) and the difference between the IgG level after 2 weeks and pr‐treatment IgG level (∆IgG). Groups are based on FcRn promoter alpha‐chain genotype, variable number of tandem repeats (VNTR). Data presented as mean and whiskers according to Tukey. * denotes a significant difference.
The group of 247 patients (VNTR 3/3 and 3/2) was used to determine whether there was a relation between the VNTR genotype and the clinical course and outcome. The GBS disability scores and MRC sum scores did not differ between patients with various VNTR genotypes at entry, nadir, or at the end of follow‐up (Table 1), or at important clinical assessment points (2 and 4 weeks, Table S1). In addition, there was no association between VNTR genotype and the time to regain the ability to walk independently or to improve 1 point on the GBS disability scale (Fig. S2).
Discussion
IVIg is used in a broad spectrum of disorders, either for replacement or immune modulation, and the pharmacokinetics of IVIg are important to facilitate personalized medicine. Metabolism of IgG is governed by FcRn and a polymorphism in the promoter region influences the expression of this receptor.7 This study assessed whether a particular FcRn polymorphism in GBS patients treated with IVIg has an effect on the degree of increase in the IgG level; and whether it influences the course of disease in GBS. We found no association between this functional genetic polymorphism in the promoter region of the FcRn gene and the pharmacokinetics of IVIg, or with the clinical outcome after IVIg in patients with GBS.
The current knowledge of the pharmacokinetics of IVIg is largely based on studies in patients with immunodeficiencies receiving replacement therapy.15 Pretreatment IgG levels (ranging from <1.5 g/L to <6 g/L) are important in these conditions for subsequent IVIg dosing.15, 16 However, this cannot be extrapolated to GBS patients with normal serum IgG levels. A second difference is the treatment regimen, as most patients with immune deficiencies receive low dosages at regular intervals (~0.4 g/kg/month).16 In contrast, high‐dose IVIg in GBS, superimposed on the normal endogenous IgG serum level, increases IgG by up to five times the normal serum level.17 The concentration‐dependent clearance of IgG and the role of FcRn herein is probably very different. A recent small study in multifocal motor neuropathy (18 patients were genotyped) treated with IVIg (same regimen as in GBS) found similar results to this work: no association between the polymorphism in the FcRn gene and the serum IgG levels.18 However, we cannot completely rule out a small effect of FcRn polymorphism on IVIg pharmacokinetics. The 2‐week interval between the pre‐ and posttreatment IgG‐level measurement could mask more subtle differences, as found previously for immune‐deficient patients requiring replacement therapy.8, 16 Whether this possible small effect on IgG levels after treatment has any impact on clinical outcome is, however, unlikely, as we found no association between the FcRn polymorphism and clinical outcome in GBS. A study in lupus nephritis, a disease in which autoantibodies play a dominant role in the pathogenesis, also reported no association of the FcRn polymorphism with outcome.19
This study was conducted in a representative cohort of patients with GBS who were treated with a standard course of IVIg. The distribution of the frequency of the various VNTR genotypes in the group of GBS patients was similar to that previously reported in healthy controls, indicating that this genetic polymorphism does not generally predispose to GBS.7, 8 Although clinical data were available for the full cohort, IgG data were limited, and the apparent differences found at 3 and 6 months may very well be due to this limited group size.
Other factors influencing the turnover of antibodies or affecting FcRn expression could play a role: for instance, serum proteases degrading the IgG molecules or cytokine‐induced regulation of FcRn expression, especially under the inflammatory conditions in GBS, could overrule the underlying genotype;20, 21, 22 providing an explanation as to why the FcRn promoter gene polymorphism influences IVIg pharmacokinetics in immunodeficient patients on replacement therapy, but not patients with this inflammatory disease of the peripheral nerves.
Author contribution
B.C.J. and W.J.R.F. designed the study. A.E.G.H., A.P.T‐G., W.vR., and W.J.R.F. performed the experiments. W.J.R.F. and A.E.G.H. analyzed the data. W.J.R.F., A.E.G.H., R.H.G.H., P.A.vD, and B.J.C. drafted the manuscript. B.C.J. supervised this study. All authors read and approved the final version of the manuscript.
Conflicts of Interest
Dr. Jacobs reports grants from Prinses Beatrix Spierfonds, during the conduct of the study; grants from Netherlands Organization for Health Research and Development, Erasmus Medical Center, Prinses Beatrix Spierfonds, GBS‐CIDP Foundation International, Baxter Biopharmaceutics, CSL‐Behring, Grifols, outside the submitted work. Dr. van Doorn reports grants from Sanguin blood supply, Baxalta, Grifols, Prinses Beatrix Spierfonds, Janivo Foundation, outside the submitted work. Dr. Huizinga reports grants from the Dutch Prinses Beatrix Spierfonds, during the conduct of the study; grants from the Dutch Prinses Beatrix Spierfonds, grants from the GBS‐CIDP Foundation International, outside the submitted work. Dr. Fokkink reports grants from Prinses Beatrix Spierfonds, during the conduct of the study.
Supporting information
Figure S1. Fragment length analysis by Genescan of VNTR of the FcRn promoter region. Confirming the genotypes found by PCR gel electrophoresis: VNTR 5/3, VNTR 4/3, VNTR 4/2, VNTR 3/3, VNTR 3/2, VNTR 3/1, and VNTR 2/2. Red lines represent the size standard reference lines, used to size the PCR products and create the x‐axis. Blue peaks indicate VNTR PCR product and the peak position on the x‐axis indicates base pair size of product, from which the according VNTR genotype can be derived.
Figure S2. Cumulative proportion of patients who improve 1 point at the GBS disability scale (F‐score) or regaining of the ability to walk unaided during follow‐up.
Table S1. Clinical severity as scored at the end of the 1st, 2nd, and 4th week of 141 GBS patients based on variable number of tandem repeats (VNTR) in the promoter of the FcRn alpha‐chain gene. Data are presented as mean (sd) or as number (percentage). ns= not significant; IVIg= intravenous immunoglobulin
Acknowledgment
This work has been supported by a grant from the Prinses Beatrix Spierfonds (grant W.OR11‐27).
References
- 1. van den Berg B, Walgaard C, Drenthen J, et al. Guillain‐Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol 2014;10:469–482. [DOI] [PubMed] [Google Scholar]
- 2. Kuitwaard K, de Gelder J, Tio‐Gillen AP, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain‐Barre syndrome. Ann Neurol 2009;66:597–603. [DOI] [PubMed] [Google Scholar]
- 3. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007;7:715–725. [DOI] [PubMed] [Google Scholar]
- 4. Tesar DB, Bjorkman PJ. An intracellular traffic jam: Fc receptor‐mediated transport of immunoglobulin G. Curr Opin Struct Biol 2010;20:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rath T, Baker K, Pyzik M, Blumberg RS. Regulation of immune responses by the neonatal fc receptor and its therapeutic implications. Front Immunol 2014; 5:664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mikulska JE, Pablo L, Canel J, Simister NE. Cloning and analysis of the gene encoding the human neonatal Fc receptor. Eur J Immunogenet 2000;27:231–240. [DOI] [PubMed] [Google Scholar]
- 7. Sachs UJ, Socher I, Braeunlich CG, et al. A variable number of tandem repeats polymorphism influences the transcriptional activity of the neonatal Fc receptor alpha‐chain promoter. Immunology 2006;119:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gouilleux‐Gruart V, Chapel H, Chevret S, et al. Efficiency of immunoglobulin G replacement therapy in common variable immunodeficiency: correlations with clinical phenotype and polymorphism of the neonatal Fc receptor. Clin Exp Immunol 2013;171:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Meche FG. Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain‐Barre syndrome. Dutch Guillain‐Barre Study Group. N Engl J Med 1992;326:1123–1129. [DOI] [PubMed] [Google Scholar]
- 10. van Koningsveld R, Schmitz PI, Meche FG, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain‐Barre syndrome: randomised trial. Lancet 2004;17:192–196. [DOI] [PubMed] [Google Scholar]
- 11. Ruts L, Drenthen J, Jongen JL, et al. Pain in Guillain‐Barre syndrome: a long‐term follow‐up study. Neurology 2010;19:1439–1447. [DOI] [PubMed] [Google Scholar]
- 12. Geleijns K, Jacobs BC, Van Rijs W, et al. Functional polymorphisms in LPS receptors CD14 and TLR4 are not associated with disease susceptibility or Campylobacter jejuni infection in Guillain‐Barre patients. J Neuroimmunol 2004;150(1–2):132–138. [DOI] [PubMed] [Google Scholar]
- 13. Hughes RA, Newsom‐Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet 1978;2:750–753. [DOI] [PubMed] [Google Scholar]
- 14. Kleyweg RP, van der Meche FG, Schmitz PI. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain‐Barre syndrome. Muscle Nerve 1991;14:1103–1109. [DOI] [PubMed] [Google Scholar]
- 15. Bonilla FA. Pharmacokinetics of immunoglobulin administered via intravenous or subcutaneous routes. Immunol Allergy Clin North Am 2008;28:803–819. , ix. [DOI] [PubMed] [Google Scholar]
- 16. Cunningham‐Rundles C. How I treat common variable immune deficiency. Blood 2010;8:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dalakas MC. High‐dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology 1994;44:223–226. [DOI] [PubMed] [Google Scholar]
- 18. Vlam L, Cats EA, Willemse E, et al. Pharmacokinetics of intravenous immunoglobulin in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry 2014;85:1145–1148. [DOI] [PubMed] [Google Scholar]
- 19. Zhou XJ, Yu L, Zhu L, et al. Association between polymorphisms in the FCGRT gene and lupus nephritis in Chinese patients. Clin Exp Rheumatol 2009;27:609–614. [PubMed] [Google Scholar]
- 20. Liu X, Ye L, Christianson GJ, et al. NF‐kappaB signaling regulates functional expression of the MHC class I‐related neonatal Fc receptor for IgG via intronic binding sequences. J Immunol 2007;179:2999–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu X, Ye L, Bai Y, et al. Activation of the JAK/STAT‐1 signaling pathway by IFN‐gamma can down‐regulate functional expression of the MHC class I‐related neonatal Fc receptor for IgG. J Immunol 2008;181:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiao SW, Lencer WI, Blumberg RS. How the controller is controlled ‐ neonatal Fc receptor expression and immunoglobulin G homeostasis. Immunology 2007;120:145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fragment length analysis by Genescan of VNTR of the FcRn promoter region. Confirming the genotypes found by PCR gel electrophoresis: VNTR 5/3, VNTR 4/3, VNTR 4/2, VNTR 3/3, VNTR 3/2, VNTR 3/1, and VNTR 2/2. Red lines represent the size standard reference lines, used to size the PCR products and create the x‐axis. Blue peaks indicate VNTR PCR product and the peak position on the x‐axis indicates base pair size of product, from which the according VNTR genotype can be derived.
Figure S2. Cumulative proportion of patients who improve 1 point at the GBS disability scale (F‐score) or regaining of the ability to walk unaided during follow‐up.
Table S1. Clinical severity as scored at the end of the 1st, 2nd, and 4th week of 141 GBS patients based on variable number of tandem repeats (VNTR) in the promoter of the FcRn alpha‐chain gene. Data are presented as mean (sd) or as number (percentage). ns= not significant; IVIg= intravenous immunoglobulin