

Abstract
Celastrol, a natural compound derived from the Chinese herb Tripterygium wilfordii Hook F, has been proven to inhibit heat shock protein 90 (HSP90) activity and has attracted much attention because of its promising effects in cancer treatment and in ameliorating degenerative neuron diseases. However, the HSP90 structure involved in celastrol interaction is not known. Here, we report a novel celastrol‐binding pocket in the HSP90 dimer, predicted by molecular docking. Mutation of the two key binding pocket amino acids (Lys546 and Tyr493) disrupted the binding of celastrol to HSP90 dimers, as detected by isothermal titration calorimetry (ITC). Interestingly, such mutations also reduced binding between HSP90 and the cochaperone Cdc37, thus providing a new explanation for reported findings that celastrol shows more obvious effects in disrupting binding between HSP90 and Cdc37 than between HSP90 and other cochaperones. In short, our work discloses a novel binding pocket in HSP90 dimer for celastrol and provides an explanation as to why celastrol has a strong effect on HSP90 and Cdc37 binding.
Keywords: Cdc37, celastrol, heat shock protein 90, molecular modeling, protein drug interaction
Abbreviations
- Cdk4
cyclin‐dependent kinase4
- HSF‐1
heat shock factor‐1
- HSP70
heat shock protein 70
- Hsp90
heat shock protein 90
- ITC
isothermal titration calorimetry
The compound celastrol, an extraction from traditional Chinese medicine Tripterygium wilfordii, exhibits inhibitory effects toward several cancer cells of different origins, such as hepatoma carcinoma cells 1, glioblastoma cells 2, lung cancer cells 3, and melanoma cells 4; this action is attributed to HSP90 inhibition. It has been observed that in celastrol's presence, the binding affinities of HSP90 to its client proteins, as well as to cochaperones, decrease with HSP90–Cdc37 binding showing the most significant decrease 5. It has been proven that celastrol can directly bind to HSP90 and thus disrupt the association of HSP90 and Cdc37 6; yet the chemical basis for celastrol's interaction with HSP90 remains unclear.
It has been about 10 years since celastrol's ability to inhibit HSP90 was discovered, and the understanding of how this inhibition takes place has since developed: First, Westerheide et al. 7 reported that celastrol could induce heat shock response by activating HSF‐1, providing indirect evidence that celastrol could inhibit HSP90, as HSF‐1 is always activated when HSP90 is inhibited. Then, by comparing gene expression signatures among different agents, Hieronymus et al. 8 found that celastrol inhibits HSP90 by binding to a pocket other than the known ATP‐binding site in HSP90's N terminus, meaning celastrol is a novel type of HSP90 inhibitor. Zhang et al. 5 then reported that celastrol disrupted HSP90–Cdc37 interaction in the super chaperone complex to exhibit antitumor activity in vitro and in vivo, with the molecular docking studies demonstrating that celastrol disrupts a critical interaction between the Glu33 side chain of HSP90 N terminus and Arg167 side chain of Cdc37. However, Sreeramulu et al. 9, via NMR technology, found that celastrol does not directly bind to the N‐terminal ATP‐binding pocket of HSP90, but rather targets Cdc37 through a nucleophile reaction. Consistent with this finding, in another study, Zhang et al. 6, who had previously suggested that celastrol targets the HSP90/Cdc37 complex in the N terminus of HSP90, reported that celastrol directly binds to the C‐terminal region to disrupt HSP90–Cdc37 binding. That celastrol can directly affect HSP90 is also supported by our findings that celastrol could reduce HSP90's intrinsic ATP activity 10. While much more is now understood about celastrol's actions, the chemical basis behind celastrol's interaction with HSP90 has not yet been fully explained.
In this work, we used molecular docking analysis and found a potential celastrol‐binding pocket on HSP90 dimer in the nucleotide‐free apo form. We then mutated two amino acids key to HSP90–celastrol binding at this site. Through isothermal titration calorimetry (ITC) experiments, we found that celastrol can bind to wild‐type, but not mutant HSP90. Interestingly, our results also indicate that these two amino acids are important to binding between Cdc37 and HSP90, by observing the direct effect of celastrol on wild‐type or mutant HSP90 complex in cell‐free system.
Results and Discussion
Molecular docking studies are a powerful tool to discover the potential binding sites of proteins to their ligands, but such studies rely heavily on the initial model's accuracy. Recent progress in HSP90 structure analysis has confirmed that HSP90 forms dimers, and that the dimeric conformation changes dramatically along the ATP‐driven HSP90 cycle. Four types of HSP90 dimeric structures have been suggested: Nucleotide‐free Apo, Compact extended ATP (HSP90: C: ATP), Closed (HSP90: C: ATP/ADP+Pi), and Extended ADP‐bound (HSP90: ADP) 11. We thought previous studies' inability to identify the binding sites on HSP90 might be due to a lack of accurate information about HSP90's structure, so for this study we reworked molecular docking studies for celastrol–HSP90 binding using all four of the above conformations.
Our molecular docking studies demonstrated that in the Nucleotide‐free Apo conformation, a celastrol‐binding pocket consisting of a C‐terminal segment from one monomer and the middle region from another monomer was identified. The cavity was about 18.0 Å in length and 8.0 Å in depth. When situated in this cavity, the two oxygen atoms of celastrol's carboxyl group could form two salt bridges, one with Lys546 (LYS535) (from one monomer) and one with Lys582 (LYS1292) (from another monomer). These two bridges enhanced the binding between celastrol and HSP90 dimers (Fig. 1).
Figure 1.
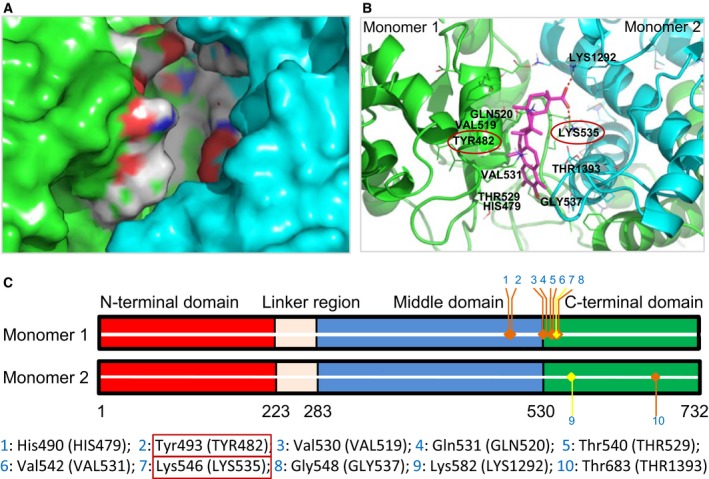
Binding of celastrol to HSP90α dimer by molecular docking. (A) Celastrol's binding pocket in the HSP90α dimer. (B) The amino acids comprising the celastrol‐binding pocket on HSP90α dimer. The mutated amino acids are circled. (C) HSP90α dimer scheme. 1–10 refer to the amino acid sites in celastrol's binding pocket.
To verify the model, we prepared mutant K546D (disrupting one of the predicated salt bridges) and Y493G (damaging one wall of the celastrol‐binding cavity). Using ITC, we found that the mutant proteins lost their ability to bind to celastrol (Fig. 2). On the contrary, both wild and mutant HSP90 could bind to 17‐AAG, which is known to bind to the ATP‐binding site in HSP90's N terminus 12, 13. Through cell‐free experiments with wild or mutant HSP90 complex coimmunoprecipitated with beads, we additionally found that the mutant proteins had reduced ability to bind to Cdc37 while retaining the ability to bind with Cdk4 and HSP70 (Fig. 3). Moreover, celastrol could reduce binding between HSP90 and Cdc37, Cdk4, and HSP70 in wild HSP90 complex, while such effects disappeared in the mutant HSP90 complex (Fig. 3B,C), providing additional evidence that the mutant sites are important to celastrol's effects on HSP90 function.
Figure 2.
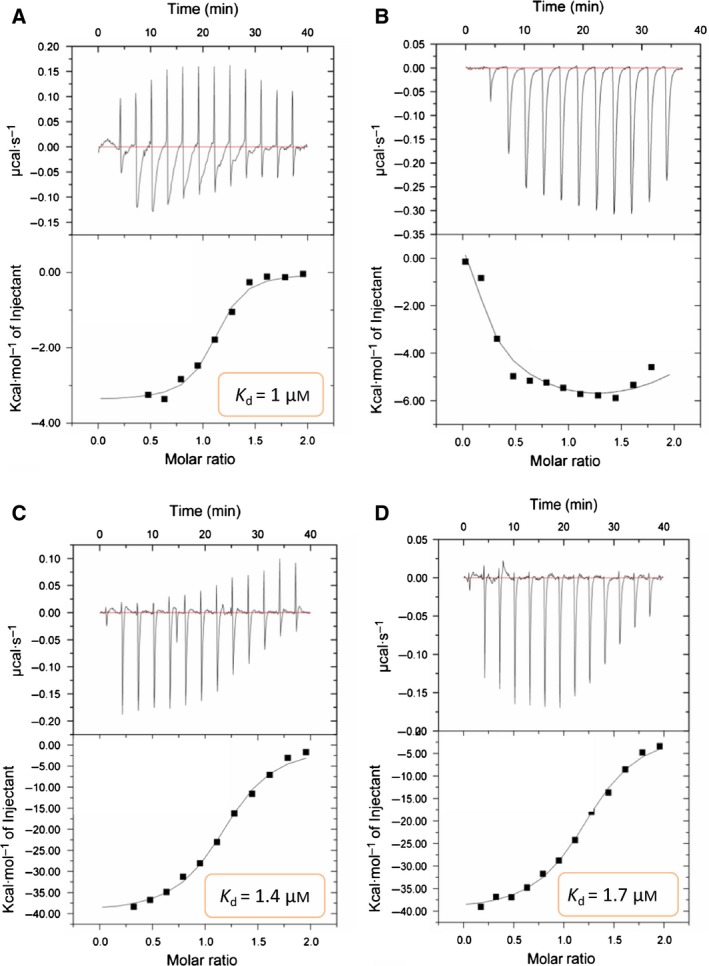
Binding of celastrol or 17‐AAG to wild or mutant human HSP90α, identified by ITC. Celastrol and wild HSP90α (A) or mutant HSP90α (B), tested by ITC (1 : 10 mixture of celastrol/HSP90α). 17‐AAG and wild HSP90α (C) or mutant HSP90α (D), tested by ITC (1 : 10 mixture of 17‐AAG/HSP90α).
Figure 3.
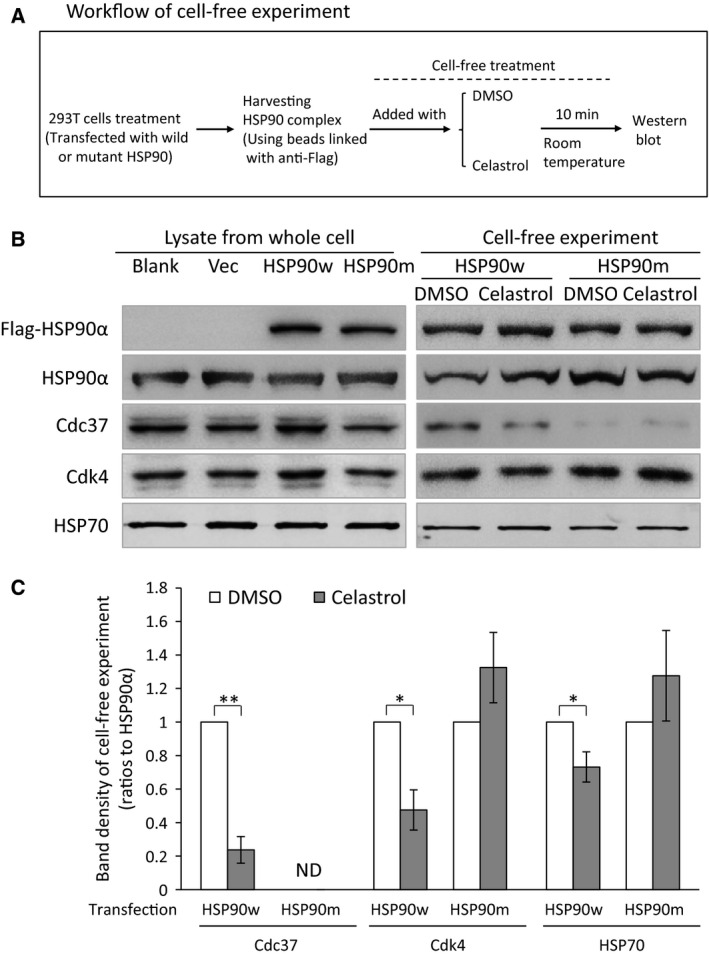
Effects of celastrol on wild‐type or mutant HSP90 complex in cell‐free system. (A) The flowchart for cell‐free experiment. The cells transfected with wild or mutant HSP90 were lysed and the whole proteins were extracted. Wild or mutant HSP90 complexes were captured by incubation of protein extracts with Protein A/G plus agarose and anti‐Flag antibody. The beads with HSP90 complex were incubated with celastrol or DMSO for 10 min before being heated to collect proteins. The proteins were subjected to western blot. (B) Western blot of the related proteins. The left panels show proteins from whole cells, while the right panels show proteins harvested from beads after cell‐free treatment. (C) The histogram and statistical results of band densities from western blot of proteins in beads after cell‐free treatments (as shown in the right panel of B). Band densities of Cdc37, Cdk4, and HSP70 were adjusted using that of HSP90α bands, and the calibrated value of each protein from the DMSO group was set as 1.0. Blank: cells without transfection. Vec: cells transfected with vector. HSP90w: cells transfected with HSP90–wild plasmid. HSP90m: cells transfected with HSP90–mutant plasmid. ND: densities were not detectable by scanning. The data are presented as mean ± standard deviation, n = 3. *P < 0.05; **P < 0.01.
Our findings are consistent with previous reports that celastrol interacts with HSP90's C terminus 6, 14. Mutation in the celastrol‐binding region reduced HSP90's ability to bind to Cdc37, indicating that this region is important for Cdc37 binding. This is also consistent with the belief that celastrol's more prominent effects result from disruption of HSP90/Cdc37 binding 5.
Conclusions
In conclusion, based on molecular docking, ITC determination, and the observed effects on HSP90 complex, our work provides novel evidence that celastrol can directly bind to HSP90 dimers, and that some residues in the C terminus of one monomer and in the middle region of another monomer of HSP90 dimers formed a celastrol‐binding pocket. Two oxygen atoms from celastrol's carboxyl group could form two salt bridges, one with Lys546 and one with Tyr493 on HSP90. This celastrol‐binding pocket is the key to HSP90–Cdc37 binding.
Experimental procedures
Molecular docking
HSP90 dimers may exist in four states, i.e., nucleotide‐free apo, ATP‐bound compact extended, ATP‐bound closed, and ADP‐bound extended states. The coordinates for each of these models were generous gifts from R.L. Matts (Oklahoma State University), whose group recently constructed these models 11. Initial inspection of the three‐dimensional structures in full‐length HSP90 dimer in the four above states suggests that the nucleotide‐free apo form is most likely to bind to the celastrol molecule; the other forms do not provide cavities large enough for small molecule binding in either the M‐ or C‐terminal domain. Thus, the nucleotide‐free apo form of HSP90 was used for our molecular docking studies with celastrol.
Molecular docking was carried out using Schrodinger Suite 2009. The ligand (celastrol) was prepared using the ligprep module, and the dimeric molecules of full‐length HSP90 were preprocessed using the protein preparation wizard before docking. The prepared ligand was docked into the nucleotide‐free apo form of HSP90 using the Glide module at standard precision, and the conformation with the best docking score was selected as our starting model for subsequent induced fit docking (IFD). IFD was carried out using the suite's IFD workflow, in which side chains within 5 Å of celastrol were softened and refined during induced fit docking. No positional or hydrophobic constraint was used.
Plasmid construction and site‐directed mutagenesis
Prokaryotic expression plasmid pET15b‐hHSP90α for human full‐length His‐HSP90α protein was kindly gifted by T. Ratajczak (University of Western Australia, Australia). By cloning, HSP90α (NCBI accession number: NP_005339) was inserted into prokaryotic or eukaryotic expression vector pSUMO‐Mut or pCMV‐tag2a for site‐directed mutagenesis and protein expression or for transient transfection (Fig. S1).
Mutants containing two sites (Y493G and K546D) of HSP90α were created by QuickChange kit (Stratagene, Santa Clara, CA, USA), and sequenced to confirm the correct incorporation of the mutations.
Protein expression and purification
Wild‐type human HSP90α and mutant proteins were expressed in E. coli strain BL21 (DE3) and purified by nickel affinity chromatography. After the purification, the proteins were dissolved in a buffer containing 10 mm Hepes pH 7.4, 150 mm NaCl, and 0.5 mm EDTA, and stored at −80 °C. The purified proteins were confirmed by gel electrophoresis (Fig. S2).
Isothermal titration calorimetry
The purified wild or mutant HSP90α proteins were dialyzed against in the buffer containing 10 mm Hepes pH 7.4, 150 mm NaCl, and 0.5 mm EDTA using Slide‐A‐Lyzer™Dialysis Cassettes (Thermo Fisher Scientific Inc., Rockford, lL, USA) three times 15. Then, the dialyzed proteins were loaded with celastrol or DMSO by MicroCal iTC200 (GE Healthcare, Marlborough, MA, USA). The cells were thermostated at 25 °C. The reaction between wild or mutant HSP90α proteins and celastrol was conducted with 60 μm protein in the cell and 600 μm ligand in the syringe, while the reaction between proteins and 17‐AAG was performed with 30 μm protein in the cell and 600 μm ligand in the syringe. The experiment was conducted with 13 injections, with a volume of 3 μL and 180 s spacing.
Transient transfection, coimmunoprecipitation, cell‐free treatment, and western blot
Transient transfection was performed into 293T cells according to the manufacturer's instructions (lipofectamine 2000; Invitrogen, Carlsbad, CA, USA).
For coimmunoprecipitation, transfected cells were incubated in lysis buffer (20 mm Tris/HCl, 25 mm NaCl, 0.1% NP40, 2 mm DTT, 20 mm Na2MoO4, and protease inhibitor cocktail, pH 7.4). Two milligrams of proteins were incubated with 4 μg of anti‐Flag antibody, and then 60 μL Protein A/G plus agarose was added. Beads were washed three times with PBS.
The wild or mutant HSP90 complexes captured by agarose beads were used for cell‐free treatment. The beads were divided into two groups, and incubated with DMSO or celastrol (final concentration of 12.5 μm) in PBS, respectively. After reaction at room temperature for 10 min, the beads were heated and the proteins collected for western blot, which was carried out according to routine protocol.
Author contributions
BP carried out ITC and cell experiments, participated in the design of the study, and drafted the manuscript. YG carried out molecular docking and participated in drafting the manuscript. YW carried out western blot. FC participated in cell culture. XZ carried out ITC. DZ and JH conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors have read and approved the final manuscript.
Supporting information
Fig. S1. Construction and gel verification of mutant or wild HSP90 expression vector.
Fig. S2. Protein expression and purification.
Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 81102349 for BP); Shanghai Pudong Youth Talent Project in Medicine (No. PWRq2011‐11 for BP); Shanghai Pudong Medical Project (No. PKJ2011‐Y13 for BP); and Shanghai Excellent Academic Leader in Medicine (No. XBR2011054, for DZ). Thanks to Mr. Steven Schwab for tidying up the English expressions and grammar. Thanks to Prof. Robert L. Matts (Department of Biochemistry and Molecular Biology, 246 Noble Research Center, Oklahoma State University, USA) for providing the four conformation models of human HSP90 dimers, and to Dr. Thomas Ratajczak (University of Western Australia, Australia) for gifting prokaryotic expression plasmid pET15b‐hHSP90α for human full‐length His‐HSP90α protein.
References
- 1. Han X, Sun S, Zhao M, Cheng X, Chen G, Lin S, Guan Y and Yu X (2014) Celastrol stimulates hypoxia‐inducible factor‐1 activity in tumor cells by initiating the ROS/Akt/p70S6K signaling pathway and enhancing hypoxia‐inducible factor‐1alpha protein synthesis. PLoS One 9, e112470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boridy S, Le PU, Petrecca K and Maysinger D (2014) Celastrol targets proteostasis and acts synergistically with a heat‐shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis 5, e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fan XX, Li N, Wu JL, Zhou YL, He JX, Liu L and Leung EL (2014) Celastrol induces apoptosis in gefitinib‐resistant non‐small cell lung cancer cells via caspases‐dependent pathways and Hsp90 client protein degradation. Molecules 19, 3508–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davis AL, Cabello CM, Qiao S, Azimian S and Wondrak GT (2013) Phenotypic identification of the redox dye methylene blue as an antagonist of heat shock response gene expression in metastatic melanoma cells. Int J Mol Sci 14, 4185–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG and Sun D (2008) A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther 7, 162–170. [DOI] [PubMed] [Google Scholar]
- 6. Zhang T, Li Y, Yu Y, Zou P, Jiang Y and Sun D (2009) Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 284, 35381–35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB and Morimoto RI (2004) Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 279, 56053–56060. [DOI] [PubMed] [Google Scholar]
- 8. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM et al (2006) Gene expression signature‐based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330. [DOI] [PubMed] [Google Scholar]
- 9. Sreeramulu S, Gande SL, Gobel M and Schwalbe H (2009) Molecular mechanism of inhibition of the human protein complex Hsp90‐Cdc37, a kinome chaperone‐cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl 48, 5853–5855. [DOI] [PubMed] [Google Scholar]
- 10. Peng B, Xu L, Cao F, Wei T, Yang C, Uzan G and Zhang D (2010) HSP90 inhibitor, celastrol, arrests human monocytic leukemia cell U937 at G0/G1 in thiol‐containing agents reversible way. Mol Cancer 9, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matts RL and Manjarrez JR (2009) Assays for identification of Hsp90 inhibitors and biochemical methods for discriminating their mechanism of action. Curr Top Med Chem 9, 1462–1478. [DOI] [PubMed] [Google Scholar]
- 12. Collins CB, Aherne CM, Yeckes A, Pound K, Eltzschig HK, Jedlicka P and de Zoeten EF (2013) Inhibition of N‐terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. Mucosal Immunol 6, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neckers L and Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zanphorlin LM, Alves FR and Ramos CH (2014) The effect of celastrol, a triterpene with antitumorigenic activity, on conformational and functional aspects of the human 90 kDa heat shock protein Hsp90alpha, a chaperone implicated in the stabilization of the tumor phenotype. Biochim Biophys Acta 1840, 3145–3152. [DOI] [PubMed] [Google Scholar]
- 15. Meiby E, Simmonite H, le Strat L, Davis B, Matassova N, Moore JD, Mrosek M, Murray J, Hubbard RE and Ohlson S (2013) Fragment screening by weak affinity chromatography: comparison with established techniques for screening against HSP90. Anal Chem 85, 6756–6766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Construction and gel verification of mutant or wild HSP90 expression vector.
Fig. S2. Protein expression and purification.