

Abstract
Objectives
Independent epidemiological studies have evaluated the association between markers of glucose metabolism (including fasting glucose, fasting insulin, homeostasis model of risk assessment-insulin resistance (HOMA-IR), glycated haemoglobin (HbA1c) and C peptide) and the risk of colorectal cancer (CRC). However, such associations have not been systematically analysed and no clear conclusions have been drawn. Therefore, we addressed this issue using a meta-analysis approach.
Design
Systematic review and meta-analysis.
Data sources
PubMed and EMBASE were searched up to May 2015.
Primary and secondary outcome measures
Either a fixed-effects or random-effects model was adopted to estimate overall ORs for the association between markers of glucose metabolism and the risk of CRC. In addition, dose–response, meta-regression, subgroup and publication bias analyses were conducted.
Results
35 studies involving 25 566 patients and 5 706 361 participants were included. Higher levels of fasting glucose, fasting insulin, HOMA-IR, HbA1c and C peptide were all significantly associated with increased risk of CRC (fasting glucose, pooled OR=1.12, 95% CI 1.06 to 1.18; fasting insulin, pooled OR=1.42, 95% CI 1.19 to 1.69; HOMA-IR, pooled OR=1.47, 95% CI 1.24 to 1.74; HbA1c, pooled OR=1.22, 95% CI 1.02 to 1.47 (with borderline significance); C peptide, pooled OR=1.27, 95% CI 1.08 to 1.49). Subgroup analysis suggested that a higher HOMA-IR value was significantly associated with CRC risk in all subgroups, including gender, study design and geographic region. For the relative long-term markers, the association was significant for HbA1c in case–control studies, while C peptide was significantly associated with CRC risk in both the male group and colon cancer.
Conclusions
The real-time composite index HOMA-IR is a better indicator for CRC risk than are fasting glucose and fasting insulin. The relative long-term markers, HbA1c and C peptide, are also valid predictors for CRC risk. Considering the included case–control studies in the current analysis, more cohort studies are warranted to enhance future analysis.
Keywords: fasting glucose, fasting insulin, HOMA-IR, HbA1c, C-peptide, colorectal cancer
Strengths and limitations of this study.
Higher levels of fasting glucose, fasting insulin, homeostasis model of risk assessment-insulin resistance (HOMA-IR), glycated haemoglobin (HbA1c) and C peptide were associated with increased risk of colorectal cancer (CRC). The real-time composite index HOMA-IR is a better indicator for CRC risk than are fasting glucose and fasting insulin.
The number of total participants was substantial, and meta-regression, subgroup analysis and dose–response analyses were applied, which enhanced the statistical power.
The range of the cut-off points between the lowest and the highest categories for fasting glucose, fasting insulin, HOMA-IR, HbA1c and C peptide levels differed between studies, which could influence the outcome of the current analyses.
Introduction
Diabetes mellitus and colorectal cancer (CRC) are both major causes of morbidity and mortality worldwide,1–4 and it has been suggested that there is a statistically significant association between diabetes and increased risk of CRC.5 6 The possible mechanisms underlying this association may be related to insulin resistance/hyperinsulinaemia, hyperglycaemia and inflammation.7 8 Insulin has pro-proliferative properties, and it can reduce apoptosis and promote the growth of CRC cell lines.9–11 Insulin resistance is a subnormal glycaemic response to endogenous insulin, which commonly precedes hyperinsulinaemia12 and can be quantitatively measured by the homeostasis model of risk assessment-insulin resistance (HOMA-IR) index.13 As a new composite index derived from fasting glucose and fasting insulin concentrations, HOMA-IR is primarily used in large-scale or epidemiological studies in the year 2000,13 although its accuracy is still under debate.
In addition to fasting glucose, fasting insulin and HOMA-IR, other markers of the glucose metabolism have also been used to monitor patients with diabetes. Glycated haemoglobin (HbA1c) is a form of haemoglobin in which a molecule of glucose is attached to its β-chain after exposure to high plasma levels of glucose. As such, it functions as an integrated indicator of average blood glucose concentrations over the period of 6–8 weeks,14 thereby providing a more stable measure of serum glucose as compared with that obtained from a one-time measurement. In addition, C peptide, which is secreted by β cells on an equal molar basis with insulin, has a relatively long half-life in the peripheral circulation,15 providing another valid indicator of insulin production.16
Many independent epidemiological studies have evaluated the association between one or two of the markers mentioned above and the risk of CRC;17–51 however, until now, no study has systematically analysed the association between all these markers and the risk of CRC. Thus, the aim of this study was to provide a quantitative assessment of the association between markers of glucose metabolism, including real-time markers (fasting glucose, fasting insulin and the composite index HOMA-IR) and relative long-term markers (HbA1c and C peptide), and the risk of CRC by a meta-analysis approach and to compare the efficacy of these markers.
Materials and methods
Data sources, search strategy and selection criteria
Systematic literature searches were conducted in PubMed and EMBASE (to May 2015) to identify eligible studies. The following terms were applied: ‘glucose’ OR ‘blood sugar’; ‘insulin’; ‘HOMA’ OR ‘homeostasis model assessment’; ‘c-peptide’ OR ‘c peptide’; ‘hemoglobin’ OR ‘haemoglobin’ OR ‘hematocrystallin’ OR ‘HbA1c’; ‘colon’ OR ‘rectal’ OR ‘colonic’ OR ‘colorectal’; ‘cancer’ OR ‘tumor’ OR ‘carcinoma’ OR ‘neoplasm’ OR ‘adenoma’. References of relevant articles were also scanned for potentially missing studies. Titles and abstracts were first scanned, and then full articles of potentially eligible studies were reviewed. References of relevant articles and reviews were also scanned for potentially missing studies. The retrieved studies were carefully examined to exclude potential duplicates or overlapping data. This meta-analysis was designed, conducted and reported according to PRISMA and MOOSE statements.52 53
Articles were included if they met all of the following criteria: (1) case–control or cohort studies evaluated the association between markers of glucose metabolism and the risk of CRC, with the markers including fasting glucose, fasting insulin, HOMA-IR, HbA1c and C peptide; (2) relative risk (RR) or OR estimates and their 95% CIs were given or could be calculated (if only case numbers of different categories were provided, we recalculated the risk ratio by a fourfold table χ2 method) and (3) articles were published as full papers in English. Reviews, letters, editorials, meeting abstracts and case reports were excluded.
Data extraction and quality assessment
Two reviewers (JX and YW) independently collected data using standardised forms and discrepancies were resolved by a third investigator (YY). We extracted the following information from each study: first author, year of publication, origin of the study population, study design (prospective or retrospective), patient characteristics (sample size, age and gender), variables adjusted for in the analysis from each study, and the reported RR (OR) with 95% CIs for the highest versus lowest categories of markers of glucose metabolism. HOMA-IR was derived as fasting glucose (mmol/L)×fasting insulin (mIU/L)/22.5, as described by Matthews et al.54
The quality of each study was assessed according to the Newcastle-Ottawa quality assessment.55
Data synthesis and statistical analysis
Either a fixed-effect or random-effect model was adopted to pool the study-specific RRs (ORs), according to the extent of heterogeneity. A random-effect model56 was used when the heterogeneity was significant (p≤0.10 and/or I2>50%). Otherwise, a fixed-effect model was applied.57 The significance of the pooled RR was determined by Z test (p<0.05 was considered to be significant). The χ2 and I2 test58 were used to assess the heterogeneity across studies. Meta-regression59 and subgroup analyses were applied to evaluate the potential effect of modification of variables, including cancer subtype, gender, geographic region and study design. Funnel plots were constructed60 and Begg's and Egger's tests were performed to assess the publication bias (p≤0.10 was considered to be significant). Egger’s method had stronger statistical power than Begg’s method.61 Please find the detailed information in the online supplementary methods part.
We analysed the dose–response relationship using linear, first-order and second-order fractional polynomial regression of the inverse variance-weighted data to estimate a curve of best fit. Best-fit curves were selected using decreased deviance compared with the reference model.62 The units of fasting glucose, fasting insulin, HbA1c and C peptide were standardised as ‘mg/dL’, ‘uIU/mL’, ‘%’ and ‘ng/mL’, respectively. Comparisons of curves to determine the best fit were done using a χ2 distribution. All analyses were conducted using Stata software (V.12.0; StatCorp, College Station, Texas, USA).
bmjopen-2016-011430supp.pdf (521.3KB, pdf)
Results
Study characteristics and data quality
After searching PubMed and EMBASE, 9648 articles were identified. Of these, 2453 duplicate papers were removed, leaving 7195 articles for assessment. A review of titles and abstracts resulted in the exclusion of 6901 articles. Of the remaining 294 articles, 259 were excluded for the following reasons: insufficient data (n=115); not on the right topic or targeted population (n=137); not an original article (n=5); and duplicate data (n=2).
The remaining 35 studies,17–51 which provided data on 25 566 patients and 5 706 361 participants, were included in this meta-analysis. The selection process is shown in figure 1, and the characteristics of the included studies are shown in online supplementary table S1. Among the included studies, 18 examined fasting glucose levels,17 19 26 29–31 34 36 37 39 40 44–47 49–51 10 examined fasting insulin levels,19 21 30 34 39 42–44 47 49 8 examined HOMA-IR values,30 34 39 42 44 47–49 8 examined HbA1c levels,18 22 23 27 28 35 36 47 and 9 examined C peptide levels.20 24 25 28 32 33 36 38 41 The results of our quality assessment are shown in online supplementary tables S2 and S3. The PRISMA checklist and flow diagram are shown in online supplementary table S4.
Figure 1.

Flow diagram of study selection process.
Association between fasting glucose, fasting insulin, HOMA-IR and the risk of CRC
Fasting glucose and CRC: A total of 18 studies involving 17 764 patients and 3 805 861 participants assessed the association between fasting glucose levels and the risk of CRC. The pooled OR of CRC risk for the highest versus lowest categories of fasting glucose level was 1.12 (95% CI 1.06 to 1.18; p<0.001), indicating a significantly positive association (figure 2A). There was moderate heterogeneity among the included studies (I2=45.4%, p=0.007), as shown in figure 2A. The results of the dose–response analysis indicated a positive correlation between fasting glucose levels and the risk of CRC, as shown in figure 3A.
Figure 2.

The association between real-time markers of glucose metabolism and the risk of CRC. Pooled ORs of CRC for the highest versus lowest categories of (A) fasting glucose level; (B) fasting insulin level and (C) HOMA-IR value. A, advanced cancer; CC, colon cancer; CRC, colorectal cancer; E, early cancer; F, female; HOMA-IR, homeostasis model of risk assessment-insulin resistance; M, male; RC, rectal cancer; RR, relative risk.
Figure 3.
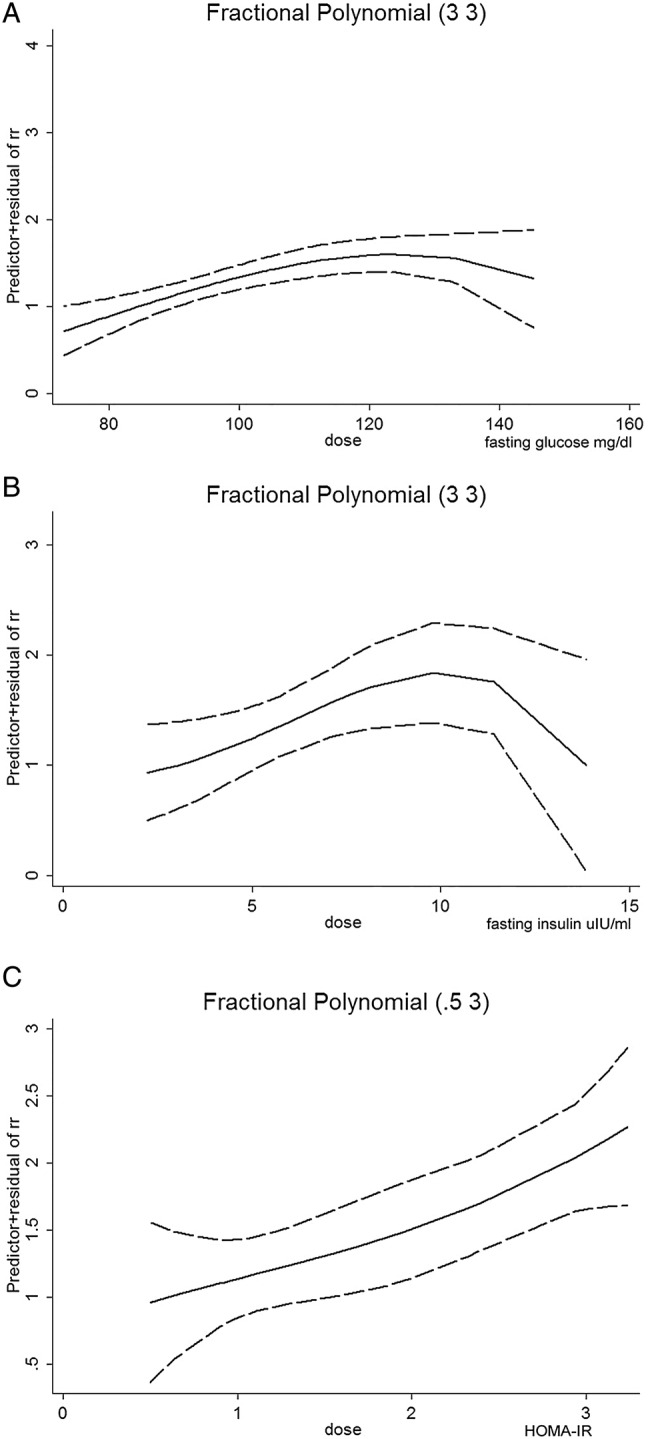
Dose–response relationship between real-time markers of glucose metabolism and the risk of CRC. Dose–response relationship between (A) fasting glucose level and the risk of CRC; (B) fasting insulin level and the risk of CRC and (C) HOMA-IR value and the risk of CRC. The solid line represents the pooled RRs, and the dotted line represents the 95% CIs of the RRs. CRC, colorectal cancer; HOMA-IR, homeostasis model of risk assessment-insulin resistance; RR, relative risk.
Subgroup analyses were conducted according to gender, geographic region, study design and cancer subtype, as shown in table 1. The pooled RR of CRC for the highest versus lowest categories of fasting glucose level was 1.13 (95% CI 1.04 to 1.22) in males and 1.07 (95% CI 0.97 to 1.19) in females, suggesting that the fasting glucose level was significantly associated with CRC risk in males but not in females. For study design, the fasting glucose level was found to be statistically associated with the risk of CRC in both case–control (pooled RR=1.22, 95% CI 1.02 to 1.46) and cohort (pooled RR=1.11, 95% CI 1.05 to 1.17) studies. When stratifying the studies according to geographic region, fasting glucose level was found to be significantly associated with CRC risk in the American and European (pooled RR=1.09, 95% CI 1.03 to 1.16) and Asian (pooled RR=1.20, 95% CI 1.08 to 1.34) populations. For CRC subtypes, the fasting glucose level was found to be significantly associated with colon cancer (pooled RR=1.08, 95% CI 1.01 to 1.16) but not rectal cancer (pooled RR=1.07, 95% CI 0.97 to 1.17). Meta-regression and subgroup analyses suggested that heterogeneity was partly influenced by study design, geographic region and cancer subtypes (table 1).
Table 1.
Meta-regression and subgroup analysis between markers of glucose metabolism and risk of CRC
| Gender |
Study design |
Geographic region |
Cancer subtypes |
|||||
|---|---|---|---|---|---|---|---|---|
| Female | Male | (Nested) case–control | Cohort | America and Europe | Asia | Colon cancer | Rectal cancer | |
| Fasting glucose | ||||||||
| Sample size (cases/controls/cohort) | 4419/5637/1 570 964 | 6346/399/1 878 566 | 3203/10 087/447 172 | 14 561/–/3 358 689 | 16 523/9218/2 331 799 | 1241/869/1 474 062 | 8052/4821/1 944 129 | 4604/–/1 909 227 |
| RR (95% CI) | 1.07 (0.97 to 1.19) | 1.13 (1.04 to 1.22) | 1.22 (1.02 to 1.46) | 1.11 (1.05 to 1.17) | 1.09 (1.03 to 1.16) | 1.20 (1.08 to 1.34) | 1.08 (1.01 to 1.16) | 1.07 (0.97 to 1.17) |
| p for trend | 0.193 | 0.004 | 0.032 | <0.001 | 0.005 | 0.001 | 0.025 | 0.162 |
| Heterogeneity (p/I2) | 0.591/0.0% | 0.385/5.6% | 0.094/41% | 0.012/48.8% | 0.019/44.8% | 0.091/45.1% | 0.116/37.9% | 0.191/31.1% |
| Fasting insulin | ||||||||
| Sample size (cases/controls/cohort) | 519/5637/98 578 | 304/501/29 133 | 2957/9127/342 711 | 282/–/12 159 | 3052/8959/354 870 | 187/168/– | – | – |
| RR (95% CI) | 1.53 (0.92 to 2.55) | 1.67 (0.92 to 3.05) | 1.45 (1.18 to 1.78) | 1.32 (0.92 to 1.89) | 1.41 (1.17 to 1.69) | 1.53 (0.73 to 3.20) | – | – |
| p for trend | 0.1 | 0.093 | <0.001 | 0.132 | <0.001 | 0.262 | – | – |
| Heterogeneity (p/I2) | 0.132/55.9% | 0.901/0.0% | 0.708/0.0% | 0.150/47.4% | 0.307/15.7% | 0.881/0.0% | – | – |
| HOMA-IR | ||||||||
| Sample size (cases/controls/cohort) | 519/5637/98 578 | 304/501/9133 | 2820/8821/342 711 | 136/–/4615 | 2769/8653/134 476 | 187/168/– | – | – |
| RR (95% CI) | 1.71 (1.26 to 2.32) | 1.99 (1.13 to 3.50) | 1.39 (1.17 to 1.66) | 2.36 (1.40 to 4.00) | 1.43 (1.21 to 1.7) | 2.3 (1.12 to 4.27) | – | – |
| p for trend | 0.001 | 0.018 | <0.001 | – | <0.001 | 0.024 | – | – |
| Heterogeneity (p/I2) | 0.394/0.0% | 0.749/0.0% | 0.320/14.0% | – | 0.082/48.8% | 0.860/0.0% | – | – |
| HbA1c | ||||||||
| Sample size (cases/controls/cohort) | 925/971/557 246 | 597/561/154 445 | 1766/2473/778 987 | 371/–/41 330 | – | – | 1093/1354/579 936 | 460/728/547 110 |
| RR (95% CI) | 1.03 (0.78 to 1.35) | 1.97 (0.54 to 7.21) | 1.23 (1.00 to 1.52) | 1.53 (0.68 to 3.43) | – | – | 1.18 (0.93 to 1.52) | 1.19 (0.79 to 1.80) |
| p For trend | 0.837 | 0.308 | 0.048 | 0.306 | – | – | 0.179 | 0.404 |
| Heterogeneity (p/I2) | 0.569/0.0% | 0.117/59.4% | 0.617/0.0% | 0.036/69.8% | – | – | 0.218/30.6% | 0.336/8.3% |
| C peptide | ||||||||
| Sample size (cases/controls/cohort) | 459/888/85 474 | 1233/2023/704 418 | – | – | 2721/3987/1 337 607 | 470/812/38 373 | 1731/2892/1 256 603 | 666/1532/1 158 373 |
| RR (95% CI) | 1.34 (0.67 to 2.70) | 1.52 (1.21 to 1.92) | – | – | 1.32 (1.10 to 1.59) | 1.27 (0.63 to 2.58) | 1.68 (1.32 to 2.12) | 0.85 (0.44 to 1.67) |
| p For trend | 0.412 | <0.001 | – | – | 0.003 | 0.506 | <0.001 | 0.643 |
| Heterogeneity (p/I2) | 0.060/64.4% | 0.109/47.1% | – | – | 0.135/36.8% | 0.026/72.6% | 0.225/26.7% | 0.542/0.0% |
CRC, colorectal cancer; HbA1c, glycated haemoglobin; HOMA-IR, homeostasis model of risk assessment-insulin resistance; RR, relative risk.
Fasting insulin and CRC: 10 studies involving 3239 patients with CRC and 354 870 participants assessed the association between fasting insulin level and the risk of CRC. Compared with the lowest categories of fasting insulin level, the pooled OR of CRC was 1.42 (95% CI 1.19 to 1.69; p<0.001) for the highest categories (figure 2B), indicating a significantly positive association. There was no significant heterogeneity across the included studies (I2=0.0%, p=0.571), as shown in figure 2B. The results of the dose–response analysis indicated a positive correlation between fasting insulin levels and the risk of CRC, which was an inverted U-shaped relationship (figure 3B).
Meta-regression and subgroup analysis (table 1) showed that the fasting insulin level was not significantly associated with CRC risk in either males (pooled RR=1.67, 95% CI 0.92 to 3.05) or females (pooled RR=1.53, 95% CI 0.92 to 2.55). For design types, fasting insulin level was found to be significantly associated with the risk of CRC in case–control (pooled RR=1.45, 95% CI 1.18 to 1.78) studies but not in cohort (pooled RR=1.32, 95% CI 0.92 to 1.89) studies. When stratifying the studies according to geographic region, the fasting insulin level was found to be significantly associated with CRC risk in American and European (pooled RR=1.41, 95% CI 1.17 to 1.69) but not Asian (pooled RR=1.53, 95% CI 0.73 to 3.20) populations.
HOMA-IR and CRC: Eight studies involving 2956 patients and 347 326 participants assessed the association between HOMA-IR and the risk of CRC. As compared with cancer risk for those with the lowest categories of HOMA-IR values, the pooled OR of CRC was 1.47 (95% CI 1.24 to 1.74; p<0.001) for those in the highest categories (figure 2C), indicating a significantly positive association. There was no significant heterogeneity across the included studies (I2=31.2%, p=0.169), as shown in figure 2C. The results of the dose–response analysis indicated a positive correlation between HOMA-IR value and the risk of CRC (figure 3C).
The meta-regression and subgroup analysis (table 1) showed that the HOMA-IR value was significantly associated with CRC risk in each subgroup for which it was calculated. Specifically, the HOMA-IR value was significantly associated with the risk of CRC in males (pooled RR=1.99, 95% CI 1.13 to 3.50) and females (pooled RR=1.71, 95% CI 1.26 to 2.32); in case–control (pooled RR=1.39, 95% CI 1.17 to 1.66) studies; and in American and European (pooled RR=1.43, 95% CI 1.21 to 1.70) and Asian (pooled RR=2.30, 95% CI 1.12 to 4.27) populations. These results indicated that as a composite index of fasting glucose and fasting insulin, HOMA-IR was a more reliable biomarker to evaluate the risk of CRC incidence than either of the individual biomarkers.
Association between HbA1c level and the risk of CRC
Eight studies including 2137 patients and 820 317 participants assessed the association between HbA1c levels and the risk of CRC. The pooled RR of CRC for the highest versus lowest categories of HbA1c level was 1.22 (95% CI 1.02 to 1.47; p=0.030), suggesting that the HbA1c level was associated with the risk of CRC with borderline significance (figure 4A). There was no significant heterogeneity across the included studies (I2=24.8%, p=0.232), as shown in figure 4A. The results of the dose–response analysis indicated a U-shaped relationship between HbA1c levels and the risk of CRC, as shown in figure 5A.
Figure 4.

The association between relative long-term markers of glucose metabolism and the risk of CRC. Pooled ORs of CRC for the highest versus lowest categories of (A) HbA1c level and (B) C peptide level. CC, colon cancer; CRC, colorectal cancer; F, female; HbA1c, glycated haemoglobin; M, male; RC, rectal cancer; RR, relative risk.
Figure 5.
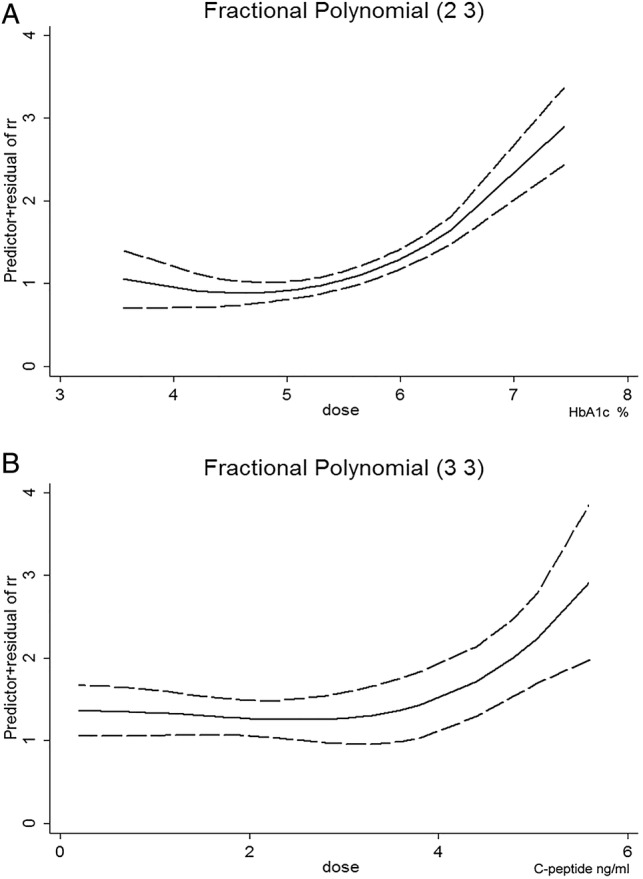
Dose–response relationship between long-term markers of glucose metabolism and the risk of CRC. Dose–response relationship between (A) HbA1c level and the risk of CRC and (B) C peptide level and the risk of CRC. The solid line represents the pooled RRs, and the dotted line represents the 95% CIs of the RRs. CRC, colorectal cancer; HbA1c, glycated haemoglobin; RR, relative risk.
Through a meta-regression and subgroup analysis (table 1), we found that the HbA1c level was not significantly associated with CRC risk in either males (pooled RR=1.97, 95% CI 0.54 to 7.21) and females (pooled RR=1.03, 95% CI 0.78 to 1.35). For study design subgroups, the HbA1c level was associated with CRC risk in case–control (pooled RR=1.23, 95% CI 1.00 to 1.52) studies with borderline significance but not in cohort (pooled RR=1.53, 95% CI 0.68 to 3.43) studies. For CRC subtypes, HbA1c level was not significantly associated with colon cancer (pooled RR=1.18, 95% CI 0.93 to 1.52) or rectal cancer (pooled RR=1.19, 95% CI 0.79 to 1.80).
Association between C peptide level and the risk of CRC
Nine studies involving 3191 patients and 1 375 980 participants assessed the association between C peptide level and the risk of CRC. The pooled RR of CRC for the highest versus lowest categories of C peptide level was 1.27 (95% CI 1.08 to 1.49; p=0.004), suggesting that the C peptide level was statistically associated with the risk of CRC (figure 4B). There was moderate heterogeneity across the included studies (I2=48.0%, p=0.037), as shown in figure 4B. The results of the dose–response analysis indicated a U-shaped relationship between C peptide levels and the risk of CRC, as shown in figure 5B.
The meta-regression and subgroup analysis (table 1) showed that C peptide level was significantly associated with CRC risk in males (pooled RR=1.52, 95% CI 1.21 to 1.92), but not in females (pooled RR=1.34, 95% CI 0.67 to 2.70). For cancer subtypes, C peptide level was significantly associated with colon cancer (pooled RR=1.68, 95% CI 1.32 to 2.12) but not rectal cancer (pooled RR=0.85, 95% CI 0.44 to 1.67). For geographic region, C peptide level was significantly associated with CRC in American and European (pooled RR=1.32, 95% CI 1.10 to 1.59) but not Asian (pooled RR=1.27, 95% CI 0.63 to 2.58) populations.
Publication bias
The PBegg and PEgger for fasting glucose were 0.012 and 0.004, respectively, which indicated significant publication bias, while there was no publication bias for fasting insulin (PBegg=0.876 and PEgger=0.698), HOMA-IR (PBegg=0.466 and PEgger=0.151), HbA1c (PBegg=0.386 and PEgger=0.508) and C peptide (PBegg=0.213 and PEgger=0.539). The funnel plots were shown in the online supplementary figures S1–S5.
We conducted further bias analysis according to the study design for fasting glucose. The publication bias results of cohort studies were PBegg=0.387 and PEgger=0.110, and the results of case–control studies were PBegg=0.009 and PEgger=0.001, which indicated that the main publication bias was from case–control studies.
bmjopen-2016-011430supp_figures.pdf (573.8KB, pdf)
bmjopen-2016-011430supp_table.pdf (642.7KB, pdf)
Discussion
Impaired glucose metabolism is one of the most common lifestyle-related disorders and has been linked to various chronic diseases including CRC.45 Therefore, the relationship between diabetes and the risk of CRC is biologically plausible.5 The current meta-analysis summarised the data from 35 studies on this topic that evaluated the association between markers of glucose metabolism and CRC risk in 25 566 patients and 5 706 361 participants. The results obtained indicated that the real-time composite index HOMA-IR has a stronger association with CRC risk than does fasting glucose or fasting insulin. Furthermore, HbA1c and C peptide, the relative long-term markers, were also valid predictors of CRC risk. Therefore, HOMA-IR levels may be a potential marker of CRC and early detection and intervention for controlling elevated glucose levels may be indicated as a way to prevent carcinogenesis.
Hyperglycaemia and circulating insulin concentration have been evaluated as potential CRC risk factors by many epidemiological studies. HOMA-IR is derived from paired serum insulin and glucose values, and this composite index may serve as an early indicator for evolving hyperinsulinaemia and/or hyperglycaemia.30 Meta-regression and subgroup analyses suggested that HOMA-IR was significantly associated with CRC risk in every subgroup that could be calculated, including gender, study designs and geographic region. This characteristic reflects the stability of the index and it may be appropriate for people from different countries to detect CRC occurrence. In contrast, fasting glucose was positively associated with risk of CRC in males, both in the case–control and cohort subgroups, in American and European and Asian populations and in colon cancer, while fasting insulin was only positively associated with the risk of CRC in case–control cohort studies and in American and European populations. Taken together, such results suggested that HOMA-IR is a more reliable composite index than the individual measurements with which to evaluate the association with CRC incidence. This conclusion was further strengthened by the dose–response analysis. Thus, it is recommended that HOMA-IR can be used as a biomarker for CRC risk in a clinical setting.
HbA1c is an integrated indicator of average blood glucose concentrations over a period of 6–8 weeks,14 which provides a more stable assessment as compared with serum glucose. In addition, C peptide has a relatively long half-life in the peripheral circulation and thus may also provide a more accurate assessment of overall insulin exposure.15 For these two relatively long-term markers of glucose metabolism, the pooled RRs for HbA1c and C peptide were 1.22 (95% CI 1.02 to 1.47; p=0.030) and 1.27 (95% CI 1.08 to 1.49; p=0.004), respectively. These results were consistent with those from real-time makers. Meta-regression and subgroup analysis showed that HbA1c was positively associated with the risk of CRC in case–control studies, while C peptide was significantly associated with CRC risk in males but not females, and in the colon cancer but not rectal cancer subgroup. The dose–response analyses also indicated positive correlations between these two markers and the risk of CRC.
Several pathophysiological and molecular mechanisms underlie the association between markers of glucose metabolism and colorectal carcinogenesis. Insulin resistance/hyperinsulinaemia and hyperglycaemia may induce colorectal carcinogenesis via inflammatory, oxidative stress and proliferative pathways,7 63 as shown in online supplementary figure S6.
Hyperglycaemia is the most prominent clinical symptom of diabetes. The interaction of hyperglycaemia-associated advanced glycation end products (AGEs) with their receptors leads to oxidative stress and inflammation.64 As a consequence, the inflammatory cytokines released, such as interleukin 6 and tumour necrosis factor-α, and transcription factors activated, including nuclear factor-κB and the signal transducer and activator of transcription (STAT) 3, all have the potential to promote colorectal carcinogenesis.65 66 In addition, inflammation-induced reactive oxygen species can damage cellular components such as DNA, proteins and lipids, all of which can directly or indirectly contribute to malignant cell transformation.65 67 Some novel regulators of gene expression, such as microRNAs, may also be involved in this process. For example, hyperglycaemia may cause endothelial dysfunction and angiogenesis in tumours by upregulating microRNA-467, resulting in tumour neoangiogenesis and malignant growth.68
Insulin exerts both metabolic and mitogenic effects through its interactions with the insulin receptor and the cognate type 1 insulin-like growth factor receptor (IGF-1R).69 The metabolic processes and glucose uptake functions of insulin proceed primarily through the PI3K/Akt pathway, while the mitogenic activity of insulin occurs primarily via the mitogen-activated protein kinases (MAPK) pathway.70 Insulin resistance and the consequently compensatory hyperinsulinaemia affect the metabolic pathways, overstimulate the mitogenic pathway and activate IGF-1R.71 Insulin-induced oncogenic activities include the activation of pathways involving multiple neoplastic protein kinases, including MAPK, extracellular signal-regulated kinase (ERK), PI3K/Akt and the mammalian target of rapamycin (mTOR), etc.72 In addition, insulin and IGF-1 can also activate Ras, which can then activate the mTOR and Wnt signalling pathway,73 74 leading to the increased sensitivity of colon cells to growth factors and accelerated progression from adenoma to carcinoma.72 As a result, the IGF-hyperinsulinaemia theory suggests that elevated insulin and free IGF-1 regulate metabolism and stimulate cellular growth and proliferation, reduce apoptosis and increase vascular endothelial growth factor levels as they do to CRC cell lines in vitro,75 76 thereby leading to a survival benefit and promoting colorectal carcinogenesis.72 77 Another possible molecular mechanism for the connection between insulin levels and CRC risk involves the hormone glucagon-like peptide 1 receptor (GLP-1), which is secreted by the intestinal endocrine L cells. Owing to insulin resistance, reduction of GLP-1 secretion causes compensatory activation of the Wnt pathway in combination with increased expression of oncogenes, such as c-Myc and cyclin D1, resulting in intestinal cell proliferation and CRC development.78
Determining the sources of heterogeneity is an important goal of meta-analysis. Meta-regression and subgroup analyses suggested that gender, study design, geographic region and cancer subtype can partially explain heterogeneity across the studies. However, for each study, only the median, midpoint or mean level of fasting glucose, fasting insulin, HOMA-IR, C peptide and HbA1c were obtained, and the related individual data were not available. Thus, a future individual participant data meta-analysis might help to further address this problem. The cut-off value and sample size of different studies and between different subgroups may also contribute to the heterogeneity. For example, in the subgroup analysis, the sample size for fasting glucose and C peptide in males was larger than in females, but it was the opposite for fasting insulin, HbA1c and HOMA-IR. The subgroup analysis results suggested that the gender difference was observed for fasting glucose and C peptide, while it was significant for HOMA-IR in men and women. Thus, the combined index HOMA-IR may be more stable than the individual markers, which further supported our conclusion.
Our study has several strengths. First, we adopted real-time markers (fasting glucose and fasting insulin), the composite marker (HOMA-IR) and long-term markers (HbA1c and C peptide) of glucose metabolism to thoroughly assess the association between markers of glucose metabolism with the risk of CRC. Second, the number of total participants was substantial, which enhanced the statistical power. Third, meta-regression, subgroup analysis and dose–response analyses were applied, which further strengthened the conclusions.
This study also has some limitations. For instance, the range of the cut-off points between the lowest and highest categories for fasting glucose, fasting insulin, HOMA-IR, HbA1c and C peptide levels differed between studies, which could influence the outcome of the current analyses. Besides, most of the included studies were case–control studies, which rely on recall for exposure measurement and are thus prone to bias. Thus, the inclusion of additional cohort studies has the potential to enhance future analysis. In addition, publication bias was observed in the fasting glucose analysis and it was caused by case–control studies. Thus, more cohort studies are warranted to enhance future analysis.
In conclusion, our findings demonstrate conclusively that markers of glucose metabolism are quantitatively associated with the risk of CRC. Furthermore, the real-time composite index HOMA-IR is more strongly associated with CRC risk than are either of the two individual markers (fasting glucose and fasting insulin). Considering the included case–control studies in the current analysis, more cohort studies are warranted to enhance future analysis.
Footnotes
Contributors: YW and DX contributed to the conception and design of the study. JX and YY contributed to the conception, design and editing of the manuscript. HW and HZ contributed to the statistical analysis. PL and JY contributed to the data acquisition, analysis and interpretation of the data. JH and PD-H contributed to the conception, design and editing of the manuscript.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Jemal A, Bray F, Center MM et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90. 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2.Whiting DR, Guariguata L, Weil C et al. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract 2011;94:311–21. 10.1016/j.diabres.2011.10.029 [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012;62:10–29. 10.3322/caac.20138 [DOI] [PubMed] [Google Scholar]
- 4.Beagley J, Guariguata L, Weil C et al. Global estimates of undiagnosed diabetes in adults. Diabetes Res Clin Pract 2014;103:150–60. 10.1016/j.diabres.2013.11.001 [DOI] [PubMed] [Google Scholar]
- 5.Larsson SC, Orsini N, Wolk A. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst 2005;97:1679–87. 10.1093/jnci/dji375 [DOI] [PubMed] [Google Scholar]
- 6.Deng L, Gui Z, Zhao L et al. Diabetes mellitus and the incidence of colorectal cancer: an updated systematic review and meta-analysis. Dig Dis Sci 2012;57:1576–85. 10.1007/s10620-012-2055-1 [DOI] [PubMed] [Google Scholar]
- 7.Onitilo AA, Engel JM, Glurich I et al. Diabetes and cancer II: role of diabetes medications and influence of shared risk factors. Cancer Causes Control 2012;23:991–1008. 10.1007/s10552-012-9971-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shikata K, Ninomiya T, Kiyohara Y. Diabetes mellitus and cancer risk: review of the epidemiological evidence. Cancer Sci 2013;104:9–14. 10.1111/cas.12043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giouleme O, Diamantidis MD, Katsaros MG. Is diabetes a causal agent for colorectal cancer? Pathophysiological and molecular mechanisms. World J Gastroenterol 2011;17:444–8. 10.3748/wjg.v17.i4.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran TT, Medline A, Bruce WR. Insulin promotion of colon tumors in rats. Cancer Epidemiol Biomarkers Prev 1996;5:1013–15. [PubMed] [Google Scholar]
- 11.Corpet DE, Jacquinet C, Peiffer G et al. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr Cancer 1997;27:316–20. 10.1080/01635589709514543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004;88:787–835, ix 10.1016/j.mcna.2004.04.013 [DOI] [PubMed] [Google Scholar]
- 13.Bonora E, Targher G, Alberiche M et al. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000;23:57–63. 10.2337/diacare.23.1.57 [DOI] [PubMed] [Google Scholar]
- 14.Bunn HF, Gabbay KH, Gallop PM. The glycosylation of hemoglobin: relevance to diabetes mellitus. Science 1978;200:21–7. 10.1126/science.635569 [DOI] [PubMed] [Google Scholar]
- 15.Van Cauter E, Mestrez F, Sturis J et al. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992;41:368–77. [DOI] [PubMed] [Google Scholar]
- 16.Bonser AM, Garcia-Webb P. C-peptide measurement: methods and clinical utility. Crit Rev Clin Lab Sci 1984;19:297–352. 10.3109/10408368409165766 [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Araki S, Tamura M et al. Relation of serum total cholesterol, serum triglycerides and fasting plasma glucose to colorectal carcinoma in situ. Int J Epidemiol 1998;27:794–8. 10.1093/ije/27.5.794 [DOI] [PubMed] [Google Scholar]
- 18.Platz EA, Hankinson SE, Rifai N et al. Glycosylated hemoglobin and risk of colorectal cancer and adenoma (United States). Cancer Causes Control 1999;10:379–86. 10.1023/A:1008953611657 [DOI] [PubMed] [Google Scholar]
- 19.Schoen RE, Tangen CM, Kuller LH et al. Increased blood glucose and insulin, body size, and incident colorectal cancer. J Natl Cancer Inst 1999;91:1147–54. 10.1093/jnci/91.13.1147 [DOI] [PubMed] [Google Scholar]
- 20.Kaaks R, Toniolo P, Akhmedkhanov A et al. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding proteins, and colorectal cancer risk in women. J Natl Cancer Inst 2000;92:1592–600. 10.1093/jnci/92.19.1592 [DOI] [PubMed] [Google Scholar]
- 21.Palmqvist R, Stattin P, Rinaldi S et al. Plasma insulin, IGF-binding proteins-1 and -2 and risk of colorectal cancer: a prospective study in northern Sweden. Int J Cancer 2003;107:89–93. 10.1002/ijc.11362 [DOI] [PubMed] [Google Scholar]
- 22.Saydah SH, Platz EA, Rifai N et al. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 2003;12:412–18. [PubMed] [Google Scholar]
- 23.Khaw KT, Wareham N, Bingham S et al. Preliminary communication: glycated hemoglobin, diabetes, and incident colorectal cancer in men and women: a prospective analysis from the European prospective investigation into cancer-Norfolk study. Cancer Epidemiol Biomarkers Prev 2004;13:915–19. [PubMed] [Google Scholar]
- 24.Ma J, Giovannucci E, Pollak M et al. A prospective study of plasma C-peptide and colorectal cancer risk in men. J Natl Cancer Inst 2004;96:546–53. 10.1093/jnci/djh082 [DOI] [PubMed] [Google Scholar]
- 25.Stattin P, Lukanova A, Biessy C et al. Obesity and colon cancer: does leptin provide a link? Int J Cancer 2004;109:149–52. 10.1002/ijc.11668 [DOI] [PubMed] [Google Scholar]
- 26.Jee SH, Ohrr H, Sull JW et al. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 2005;293:194–202. 10.1001/jama.293.2.194 [DOI] [PubMed] [Google Scholar]
- 27.Lin J, Ridker PM, Pradhan A et al. Hemoglobin A1c concentrations and risk of colorectal cancer in women. Cancer Epidemiol Biomarkers Prev 2005;14:3010–12. 10.1158/1055-9965.EPI-05-0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei EK, Ma J, Pollak MN et al. A prospective study of C-peptide, insulin-like growth factor-I, insulin-like growth factor binding protein-1, and the risk of colorectal cancer in women. Cancer Epidemiol Biomarkers Prev 2005;14:850–5. 10.1158/1055-9965.EPI-04-0661 [DOI] [PubMed] [Google Scholar]
- 29.Chung YW, Han DS, Park YK et al. Association of obesity, serum glucose and lipids with the risk of advanced colorectal adenoma and cancer: a case-control study in Korea. Dig Liver Dis 2006;38:668–72. 10.1016/j.dld.2006.05.014 [DOI] [PubMed] [Google Scholar]
- 30.Limburg PJ, Stolzenberg-Solomon RZ, Vierkant RA et al. Insulin, glucose, insulin resistance, and incident colorectal cancer in male smokers. Clin Gastroenterol Hepatol 2006;4:1514–21. 10.1016/j.cgh.2006.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rapp K, Schroeder J, Klenk J et al. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia 2006;49:945–52. 10.1007/s00125-006-0207-6 [DOI] [PubMed] [Google Scholar]
- 32.Jenab M, Riboli E, Cleveland RJ et al. Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer 2007;121:368–76. 10.1002/ijc.22697 [DOI] [PubMed] [Google Scholar]
- 33.Otani T, Iwasaki M, Sasazuki S et al. Plasma C-peptide, insulin-like growth factor-I, insulin-like growth factor binding proteins and risk of colorectal cancer in a nested case-control study: the Japan public health center-based prospective study. Int J Cancer 2007;120:2007–12. 10.1002/ijc.22556 [DOI] [PubMed] [Google Scholar]
- 34.Gunter MJ, Hoover DR, Yu H et al. Insulin, insulin-like growth factor-I, endogenous estradiol, and risk of colorectal cancer in postmenopausal women. Cancer Res 2008;68:329–37. 10.1158/0008-5472.CAN-07-2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rinaldi S, Rohrmann S, Jenab M et al. Glycosylated hemoglobin and risk of colorectal cancer in men and women, the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev 2008;17:3108–15. 10.1158/1055-9965.EPI-08-0495 [DOI] [PubMed] [Google Scholar]
- 36.Stocks T, Lukanova A, Johansson M et al. Components of the metabolic syndrome and colorectal cancer risk; a prospective study. Int J Obes (Lond) 2008;32:304–14. 10.1038/sj.ijo.0803713 [DOI] [PubMed] [Google Scholar]
- 37.Stocks T, Rapp K, Bjørge T et al. Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): analysis of six prospective cohorts. PLoS Med 2009;6:e1000201 10.1371/journal.pmed.1000201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakajima TE, Yamada Y, Hamano T et al. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci 2010;101:1286–91. 10.1111/j.1349-7006.2010.01518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto S, Nakagawa T, Matsushita Y et al. Visceral fat area and markers of insulin resistance in relation to colorectal neoplasia. Diabetes Care 2010;33:184–9. 10.2337/dc09-1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stocks T, Lukanova A, Bjørge T et al. Metabolic factors and the risk of colorectal cancer in 580,000 men and women in the metabolic syndrome and cancer project (Me-Can). Cancer 2011;117:2398–407. 10.1002/cncr.25772 [DOI] [PubMed] [Google Scholar]
- 41.Wu K, Feskanich D, Fuchs CS et al. Interactions between plasma levels of 25-hydroxyvitamin D, insulin-like growth factor (IGF)-1 and C-peptide with risk of colorectal cancer. PLoS ONE 2011;6:e28520 10.1371/journal.pone.0028520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen MW, Ye S, Zhao LL et al. Association of plasma total and high-molecular-weight adiponectin with risk of colorectal cancer: an observational study in Chinese male. Med Oncol 2012;29:3129–35. 10.1007/s12032-012-0280-2 [DOI] [PubMed] [Google Scholar]
- 43.Dankner R, Shanik MH, Keinan-Boker L et al. Effect of elevated basal insulin on cancer incidence and mortality in cancer incident patients: the Israel GOH 29-year follow-up study. Diabetes Care 2012;35:1538–43. 10.2337/dc11-1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ollberding NJ, Cheng I, Wilkens LR et al. Genetic variants, prediagnostic circulating levels of insulin-like growth factors, insulin, and glucose and the risk of colorectal cancer: the Multiethnic Cohort study. Cancer Epidemiol Biomarkers Prev 2012;21:810–20. 10.1158/1055-9965.EPI-11-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wulaningsih W, Garmo H, Holmberg L et al. Serum lipids and the risk of gastrointestinal malignancies in the Swedish AMORIS Study. J Cancer Epidemiol 2012;2012:792034 10.1155/2012/792034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hakozaki Y, Mitani K, Kouichi T et al. Association of laboratory data on lifestyle-related factors with the risk of colorectal adenoma and cancer: a retrospective cohort study with long-term follow-up total colonoscopy. J Gastroenterol Hepatol (Hong Kong) 2013;2:618–22. [Google Scholar]
- 47.Parekh N, Lin Y, Vadiveloo M et al. Metabolic dysregulation of the insulin-glucose axis and risk of obesity-related cancers in the Framingham heart study-offspring cohort (1971–2008). Cancer Epidemiol Biomarkers Prev 2013;22:1825–36. 10.1158/1055-9965.EPI-13-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erarslan E, Coşkun Y, Türkay C et al. IGF-I levels and visceral fat accumulation in colonic neoplasia. Clin Res Hepatol Gastroenterol 2014;38:99–105. 10.1016/j.clinre.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 49.Kabat GC, Kim MY, Strickler HD et al. A longitudinal study of serum insulin and glucose levels in relation to colorectal cancer risk among postmenopausal women. Br J Cancer 2012;106:227–32. 10.1038/bjc.2011.512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stattin P, Björ O, Ferrari P et al. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007;30:561–7. 10.2337/dc06-0922 [DOI] [PubMed] [Google Scholar]
- 51.Shin HY, Jung KJ, Linton JA et al. Association between fasting serum glucose levels and incidence of colorectal cancer in Korean men: the Korean Cancer Prevention Study-II. Metab Clin Exp 2014;63:1250–6. 10.1016/j.metabol.2014.07.006 [DOI] [PubMed] [Google Scholar]
- 52.Stroup DF, Berlin JA, Morton SC et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000;283:2008–12. [DOI] [PubMed] [Google Scholar]
- 53.Moher D, Liberati A, Tetzlaff J et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009;6:e1000097 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matthews DR, Hosker JP, Rudenski AS et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–19. 10.1007/BF00280883 [DOI] [PubMed] [Google Scholar]
- 55.Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol 2010;25:603–5. 10.1007/s10654-010-9491-z [DOI] [PubMed] [Google Scholar]
- 56.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–88. 10.1016/0197-2456(86)90046-2 [DOI] [PubMed] [Google Scholar]
- 57.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–58. 10.1002/sim.1186 [DOI] [PubMed] [Google Scholar]
- 58.Higgins JP, Thompson SG, Deeks JJ et al. Measuring inconsistency in meta-analyses. BMJ 2003;327:557–60. 10.1136/bmj.327.7414.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higgins JP, Thompson SG. Controlling the risk of spurious findings from meta-regression. Stat Med 2004;23:1663–82. 10.1002/sim.1752 [DOI] [PubMed] [Google Scholar]
- 60.Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50:1088–101. 10.2307/2533446 [DOI] [PubMed] [Google Scholar]
- 61.Hayashino Y, Noguchi Y, Fukui T. Systematic evaluation and comparison of statistical tests for publication bias. J Epidemiol 2005;15:235–43. 10.2188/jea.15.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Royston P. A strategy for modelling the effect of a continuous covariate in medicine and epidemiology. Stat Med 2000;19:1831–47. [DOI] [PubMed] [Google Scholar]
- 63.Vigneri P, Frasca F, Sciacca L et al. Diabetes and cancer. Endocr Relat Cancer 2009;16:1103–23. 10.1677/ERC-09-0087 [DOI] [PubMed] [Google Scholar]
- 64.Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: the role of multiligand/RAGE axis. Carcinogenesis 2010;31:334–41. 10.1093/carcin/bgp322 [DOI] [PubMed] [Google Scholar]
- 65.Mantovani A, Allavena P, Sica A et al. Cancer-related inflammation. Nature 2008;454:436–44. 10.1038/nature07205 [DOI] [PubMed] [Google Scholar]
- 66.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009;9:798–809. 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle 2009;8:3267–73. 10.4161/cc.8.20.9699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhattacharyya S, Sul K, Krukovets I et al. Novel tissue-specific mechanism of regulation of angiogenesis and cancer growth in response to hyperglycemia. J Am Heart Assoc 2012;1:e005967 10.1161/JAHA.112.005967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Menting JG, Whittaker J, Margetts MB et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013;493:241–5. 10.1038/nature11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sciacca L, Vigneri R, Tumminia A et al. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis 2013;23:808–15. 10.1016/j.numecd.2013.05.006 [DOI] [PubMed] [Google Scholar]
- 71.Cusi K, Maezono K, Osman A et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest 2000;105:311–20. 10.1172/JCI7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berster JM, Göke B. Type 2 diabetes mellitus as risk factor for colorectal cancer. Arch Physiol Biochem 2008;114:84–98. 10.1080/13813450802008455 [DOI] [PubMed] [Google Scholar]
- 73.Sun J, Jin T. Both Wnt and mTOR signaling pathways are involved in insulin-stimulated proto-oncogene expression in intestinal cells. Cell Signal 2008;20:219–29. 10.1016/j.cellsig.2007.10.010 [DOI] [PubMed] [Google Scholar]
- 74.Yi F, Sun J, Lim GE et al. Cross talk between the insulin and Wnt signaling pathways: evidence from intestinal endocrine L cells. Endocrinology 2008;149:2341–51. 10.1210/en.2007-1142 [DOI] [PubMed] [Google Scholar]
- 75.Björk J, Nilsson J, Hultcrantz R et al. Growth-regulatory effects of sensory neuropeptides, epidermal growth factor, insulin, and somatostatin on the non-transformed intestinal epithelial cell line IEC-6 and the colon cancer cell line HT 29. Scand J Gastroenterol 1993;28:879–84. 10.3109/00365529309103129 [DOI] [PubMed] [Google Scholar]
- 76.Giovannucci E. Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr 2001;131(11 Suppl): 3109S–20S. [DOI] [PubMed] [Google Scholar]
- 77.Smith CJ, McKay GA, Fisher M. Diabetes, colorectal cancer and cyclooxygenase 2 inhibition. Int J Clin Pract 2008;62:810–15. 10.1111/j.1742-1241.2008.01726.x [DOI] [PubMed] [Google Scholar]
- 78.Jin T. Why diabetes patients are more prone to the development of colon cancer? Med Hypotheses 2008;71:241–4. 10.1016/j.mehy.2008.03.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2016-011430supp.pdf (521.3KB, pdf)
bmjopen-2016-011430supp_figures.pdf (573.8KB, pdf)
bmjopen-2016-011430supp_table.pdf (642.7KB, pdf)