

Abstract
The increasing trend of antibiotic resistance in Acinetobacter drastically limits the range of therapeutic agents required to treat multidrug resistant (MDR) infections. This study focused on analysis of novel Acinetobacter strains using a genomics and systems biology approach. Here we used a network theory method for pathogenic and non-pathogenic Acinetobacter spp. to identify the key regulatory proteins (hubs) in each strain. We identified nine key regulatory proteins, guaA, guaB, rpsB, rpsI, rpsL, rpsE, rpsC, rplM and trmD, which have functional roles as hubs in a hierarchical scale-free fractal protein-protein interaction network. Two key hubs (guaA and guaB) were important for insect-associated strains, and comparative analysis identified guaA as more important than guaB due to its role in effective module regulation. rpsI played a significant role in all the novel strains, while rplM was unique to sheep-associated strains. rpsM, rpsB and rpsI were involved in the regulation of overall network topology across all Acinetobacter strains analyzed in this study. Future analysis will investigate whether these hubs are useful as drug targets for treating Acinetobacter infections.
Acinetobacter is a Gram negative nosocomial pathogen1 that causes a variety of infections in humans ranging from respiratory failure, ventilator associated pneumonia, bacteremia and wound infections2. The major species of Acinetobacter associated with nosocomial infections are A. baumannii, A. nosocomialis, A. pittii A. johnsonii and A. lwoffii3. Systems biology is the study of an organism, viewed as an integrated and interacting network of genes, proteins and biochemical reactions, that form the functional units capable of operations needed for cell and tissue/organ level physiological function4. Protein-protein interaction (PPIs) network analysis is a valuable systems biology tool for identifying drug targets and functional mechanisms5. PPIs can be used to elucidate the cellular events that maintain physiological stability and integrity. Using whole genome data, we have constructed protein-protein interaction networks for four strains of Acinetobacter spp. isolated from different animal intestines to determine the how these networks vary across environments. To delineate differences, we employed hierarchical network theory to quantify the structural properties of each network, such as the emergence of modules/communities and sparsely distributed hubs6,7, and self-organized working principle8. The emergence of modules/communities may correspond to independent functions obeying their own laws, with activities being nonlinear in nature9. The sparsely distributed hubs may interfere and control network stability within the community9 as well as other communities. Hubs and highly connected proteins play a crucial role in biological networks10.
We have sequenced and assembled the genomes of 3 Acinetobacter spp. strains (SFA, SFB and SFC) isolated from sheep feces, and one strain (HA) isolated from the gut of a 5th instar larva of polyphagous insect, Helicoverpa armigera. A hierarchical protein-protein interaction network (PPI) was constructed, and subnetwork/modules analyzed, to identify regulatory proteins important for cellular physiological processes. Key proteins are defined as randomly placed, with important functional roles and a high degree of interactions11,12 in each isolated strain. The STRING v10 database for A. lwoffi and A. johnsonni was used to as a basis for building the PPI network of the four novel strains.
Material and Methods
Isolation and culturing of strains
Acinetobacter strains were isolated from sheep feces, and designated SFA, SFB and SFC. All isolates grew well at 26 °C on both Mac-Conkey agar and blood agar plates. A fourth Acinetobacter strain, HA, was isolated from the 5th instar larva of Helicoverpa armigera (polyphgous pest) from an agricultural field in Maharashtra, India. Acinetobacter genus and species level identification was achieved by 16S rRNA gene amplification and sequencing using the universal primers 8F and 1546R, and by rpoB PCR and sequencing13.
Whole genome sequencing and assembly
The isolated Acinetobacter strains were grown at 26 °C on both Mac-Conkey agar and blood agar plates until mid-log phase with shaking at 250 rpm. Whole genomic DNA extraction was performed according to the manufacturer’s instructions using the Promega Wizard Genomic DNA purification kit (Promega, Madison, WI). The concentration of DNA was determined by picogreen assay. DNA was used to construct TruSeq DNA libraries with manufacturer’s defaults, which were then sequenced on an Illumina HiSeq2000 platform with 100 base paired-end sequencing. The FASTQ paired-end reads were assembled using Velvet de-novo assembler14, coverage was typically 30x and assembled genome size approximately 3 Mb. Genome assemblies were validated for the misassembled and low coverage regions using BWA15 and Tablet16 software packages. Quality filtered contigs were further extended using paired-end criterion.
Genome Annotation and Phylogenetic affiliation
Final assemblies were checked for the percentage completeness using with 31 protein encoding phylogenetic marker genes17, and 107 single copy marker genes18. Each genome revealed presence of all 31/31 and 107/107 genes, which suggests completeness. Open reading frames (ORFs) were called for each genome using FragGeneScan v1.1619. Predicted ORF’s were annotated by KAAS (KEGG Automatic Annotation Server)20 to assign KEGG orthologs (KO) identifiers to the query ORFs sequences using GHOSTXx21 algorithm against KEGG GENES database22. For automatic genome annotations, the Acinetobacter spp. SFA, SFB, SFC and HA genome assemblies were submitted to Rapid Annotation using Subsystems Technology (RAST) Server23. Annotated genomes are accessible from the RAST server by logging in with the guest account with the accession numbers (RAST-ID) 258824, 258827, 258830 & 262612 for SFA, SFB SFC and HA respectively. Assembled genomes were phylogenetically delineated using two way ANI script in PYANI Master Pipeline using percentage identity algorithm at default parameter. Reference genomes were adopted from the list of all representative species of Acinetobacter maintained at the Broad Institute https://olive.broadinstitute.org/collections/acinetobacter.5/strains on 14/10/2015.
Construction of protein-protein interaction network of Acinetobacter strains based on phylogenetic cluster analysis
To construct protein-protein interactions for the novel strains we employed the STRING Database (v10)24 which is the most comprehensive Acinetobacter protein interaction resource. A. lwoffi and A. johnsonni were selected for PPI construction, as these were phylogenetically closer to the novel strains. The STRING v10 database consists of known and predicted PPIs, which included direct (physical) and indirect (functional) associations. Here we provided the protein sequences of novel Acinetobacter strains as an input and queried against A. lwoffi and A. johnsonii PPIs. The interaction associations were integrated with different sources such as genomic context, high-throughput experimental data, database and literature mining, and analysis of co-expressed genes. The PPI networks were visualized using Cytoscape Version 3.0.125.
Identification of highly regulating nodes in the network
The protein-protein interaction networks focused on finding hubs which highly connected proteins were considered to play a crucial role in biological networks. Hubs are proteins having a high degree of interactions/edges and are randomly placed in the network, having important functional roles26. In our study, using network analyzer, the plug-in of Cytoscape v 3.0.1 and Perl programming version 5.18.2.2, we identified that the hub proteins communicated with many other significant proteins involved in the network.
Statistical analysis of the Network
The statistical and functional significance of the network, is proposed to be measured using various statistical parameters, namely in the proposed case, probability of degree distribution, average clustering co-efficient and average neighborhood connectivity27. The network is constructed to find if it obeyed power law,
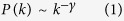 |
indicating the scale free nature of the network, where, γ is an order parameter which identified the different topological structure of a scale free network. The clustering co-efficient C (k), which is defined by
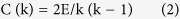 |
and is the ratio of the number of edges E of the node having a k degree with neighbors to the total possible number of such edges,
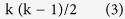 |
is a measure of the topological structure of the network28. The neighborhood connectivity of a node is the number of connected neighbors with it and characterizes the correlation pattern of connectivity of interacting nodes in the network. This connectivity correlation would be measured by defining a conditional probability
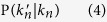 |
which is the probability of making a link from a node having degree kn to another node of degree k′n9. Then the average neighbourhood connectivity of nodes with connectivity kn is given by9,
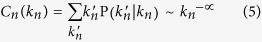 |
following a power law scaling behaviour with α < 1 for most of the real networks (Maslov and Sneppen, 2002; Pastor-Satorras et al., 2001). If Cn(kn) is an increasing function of kn (for negative values of α) then the topology of the network show assortive mixing29 where high degree (the number of edges per node) nodes have affinity to connect to other high degree nodes in the network. However,
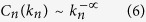 |
with positive values of α, is the signature of the network having hierarchical structure29, where low degree nodes tend to connect high degree hubs29 and the few high degree hubs present in the network try to control the low degree nodes. The two-centrality measurements (Betweenness centrality & Closeness centrality) were also calculated.
The centrality measurement of the network
We considered two centrality measurements to analyze our network as described below.
Betweenness centrality
The betweenness centrality (CB) quantifies a node, occurring a number of times to bridge along the shortest path between two other nodes6, which could be calculated by,
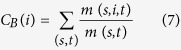 |
where, m (s,i,t) is the number of shortest path, connecting s and t that pass through node i; and m (s,t) is the number of shortest paths in-between nodes s and t. The sum is to be taken of all pairs (s,t) of distinct nodes. In a complex network, the nodes which have high value of CB lie on paths between many other nodes, and have high influencing capability of information spreading within the network30.
Closeness centrality
Closeness centrality (CC) can be established in terms of “shortest path lengths” between pairs of nodes31. The farness of a node can be estimated by the sum of its distances to all other nodes in the network; and closeness is measured as the inverse of this farness32. The closeness centrality of node is defined by,
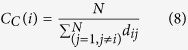 |
where, dij is the shortest distance between node i and j, and N is the size of the network. The CC of a node in a network describes the efficiency of the node for information propagation in the network33. The high CC valued nodes in the complex network have higher efficiency to propagate information in the entire network34; whereas, nodes having low CC values have higher receiving capabilities of information32.
Module and its functional enrichment analysis
Modules of large PPI network are defined as the set of statistics and functionally significant interacting genes26. MCODE35, the plug-in of Cytoscape, identifies the clusters that are highly interconnected regions in a network. We used default setting of MCODE, which analyzed networks, using Scoring [include loops, degree cutoff (2)] and Finding [node score cutoff (0.2), haircut, node density cutoff (0.1), K-core (2), Maximum Depth (100)] parameters that were optimized to produce the best results for the network. The potential clusters were identified by a search method, estimating their significance scores with a high score (>1) and a decent number of nodes and edges36. The extracted clusters were ranked by scoring through density and size. Once the nodes in a cluster were identified, one could intuitively reduce the complexity of the network by replacing the individual nodes with one large parent node, which allowed focusing on the interactions with the cluster. To understand the functional role of proteins involved in top three modules of each strain, we subjected the module proteins for GO annotation. Because modules tend to have a similar function, we over-represented the Gene Ontology categories (Molecular function, Biological process, Cellular Components) for modules in each strains network. The major categories were considered based on the percentage of each set of nodes to construct pie diagrams that allowed better visualization of the functional categories.
Network motif
In biological networks, these motifs are suggested to be recurring circuit elements that carry out key information processing tasks37. To understand these complex networks, we sought to break down such networks into basic building blocks. A network motif was defined based on the criterion that the number of occurrences must be at least five, and also must be significantly higher than that used in randomized networks. We applied FANMOD38 on the complete network, to select network motifs. The significance test was carried out on 1000 randomized networks, and a pattern with P < 0.05 was considered statistically significant. Clusters were analyzed for three node motif, using MCODE, from which we identified the motif within highly clustered nodes.
Results
Whole genome sequencing and phylogenomic analysis of Acinetobacter strains
Approximately 1 Gbp of sequence was generated for each Acinetobacter strain (SFA, SFB, SFC and HA). Draft genomes were de novo assembled - SFA (25 contigs), SFB (26 contigs), SFC (121 contigs) and HA (102 contigs) - with an average genome size of 3.0 Mb and the average G+C content was 40% (Table 1). Two way ANI using genome similarity identity % demarcated A. lwoffi as the closest relative for SFA (identity % 89), while SFB and SFC were closer to A. johnssonii (83% & 84% identity, respectively). The HA strain, isolated from insect gut, was most closely related to A. schindleri (97% identity; Fig. 1). Therefore, SFA, SFB and SFC were most closely related to generally non-pathogenic or opportunistic pathogens such as A. lwoffi and A. johnssonii, which have previously been isolated from bacon, eggs, fish, and frozen food, and show resistance to desiccation and disinfectants39. A. lwoffi is commonly associated with human skin, but has been linked to bacteremia, cancer and systemic lupus erythematosus40. A. johnssonii is mostly associated with the environment and has occasionally been linked to infections41. A. schindleri has been suggested to be a misidentified opportunistic pathogen in patients with underlying predisposition42.
Table 1. General genomic features of Acinetobacter strains for comparative analysis.
| Genome | Estimated genome size | Average GC content | CDS | Coding density | Total rRNA | tRNA | rRNA | Status | Source | Accession No. |
|---|---|---|---|---|---|---|---|---|---|---|
| Acinetobacter sp SFA | 3.13 | 42 | 3140 | 86.38 | 80 | 74 | 6 | Draft | Sheep | LSZI00000000 |
| Acinetobacter sp SFB | 3.30 | 38 | 3231 | 85.11 | 72 | 68 | 4 | Draft | Sheep | LSZH00000000 |
| Acinetobacter sp SFC | 3.32 | 38 | 3268 | 85.52 | 75 | 69 | 6 | Draft | Sheep | LSZG00000000 |
| Acinetobacter sp HA | 3.12 | 41 | 3140 | 85.91 | 77 | 64 | 13 | Draft | Insect | AJXD00000000 |
Figure 1.

Dual dendrogram of novel Acinetobacter strains (A) Genome to genome alignment coverage (%). (B) Genome-genome similarity identity (%) of SFA, SFB, SFC and HA with respect to 119 reference genotypes. Color bar predicted correlation coefficients (0 to 1.0) are shown with the color scale on the basis of respective percentage identity.
Characterization of protein-protein interaction network in four Acinetobacter strains
Cataloguing the stable and transient PPIs in a cell can facilitate functional annotation of gene products, providing insights into the organization of the proteome. Following removal of redundant interactions and protein nodes, the resulting network had 2693 interactions, involving 422 proteins for SFA, 2620 interactions involving 414 proteins for SFB, 2401 interactions involving 426 proteins for SFC, and 2638 interactions involving 420 proteins for HA (Fig. 2).
Figure 2. The PPI network of four novel Acinetobacter strains.

Expanded view of the network imported from Cytoscape, where nodes represent proteins and edges the physical interaction. (A) All nodes and edges of SFA strain PPI are filled circles (cyan) and lines (gray), respectively. (B) All nodes and edges of SFB strain PPI are filled circles (yellow) and lines (gray), respectively. (C) All nodes and edges of SFC strain PPI are filled circles (green) and lines (gray), respectively. (D) All nodes and edges of HA strain PPI is filled circles (orange) and lines (gray), respectively.
The topological properties of each PPI network were parameterized with probability of degree distribution P (k), which suggested that each network followed a power law scaling behavior
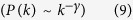 |
with the value of the degree exponent γ ~ 0.6 in all the four strains (Fig. 3A–D). A straight-line fitted to the data curve with a correlation co-efficient value of ~0.8 in all the four stains. The small value of γ (γ < 2) indicated that the network was hierarchical8, signifying the emergence of hierarchical modules and/or communities7, with a sparse distribution of highly connected hubs. That the few highly-connected hubs are connected to many low-degree nodes is indicative of a regulatory power of the hubs over these nodes. In confirmation of a hierarchical network7, the average clustering co-efficient C(kn), calculated as a function of number of neighbors kn, again followed the power scaling law given by
Figure 3.

The topological properties of four novel Acinetobacter strains network depicting with correlation coefficient values (r2): (A) probability of degree distribution P(k), (B) average clustering coefficient, (C) average neighborhood connectivity, (D) Betweenness centrality and (E) Closeness centrality of the PPI network. All these properties follow the power law distribution and show the nature of scale-free network, suggesting a hierarchical organization in the network.
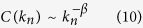 |
with β = ~0.1 in all the four strains. The straight line is the fitted curve with correlation coefficient value of ~0.5 in all these four strains (Fig. 3A–D).
Average neighborhood connectivity Cn (kn), constructed as a function of kn, also followed a power scaling law given by,
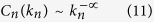 |
with α = ~0.5 for all the four strains (Fig. 3A–D), also supporting that the network is hierarchical7, where the straight line is the fitted curve with correlation coefficient of ~0.8 to the data points. The number of degree (edges per node) was calculated based on each PPI network, and we list here the main hubs for each network with their degrees: rpsB (106), rpsI (105), rplm (103) and rpsL (90) (SFA - Fig. 2A), rpsI (116), rplm (114), rpsC (87) and rpsE (87) (SFB Fig. 2B), rplm (104), rpsI (102), rpsB (82) and trmD (78) (SFC Fig. 2C) and guaA (148), rpsI (110), guaB (97) and rpsB (93) (HA Fig. 2D). The four genes in each strain network are likely indicative of key regulatory functions in each genome33. The modular topological structure of the network demonstrated the existence of various functional modules or sub-networks, and also the organization among these modules7.
For the networks, CB and CC followed power law scaling behavior with k, 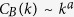 , and
, and 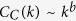 , where a and b are positive values (Fig. 3D,E). It was also found that the fitted straight lines on the network data of these two centrality measurements were approximately parallel with the average value of “a” equal to 0.51 (SFA), 0.41 (SFB) 0.54 (SFC) and 0.90 (HA), and “b” equal to 0.02 (SFA), 0.09 (SFB) 0.03 (SFC) and 0.04 (HA). The increasing value of CB with k indicated that high degree nodes have the greatest information spreading capability in the network. Further, CC analysis shows that high degree nodes rapidly disseminate signal information to low degree nodes. Therefore, the four hubs in each PPI network are the main signal propagating nodes in both the network, and their respective modules.
, where a and b are positive values (Fig. 3D,E). It was also found that the fitted straight lines on the network data of these two centrality measurements were approximately parallel with the average value of “a” equal to 0.51 (SFA), 0.41 (SFB) 0.54 (SFC) and 0.90 (HA), and “b” equal to 0.02 (SFA), 0.09 (SFB) 0.03 (SFC) and 0.04 (HA). The increasing value of CB with k indicated that high degree nodes have the greatest information spreading capability in the network. Further, CC analysis shows that high degree nodes rapidly disseminate signal information to low degree nodes. Therefore, the four hubs in each PPI network are the main signal propagating nodes in both the network, and their respective modules.
Modules and functional enrichment analysis
We identified significant modules (Fig. 4) in each network that could have distinct biological functions, and were functionally separable. Three such significant clusters were identified for each strain (SFA, SFB, SFC and AHA; Fig. 4A–C). Each module was ranked based on the MCODE network score43. The details of each module were represented in Table 2. Module 1 of SFB and SFC includes four of its hubs (SFB: rpsI, rplM, rpsE, rpsC & SFC: rplM, rpsI, rpsB and trmD), while HA has three hubs (guaA, rpsB and rpsI), and SFA had two hubs (rpsL and rpsB; Fig. 4A). This indicated that most of the significantly large hubs did not only interfere in the internal regulation of their own modules in the network, but also affected other modules. However, hubs like trmD (SFC), guaA, and guaB (HA) were not present in any of the other modules, suggesting that these hubs indirectly interfered with modular properties and activities. These modules were found to be linked via sparsely distributed nodes, which can mediate cross talk among the modules44. Functional enrichment analysis for molecular, biological and cellular components was performed in the three modules for each strain (Fig. 5A–D). Module proteins were performing similar functions in each strain isolated from sheep. Module-1 of all strains was majorly involved in structural molecular activity, whereas module-2 and 3 were involved in catalytic activity. Metabolic pathway is the common and highly involved biological process by all the proteins present in each module of all the four strains.
Figure 4. Skeletal structure of the modules in the novel Acinetobacter strains PPI network.

All the modules 1–3 are constructed and analyzed using MCODE. (A) In module 1 all the nodes are in filled circles (orange), with scoring value 48.85 (SFA), 44.54 (SFB), 43.62 (SFC) and 39.65 (HA); (B) modules 2 all the nodes are in filled circles (green), with scoring value 8 (SFA), 7.38 (SFB), 7.15 (SFC) and 8 (HA), (C) module 3 all the nodes are in filled circles (pink), with scoring value 8 (SFA), 7.14 (SFB), 5.62 (SFC) and 7.71 (HA) with the corresponding edges in grey lines.
Table 2. Modules in four novel Acinetobacter strains.
| Strains | Module-1 | Module-2 | Module-3 |
|---|---|---|---|
| SFA-Nodes | 50 | 8 | 8 |
| SFA-Edges | 1197 | 28 | 28 |
| SFA-Scores | 48.857 | 8 | 8 |
| SFB-Nodes | 45 | 14 | 15 |
| SFB-Edges | 980 | 48 | 50 |
| SFB-Scores | 44.545 | 7.385 | 7.143 |
| SFC-Nodes | 44 | 20 | 17 |
| SFC-Edges | 938 | 68 | 45 |
| SFC-Scores | 43.628 | 7.158 | 5.625 |
| HA-Nodes | 41 | 13 | 15 |
| HA-Edges | 793 | 48 | 54 |
| HA-Scores | 39.65 | 8 | 7.714 |
Figure 5. Functional annotation of three modules depicting biological processes, cellular component and molecular function represented in pie charts.

(A) Modules of PPI in HA strains. (B) Modules of PPI in strain SFA. (C) Modules of PPI in strain SFB. (D) Modules of PPI in strain SFC.
Network Motif
A statistically significant basic skeleton of three node motifs were identified with frequencies of 46%, 22%, 44%, 30% (P < 0.05) in SFA, SFB, SFC and HA, respectively (Fig. 6A–D). This motif appeared in at least 5 out of 1000 random permutations of the PPI networks (FANMOD).
Figure 6. Hub motifs in the Acinetobacter strains PPI network.

(A) Hub-node motifs of SFA strains showing four motifs (green); (B) Hub-node motifs of SFB strains showing four motifs (blue); (C) Hub-node motifs of SFC strains showing four motifs (pink); (D) Hub-node motifs of HA strains showing two motifs (orange).
MCODE was used to further identify additional significant motifs in the four PPI networks, and 3 node motifs were found in all four strains, whereas four node motifs were only identified in SFB network. We also found that hubs nodes were interacting with each other and forming a three-node motif, which was similar to the pattern of motif identified by FANMOD. The HA hub node motif interaction was different to the other strains. In SFA, SFB and SFC all the hubs were interacting with each other (total six edges in each strain) (Fig. 6A–C); whereas HA hubs protein were interacting each other with possible five edges (Fig. 6D). The four hub motifs in HA were further classified into 2 different three-node motifs (Fig. 6D), whereas in SFA, SFB and SFC the four hub node motifs were further classified into 4 different three-node motifs (Fig. 6A–C).
Discussion
Understanding signaling processes and identifying interacting proteins could be essential in identifying novel drug targets in Acinetobacter. Mathematical models such as Bayesian networks, ordinary differential equations, boolean network and Petri nets have previously been used to try and pinpoint proteins that are important in networks45,46. The network-based approach applied here, using experimentally observed and literature-available data, enables the construction of preliminary models to understand system regulation in Acinetobacter. Each module/sub-network was cohesive to the sub-subnetwork as reported earlier47, and each sub-subnetwork (module) was reduced to a cluster of connected triangles (basic motifs), corresponding to significant positive and negative feedback loops47. Further we found that each network comprised three smaller modules, with different functional components. The three modules in each network exerted the most influence over the main regulating network, with the main hubs acting as rapid signal dissemination nodes throughout the functional modules. These main hubs were mostly found in the main module-1, because it had the capacity to influence the other modules. Further splitting modules to smaller level i.e. hubs which are seed genes of the whole interacting network, we identified nine genes as important network regulators (guaA, rpsI, rpsB, guaB, rpsL, rplM, rpsC, rpsE and trmD), where rpsI was found to be important as it was present in all the four strains. Identified hubs genes were mainly associated to the translation process. The genes rpsB, rpsI, rpsL, rpsE and rpsC encode the bacterial ribosome protein 30S subunit (small ribosomal subunit of prokaryotic system), whereas rplM is encodes the 50S (large ribosomal subunit). The guaA encodes for GMP synthase (EC: 6.3.5.2) whereas guaB encodes an IMP dehydrogenase (EC: 1.1.1.205). Our studies indicate that the GuaA and GuaB proteins are critical for the survival of bacteria and could play an important role in the infection cycle of Acinetobacter, as shown earlier in the case of tick borne pathogen B. burgdorferi48 Human IMP dehydrogenase inhibitors are validated targets for immunosuppressive, antiviral and anticancer drugs, but the potential of microbial IMP dehydrogenase inhibitors has yet to be exploited in antimicrobial chemotherapy49. The trmD gene which is essential for bacterial growth is a tRNA modification enzyme encoding the enzyme tRNA (guanine37-N1)-methyltransferase D (EC 2.1.1.228) responsible for converting G37 to m(1)G37 on the 3′ side of the tRNA anticodon. This enzyme is responsible for one carbon group methyl transferase50. More specifically for the transfer of the N1-methyl group on the upcoming tRNA from S-adenosyl-L-methionine51. This methylates the methionine which is the first amino acid in translation step. The reaction involves methyl transfer from S-adenosyl methionine and is critical to minimize tRNA frameshift errors on the ribosome.
The network for strain SFA comprised 4 hubs (Fig. 6A) with one large ribosomal rplM, which is largest of all subunits (L13) along with the three smaller ribosomal subunits rpsI, rps and rpsB. Strain SFB motif network was including the almost same hubs (Fig. 6B) with large subunit, rplM (L13) along with the smaller subunits rpsI, rpsE and rpsC. Their close interaction indicates the predominance of translational process for the cell system maintenance. Another gut isolate, SFC shared the same hubs (Fig. 6C) of large and small subunits of ribosomal assembly along with trmD. The insect gut isolate, strain HA comprised of two smaller subunits of ribosomal assembly motifs rpsI and rpsB, along with two other hubs guaA and gubB (Fig. 6D). The genes that encoded them are involved in de-novo purine biosynthesis and its metabolism. They carry out the two step reaction involving the conversion of inosine monophosphate (IMP) to xanthosine monophosphate (XMP) by inosine 5′-monophosphate dehydrogenase44 coded by gene guaB. Second step is the conversion of the XMP to guanosine monophosphate (GMP) by Guanosine monophosphate synthetase coded by guaA48. The first step is the rate limiting step which ultimately determines the denovo synthesis of guanine nucleotide whereas the second step is the branching point of pathway where synthesis of guanosine or adenine is diverged in de novo purine synthesis. The motif analysis demonstrated that the interaction between rplM, rpsB and rpsI was crucial, with a regulatory function in SFA and SFC. The rpsI gene is present in all Acinetobacter strains as a regulatory gene. Even guaB in the HA network, which was not present in any of the three modules, acted as a mediator to cross talk among the modules and also indirectly interfered with modular properties and activities. Network motif analysis also suggests guaA and guaB are key regulatory components for the pathogenic strains of Acinetobacter spp.
On the basis of whole genome phylogeny we propose the names Acinetobacter aries sp. nov. for strain SFA, Acinetobacter ovis sp. nov. for the strains SFB and SFC and Acinetobacter armigera sp. nov. for strain HA. Although, in silico predictions warrant further experimental confirmation of the key regulators, the current study lays the foundation in order to understand the role of key regulators in Acinetobacter. Targeting functional genes involved in regulation of hierarchical protein networks provide us with an alternative way in treating infections caused by Acinetobacter spp.
Additional Information
Accession codes: The accession numbers for the Acinetobacter sp. of SFA, SFB SFC and HA were LSZI00000000, LSZH00000000, LSZG00000000 & AJXD00000000 respectively.
How to cite this article: Gupta, V. et al. Comparative genomic analysis of novel Acinetobacter symbionts: A combined systems biology and genomics approach. Sci. Rep. 6, 29043; doi: 10.1038/srep29043 (2016).
Acknowledgments
This work was supported by University of Delhi, Department of Science and Technology- Promotion of University Research and Scientific Excellence (DST-PURSE). V.G., S.H. and U.S. gratefully acknowledge the Council for Scientific and Industrial Research (CSIR), University Grant Commission (UGC) and Department of Biotechnology (DBT) for providing research fellowship.
Footnotes
Author Contributions R.L. and B.S.L. conceived the idea. M.R. assisted in lab work and K.F. helped in sequencing the genomes. PPI networking was done by S.H. Manuscript was mainly written by V.G. and S.H. U.S. did the phylogenetic classification work. J.A.G. and Y.S. did the editing and proof reading part in the manuscript. All authors discussed the results and commented on the preliminary drafts, read and approved the final version of the manuscript.
References
- Bergogne-Berezin E. & Towner K. J. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clinical microbiology reviews 9, 148–165 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Multidrug resistance in bacteria. Annual review of biochemistry 78, 119–146, 10.1146/annurev.biochem.78.082907.145923 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visca P., Seifert H. & Towner K. J. Acinetobacter infection–an emerging threat to human health. IUBMB life 63, 1048–1054, 10.1002/iub.534 (2011). [DOI] [PubMed] [Google Scholar]
- Kitano H. Perspectives on systems biology. New Generation Computing 18, 199–216, 10.1007/bf03037529 (2000). [DOI] [Google Scholar]
- Pedamallu C. S. & Posfai J. Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source code for biology and medicine 5, 8, 10.1186/1751-0473-5-8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan M. & Newman M. E. Community structure in social and biological networks. Proceedings of the National Academy of Sciences of the United States of America 99, 7821–7826, 10.1073/pnas.122653799 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasz E., Somera A. L., Mongru D. A., Oltvai Z. N. & Barabasi A. L. Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555, 10.1126/science.1073374 (2002). [DOI] [PubMed] [Google Scholar]
- Barabasi A. L. & Oltvai Z. N. Network biology: understanding the cell’s functional organization. Nature reviews. Genetics 5, 101–113, 10.1038/nrg1272 (2004). [DOI] [PubMed] [Google Scholar]
- Barzel B. & Barabasi A.-L. Network link prediction by global silencing of indirect correlations. Nat Biotech 31, 720–725, 10.1038/nbt.2601 http://www.nature.com/nbt/journal/v31/n8/abs/nbt.2601.html#supplementary-information (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadhal E., Gamieldien J. & Mwambene E. C. Protein interaction networks as metric spaces: a novel perspective on distribution of hubs. BMC Systems Biology 8, 1–11, 10.1186/1752-0509-8-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. & Zhang J. Toward a molecular understanding of pleiotropy. Genetics 173, 1885–1891, 10.1534/genetics.106.060269 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhajosyula R. R., Chakravarti D., Lutfeali S., Ray A. & Raval A. Identifying hubs in protein interaction networks. PloS one 4, e5344, 10.1371/journal.pone.0005344 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B., Gundi V. A., Khamis A. & Raoult D. Sequencing of the rpoB gene and flanking spacers for molecular identification of Acinetobacter species. Journal of clinical microbiology 44, 827–832, 10.1128/JCM.44.3.827-832.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D. R. & Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18, 821–829, 10.1101/gr.074492.107 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, 10.1093/bioinformatics/btp698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I. et al. Tablet–next generation sequence assembly visualization. Bioinformatics 26, 401–402, 10.1093/bioinformatics/btp666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F. D. et al. Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287, 10.1126/science.1123061 (2006). [DOI] [PubMed] [Google Scholar]
- Dupont C. L. et al. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. The ISME journal 6, 1186–1199, 10.1038/ismej.2011.189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho M., Tang H. & Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic acids research 38, e191, 10.1093/nar/gkq747 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A. C. & Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic acids research 35, W182–185, 10.1093/nar/gkm321 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S., Kakuta M., Ishida T. & Akiyama Y. GHOSTX: an improved sequence homology search algorithm using a query suffix array and a database suffix array. PloS one 9, e103833, 10.1371/journal.pone.0103833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Kawashima S., Okuno Y. & Hattori M. The KEGG resource for deciphering the genome. Nucleic acids research 32, D277–D280 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics 9, 75, 10.1186/1471-2164-9-75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research 43, D447–452, 10.1093/nar/gku1003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504, 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U., Ukil S., Dimitrova N. & Agrawal S. Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PloS one 4, e8100, 10.1371/journal.pone.0008100 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert R. & Barabási A.-L. Statistical mechanics of complex networks. Reviews of Modern Physics 74, 47–97 (2002). [Google Scholar]
- Watts D. J. & Strogatz S. H. Collective dynamics of /‘small-world/’ networks. Nature 393, 440–442 (1998). [DOI] [PubMed] [Google Scholar]
- Almaas E. Biological impacts and context of network theory. The Journal of experimental biology 210, 1548–1558, 10.1242/jeb.003731 (2007). [DOI] [PubMed] [Google Scholar]
- Freeman L. C. Centrality in social networks conceptual clarification. Social Networks 1, 215–239, http://dx.doi.org/10.1016/0378-8733(78)90021-7 (1978). [Google Scholar]
- Sabidussi G. The centrality of a graph. Psychometrika 31, 581–603 (1966). [DOI] [PubMed] [Google Scholar]
- Newman M. E. The structure of scientific collaboration networks. Proceedings of the National Academy of Sciences of the United States of America 98, 404–409, 10.1073/pnas.021544898 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M. C., Zalatan J. G. & Lim W. A. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686, 10.1126/science.1198701 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kas M., Carley K. M. & Carley L. R. In Advances in Social Networks Analysis and Mining (ASONAM), 2013 IEEE/ACM International Conference on. 1250–1258.
- Bader G. D. & Hogue C. W. V. An automated method for finding molecular complexes in large protein interaction networks. BMC bioinformatics 4, 1–27, 10.1186/1471-2105-4-2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki N., Berengueres J. & Efimov D. Detection of protein complexes using a protein ranking algorithm. Proteins 80, 2459–2468, 10.1002/prot.24130 (2012). [DOI] [PubMed] [Google Scholar]
- Milo R. et al. Superfamilies of evolved and designed networks. Science 303, 1538–1542, 10.1126/science.1089167 (2004). [DOI] [PubMed] [Google Scholar]
- Wernicke S. & Rasche F. FANMOD: a tool for fast network motif detection. Bioinformatics 22, 1152–1153, 10.1093/bioinformatics/btl038 (2006). [DOI] [PubMed] [Google Scholar]
- Rathinavelu S., Zavros Y. & Merchant J. L. Acinetobacter lwoffii infection and gastritis. Microbes and infection/Institut Pasteur 5, 651–657 (2003). [DOI] [PubMed] [Google Scholar]
- Ku C. S., Hsueh R. P., Yang C. P. & Luh T. K. Clinical and Microbiological Characteristics of Bacteremia Caused by Acinetobacter lwoffii. European Journal of Clinical Microbiology and Infectious Diseases 19, 501–505, 10.1007/s100960000315. [DOI] [PubMed] [Google Scholar]
- Feng Y., Yang P., Wang X. & Zong Z. Characterization of Acinetobacter johnsonii isolate XBB1 carrying nine plasmids and encoding NDM-1, OXA-58 and PER-1 by genome sequencing. The Journal of antimicrobial chemotherapy 71, 71–75, 10.1093/jac/dkv324 (2016). [DOI] [PubMed] [Google Scholar]
- Dortet L., Legrand P., Soussy C. J. & Cattoir V. Bacterial identification, clinical significance, and antimicrobial susceptibilities of Acinetobacter ursingii and Acinetobacter schindleri, two frequently misidentified opportunistic pathogens. Journal of clinical microbiology 44, 4471–4478, 10.1128/JCM.01535-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G. D. & Hogue C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC bioinformatics 4, 2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R. et al. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry 38, 4691–4700, 10.1021/bi982858v (1999). [DOI] [PubMed] [Google Scholar]
- Aldridge B. B., Burke J. M., Lauffenburger D. A. & Sorger P. K. Physicochemical modelling of cell signalling pathways. Nature cell biology 8, 1195–1203, 10.1038/ncb1497 (2006). [DOI] [PubMed] [Google Scholar]
- Calzone L. et al. Mathematical modelling of cell-fate decision in response to death receptor engagement. PLoS computational biology 6, e1000702, 10.1371/journal.pcbi.1000702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert R. Scale-free networks in cell biology. Journal of cell science 118, 4947–4957, 10.1242/jcs.02714 (2005). [DOI] [PubMed] [Google Scholar]
- Tesmer J. J., Klem T. J., Deras M. L., Davisson V. J. & Smith J. L. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nature structural biology 3, 74–86 (1996). [DOI] [PubMed] [Google Scholar]
- Hedstrom L., Liechti G., Goldberg J. B. & Gollapalli D. R. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr Med Chem. 13, 1909–1918 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz J., Gold M. & Anders M. The Enzymatic Methylation of Ribonucleic Acid and Deoxyribonucleic Acid. Iv. The Properties of the Soluble Ribonucleic Acid-Methylating Enzymes. The Journal of biological chemistry 239, 3474–3482 (1964). [PubMed] [Google Scholar]
- Smolar N., Hellman U. & Svensson I. Two transfer RNA (1-methylguanine) methylases from yeast. Nucleic acids research 2, 993–1004 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]