

Abstract
A 61-year-old woman presented with longstanding cough and progressive dyspnoea. She underwent an extensive evaluation and was diagnosed with common variable immunodeficiency (CVID) with granulomatous lymphocytic interstitial lung disease (GLILD). She was initially treated with subcutaneous immunoglobulin therapy, having declined intravenous immunoglobulin (IVIG) therapy. She also declined treatment with oral glucocorticoids. Over several months, she became increasingly symptomatic and developed increased pulmonary infiltrates, pleural effusions, mediastinal adenopathy, splenomegaly, pancytopenia and ascites. An interdisciplinary team composed of an immunologist, pulmonologist and haematologist deliberated over a therapeutic management approach. The patient received a recently reported immunotherapy regimen with azathioprine and rituximab. The therapy led to rapid improvement of her constitutional and respiratory symptoms, with clinical and radiographic improvement in her interstitial lung disease, lymphadenopathy, pleural effusions and ascites. This case report reviews the literature surrounding the diagnosis and management of GLILD.
Background
Common variable immune deficiency (CVID) results in low levels of most or all immunoglobulin classes, a defective host antibody response and frequent bacterial infections.1 About one-third of patients with CVID develop granulomatous lymphocytic interstitial lung disease (GLILD).2 GLILD is a rare, non-infectious complication that significantly increases mortality risk in patients who have common variable immunodeficiency.3 There are limited data describing appropriate management strategies in patients who develop this condition.1 There have been mixed reports about the utility of intravenous immunoglobulin therapy to help control granulomatous changes.4 5 Proposed management strategies have included immunosuppressant medications such as corticosteroids, however, there is no clear standard of treatment.6 A retrospective review of seven patients demonstrates improvement with the use of rituximab and azathioprine.1 We report the clinical course of a patient with GLILD treated with rituximab and azathioprine, and review the current literature about the condition.
Case presentation
A 61-year-old non-smoking woman with a history of right-sided breast cancer (stage 1A treated with right mastectomy and elective left mastectomy followed by doxorubicin and cyclophosphamide 13 years prior) had been diagnosed with CVID and GLILD ∼1 year earlier. She presented for follow-up in clinic, with gradually worsening fatigue, shortness of breath, night sweats, pancytopenia and abdominal distension, despite immunoglobulin replacement therapy for 1 year. Physical examination revealed diffuse bilateral crackles, abdominal distention with a fluid shift and splenomegaly.
Investigations
The patient had undergone an extensive evaluation on initial diagnosis of CVID ∼1 year prior to her visit. Her laboratory studies at that time had demonstrated markedly reduced levels of IgG=58 mg/dL (normal 716–1554 mg/dL), IgM=7 mg/dL (normal 45–259 mg/dL) and IgA <5 mg/dL (normal 71–377 mg/dL). CT of her chest with intravenous contrast revealed diffuse scattered ground glass and nodular consolidations, bronchiectasis and multiple enlarged mediastinal and hilar lymph nodes bilaterally. Transbronchial lung biopsy was performed demonstrating lymphocytic bronchiolitis, interstitial pneumonitis and focal organising pneumonia, and minute granulomas (figures 1–4). Fungal, viral and acid fast cultures were negative. There was no evidence of malignancy. Polyclonal B cells were noted. Pulmonary function tests revealed a mild restrictive pattern with a moderately reduced diffusing capacity.
Figure 1.
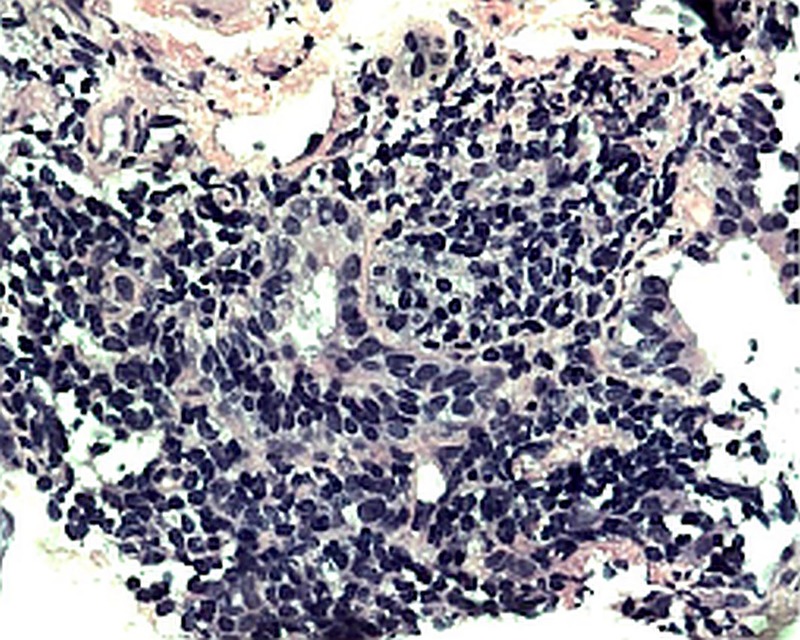
Mild lymphocytic infiltrate involving small airways and swollen alveolar septae consistent with lymphocytic bronchiolitis.
Figure 2.
Inflammatory changes in the interstitium consistent with interstitial pneumonitis.
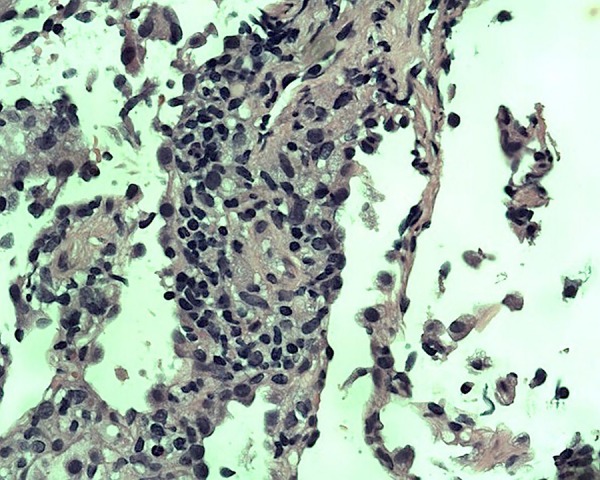
Figure 3.
Presence of organising pneumonia due to granulation tissue that fills bronchiolar and alveolar lumen.
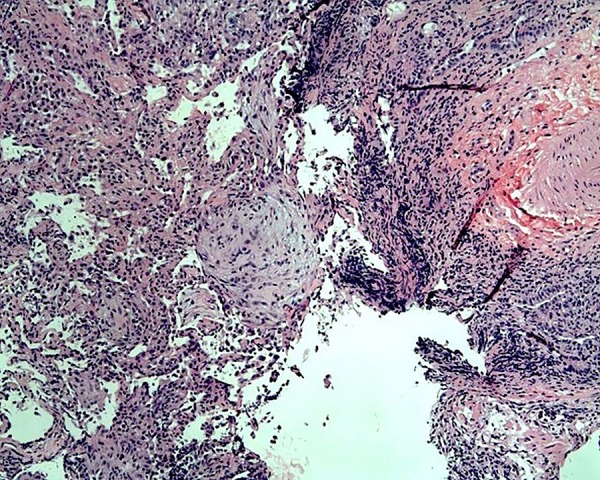
Figure 4.

Organised collection of macrophages composing minute granulomata.
Owing to the recent worsening of her symptoms, the patient was admitted to the hospital for further evaluation. Her laboratory studies demonstrated normal IgG levels after IVIG therapy. She continued to have low IgM and IgA levels. She also had significant leucopenia with a white blood cell count of 1800 cells/mL and thrombocytopenia with a platelet count of 52 000 cells/mL.
CT of the chest, abdomen and pelvis with intravenous contrast revealed worsening cervical, mediastinal and hilar lymphadenopathy, new bilateral pleural effusions and bilateral lower lobe bronchiectasis with peribronchial thickening (figure 5). There was patchy ground glass appearing foci and solid parenchymal nodules in the lung bases. A new hypoenhancing lesion was also noted in the liver. There was marked splenomegaly (23 cm) and worsening retroperitoneal, periaortic, pelvic and inguinal lymphadenopathy. The imaging also demonstrated new ascites (figure 6).
Figure 5.
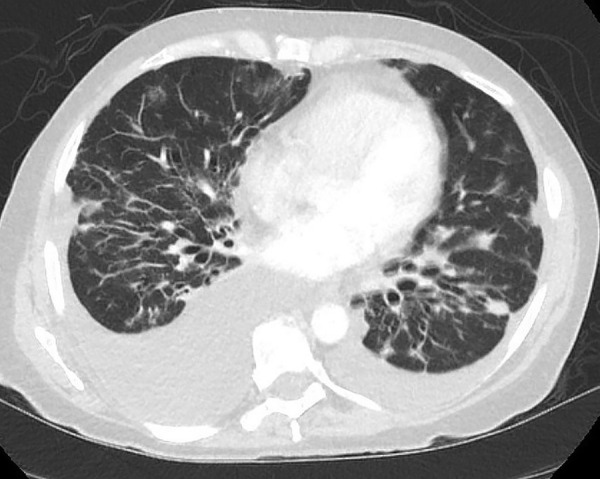
CT of the chest demonstrating bilateral pleural effusions and peribronchial thickening.
Figure 6.
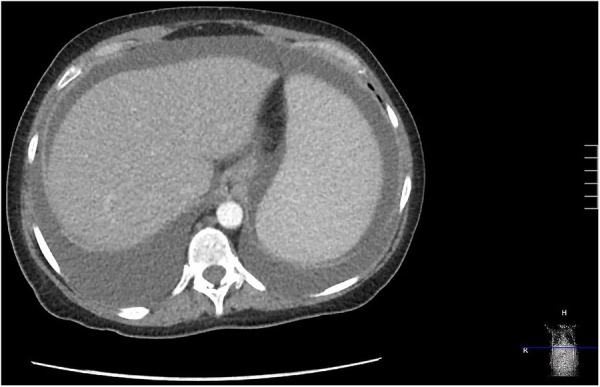
CT of the abdomen demonstrating splenomegaly and ascites.
Differential diagnosis
Given the new onset of ascites, pleural effusions and night sweats, there was concern for malignancy, particularly lymphoma. Patients with CVID have demonstrated increased risk for the development of lymphoma.7 Several additional studies and procedures were completed in our patient to evaluate for lymphoproliferative disease. Repeat transbronchial subcarinal node biopsy showed some atypical B cells, but cytogenetic studies were negative for lymphoma. Flow cytometry did not suggest a lymphoproliferative disorder. Bone marrow biopsy demonstrated normocellular bone marrow with no overt evidence of acute leukaemia or lymphoproliferative disorder and normal cytogenetics. Ascitic fluid revealed a small percentage of a B cell clone of uncertain significance, but cytogenetic studies were negative for lymphoma. The serum-ascites albumin gradient was elevated at 1.9, consistent with a non-peritoneal cause of ascites. We suspected the development of the ascites was due to hepatic involvement of this granulomatous process. Tissue was not obtained because the patient declined a liver biopsy. Pleural fluid did not demonstrate any clonal cell population. Bronchoalveolar lavage was negative for infection and malignant cells. Left retroperitoneal lymph node biopsy was also negative for lymphoma and demonstrated scattered non-caseating granulomata. This thorough evaluation reassured the management team that this patient did not have a lymphoproliferative disease contributing to her condition.
The diagnosis of GLILD is based on a constellation of certain clinical and histological characteristics. However, some of these features are shared with other conditions that must be excluded when considering the diagnosis of GLILD. Infection may be excluded from the differential diagnosis with laboratory studies and complete culture analysis. Bronchiolitis obliterans organising pneumonia is a complication of graft versus host disease, but can develop in CVID for unclear reasons.8 It presents with fatigue, fever, cough and new areas of consolidation on thoracic imaging, and improves with corticosteroids.9 GLILD may also be confused with sarcoidosis; the median age for both conditions occurs in the fourth decade. Non-necrotising granulomas are characteristic for both conditions; however, lymphoproliferative pneumonitis and follicular bronchiolitis are distinct components of GLILD.10
Treatment
The patient was started on daily oral azathioprine therapy. Owing to her thrombocytopenia, she was started on half the dose of azathioprine, which was then increased after 2 months to 1.5 mg/kg. She was also started on rituximab infusions at 375 mg/m2/infusion, administered each week for 4 weeks every 4–6 months.
Outcome and follow-up
The patient experienced dramatic improvement in her symptoms since initiation of her therapy and did not require hospitalisation in the following year. Imaging after 3 months demonstrated resolution of her pleural effusions, reduction in retroperitoneal lymphadenopathy and stable splenomegaly (figures 7 and 8). Lateral views of chest radiographs before and after therapy demonstrate remarkable improvement (figure 9A,B). While a repeat CT chest would have satisfied academic curiosity, the authors determined this was not necessary for management since the patient's clinical condition and chest X-rays demonstrated significant improvement. Her laboratory work also showed significant improvement in her thrombocytopenia and stable leucopenia.
Figure 7.
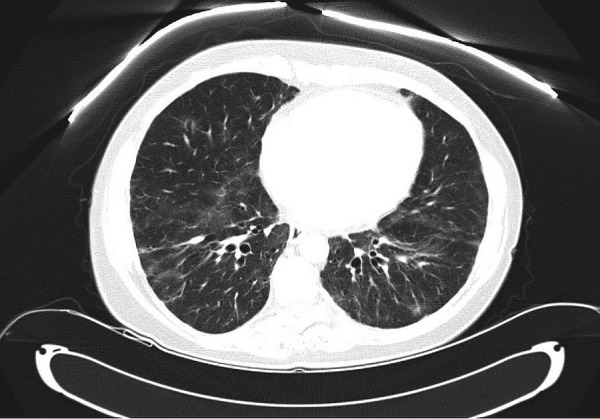
CT of the chest demonstrating resolution of bilateral pleural effusions 3 months after initiation of rituximab and azathioprine.
Figure 8.
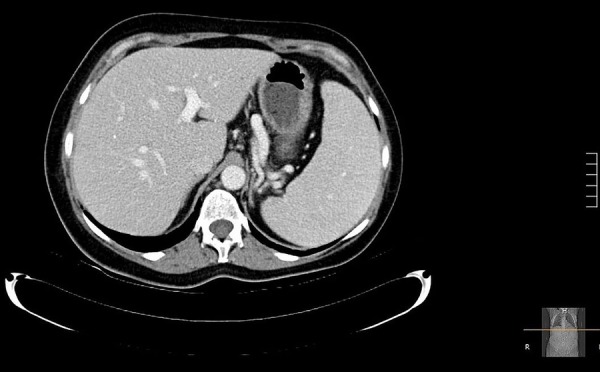
CT of the abdomen with contrast demonstrating resolution of ascites after 3 months of therapy.
Figure 9.

(A and B) Lateral views of chest X-ray images before and after 3 months of therapy.
Discussion
CVID is a primary immunodeficiency consisting of hypogammaglobulinaemia. First recognised in 1953, CVID is due to failure of B cell differentiation and causes a decrease in the number of plasma cells, yet the aetiology of this immunological failure is unknown in most cases.11 CVID is diagnosed with low levels of IgG, IgA and/or IgM, with poor antibody response to vaccination.11 The average age of diagnosis in the USA ranges between 20 and 40 years, with ∼6–7 years of delay in diagnosis,11 while data from the European Society for Immunodeficiencies indicate that 34% of patients with CVID are diagnosed before the age of 10 years. CVID has a prevalence of 1:25 000 to 1:50 000.11 The condition is a diagnosis of exclusion and other aetiologies for hypogammaglobulinaemia must be excluded.1 Patients generally have recurrent infections, typically involving the sinopulmonary and gastrointestinal systems.11 Treatment with IVIG helps reduce the rates of infection in these patients.1 In addition to B-cell dysregulation, CVID also manifests with abnormalities in T cells.12 Lymphoproliferative disease and autoimmune conditions such as idiopathic thrombocytopenic purpura can develop in patients with CVID.13 There are several notable non-infectious complications. Risk of mortality is noted to be 11 times higher for patients with non-infectious complications such as lymphoma, functional or structural lung disease, hepatitis and gastrointestinal disease.11
Granulomatous lymphocytic interstitial lung disease
About one-third of patients with CVID will develop clinically significant interstitial lung disease.2 The diagnosis of GLILD is made with the presence of non-necrotising granulomas, lymphocytic interstitial lung disease and follicular bronchiolitis on histological examination.6 Patients also develop diffuse adenopathy, splenomegaly and non-caseating granulomas.1 GLILD reportedly causes an increase in morbidity and mortality, with the mean survival of patients with GLILD averaging half that of patients without GLILD.3 There is still no standard of care in the management of patients with GLILD.1
Management of GLILD
GLILD in CVID is an entity that warrants further study and clinical characterisation. Owing to the rarity of the disease, it is difficult to implement a prospective randomised control trial demonstrating the efficacy of a treatment protocol in any single institution. Corticosteroids have been commonly used without established standard management guidelines.6 Patients often demonstrate improvement in symptoms followed by relapse with cessation of steroids.6
In a retrospective study by Chase et al,1 seven patients were evaluated after using azathioprine and rituximab. All seven patients experienced significant symptom improvement, as well as improvement in pulmonary function and parenchymal abnormalities.1 In our patient, the use of this immunotherapy combination effectively managed symptoms and significantly improved her thrombocytopenia. A controlled study is needed using azathioprine and rituximab in patients afflicted with GLILD in the setting of CVID. A nationwide endeavour for treated patients to be observed over time would also aid in understanding the sequela of this therapy as well as development of a standardised treatment regimen for GLILD.
Patient's perspective.
I am grateful to my University of Florida (UF) Health medical team—Immunologist Juan Guarderas, MD; Haematologist Marc Zumberg, MD; and Pulmonologist Daniel Urbine, MD—for their expertise and their willingness to collaborate with each other, with their associates at other academic medical centres and with me. I am grateful for the communications technologies that facilitate these interactions. In the midst of a bewildering health condition, I continue to feel that a team of caring, committed experts is supporting me. My physicians have been open to new protocols, including my self-administered subcutaneous infusions of immunoglobulin, an option I investigated and proposed. With few guidelines available for patients with my condition, I have learned to listen to my body and to be aware, for example, of my compromised energy level, and to manage it. It is important to for me to be a participating partner on the team that makes my treatment decisions. The protocol of daily Imuran tablet and weekly intravenous infusions of Rituximab for 4 weeks every 6 months seems to be working: my lungs remain clear (uncongested). This feels like a miracle.
Learning points.
Common variable immunodeficiency (CVID) is a primary immunodeficiency that consists of hypogammaglobulinaemia and poor host antibody response. The condition has infectious and non-infectious complications, with the latter as a consequence of immune dysregulation.
IVIG is the mainstay of therapy to prevent infectious complications, but non-infectious complications need to often be managed with immunosuppressive therapy.
Granulomatous lymphocytic interstitial lung disease (GLILD) is a rare, non-infectious complication of CVID that is reportedly responding to therapy with azathioprine and rituximab.
Lymphadenopathy, cytopenias and splenomegaly are often seen in CVID and can be confused with lymphoma. Thorough evaluation is needed as non-Hodgkin’s lymphoma is also seen with increased frequency in CVID.
There is no standardisation of therapy due lack of randomised controlled studies in the management of GLILD.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Chase NM, Verbsky JW, Hintermeyer MK et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013;33:30–9. 10.1007/s10875-012-9755-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol 2014;34:642–54. 10.1007/s10875-014-0068-6 [DOI] [PubMed] [Google Scholar]
- 3.Bates CA, Ellison MC, Lynch DA et al. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004;114:415–42. 10.1016/j.jaci.2004.05.057 [DOI] [PubMed] [Google Scholar]
- 4.Ardeniz O, Cunningham-Rundles C. Granulomatous Disease in Common Variable Immunodeficiency. Clin Immunol 2009;133:198–207. 10.1016/j.clim.2009.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arish N, Eldor R, Fellg Y et al. Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax 2006;61:1096–7. 10.1136/thx.2004.029819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parks J, Levinson A. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 2010;134:97–103. 10.1016/j.clim.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 7.Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr Opin Hematol 2008;15:368–74. 10.1097/MOH.0b013e328302c7b6 [DOI] [PubMed] [Google Scholar]
- 8.Tanaka N, Kim J, Bates C et al. Lung diseases in patients with common variable immunodeficiency. J Comput Assist Tomogr 2006;30:828–38. 10.1097/01.rct.0000228163.08968.26 [DOI] [PubMed] [Google Scholar]
- 9.Kaufman J, Komorwoski R. Bronchiolitis obliterans organizing pneumonia in common variable immunodeficiency syndrome. Chest 1991;100:552–3. 10.1378/chest.100.2.552 [DOI] [PubMed] [Google Scholar]
- 10.Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med 2014;35:330–5. 10.1055/s-0034-1376862 [DOI] [PubMed] [Google Scholar]
- 11.Resnick ES, Moshier EL, Godbold JH et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012;119:1650–7. 10.1182/blood-2011-09-377945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stagg AJ, Funauchi M, Knight SC et al. Failure in antigen responses by T cells from patients with common variable immunodeficiency (CVID). Clin Exp Immunol 1994;96:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gobert D, Bussel JB, Cunningham-Rundles C et al. Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: a retrospective multicenter study on 33 patients. Br J Haematol 2011;155:498–508. 10.1111/j.1365-2141.2011.08880.x [DOI] [PMC free article] [PubMed] [Google Scholar]