

SUMMARY
Recent advances in single-particle cryoelecton microscopy (cryo-EM) are enabling generation of numerous near-atomic-resolution structures for well-ordered protein complexes with sizes ≥ ~200 kDa. Whether cryo-EM methods are equally useful for high-resolution structural analysis of smaller, dynamic protein complexes such as those involved in cellular metabolism remains an important question. Here, we present 3.8 Å resolution cryo-EM structures of isocitrate dehydrogenase (93 kDa) and identify the nature of conformational changes induced by binding of the allosteric small-molecule inhibitor ML309. We also report 2.8-Å- and 1.8-Å-resolution structures of the cancer targets lactate dehydrogenase (145 kDa) and glutamate dehydrogenase (334 kDa), respectively. With these results, two perceived barriers in single-particle cryo-EM are overcome: our tests demonstrated crossing 2 Å resolution and obtaining maps of proteins with sizes < 100 kDa, demonstrating that cryo-EM can be used to investigate a broad spectrum of drug-target interactions and dynamic conformational states.
Graphical Abstract
INTRODUCTION
Cryo-electron microscopy (cryo-EM) is now firmly established as a central tool in the arsenal of structural biology. The ability to obtain near-atomic-resolution structures using cryo-EM was shown initially almost three decades ago in the context of electron crystallographic studies of membrane proteins (Henderson et al., 1990). Continued advances in single-particle cryo-EM over the next two decades enabled resolution analysis of non-crystalline samples with high internal symmetry such as icosahedral and helical viruses (Ge and Zhou, 2011; Settembre et al., 2011; Yu et al., 2008; Zhang et al., 2010). Large and relatively stable complexes such as ribosomes also proved especially amenable to analysis using cryo-EM methods, first at medium resolution (Matadeen et al., 1999; Rawat et al., 2003) and more recently at near-atomic resolution (Amunts et al., 2014; Fischer et al., 2015; Jomaa et al., 2016; Wong et al., 2014). These successes have now been extended to a wide spectrum of protein complexes, including several integral membrane proteins (Bai et al., 2015b; Du et al., 2015; Liao et al., 2013; Matthies et al., 2016). Structures determined by cryo-EM can now reach resolutions as high as 2.2 Å and 2.3 Å, as exemplified by structures of the 465 kDa β-galactosidase (Bartesaghi et al., 2015) and the 540 kDa AAA ATPase p97 (Banerjee et al., 2016). However, all of the near-atomic-resolution structures reported have been of proteins with sizes in the range of ~200 kDa or larger, and an informal opinion in the field is that cryo-EM technology is primarily suited for analysis of relatively stable proteins with sizes >150 kDa (Thompson et al., 2016). The smallest protein for which a cryo-EM structure has been reported using single particle cryo-EM is that of the 135 kDa ABC exporter TmrAB, at ~10 Å resolution (Kim et al., 2015), and the challenges in achieving near-atomic resolution for small proteins, even with sizes as large as ~300 kDa have been noted (Skiniotis and Southworth, 2016; Cheng, 2015; Belnap, 2015). Crystallographic analyses are generally very powerful for proteins in this size range but usually require that flexible regions are either deleted or altered. Given that small, dynamic protein complexes are implicated in numerous cellular processes, there is considerable interest in determining whether cryo-EM methods can be also applicable for structural analysis of this class of proteins under near-native conditions and at near-atomic resolution.
A principal reason why small proteins such as those with sizes <150 kDa have been considered intractable for analysis by cryo-EM is that the errors in alignment of individual projection images become progressively higher as the size of the scattering entity gets smaller (Henderson et al., 2011). In principle, with a perfect detector that displays minimal falloff in detective quantum efficiency (DQE) even at Nyquist frequency (Henderson, 1995; McMullan et al., 2014), it should be possible to achieve accurate alignment of projection images of smaller proteins, but all currently available detectors still show a significant drop in DQE at higher frequencies. The use of phase plates is an option that may help partially alleviate the problem of image contrast (Danev and Baumeister, 2016), but these developments are still at an early stage. One way to compensate for the falloff in DQE at higher spatial frequencies is to collect data at higher magnification. This strategy, however, lowers contrast and makes the alignment of individual frames collected in movie mode of data collection more challenging. Experimental approaches to optimize specimen preparation provide an alternative route to improve image contrast: it can be easier to minimize the background scattering from the ice layer for smaller proteins because the lower the molecular weight, the thinner the ice layer that is required to surround the protein with an aqueous layer.
To further test the limits of what is possible with present-day cryo-EM technology, we have analyzed structures of two small, soluble enzymes implicated in cancer metabolism: the 145 kDa lactate dehydrogenase (LDH B, a tetramer composed of four identical ~36 kDa subunits) and the 93 kDa isocitrate dehydrogenase (IDH1, a dimer composed of two identical ~47 kDa subunits). In both cases, we tested whether structures can be obtained at high enough resolution to localize bound small-molecule ligands and to determine the structures of ligand-bound complexes. We also carried out structural analysis of the conformationally dynamic enzyme glutamate dehydrogenase (GDH). Earlier crystallographic analyses of apo-GDH, a 334 kDa hexameric protein composed of six identical ~56 kDa subunits, have been limited to a resolution of 2.7 Å, possibly because some regions of the protein may display a continuum of conformational states even in the context of a crystal lattice. This is a feature that may well be shared by a large majority of protein complexes involved in signaling and metabolism. While it is expected that image averaging will limit resolutions that can be achieved for the conformationally flexible components, it is interesting to ascertain whether cryo-EM analyses can be used to obtain near-atomic resolution for the ordered regions of these types of protein complexes.
RESULTS
Structural Studies of Lactate Dehydrogenase
Cancer cells catalyze the production of lactate from pyruvate, the final metabolite generated from glucose during glycolysis. In non-malignant cells, pyruvate enters mitochondria to participate in the Krebs cycle and in oxidative phosphorylation. Under anaerobic conditions, such as that found in muscle cells, tetrameric LDH mediates the formation of lactate from pyruvate using NADH as a cofactor. However, in cancer cells, irrespective of oxygen availability, glycolysis followed by production of lactate through LDH is the preferred pathway, enhancing the production of metabolic precursors required for biosynthesis of cellular macromolecules. The recognition in a growing number of cancers of the central role of both LDH A and B isoforms (Fiume et al., 2014; McCleland et al., 2013; Rodriguez et al., 2003) has driven the identification of small-molecule inhibitors, including mercaptocyclohex-2-enone derivatives that bind away from the NAD-binding pocket and are not competitive inhibitors (Dragovich et al., 2014), as well as sulfamoylquinoline benzoic acid derivatives, which compete with NAD binding to LDH (Billiard et al., 2013). To evaluate the potential of cryo-EM to determine structures of a small protein complex such as LDH and localize the binding sites of potential small-molecule inhibitors, we carried out cryo-EM analysis of LDH B in complex with GSK2837808A, a 650 Dalton compound in the quinoline 3-sulfonamide series (Billiard et al., 2013). These compounds potently inhibit activity of LDH A and B, as well as isoforms with mixtures of LDH A and B, making them attractive candidates for use in treatment of solid tumors that rely principally on aerobic glycolysis for survival.
The cryo-EM structure of the LDH B-GSK2837808A complex at an overall resolution of 2.8 Å (Figure S1A; Table 1) enables delineation of the binding site of the inhibitor (Figures 1A–1C). The location of the compound at the periphery of the protein is in excellent agreement with the placement of inhibitors in the two X-ray structures (4QSM and 4QT0 at resolutions of 3.0 Å and 3.2 Å, respectively) that are available for LDH A in complex with quinoline 3-sulfonamides (Figures S1B and S1C). The density for the inhibitor is not adequate to model in a specific conformation, which could be a consequence of high conformational flexibility coupled with its location at the outer edge of the protein with a high degree of solvent exposure. Nevertheless, most regions of the polypeptide display density that enables unambiguous construction of an atomic model for the protein (Figures 1D and 1E), including localization of numerous water molecules in the density map (Figures S1D and 1E). The fact that the overall quality and resolution of the structure is comparable to, if not better than, that obtained from X-ray crystallography, validates the potential of cryo-EM to obtain near-atomic-resolution structures of <150 kDa complexes of this kind, without the need for crystallization.
Table 1.
Summary of Data Collection and Image-Processing Parameters for the Density Maps of LDH, IDH1, and GDH Reported in this Manuscript
| Total number of micrographs | Defocus range (microns) | Starting number of particles | Particles used in final map | Frames used in final map | Bfactor for sharpening (Å2) | Final resolution (Å) | |
|---|---|---|---|---|---|---|---|
| LDH-inh | 1,707 | 0.8–2.2 | 508,402 | 50,865 (10%) | 0–29 | −150 | 2.8 |
| IDH-apo | 1,506 | 0.7–3.0 | 499,184 | 49,936 (10%) | 0–29 | −180 | 3.8 |
| IDH-inh | 820 | 0.7–2.6 | 232,343 | 46,483 (20%) | 0–29 | −180 | 3.8 |
| GDH | 232 | 0.8–2.1 | 45,388 | 21,818 (48%) | 3–9 | −90 | 1.8 |
Figure 1. Cryo-EM Analysis of LDHB Bound to Inhibitor GSK2837808A.

(A) Cryo-EM density map of complex showing density for GSK2837808A (red) and selected residues in the binding pocket.
(B) Ribbon diagram of refined structure of LDHB showing the location of the bound inhibitor (marked by arrows) on the periphery of the tetramer.
(C) Structure of GSK2837808A.
(D and E) Visualization of side-chains in the cryo-EM density map in an α-helical segment (D) and loop region (E) of the polypeptide.
See also Figure S1.
Structural Studies of Isocitrate Dehydrogenase
The conversion of isocitrate to α-ketoglutarate (αKG) is catalyzed by NADP+-bound IDH1. IDH1 proteins with mutations at residue Arg132 are found in cancer cells such as those in glioblastoma. This residue, most often found mutated to a histidine or a cysteine, is critical for the maintenance of the active site (Dang et al., 2009; Yang et al., 2010). The R132H and R132C variants of IDH1 lose affinity for isocitrate; instead, with the cofactor NADPH, these mutants bind αKG and convert it to R(−)-2-hydroxyglutyrate (2HG), an oncometabolite. ML309 is a potent and selective inhibitor of IDH1 R132H/C, decreasing production of 2HG in glioblastoma cells (Davis et al., 2014). Although a number of structures of the R132H mutant exist, structures of IDH Arg132 mutants in complex with the ML309 inhibitor have not yet been determined. Structural analysis of IDH1 by cryo-EM is technically challenging given its small size, low symmetry (C2), and potential flexibility.
In Figure 2A, we present a cryo-EM structure for the R132C mutant of IDH1 that shows the characteristic butterfly fold of the dimer at a nominal resolution of 3.8 Å (Figure S2A). The polypeptide backbone and densities for the larger side chains are resolved in many regions (Figures 2B and 2C), enabling placing of the polypeptide chain into the density map using prior crystal structures of IDH1 as a guide. The quality of the map is variable, with the expected gradient from regions that are in the well-packed interior at higher resolution, to those on the periphery with greater solvent exposure at lower resolution. Comparison of the structure of apo-IDH1 with that of ML309-bound IDH1 enables localization of ML309 in the structure (Figures 2A and 2D). The location of the inhibitor in the cryo-EM structure (Figure 2D) in a solvent-exposed region close to the 2-fold axis and near the catalytic site is similar to that seen for many IDH1-inhibitor complexes that have been amenable to crystallography (Deng et al., 2015; Okoye-Okafor et al., 2015; Wu et al., 2015; Zheng et al., 2013).
Figure 2. Cryo-EM Analysis of IDH1 in the Absence and Presence of the Inhibitor ML309.

(A) Cryo-EM map of the apo-IDH1 (isocitrate dehydrogenase) dimer, colored by subunit.
(B and C) Selected regions of the IDH1 map demonstrating density for side-chains in a α-helical region (B) and a β sheet region (C).
(D) Cryo-EM map of IDH1 in complex with ML309 showing density (red) for the inhibitor (inset) close to the dimer interface.
(E) Superposition of the apo-(gray) and ML309-bound (yellow/blue) IDH1 structures shows the outward movement of the subunits with ML309 binding (density shown in red). The black arrows indicate the direction of the changes in tertiary structure while the yellow and blue arrows show the overall movements at the level of quaternary structure.
See also Figures S2 and S3, and Movie S1.
Despite the modest resolution of the maps, the structures provide new insights into structural aspects of IDH1 and its interaction with the inhibitor. Cryo-EM reconstructions carried out without imposition of 2-fold symmetry resulted in maps at a resolution of ~4.5 Å, similar to those obtained with the use of 2-fold symmetry (Figure S2B), indicating that the structure of the IDH1 dimer in solution is symmetric at least at this resolution. This is a different result than that obtained from crystal structures of IDH1, which are mostly asymmetric with regions in the two protomers exhibiting different conformations (Xu et al., 2004; Yang et al., 2010). In our structures, both polypeptide chains that form the IDH1 dimer appear to be equivalent, suggesting that the apparent asymmetry is a likely consequence of crystal lattice formation and is not an intrinsic property of the dimer, as confirmed by inspection of lattice packing (Figure S3A). Further, comparison of the apo- and inhibitor-bound structures shows that ML309 binding essentially drives a wedge between the two protomers, leading to an outward movement of the peripheral domains (Figure 2E) that is clearly evident even when comparing reconstructions obtained without imposition of symmetry (Movie S1). This structural change includes displacement of a stretch of polypeptides spanning residues 270–281. The density for bound NADPH and the surrounding residues in the pocket is adequate for placement of the cofactor in the pocket (Figure S3B), but the low 3.8 Å resolution of the maps precludes identification of the detailed interactions within the binding pocket. Inspection of the asymmetrically reconstructed density map also enables the unambiguous identification of additional density consistent with the binding of a single molecule of ML309 at the dimer interface (Figure S3C), although its precise orientation cannot be determined. Nevertheless, the location of the catalytic site between the NADPH binding site and the site of bound ML309 potentially explains how ML309 binding can cause a profound change in the catalytic activity of the enzyme. These results show that cryo-EM density maps from small, <100 kDa dynamic proteins can not only help localize binding sites for small molecule inhibitors, but also potentially predict changes in activity that can complement information obtained from crystallographic studies.
Cryo-EM Analysis of Glutamate Dehydrogenase
The smaller size, lower symmetry, and conformational flexibility may all be contributing factors that limit resolution of the IDH1 structures obtained using cryo-EM methods. Intrinsic flexibility of proteins also poses a challenge for determining high-resolution structures even with X-ray crystallography. In many cases, crystal contacts can freeze specific protein conformations, enabling structure determination at resolutions of 2 Å or better, but in other instances, local flexibility can be high enough to limit overall resolution. The cancer target and hexameric enzyme GDH is a good example of this problem. GDH displays closed and open conformations involving large movements of the nucleotide binding domain (NBD), but even when trapped in closed or open conformations, the highest resolutions reported so far from crystallographic studies and cryo-EM analyses of the open conformation are ~2.7 Å (Smith et al., 2002) and ~3.3 Å (Borgnia et al., 2016), respectively. We therefore used GDH to evaluate whether we could further optimize cryo-EM methods to achieve resolutions of ~2 Å or better for the more ordered regions, despite the presence of significant conformational flexibility in other areas of the protein.
Using cryo-EM projection images that were selected for reduced beam-induced drift, we determined a cryo-EM structure for GDH in the open state using <22,000 molecular images (Figures 3, S4, S5; Table 1; Movie S3). Irrespective of whether the maps were divided into one, two, or more classes, the density maps displayed a clear difference between well-ordered central regions covering the central half of the complex and “fuzzy” outer regions for the rest, as illustrated by slices through projection images of the density map (Figures 3A and 3B). Based on comparison of the extent of movement determined by classifying the data to produce two discrete 3D structures, the range of motion in the peripheral NBD (Figure 3C; Movie S2) is seen to be small in comparison to the large domain movements observed for transition from open to closed conformations (Figure 3C) (Borgnia et al., 2016).
Figure 3. Cryo-EM Analysis of GDH.

(A and B) Projection views of two 3D classes from cryo-EM analysis of GDH (glutamate dehydrogenase). In both classes, there is well-defined density at the core, but it is weakly defined at the peripheral nucleotide binding domain (NBD). The two classes display similar structures in the interior but differ slightly in the peripheral NBD. The two classes are likely to be subsets of a continuum of states with varying orientations for the outer domain relative to the core.
(C) Ribbon diagrams of the open and closed structures (Borgnia et al., 2016) demonstrating the more extensive NBD movement associated with substrate binding and catalytic cleft closure.
(D) A selected region of the cryo-EM map of the GDH structure, highlighting high-resolution features such as “holes” in the aromatic rings of Tyr382, Phe383, and Trp385, water molecules (shaded yellow), and well-resolved densities for carbonyl bonds.
The binding of GTP (an inhibitor) and ADP (an activator) to two allosteric sites in each protomer modulates the transition between “closed” and “open” states of GDH. These allosteric modulators tightly control GDH function in vivo. The two states we present in Figures 3A and 3B are both in the “open” conformation with differences likely due to small perturbations arising from the unhindered sampling of conformations due to the twisting motions of the pivot helix in the absence of either ADP or GTP in solution. Despite the presence of the flexible outer domains (Figures 3A and 3B), most of the interior of GDH can nevertheless be visualized at resolutions better than 2 Å, well beyond the 2.7 Å resolution obtained with X-ray crystallographic analysis, in which the flexible regions presumably limit the order of the crystals obtained. In comparison to our earlier work with β-galactosidase (Bartesaghi et al., 2015), in which we reported an average resolution of 2.2 Å, the density map for GDH at 1.8 Å shows better-defined and more uniform density over most of the ordered central core, spanning the subunit interface (Figure 3D), as well as clear density for carbonyl bonds and several H-bonded water molecules and well-defined density for extended Lys side chains (Figures S4B–S4E).
DISCUSSION
We have previously shown that it is possible to achieve atomic resolution information from an inhibitor-bound protein complex such as the 465 kDa β-galactosidase (Banerjee et al., 2016; Bartesaghi et al., 2015). The inhibitor-bound state of β-galactosidase is relatively stable, with only a small gradient in temperature factor between the central and peripheral regions, making it possible to achieve high resolution across the breadth of the protein. However, many proteins can have flexible domains, as in the case of GDH, even when they are expected to be in a single conformational state. While cryo-EM can be used to effectively mask out flexible or disordered areas of a protein during steps in image processing (Amunts et al., 2014; Bai et al., 2015a), it has remained an open question whether the presence of a large fraction of the protein that is conformationally mobile may preclude achieving atomic resolution for the more structured or stable regions of the complex. The fact that we can visualize densities for each of the 20 amino acids in the GDH cryo-EM map (Figure 4), without 3D masking during refinement, addresses this question definitively while also enabling the crossing of the 2 Å resolution mark in single particle cryo-EM. These cryo-EM densities are easily comparable to those obtained by X-ray crystallography at a similar resolution (Figure S5).
Figure 4. Density Representations for a Representative Example of Each of the 20 Amino Acid Types from the 1.8 Å Resolution Cryo-EM Structure of apo-GDH.

Features such as holes in aromatic rings, as well as the “zigzag” structure of extended sidechains such as Arg and Lys, are visible in the density maps. See also Figure S5 and Movie S3.
The maps we have obtained for LDH display 2.8 Å resolution in the protein component and densities for numerous water molecules (Figures S1D and S1E), but density for the bound ligand is at lower resolution. One reason that the density for the bound ligand is less well-defined than that of the polypeptide may be because small molecule ligands bound to the protein periphery are likely to sample far more conformations in aqueous solution than what is possible in the context of a crystal lattice. Most solvents used for protein crystallization invariably include additives such as PEG that dehydrate the crystal by removing excess solvent and tightening the packing of protein molecules while reducing the size of the solvent channels around the protein. As a result, these additives improve crystal order and diffraction resolution, but at the same time, also affect ligand-protein interactions, as dehydration reduces the number of water molecules in the binding pocket and therefore minimizes the mobility of the ligand. Thus, cryo-EM studies in aqueous dispersions and X-ray crystallographic studies in ordered 3D crystals provide potentially complementary snapshots of ligand-protein interactions that are likely to occur in cellular environments.
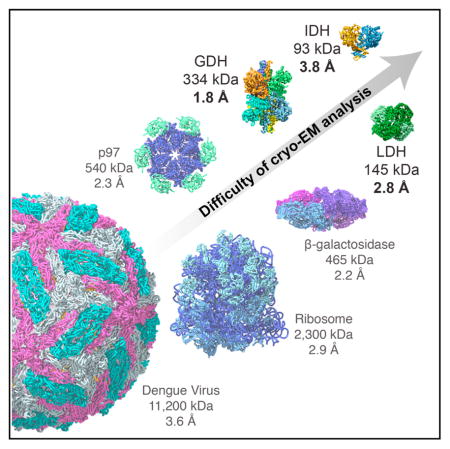
Finding the correct orientation of each projection image is central for the success of the single particle cryo-EM reconstruction process. The practical size limitation for high-resolution structure determination is currently a question of considerable interest within the cryo-EM field. Figure 5 provides a comparison of the relative sizes of complexes whose structures have been studied at near-atomic resolution, including those reported in the present study. Given the present state of detector and microscope technology, a figure of 150 kDa is thought to be a practical lower limit even for medium resolutions (Thompson et al., 2016), even though theoretical arguments have been advanced for why much smaller sizes should be accessible (Glaeser and Hall, 2011; Henderson, 1995). As instrumentation and methods for data analysis have improved, our ability to overcome the handicaps of small size, low symmetry, and high flexibility have likewise improved, allowing the reconstruction of not just large, highly symmetric complexes such as icosahedral viruses, but now also of smaller, lower symmetry complexes as well (Figure 5A). In this study, we have shown that not only is it possible to determine the structure of a protein smaller than 150kDa to ~3 Å resolution, it is also possible to determine the structure of a protein <100 kDa to near-atomic (<4 Å) resolution, coupled with the visualization of the location of bound small-molecule inhibitors in both cases. From an analysis of all of the parameters that influenced final map quality, our conclusion is that selecting for specimens with thin ice, retaining only those images that display the lowest beam-induced motion, and using a sufficiently small pixel size and the improved XP sensor with higher DQE performance were the key factors that enabled us to obtain high resolution with these smaller protein complexes.
Figure 5. Relative Sizes of Small Metabolic Complexes Compared to Other Structures Solved by Cryo-EM and Variations in Flexibility across Different Regions.

(A) Relative sizes of GDH, LDHB, and IDH1 in comparison to a representative set of other structures that have been solved by cryo-EM to near-atomic resolution including icosahedral viruses, ribosomes, and protein complexes such as β-galactosidase and the AAA ATPase p97.
(B–D) Ribbon diagram of hexameric GDH (B), tetrameric LDHB (C), and dimeric IDH1 (D), colored to show B-factor variation from blue (lowest) to red (highest) B factors, respectively.
See also Movie S3.
As more examples of cryo-EM structures are reported, there are beginning to be noticeable patterns in the differences between the visual appearance of cryo-EM density maps and those obtained using X-ray crystallography. In visualizing cryo-EM density maps, different regions of the map are typically explored by implementing B-factor sharpening specific to the different segments of the map, but deposited maps and figures generally show density maps sharpened with a single value for B factor for the entire protein, which poses a challenge for capturing all of the information in a single view. Nevertheless, it is clear that in all three of the protein complexes that we have analyzed, the outer regions are significantly more flexible than the core of the complex (Figures 5B–5D). Since cryo-EM analyses report on the structure of the proteins in solution, they can provide information on quaternary structures complementary to that obtained from crystal structures, which may be influenced by contacts within the crystal lattice. A common feature of the cryo-EM density maps we have observed in structures of complexes of ligands with diverse proteins such as LDH, IDH1, β-galactosidase, and p97 is that the density for bound ligands, especially in the peripheral regions, is generally somewhat weaker and at lower resolution than that of residues in the binding pocket. As noted earlier in the discussion, this is perhaps not surprising given the presence of numerous single bonds around which rotations are possible, coupled with poor steric and electrostatic fits between ligand and protein. Even in regions where the density map of the polypeptide is poorly defined in small proteins such as IDH1, crystallographic information can provide an invaluable framework to interpret the cryo-EM structures, enabling identification of sites of inhibitor binding.
Size, symmetry, and intrinsic flexibility are all parameters that play a role in determining the resolutions that can be achieved using cryo-EM. LDH and β-galactosidase are both tetramers with the same symmetry (D2), but LDH is about a third of the size of β-galactosidase. The lower resolutions currently achieved with LDH are most likely from the reduced accuracy of alignment of projection images, and this comparison is thus a good way to assess effect of size on resolution with presently available technology. The further drop in resolution observed for IDH1 could be due to reduction in size, lower symmetry, and potentially greater flexibility of the dimeric enzyme. Reconstruction of IDH1 without application of symmetry resulted in only a slightly lower resolution (~4.5 Å), indicating that our results are likely general in nature. It is possible that, for these small enzymes, improvements in resolution could be obtained by the use of one or two bound Fab fragments (Wu et al., 2012). Fab fragments not only increase the size of a complex, but also assist with orientation assignment and can stabilize certain conformations of highly flexible complexes (Kim et al., 2015; Lyumkis et al., 2013b). Since Fab fragments are ~50 kDa in size, an important consequence of our results is that the presence of one or two bound Fab fragments may bring almost any protein into a size range suitable for analysis by cryo-EM. Although we have not yet achieved ~2 Å resolution with small complexes like IDH and LDH, it is likely that further advances in detector technology will contribute toward achieving this goal. Ongoing hardware developments such as phase plates (Danev and Baumeister, 2016), as well as improvements in software for image alignment and reconstruction of heterogeneous complexes, will also undoubtedly be necessary to advance cryo-EM frontiers to determine high resolution structures of even smaller and more challenging complexes.
EXPERIMENTAL PROCEDURES
Proteins and Grid Preparation
The chicken heart LDH (catalog #59747, Sigma-Aldrich, St. Louis) lyophilized sample was suspended in 550 μl 1 × PBS buffer and centrifuged at 20,000 × g for 30 min. Thereafter, it was subjected to gel filtration on a Superdex-200 size-exclusion chromatography column connected to a ÄKTA FPLC apparatus (GE Healthcare Bio-Sciences, Piscataway) with an elution buffer comprised of 1×PBS. The ligand GSK 2837808A (catalog #5189 Tocris Bioscience) was dissolved in 100% DMSO and added to LDH (~1.5 mg/ml) to a final inhibitor concentration of 75 μM and 0.5% DMSO.
Human IDH1 R132C mutant protein was purified following the published protocol (Davis et al., 2014). ML309 was dissolved in 100% DMSO and added to IDH1 (2.8 mg/ml) to a final inhibitor concentration of 50 μM and 0.5% DMSO.
GDH (catalog #G2626, Sigma-Aldrich, St. Louis) was dialyzed overnight against gel-filtration buffer (100 mM potassium phosphate [pH 6.8]) prior to purification by size-exclusion chromatography (SEC) using a Superdex-200 size-exclusion chromatography column connected to a ÄKTA FPLC apparatus (GE Healthcare Bio-Sciences, Piscataway). Final protein concentration was ~3 mg/ml.
Prior to preparation of frozen-hydrated specimens, 3–4 μl of a freshly thawed aliquot of purified proteins or inhibitor-protein complexes were deposited on Quantifoil R1.2/1/3 grids and plunge frozen in liquid ethane cooled by liquid nitrogen using either a Leica plunge-freeze device (for the LDH and IDH1 samples) or a Vitrobot (for the GDH samples). For experiments with added inhibitor, the protein was pre-incubated for 30 min at room temperature under the buffer conditions indicated above for each kind of protein complex.
Data Acquisition
The grids were imaged using a Titan Krios transmission electron microscope (FEI Company, Hillsboro) aligned for parallel illumination and operated at 300 kV, with the specimen maintained at liquid nitrogen temperatures. Images were recorded on a K2 Summit camera (Gatan, Pleasanton) operated in super-resolution counting mode, placed at the end of a GIF Quantum energy filter (Gatan), functioning in zero-energy-loss mode with a slit width of 20 eV as described previously (Bartesaghi et al., 2015). Images were typically collected with a defocus range between −0.7–−3.0 μm. For LDH and IDH, a dose rate of ~5 e−/Å2/s (at the specimen plane) was used with a total exposure time of 12 s. Intermediate frames were recorded every 0.2 s giving an accumulated dose of ~60 e−/Å 2 and a total of 60 frames per image. Pixel size was set to 0.495 Å. For GDH, a dose rate of ~2.6 e−/Å2/s was used and a total exposure time of 15.2 s. Intermediate frames were recorded every 0.4 s giving an accumulated dose of ~ 40 e−/Å2 and a total of 38 frames per image with a pixel size of 0.637 Å.
Image Processing
Movie frame alignment and contrast transfer function (CTF) estimation for each micrograph were carried out as described previously (Bartesaghi et al., 2014). For the LDHB and IDH1 reconstructions, particles were picked from the original micrographs using a Gaussian disk of 42 Å in radius as the search template. Particles were extracted using a binning factor of 8 and a box size of 96 pixels and subsequently subjected to 12 rounds of Frealign refinement (Lyumkis et al., 2013a). The numbers of particle images used for each final reconstruction are shown in Table 1. The subset of particles that contributed to the reconstruction in the last iteration was re-extracted from the original micrographs using a binning factor of 4 and a box size of 192 pixels followed by eight additional iterations of local refinement in Frealign. In a similar way, particles were re-extracted and further refined two additional times using a binning factor of 2 and a box size of 768 pixels, first using the full exposure and then using only the first half of the exposure (frames 1–30), corresponding to ~30 e−/Å2. B-factor sharpening was applied to the final maps for purposes of visualization (Table 1).
Images of GDH were processed in a way similar to that done for LDHB and IDH1, this time using a Gaussian disk of 75 Å in radius as the search template for particle picking. The selection of particles that contributed to the reconstruction at each iteration was in this case done according to the bi-modal distribution of Frealign scores (Banerjee et al., 2016). Re-extraction of selected particles from the original micrographs using progressively less binning followed by local refinement was done as described above for the LDHB and IDH1 datasets. In addition, particles with a binning factor of 2 and a box size of 768 pixels were re-extracted one final time using frames 3–9 and used to produce the final reconstruction by applying the latest set of alignments (derived using the first half of the exposure) without further refinement. A B factor of ~90 Å2 was applied to the final map for purposes of visualization.
To estimate resolution, we used two approaches, both of which provided mutually consistent results. In one approach, we used the strategy implemented in Frealign to carry out refinement using only low-resolution information (to prevent over-fitting) and then measured the value at which the FSC value drops to 0.143. The highest resolution we used for refining particle orientations was 6 Å for the IDH1 datasets and 4 Å for the LDHB and GDH datasets. As a complementary method, we computed the correlation between the experimentally derived map and the refined model, and determined the value at which the correlation drops to 0.5.
Atomic Model Refinement
The starting model used for the refinement of LDH was from the coordinates from the X-ray structure 1I0Z. Changes were made to the initial model in order to incorporate the differences between the human and chicken LDH amino acid sequences. The structure of the apo LDH B was initially refined by rigid-body refinement followed by real-space refinement using the program Phenix (Adams et al., 2010). Manual adjustments were made using the program COOT (Emsley et al., 2010). Solvent molecules were added manually followed by a round of real-space refinement.
The starting model used for the refinement of NADPH-bound IDH1 was from the coordinates from the structure 3MAP. Prior to refinement, the residue Arg132 was mutated to a cysteine and the isocitrate molecule was removed from the model. The structure of the NADPH-bound IDH1 was initially refined by rigid-body refinement followed by real-space refinement using the program Phenix. Manual adjustments were made by refinement in COOT following another round of real-space refinement. For the ML-309-bound structure, the starting model was the NADPH-bound IDH1 obtained using the cryo-EM data. The refinement protocol was identical to the one used previously.
The PDB coordinates 3JCZ served as a starting point for the refinement of apo-GDH. Initially, rigid-body fitting followed by real-space refinement using the program Phenix was used to refine the model. Manual adjustments were made in COOT. Solvent molecules were added automatically using stringent thresholds followed by manual inspection. Thereafter, the model was refined again using real-space refinement as implemented in Phenix.
Supplementary Material
Highlights.
Near-atomic-resolution structure of isocitrate dehydrogenase, a < 100 kDa enzyme
1.8 Å resolution cryo-EM map of 334 kDa glutamate dehydrogenase
2.8 Å resolution cryo-EM map of lactate dehydrogenase, a 145 kDa protein
Localization of small-molecule inhibitors bound to metabolic protein complexes
Acknowledgments
This research was supported by funds from the Center for Cancer Research, National Cancer Institute, NIH in Bethesda. We thank Kieran Moynihan, Robert Mueller, and Joe Cometa for technical assistance with electron microscopy; Fred Ulmer, Paul Mooney, and Chris Booth for advice and assistance with optimizing K2 detector performance; and Rishub Jain, Jierui Fang, and Amy Jin for assistance with data analysis. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
ACCESSION NUMBERS
The accession numbers for the density maps reported in this paper is Electron Microscopy Data Bank: wwww, xxxx, yyyy, and zzzz. The accession numbers for the refined atomic models reported in this paper is PDB: wwww, xxxx, yyyy, zzzzz.
Supplemental Information includes five figures and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.05.040.
AUTHOR CONTRIBUTIONS
Specimen preparation was carried out by A.M. and P.R., data collection was carried out by A.M., image processing was carried out by A.B., data analysis, interpretation, and presentation was carried out by S.B., A.B., A.M., V.F., L.A.E., J.L.S.M., and S.S., and samples of purified IDH1 and ML309 were provided by M.D., R.P., and M.B. Final figure preparation was carried out primarily by V.F. with help from A.M. and A.B. Project conception, organization, and supervision was provided by S.S., who was also primarily responsible for writing the manuscript, with help from S.B., A.B., A.M., L.A.E., V.F., and J.L.S.M.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife. 2015a;4:e11182. doi: 10.7554/eLife.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, Shi Y. An atomic structure of human γ-secretase. Nature. 2015b;525:212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Matthies D, Banerjee S, Merk A, Subramaniam S. Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy. Proc Natl Acad Sci USA. 2014;111:11709–11714. doi: 10.1073/pnas.1402809111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, Subramaniam S. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science. 2015;348:1147–1151. doi: 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnap DM. Electron Microscopy and Image Processing: Essential Tools for Structural Analysis of Macromolecules. Curr Protoc Protein Sci. 2015;82:17.2.1–17.2.61. doi: 10.1002/0471140864.ps1702s82. [DOI] [PubMed] [Google Scholar]
- Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colón M, Dodson CS, Gilbert SA, Greshock J, Jing J, et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013;1:19. doi: 10.1186/2049-3002-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgnia MJ, Banerjee S, Merk A, Matthies D, Bartesaghi A, Rao P, Pierson J, Earl LA, Falconieri V, Subramaniam S, Milne JL. Using Cryo-EM to Map Small Ligands on Dynamic Metabolic Enzymes: Studies with Glutamate Dehydrogenase. Mol Pharmacol. 2016;89:645–651. doi: 10.1124/mol.116.103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. Single-Particle Cryo-EM at Crystallographic Resolution. Cell. 2015;161:450–457. doi: 10.1016/j.cell.2015.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Baumeister W. Cryo-EM single particle analysis with the Volta phase plate. eLife. 2016;5:e13046. doi: 10.7554/eLife.13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Gross S, Shen M, Straley KS, Pragani R, Lea WA, Popovici-Muller J, DeLaBarre B, Artin E, Thorne N, et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem. 2014;289:13717–13725. doi: 10.1074/jbc.M113.511030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Shen J, Yin M, McManus J, Mathieu M, Gee P, He T, Shi C, Bedel O, McLean LR, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovich PS, Fauber BP, Boggs J, Chen J, Corson LB, Ding CZ, Eigenbrot C, Ge H, Giannetti AM, Hunsaker T, et al. Identification of substituted 3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorg Med Chem Lett. 2014;24:3764–3771. doi: 10.1016/j.bmcl.2014.06.076. [DOI] [PubMed] [Google Scholar]
- Du J, Lü W, Wu S, Cheng Y, Gouaux E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature. 2015;526:224–229. doi: 10.1038/nature14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature. 2015;520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- Fiume L, Manerba M, Vettraino M, Di Stefano G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med Chem. 2014;6:429–445. doi: 10.4155/fmc.13.206. [DOI] [PubMed] [Google Scholar]
- Ge P, Zhou ZH. Hydrogen-bonding networks and RNA bases revealed by cryo electron microscopy suggest a triggering mechanism for calcium switches. Proc Natl Acad Sci USA. 2011;108:9637–9642. doi: 10.1073/pnas.1018104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser RM, Hall RJ. Reaching the information limit in cryo-EM of biological macromolecules: experimental aspects. Biophys J. 2011;100:2331–2337. doi: 10.1016/j.bpj.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, Rosenthal PB. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol. 2011;413:1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomaa A, Boehringer D, Leibundgut M, Ban N. Structures of the E. coli translating ribosome with SRP and its receptor and with the trans-locon. Nat Commun. 2016;7:10471. doi: 10.1038/ncomms10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM, Tampé R, Craik CS, Cheng Y. Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature. 2015;517:396–400. doi: 10.1038/nature13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryomicroscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Brilot AF, Theobald DL, Grigorieff N. Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol. 2013a;183:377–388. doi: 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013b;342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matadeen R, Patwardhan A, Gowen B, Orlova EV, Pape T, Cuff M, Mueller F, Brimacombe R, van Heel M. The Escherichia coli large ribosomal subunit at 7.5 A resolution. Structure. 1999;7:1575–1583. doi: 10.1016/s0969-2126(00)88348-3. [DOI] [PubMed] [Google Scholar]
- Matthies D, Dalmas O, Borgnia MJ, Dominik PK, Merk A, Rao P, Reddy BG, Islam S, Bartesaghi A, Perozo E, Subramaniam S. Cryo-EM Structures of the Magnesium Channel CorA Reveal Symmetry Break upon Gating. Cell. 2016;164:747–756. doi: 10.1016/j.cell.2015.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, Solon M, Tao J, Li L, Shames D, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. 2013;19:773–784. doi: 10.1158/1078-0432.CCR-12-2638. [DOI] [PubMed] [Google Scholar]
- McMullan G, Faruqi AR, Clare D, Henderson R. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy. 2014;147:156–163. doi: 10.1016/j.ultramic.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, Rominger C, Quinn C, Smallwood A, Wiggall KJ, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–886. doi: 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, Linde J, Vestergaard B, Ehrenberg M, Frank J. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature. 2003;421:87–90. doi: 10.1038/nature01224. [DOI] [PubMed] [Google Scholar]
- Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, van Gurp RJ, Oosterhuis JW, Lu YJ, Huddart R, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003;22:1880–1891. doi: 10.1038/sj.onc.1206302. [DOI] [PubMed] [Google Scholar]
- Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. Atomic model of an infectious rotavirus particle. EMBO J. 2011;30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiniotis G, Southworth DR. Single-particle cryo-electron microscopy of macromolecular complexes. Microscopy (Oxf) 2016;65:9–22. doi: 10.1093/jmicro/dfv366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Schmidt T, Fang J, Wu J, Siuzdak G, Stanley CA. The structure of apo human glutamate dehydrogenase details subunit communication and allostery. J Mol Biol. 2002;318:765–777. doi: 10.1016/S0022-2836(02)00161-4. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Walker M, Siebert CA, Muench SP, Ranson NA. An introduction to sample preparation and imaging by cryo-electron microscopy for structural biology. Methods. 2016;100:3–15. doi: 10.1016/j.ymeth.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Bai XC, Brown A, Fernandez IS, Hanssen E, Condron M, Tan YH, Baum J, Scheres SH. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 2014;3:e03080. doi: 10.7554/eLife.03080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Avila-Sakar A, Kim J, Booth DS, Greenberg CH, Rossi A, Liao M, Li X, Alian A, Griner SL, et al. Fabs enable single particle cryoEM studies of small proteins. Structure. 2012;20:582–592. doi: 10.1016/j.str.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Jiang H, Zheng B, Kogiso M, Yao Y, Zhou C, Li XN, Song Y. Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases by 2-Thiohydantoin Compounds. J Med Chem. 2015;58:6899–6908. doi: 10.1021/acs.jmedchem.5b00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- Yang B, Zhong C, Peng Y, Lai Z, Ding J. Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 2010;20:1188–1200. doi: 10.1038/cr.2010.145. [DOI] [PubMed] [Google Scholar]
- Yu X, Jin L, Zhou ZH. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell. 2010;141:472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Yao Y, Liu Z, Deng L, Anglin JL, Jiang H, Prasad BV, Song Y. Crystallographic Investigation and Selective Inhibition of Mutant Isocitrate Dehydrogenase. ACS Med Chem Lett. 2013;4:542–546. doi: 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.