

Abstract
Microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1) is an uncommon cause of microcephaly and intrauterine growth retardation in a newborn. Early identifying features include but are not limited to sloping forehead, micrognathia, sparse hair, including of eyebrows and short limbs. Immediate radiological findings may include partial or complete agenesis of the corpus callosum, interhemispheric cyst and shallow acetabula leading to dislocation. Genetic testing displaying a mutation in RNU4ATAC gene is necessary for definitive diagnosis. Early identification is important as MOPD1 is an autosomal recessive condition and could present in subsequent pregnancies. The purpose of this case is to both identify and describe some common physical findings related to MOPD1. We present a case of MOPD1 in a girl born to non-consanguineous parents that was distinct for subglottic stenosis and laryngeal cleft.
Background
Microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1) is a rare autosomal recessive bone dysplasia characterised by intrauterine and postnatal growth restriction, microcephaly, partial or complete agenesis of the corpus callosum, dwarfism and other neurological abnormalities.1 Patients with MOPD1 typically present with short limbs, dislocation of hips and elbows, sparse hair, including of eyebrows, seizures and facial abnormalities (sloping forehead, prominent eyes and micrognathia).2
Genetic research has shown that the cause of MOPD1 is a mutation in the RNU4ATAC gene, which encodes for U4atac, an RNA component of the minor spliceosome.3 Prognosis for MOPD1 is poor and life expectancy is typically <1 year.
This case highlights the identifying features and genetics related to successfully diagnosing a case of MOPD1. The rarity of this disease and overlapping features with other forms of primordial dwarfism make definitive diagnosis challenging.
Case presentation
In the pregnancy leading to the birth of our patient, ultrasonography performed at 31 weeks was notable for oligohydramnios, with some concern for both microcephaly and intrauterine growth restriction. Follow-up ultrasound 1 month later showed no further growth and the mother was referred to the local hospital for fetal non-stress testing. As the fetal non-stress test was non-reactive and umbilical artery Doppler velocimetry revealed decreased blood flow, caesarean section was performed to deliver the infant girl at 36 and 5/7 weeks.
Notable physical features at birth were low-set dysplastic ears, absence of scalp hair, eyebrows and eyelashes, microcephaly, micrognathia (figure 1), bilateral elbow and knee contractures (figure 2), ulnar deviation of fingers, short and abnormally tapered fingers (figure 3), single palmar crease, and oedema of the hands and dorsal aspect of the feet.
Figure 1.

Patient displays micrognathia, dysplastic ears and complete lack of hair.
Figure 2.
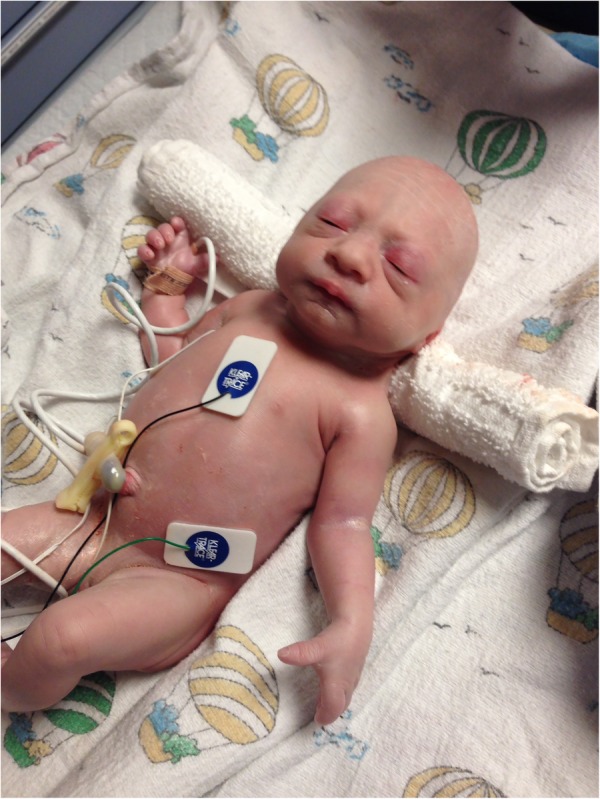
Patient displays limb contractures.
Figure 3.

Patient displays short, tapered fingers and prominent eyes.
The infant had a birth weight of 1620 g, length of 38 cm and a head circumference of 28.5 cm, placing her well below the 3rd centile in each of those categories. APGAR scores of 8 and 9 were recorded at 1 and 5 min, respectively. The patient required no supplemental oxygen or mechanical ventilation. She did develop hypoglycaemia and hyperbilirubinaemia. Owing to poor motor function and a weak swallow reflex, a nasogastric tube was placed and tube feedings followed. She was discharged 2 weeks later.
Given the physical findings, the presumptive diagnosis was primordial dwarfism of unknown subtype. Genetic testing at 11 months confirmed MOPD1 with homozygous mutation, 51 G>A, of the RNU4ATAC gene.
Investigations
MRI revealed partial agenesis of the corpus callosum (figure 4), and a small interhemispheric cyst (figure 5) that may have been communicating with the third ventricle. Abdominal ultrasound and EEG indicated no abnormalities.
Figure 4.

MRI demonstrating partial agenesis of the corpus callosum.
Figure 5.
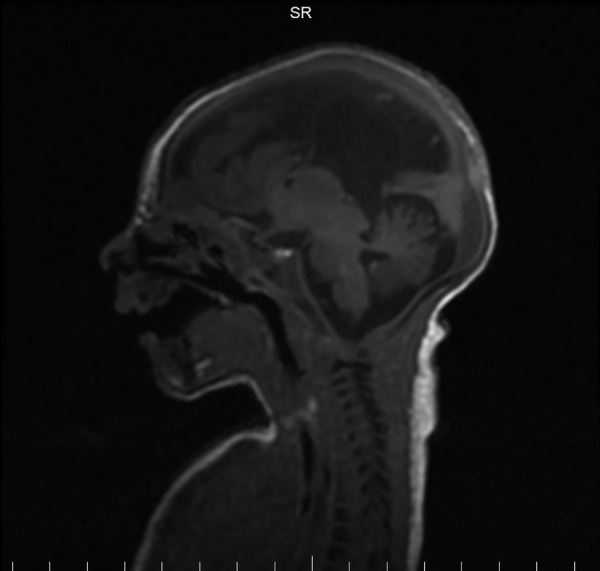
MRI demonstrating interhemispheric cyst.
Transthoracic echocardiogram showed an ostium secundum atrial septal defect. Later healthcare visits revealed bilateral femoral heads subluxated in the cephalad direction, bilateral acetabula that were abnormally shallow with blunt angles at the ilia, a laryngeal cleft type 1 and Cotton-Meyer grade 2 subglottic stenosis.
Differential diagnosis
Differential diagnosis of MOPD1 includes: Seckel dwarfism, MOPD II, Russell-Silver syndrome and Meier-Gorlin syndrome.
Treatment
Treatment for MOPD1 is supportive only. Genetic counselling for the parents is recommended before considering further pregnancies.
Outcome and follow-up
Prognosis is poor with most patients dying within the first year. Follow-up revealed our patient is currently aged 2 years.
Discussion
MOPD1 is a rare condition with an unknown incidence, but is likely <1 in 1 000 000 live births.2 This disorder is autosomal recessive and is diagnosed with an RNU4ATAC mutation on chromosome 2q14.2.3 In the few published cases, many neonates with MOPD1 were born to consanguineous parents; however, sporadic cases relating to non-consanguineous parents have been documented. The classic presentation of MOPD1 is intrauterine and postnatal growth retardation, microcephaly, partial or complete agenesis of the corpus callosum, short limbs, dislocation of hips and elbows, sparse hair, including of eyebrows, seizures and facial abnormalities.1 2 This presentation should lead the healthcare provider to seek further imaging and genetic testing for diagnosis. Cardinal brain MRI features in MOPD1 include gyration abnormalities, callosal body malformations and interhemispheric cysts.4 While the physical examination and imaging studies are important in determining the severity of dysfunction, genetic testing is currently the only definitive way to diagnose MODP1. Owing to its rarity, some cases of MOPD1 may either go undiagnosed or misdiagnosed. In our case, genetic testing at 11 months confirmed the diagnosis of MOPD1.
Given the rarity of this genetic mutation, establishing a list of frequent physical findings, leading to genetic testing, is important for early diagnosis. Our patient's presentation shows similarities with other published cases. Abdel-Salam et al4 listed 14 cases of homozygous 51 G>A mutations. These patients displayed four main neurological characteristics including gyral pattern abnormalities, corpus callosum abnormalities, cerebellar vermis hypoplasia and interhemispheric cyst. Abnormal gyral pattern was reported in 43% and another 43% reported pachygyria. Agenesis of the corpus callosum was found in 86% while 14% presented with hypogenesis of the corpus callosum. Interhemispheric cyst was present in 36% and another 36% had cerebellar vermis hypoplasia.4 Our patient presented with interhemispheric cyst and partial agenesis of the corpus callosum. Other similar cases include that by Abolila et al,5 notable for pachygyria, agenesis of the corpus callosum and interhemispheric cyst without cerebellar vermis hypoplasia; mutational analysis was not performed in that case. Edery et al6 described multiple cases in an Algerian family with a 51 G>A mutation that also presented with pachygyria, agenesis of the corpus callosum and interhemispheric cyst.
Other physical findings in MOPD1 include cerebellar vermis hypoplasia, lack of retinal pigmentation and polymicrogyria.4 6 These variations in presentation of MOPD1 and an overall paucity of information regarding the syndrome can lead to a delayed diagnosis. While early diagnosis does not change the outcome of this fatal genetic disease, early detection is essential to raise awareness and educate the parents on the risk of future children having a similar outcome.
In summary, while many of the physical findings are similar to other cases of MOPD1, our case is distinct for laryngeal cleft type 1 and Cotton-Meyer grade 2 subglottic stenosis. Aside from physical findings, genetic testing revealing a mutation of the RNU4ATAC gene is required to differentiate and diagnose MOPD1.3
Patient's perspective.
‘My daughter has been one of the greatest blessings in my life. She has taught me so many life lessons and has helped me become the person I have always wanted to be. Even though she was sent to Earth in an imperfect body, her strong personality and spirit shine through. I am grateful every day that she was sent to our family.’
Learning points.
Genetic counselling is important for future family planning.
Microcephalic osteodysplastic primordial dwarfism (MOPD1) is a rare cause of microcephaly and intrauterine growth restriction occurring most commonly in births to consanguineous parents.
MOPD1 can only be confirmed with genetic testing revealing a mutation of RNU4ATAC.
Footnotes
Contributors: SF, AJ and WP conducted the primary research and were involved in writing of this case report.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kniffin C, O'Neill M, Hamosh A et al. OMIM Entry—# 210710—MICROCEPHALIC OSTEODYSPLASTIC PRIMORDIAL DWARFISM, TYPE I; MOPD1 [Internet]. Omim.org 2016. [cited 7 March 2016]. http://www.omim.org/entry/210710#24
- 2.Orphanet: Microcephalic osteodysplastic primordial dwarfism types I and III. Orpha.net 2016. [cited 30 March 2016]. http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=2636
- 3.He H, Liyanarachchi S, Akagi K et al. Mutations in U4atac snRNA, a Component of the Minor Spliceosome, in the Developmental Disorder MOPD I. Science 2011;332:238–40. 10.1126/science.1200587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Salam G, Abdel-Hamid M, Hassan N et al. Further delineation of the clinical spectrum in RNU 4 ATAC related microcephalic osteodysplastic primordial dwarfism type I. Am J Med Genet 2013;161:1875–81. 10.1002/ajmg.a.36009 [DOI] [PubMed] [Google Scholar]
- 5.Abolila R, Alsawan R, Alrefaie M. Microcephalic osteodysplastic primordial dwarfism (MOPD) type I with lissencephaly and brain cyst. Egypt J Med Hum Genet 2012;13:363–5. 10.1016/j.ejmhg.2012.06.005 [DOI] [Google Scholar]
- 6.Edery P, Marcaillou C, Sahbatou M et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 2011;332:240–3. 10.1126/science.1202205 [DOI] [PubMed] [Google Scholar]