

Summary
Myofibroblasts (myoFb) are phenotypically transformed, contractile fibroblast-like cells expressing α-smooth muscle actin microfilaments. They are integral to collagen fibrillogenesis with scar tissue formation at sites of repair irrespective of the etiologic origins of injury or tissue involved. MyoFb can persist long after healing is complete, where their ongoing turnover of collagen accounts for a progressive structural remodeling of an organ (a.k.a. fibrosis, sclerosis or cirrhosis). Such persistent metabolic activity is derived from a secretome consisting of requisite components in the de novo generation of angiotensin (Ang) II. Autocrine and paracrine signaling induced by tissue AngII is expressed via AT1 receptor ligand binding to respectively promote: i) regulation of myoFb collagen synthesis via the fibrogenic cytokine TGF-β1-Smad pathway; and ii) dedifferentiation and protein degradation of atrophic myocytes immobilized and ensnared by fibrillar collagen at sites of scarring.
Several cardioprotective strategies in the prevention of fibrosis and involving myofibroblasts are considered. They include: inducing myoFb apoptosis through inactivation of antiapoptotic proteins; AT1 receptor antagonist to interfere with auto-/paracrine myoFb signaling or to induce counterregulatory expression of ACE2; and attacking the AngII-AT1R-TGF-β1-Smad pathway by antibody or the use of triplex-forming oligonucleotides.
Keywords: cardiac fibrosis, myofibroblast secretome, auto-/paracrine signaling, angiotensin II, cardioprotection
Introduction
Fibrosis is a fundamental component of the pathological remodeling found in the failing heart irrespective of its etiologic origins. In hypertensive heart disease, for example, a diffuse interstitial fibrosis and microscopic scarring are present in association with cardiomyocyte hypertrophy and atrophy which is found at sites of fibrosis [1, 2]. In ischemic cardiomyopathy, an important morphologic finding remote to an infarct scar is the widely scattered foci of microscopic scars, indicative of ongoing bouts of necrosis with reparative fibrosis [1, 3], and with an attendant atrophy of cardiomyocytes ensnared by fibrillar collagen. Scarring is also a feature of hypertrophic cardiomyopathy involving both the hypertrophied left and nonhypertrophied right ventricles and where heterogeneity in myocyte size is again seen [4, 5].
Myofibroblasts are responsible for scar tissue formation at every site of cardiomyocyte necrosis. Myocyte apoptosis elicits neither a wound-healing response nor scarring and therefore leaves no morphologic footprint [6]. Metabolic signaling from the myofibroblast secretome regulates ongoing fibrillar collagen production integral to the scar tissue formation and its subsequent turnover. During the early stages of wound repair, inflammatory cell-derived TGF-β1 induces the de novo expression of angiotensinogen, renin and angiotensin-converting enzyme (ACE) and the secretory phenotype of myofibroblasts and which, in turn, serves to generate angiotensin II at the site of repair (see Figure 1) [7-9]. The autocrine signaling invoked by this tissue peptide is mediated via AT1 receptor binding with the resultant expression of the fibrogenic cytokine transforming growth factor (TGF)-β1 [10]. Together with activation of downstream connective tissue growth factor and Smad-signaling pathway, the deposition of fibrillar collagen types I and III follows with scar tissue formation. An active interplay also exists between myofibroblasts and the extracellular structural protein matrix, including incorporation of latent TGF-β1 with its binding protein and its release and activation by proteases under the influence of reactive oxygen species [11]. The heterocellular paracrine signaling between myofibroblast-derived AngII and neighboring cardiomyocytes (see Figure 2) raises myocyte cytosolic [Ca2+]i to induce oxidative stress and activate redox-sensitive proteolytic ligases of the ubiquitin proteasome system (UPS) with resultant protein degradation leading to cell atrophy. Myofibroblasts also promote the dedifferentiation of these atrophic myocytes with re-expression of fetal genes, including β-myosin heavy chain and natriuretic peptides [12-20]. The re-expression of these fetal genes in atrophic myocytes, as well as in hypertrophied myocytes, is mediated by reduced intracellular thyroid hormone signaling. This localized hypothyroid state arises from the increased degradative activity of deiodinase-3 and its metabolism of T3 and T4 into inactive compounds [21-23].
Figure 1.

The myofibroblast secretory phenotype found at the site of healing. This myofibroblast secretome includes the de novo generation of angiotensin II and subsequent induction of collagen synthesis by these cells. Included in the secretome is the expression of renin, ACE and AT1 receptors. Autocrine actions of angiotensin II, mediated via AT1 receptor binding, results in expression of fibrogenic TGF-β1 and CTGF to stimulate myofibroblast production of fibronectin, which forms a provisional scaffold for type I and type III collagen fibrillogenesis. Abbreviations: ACE, angiotensin-converting enzyme; AT1, angiotensin II type 1; CTGF, connective tissue growth factor; MMPs, matrix metalloproteinases; TGF-β1, transforming growth factor β1. Adapted from Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15-26.
Figure 2.
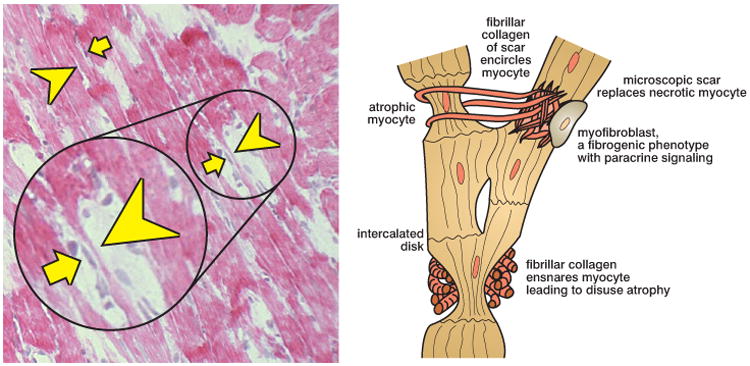
Segmental myocyte atrophy along a myofiber composed of individual myocytes joined end-to-end to form an in-series syncytium. Left panel: longitudinal perspective of several myofiber syncytia as seen by light microscopy. Arrowheads indicate atrophied cells composing this syncytia while arrows identify myofibroblasts juxtaposed to these atrophied myocytes (hematoxylin and eosin, ×200). Right panel: a schematic representation of normal and atrophic myocytes of the myofiber syncytium and where collagen fibrils emanating from scar tissue encircle myocytes. Myocytes so ensnared are smaller and subject to disuse atrophy. An activated myofibroblast with a fibrogenic phenotype is seen in proximity to an atrophied myocyte. Reprinted with permission from Al Darazi F, Zhao W, Zhao T, Sun Y, Marion TN, Ahokas RA, Bhattacharya SK, Gerling IC, Weber KT. Small dedifferentiated cardiomyocytes bordering on microdomains of fibrosis: evidence for reverse remodeling with assisted recovery. J Cardiovasc Pharmacol. 2014;64:237-246.
Given their diverse roles in cardiac remodeling, myofibroblasts and their secretome are targeted in the prevention of fibrosis. The purpose of this mini-review is to provide a perspective that addresses the role of myofibroblasts in cardiac repair, their secretome and its auto- and paracrine signaling by angiotensin II in leading to adverse myocardial remodeling, and finally several myofibroblast-directed cardioprotective strategies. A full discourse on the many aspects of myofibroblast biology and antifibrotic strategies that could be utilized in cardioprotection is beyond the scope of this report. The interested reader is referred to reviews found elsewhere [24-27].
Myofibroblasts and Tissue Repair
Myofibroblast-mediated scar tissue formation appears following cardiomyocyte necrosis, irrespective of whether such cell loss involves a segment of myocardium with a macroscopic infarct scar or as a microscopic scar with the loss of individual myocytes. The origins of these fibroblast-like cells remain uncertain. Pericytes of the microvasculature, circulating fibrocytes derived from bone marrow stem cells, and usual interstitial fibroblasts have each been implicated [24, 28]. Valvular interstitial cells, normal residents of heart valve leaflets and having a myofibroblast phenotype, are another possible source [29]. An endothelial-mesenchymal cell transition has also been suggested [30].
In response to inflammatory cell-derived TGF-β1 activation, myofibroblasts produce angiotensin II and fibronectin [31], creating a provisional scaffolding for the subsequent angiotensin II-AT1R-TGF-β1-Smad-mitogen-activated-protein-kinase-mediated deposition of fibrillar collagen types I and III [11, 32], the major structural proteins forming scar tissue [33, 34]. Stiff, cross-linked type I fibrillar collagen confers strength to resist scar deformation and, in turn, to resist myocardial thinning and ventricular chamber dilatation. And while the structural integrity of myocardium is preserved, its architecture, mechanical and electrical characteristics have been disrupted.
Myofibroblasts dominate the regulation of collagen turnover in the injured heart [35, 36]. Fibrillogenesis is self-regulated through myofibroblast expression of molecules requisite to the formation of angiotensin II and its subsequent autocrine signaling via AT1 receptor binding [10, 37-40]. At each site of scarring with their resident myofibroblasts, is the high-density expression of ACE and AT1 receptor binding involving these cells irrespective of the injured tissue or the cause of injury [37-41]. The de novo generation of angiotensin peptides has been demonstrated in α-SMA-positive valvular interstitial cells and myofibroblasts harvested from a 4-wk-old infarct scar [29, 42]. Their ACE activity and responsiveness to ACE inhibition remains intact at each site, including that of the fibrosed visceral pericardium as detected by monitoring angiotensin II in the superfusate of the isolated perfused heart [43].
Persistent Myofibroblasts and their Activity
Myofibroblasts persist for months [32] and even years [44] within an infarct scar. Their anticipated disappearance through programmed cell death when tissue repair is complete fails to occur [44]. Beyond their persistence is a secretory phenotype with ongoing scar tissue collagen turnover to confer stability and strength to the infarct scar of this muscular pump. However, the mobility of α-smooth muscle actin-containing myofibroblasts in migrating to distant sites and the soluble signals they generate at these sites invokes a progressive fibrosis of myocardium. The pathophysiological mechanisms accounting for this progressive fibrosis, however, remain unclear. Several possibilities, each driven by reactive oxygen species derived from mitochondria and/or NADPH oxidase of these cells [45-47], could be theorized. A persistent highly synthetic myofibroblast phenotype with an excessive generation of type I collagen driven by angiotensin II-induced intracellular Ca2+ overloading with oxidative stress [48]. Another is repeated episodes of nonischemic cardiomyocyte necrosis, with each bout eliciting a wound-healing response. Such a repetition of the wound-healing response would suggest myofibroblasts are never quiescent, instead remaining in an unrelenting profibrogenic phenotype.
Myofibroblast Secretome: Autocrine Signaling
The de novo generation of the angiotensin II by myofibroblasts is a self-regulating autocrine signal that can lead to ongoing fibrosis via protracted AT1 receptor binding. Also involved in this self-sustained myofibroblast collagen synthesis is TGF-β1 [49] and Smad-dependent and Smad-independent proteins with connective tissue growth factor (see Figure 1) [50]. Downstream to AT1 receptor binding signal transduction pathways involve TGF-β1, Smad proteins, and connective tissue growth factor. Interleukins 1, 6, and 13, and noncoding RNA molecules are involved in regulating myofibroblast collagen synthesis [51]. Intracellular signaling pathways regulating the myofibroblast secretome and collagen turnover are under investigation [52]. TGF-β1, for example, regulates myofibroblast expression of scleraxis, a profibrotic transcription factor which stimulates collagen synthesis via a Smad-independent pathway [53]. TGF-β1 suppresses myofibroblast gene expression of matrix metalloproteinases involved in collagen degradation [54]. Because myofibroblasts are persistent and have ongoing auto-/paracrine activity, scar tissue is metabolically active [35].
Myofibroblast Secretome: Paracrine Signaling
Fibrosis is a “crucial determinant” of the tissue heterogeneity found within the diseased myocardium of the failing heart [55]. Myocyte size is normally variable [56]; this variability is accentuated in the failing heart because tendrils of fibrillar collagen, emanating from scar tissue to secure it within this contractile organ, ensnare and immobilize neighboring cardiomyocytes (see Figure 2). In so doing, the work of these myocytes is reduced and disuse atrophy ensues [57].
Myofibroblast angiotensin II signaling has paracrine properties on neighboring myocytes again mediated by AT1 receptor binding with increments in cytosolic [Ca2+]i taking place via store-operated Ca2+ channels that induce oxidative stress and myocyte dedifferentiation with re-expression of β-MHC and ANP, a fetal gene program also found in hypertrophied myocytes. The important role of oxidative stress in promoting protein degradation with atrophy is further suggested by the response to antioxidants in its prevention [12-17, 19, 20, 58-60]. Angiotensin II and oxidative stress are similarly involved in skeletal muscle atrophy [61]. Activation of redox-sensitive, proteolytic ligases (MuRF1 and atrogin-1) of the ubiquitin-proteasome system with attendant protein degradation is essential to atrophy of immobilized myocytes ensnared by fibrillar collagen (see Figure 3) [20].
Figure 3.

Myofibroblast (myoFb) cross-talk with neighboring myocytes via paracrine signaling involving de novo angiotensin (Ang) II generation and AT1 receptor binding. Ensuing IP3 stimulation leads to the release from and subsequent fall in endoplasmic reticulum [Ca2+]er whose Ca2+ sensor, STIM1, in turn, is then activated to promote store-operated Ca2+ channel entry (SOCE) to raise cytosolic [Ca2+]i and mitochondrial [Ca2+]m. Ensuing oxidative stress and reactive oxygen species (ROS) activate proteolytic UPS ligases (MuRF1 and atrogin-1) leading to myocyte protein degradation with resultant atrophy.
The loss of cardiomyocytes through necrotic and apoptotic forms of cell death, together with atrophic cardiomyocytes contribute to the progressive nature of heart failure. Collectively, the structural remodeling of myocardium by progressive accumulation of an excessive fibrillar collagen matrix has led to the concept of interstitial heart disease [62, 63]. Molecular signaling emanating from myofibroblasts are therefore logical targets for developing cardioprotective strategies to prevent fibrosis.
Cardioprotective Strategies
Given the adverse impact of fibrosis on tissue stiffness, arrhythmogenesis, and the conversion of adaptive to pathologic hypertrophy, antifibrotic strategies are of marked interest. Several are briefly discussed.
Subsarcolemmal Mitochondria
One strategy proposed to attenuate the appearance of cardiac fibrosis is mitochondria-targeted interventions aimed at preventing cardiomyocyte necrosis, a requisite that initiates tissue repair with scarring. This mitochondria-targeted cardioprotective strategy has focused on interrupting the mitochondriocentric signal-transducer-effector pathway to necrosis by preventing subsarcolemmal mitochondrial Ca2+ overload. This includes the use of targeted antioxidants or inhibition of the opening of the mitochondrial inner membrane permeability transition pore which is less resistant in these organelles as contrasted to interfibrillar mitochondria [64-66]. Such strategies have included nutriceuticals (flavonoids) [67], pharmaceuticals (cyclosporine A or third-generation β-adrenergic-receptor antagonists) [68], inhaled hydrogen gas [69], or microRNAs [70-72].
Myofibroblast Survival
Therapies that disrupt myofibroblast survival are another potential direction in cardioprotection. Apoptotic clearance of myofibroblasts would serve to remove the ongoing cellular supply of collagen. Nuclear factor kappa B (NF-κB), is integral to fibrogenesis and myofibroblast survival. Transcriptional inhibition of NF-κB, a redox-sensitive transcription factor, could inactivate antiapoptotic proteins [73] as may epigenetic modifications of myofibroblast gene expression and survival [74]. A DNA methylation inhibitor has shown promise in the treatment of hypertension-induced hypertrophy with fibrosis [75].
Myofibroblast-Derived Angiotensin II
Angiotensin II and its AT1 receptor binding play a pivotal role in the autocrine regulation of collagen turnover and paracrine myocyte signaling. Losartan and valsartan, AT1 receptor blockers, have each proven cardioprotective [10, 40]. Other AT1 receptor antagonists likewise prevent reactive fibrosis and attenuate reparative fibrosis in the infarcted heart, in hypertensive heart disease, and in cardiomyopathy induced by rapid atrial pacing [37, 40, 76-79]. Efficacy of AT1 receptor blockade in suppressing the myofibroblast-based ACE-angiotensin II-TGF-β1 signaling axis to fibrosis also has been demonstrated in other diseased cardiovascular tissues, including systemic arterioles and aortic aneurysm [80-82].
ACE2 Expression
ACE2, a homologue of ACE, offers a counter-regulatory approach to the control of tissue angiotensin II. ACE2 hydrolyzes the octapeptide angiotensin II into angiotensin (1–7) which exerts its actions via binding to a Mas receptor. The ACE2-angiotensin (1–7)-Mas signaling axis is in equilibrium with the ACE-angiotensin II-AT1 receptor axis and angiotensin (1–7) is counter-regulatory to angiotensin II. Angiotensin (1–7) formation is dependent on angiotensin II as its substrate [83]. Increased levels of cardiac ACE2 and angiotensin (1–7)-forming activity are found in the failing heart and its cardiomyocytes [84, 85] and ACE2 activity is insensitive to ACE inhibitors [83]. AT1-receptor antagonists, on the other hand, increase cardiac expression of ACE2 [86, 87]. Loss of ACE2 augments maladaptive angiotensin II-based remodeling, including upregulation and activation of tissue matrix metalloproteinase [88]. These effects can be blocked by an AT1-receptor antagonist [88]. By contrast, overexpression of ACE2 and upregulated angiotensin (1–7) attenuate pathological remodeling [89, 90]. Chronic inhibition of ACE2 results in increased fibrosis [91].
TGF-β1 Pathway
The TGF-β1 signaling pathway to collagen synthesis can be blocked using a TGF-β1 antibody, antisense oligonucleotide, or its soluble truncated receptor [73]. The Smad 2/3 pathway can be attenuated [92]. Downstream molecular events can be blocked by triplex-forming oligonucleotides to prevent collagen gene transcription and the accumulation of scar tissue [67, 68, 71, 93-95]. Such triplex-forming sequences are present in the promoter of Col1α1 gene and form efficient triplexes with the exogenously added triplex-forming oligonucleotides and inhibit type I collagen accumulation. Their efficiency in the control of cardiac fibrosis remains to be elucidated.
Limitations
One major caveat to these cardioprotective strategies is the fact collagen turnover is common to all tissues and must not be compromised. Cardiac-targeted strategies, therefore, must be tissue specific, delivered to precise cellular and subcellular locations. Moreover, scar tissue preserves the structural integrity of the heart. Should its formation be prevented at the site of myocyte necrosis, the myocardium would be weakened and subject to rupture. Limited proteolytic digestion of interstitial fibrosis, on the other hand, would be desirable if it releases atrophic myocytes ensnared by fibrillar collagen and regression of fibrous tissue would improve diastolic stiffness. Such has been attained in hypertensive heart disease with an ACE inhibitor or angiotensin-receptor antagonist [96-99].
Summary and Conclusions
When cardiomyocyte necrosis occurs, a population of collagen-expressing myofibroblasts appears to produce scar tissue and preserve the structural integrity of injured myocardium. Persistent myofibroblasts and their secretory phenotype, however, continue their ongoing deposition of stiff, cross-linked type I fibrillar collagen. This progressive fibrosis has multiple adverse consequences on its structure and function. Indeed, fibrosis is fundamental to adverse structural remodeling of the failing heart, irrespective of its etiologic origins, and draws attention to the important role of myofibroblasts and the entity interstitial heart disease.
The myofibroblast secretome and its angiotensin II–AT1 receptor–TGF-β1-Smad autocrine signaling pathway to fibrosis, is common to sites of healing in the heart, as well as other tissues and organs. Both auto- and paracrine signaling involving myofibroblasts and myocytes, respectively, can be interrupted with an AT1-receptor antagonist. By upregulating a counter-regulatory ACE2-angiotensin (1–7)-Mas receptor axis in degrading AngII, an alternate strategy in cardioprotection could be realized. Other cardioprotective strategies include: the use of mitochondria-targeted antioxidants to prevent cardiomyocyte necrosis and subsequent scarring; the use of therapies that modify myofibroblast survival; and targeting downstream pathways using microRNAs and triplex-forming oligonucleotides to prevent type I collagen transcription.
Expert Commentary
As is the case for any organ, fibrosis disrupts architecture and function of its parenchyma. In the heart, fibrosis can preserve the structure of this hollow muscular organ when injured, but at the expense of its diastolic and systolic function. When cardiomyocyte necrosis occurs, a population of collagen-expressing myofibroblasts appear at the site of injury to produce scar tissue and repair the wound. Myofibroblasts are persistent and have secretory phenotype which continues their deposition of stiff, cross-linked type I fibrillar collagen at and remote to the site of injury with this progressive fibrosis having multiple adverse consequences on ventricular structure and function. Indeed, fibrosis is a fundamental component of the adverse structural remodeling of the failing heart and the entity interstitial heart disease. Fibrosis therefore draws attention to the important role of myofibroblasts and their active participation in adverse cardiac remodeling.
The myofibroblast secretome and its angiotensin II–AT1 receptor–TGF-β1-Smad autocrine signaling pathway to fibrosis, is common to sites of healing in the heart irrespective of the etiologic origins of injury. Autocrine signaling involving myofibroblasts and paracrine heterocellular signaling with myocytes can each be interrupted with an AT1-receptor antagonist. By upregulating a counter-regulatory ACE2-angiotensin (1–7)-Mas receptor axis in degrading AngII, an alternate strategy in cardioprotection could be realized. Other cardioprotective strategies include: the use of mitochondria-targeted antioxidants to prevent cardiomyocyte necrosis and subsequent scarring; the use of therapies that modify myofibroblast survival; and targeting downstream pathways using microRNAs and triplex-forming oligonucleotides to prevent type I collagen transcription are each under investigation. These are but several strategies that are under consideration in the prevention of cardiac fibrosis.
Five-Year View
Heart failure continues to be a worldwide health problem of ever-increasing proportions. The appearance of the congestive heart failure (CHF) syndrome with its characteristic symptoms and signs is rooted in neurohormonal activation. Accordingly, today's management of CHF focuses on pharmacologic interference with effector hormones of the renin-angiotensin-aldosterone and adrenergic nervous systems. This is a palliative approach. It does not address the pathophysiologic basis of the heart's failure as a muscular pump. The adverse structural remodeling of myocardium by a progressive fibrosis imparted by a population of persistent, metabolically active myofibroblasts offers several potential cardioprotective strategies, which are under development and implementation. They include: inducing myofibroblast apoptosis through inactivation of antiapoptotic proteins; AT1 receptor antagonist to interfere with auto-/paracrine myofibroblast signaling or to induce counterregulatory expression of ACE2; and attacking the TGF-β1-Smad pathway by antibody or antisense or triplex-forming oligonucleotides. Other potential strategies under development are beyond the scope of this brief perspective. This notwithstanding, targeting the secretory phenotype of this important nonmyocyte cell and its active participation in the appearance of interstitial heart disease offers potential in the prevention of heart failure. The next five years will determine whether this potential can be realized.
Key Issues.
When cardiomyocyte necrosis occurs, a population of collagen-expressing myofibroblasts appear to produce scar tissue and preserve the structural integrity of injured myocardium.
Myofibroblasts (myoFb) are phenotypically transformed, contractile fibroblast-like cells expressing α-smooth muscle actin microfilaments. They are integral to collagen fibrillogenesis with scar tissue formation at sites of repair irrespective of the etiologic origins of injury or tissue involved.
MyoFb can persist long after healing is complete, where their ongoing turnover of collagen accounts for a progressive structural remodeling of an organ (a.k.a. fibrosis, sclerosis or cirrhosis). Such persistent metabolic activity is derived from their secretome consisting of requisite components in the de novo generation of angiotensin (Ang) II.
Autocrine and paracrine signaling induced by tissue AngII is expressed via AT1 receptor ligand binding to respectively promote: i) regulation of myoFb collagen synthesis via the fibrogenic cytokine TGF-β1-Smad pathway; and ii) dedifferentiation and protein degradation of atrophic myocytes immobilized and ensnared by fibrillar collagen at sites of scarring.
Several cardioprotective strategies in the prevention of fibrosis are considered. They include: inducing myoFb apoptosis through deactivation of antiapoptotic proteins; AT1 receptor antagonist to interfere with auto-/paracrine myoFb signaling or to induce counterregulatory expression of ACE2; and attacking the TGF-β1-Smad pathway by antibody or the use of antisense or triplex-forming oligonucleotides.
Acknowledgments
This work was supported, in part, by NIH grants R01-HL73043 and R01-HL90867 (to KT Weber); R01-HL77668 and R01-HL96503 (to Y Sun). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Financial and competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Cotran RS, Kumar V, Robbins SL. The heart. In: Cotran RS, Kumar V, Robbins SL, editors. Robbins Pathologic Basis of Disease. 4th. Philadelphia: W B Saunders; 1989. pp. 597–656. [Google Scholar]
- 2.Pearlman ES, Weber KT, Janicki JS, Pietra GG, Fishman AP. Muscle fiber orientation and connective tissue content in the hypertrophied human heart. Lab Invest. 1982;46:158–164. [PubMed] [Google Scholar]
- 3.Beltrami CA, Finato N, Rocco M, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. **The presence of widely scattered foci of fibrosis found in the explanted failing human heart of ischemic origins. [DOI] [PubMed] [Google Scholar]
- 4.Factor SM, Butany J, Sole MJ, et al. Pathologic fibrosis and matrix connective tissue in the subaortic myocardium of patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1991;17:1343–1351. doi: 10.1016/s0735-1097(10)80145-7. [DOI] [PubMed] [Google Scholar]
- 5.Choudhury L, Mahrholdt H, Wagner A, et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;40:2156–2164. doi: 10.1016/s0735-1097(02)02602-5. [DOI] [PubMed] [Google Scholar]
- 6.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Zhang J, Zhang JQ, Weber KT. Renin expression at sites of repair in the infarcted rat heart. J Mol Cell Cardiol. 2001;33:995–1003. doi: 10.1006/jmcc.2001.1365. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y, Cleutjens JPM, Diaz-Arias AA, Weber KT. Cardiac angiotensin converting enzyme and myocardial fibrosis in the rat. Cardiovasc Res. 1994;28:1423–1432. doi: 10.1093/cvr/28.9.1423. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, Weber KT. Cells expressing angiotensin II receptors in fibrous tissue of rat heart. Cardiovasc Res. 1996;31:518–525. [PubMed] [Google Scholar]
- 10.Sun Y, Zhang JQ, Zhang J, Ramires FJA. Angiotensin II, transforming growth factor-β1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–1569. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 11.Horiguchi M, Ota M, Rifkin DB. Matrix control of transforming growth factor-β function. J Biochem. 2012;152:321–329. doi: 10.1093/jb/mvs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ausma J, Wijffels M, van Eys G, et al. Dedifferentiation of atrial cardiomyocytes as a result of chronic atrial fibrillation. Am J Pathol. 1997;151:985–997. [PMC free article] [PubMed] [Google Scholar]
- 13.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol. 2005;202:891–899. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- 14.LaFramboise WA, Scalise D, Stoodley P, et al. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–1808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 15.Rücker-Martin C, Pecker F, Godreau D, Hatem SN. Dedifferentiation of atrial myocytes during atrial fibrillation: role of fibroblast proliferation in vitro. Cardiovasc Res. 2002;55:38–52. doi: 10.1016/s0008-6363(02)00338-3. [DOI] [PubMed] [Google Scholar]
- 16.Driesen RB, Verheyen FK, Dispersyn GD, et al. Structural adaptation in adult rabbit ventricular myocytes: influence of dynamic physical interaction with fibroblasts. Cell Biochem Biophys. 2006;44:119–128. doi: 10.1385/CBB:44:1:119. [DOI] [PubMed] [Google Scholar]
- 17.Szibor M, Pöling J, Warnecke H, Kubin T, Braun T. Remodeling and dedifferentiation of adult cardiomyocytes during disease and regeneration. Cell Mol Life Sci. 2014;71:1907–1916. doi: 10.1007/s00018-013-1535-6. **Cardiomyocyte dedifferentiated with fegal gene program in the failing heart. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaglia T, Dedja A, Candiotto C, et al. Cardiac interstitial cells express GATA4 and control dedifferentiation and cell cycle re-entry of adult cardiomyocytes. J Mol Cell Cardiol. 2009;46:653–662. doi: 10.1016/j.yjmcc.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Kamalov G, Zhao W, Zhao T, et al. Atrophic cardiomyocyte signaling in hypertensive heart disease. J Cardiovasc Pharmacol. 2013;62:497–506. doi: 10.1097/FJC.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al Darazi F, Zhao W, Zhao T, et al. Small dedifferentiated cardiomyocytes bordering on microdomains of fibrosis: evidence for reverse remodeling with assisted recovery. J Cardiovasc Pharmacol. 2014;64:237–246. doi: 10.1097/FJC.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pol CJ, Muller A, Simonides WS. Cardiomyocyte-specific inactivation of thyroid hormone in pathologic ventricular hypertrophy: an adaptative response or part of the problem? Heart Fail Rev. 2010;15:133–142. doi: 10.1007/s10741-008-9133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wassen FW, Schiel AE, Kuiper GG, et al. Induction of thyroid hormone-degrading deiodinase in cardiac hypertrophy and failure. Endocrinology. 2002;143:2812–2815. doi: 10.1210/endo.143.7.8985. [DOI] [PubMed] [Google Scholar]
- 23.Pol CJ, Muller A, Zuidwijk MJ, et al. Left-ventricular remodeling after myocardial infarction is associated with a cardiomyocyte-specific hypothyroid condition. Endocrinology. 2011;152:669–679. doi: 10.1210/en.2010-0431. [DOI] [PubMed] [Google Scholar]
- 24.Dixon IMC, Wigle JT. Cardiac Fibrosis and Heart Failure: Cause or Effect? New York: Springer International; 2015. **A recent compilation of state-of-the-art reviews dealing with pathogenesis and pathophysiology of cardiac fibrosis and the role of myofibroblasts. [Google Scholar]
- 25.Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124:4673–4677. doi: 10.1172/JCI74368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neary R, Watson CJ, Baugh JA. Epigenetics and the overhealing wound: the role of DNA methylation in fibrosis. Fibrogenesis Tissue Repair. 2015;8:18. doi: 10.1186/s13069-015-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 29.Katwa LC, Tyagi SC, Campbell SE, et al. Valvular interstitial cells express angiotensinogen and cathepsin D, and generate angiotensin peptides. Int J Biochem Cell Biol. 1996;28:807–821. doi: 10.1016/1357-2725(96)00012-x. [DOI] [PubMed] [Google Scholar]
- 30.Montorfano I, Becerra A, Cerro R, et al. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab Invest. 2014;94:1068–1082. doi: 10.1038/labinvest.2014.100. [DOI] [PubMed] [Google Scholar]
- 31.Bondi CD, Manickam N, Lee DY, et al. NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cleutjens JPM, Verluyten MJA, Smits JFM, Daemen MJAP. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 33.Whittaker P. Unravelling the mysteries of collagen and cicatrix after myocardial infarction. Cardiovasc Res. 1996;31:19–27. [PubMed] [Google Scholar]
- 34.Fomovsky GM, Rouillard AD, Holmes JW. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol. 2012;52:1083–1090. doi: 10.1016/j.yjmcc.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Kiani MF, Postlethwaite AE, Weber KT. Infarct scar as living tissue. Basic Res Cardiol. 2002;97:343–347. doi: 10.1007/s00395-002-0365-8. [DOI] [PubMed] [Google Scholar]
- 36.van den Borne SW, Diez J, Blankesteijn WM, et al. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Weber KT. Angiotensin-converting enzyme and wound healing in diverse tissues of the rat. J Lab Clin Med. 1996;127:94–101. doi: 10.1016/s0022-2143(96)90170-5. *Myofibroblast secretory phenotype expressing ACE at diverse sites of fibrous tissue formation. [DOI] [PubMed] [Google Scholar]
- 38.Sun Y, Ramires FJA, Zhou G, Ganjam VK, Weber KT. Fibrous tissue and angiotensin II. J Mol Cell Cardiol. 1997;29:2001–2012. doi: 10.1006/jmcc.1997.0451. [DOI] [PubMed] [Google Scholar]
- 39.Katwa LC, Sun Y, Campbell SE, et al. Pouch tissue and angiotensin peptide generation. J Mol Cell Cardiol. 1998;30:1401–1413. doi: 10.1006/jmcc.1998.0708. [DOI] [PubMed] [Google Scholar]
- 40.Sun Y, Zhang J, Lu L, et al. Tissue angiotensin II in the regulation of inflammatory and fibrogenic components of repair in the rat heart. J Lab Clin Med. 2004;143:41–51. doi: 10.1016/j.lab.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Weber KT. Angiotensin II and aldosterone receptor binding in rat heart and kidney: response to chronic angiotensin II or aldosterone administration. J Lab Clin Med. 1993;122:404–411. [PubMed] [Google Scholar]
- 42.Katwa LC, Campbell SE, Tyagi SC, et al. Cultured myofibroblasts generate angiotensin peptides de novo. J Mol Cell Cardiol. 1997;29:1375–1386. doi: 10.1006/jmcc.1997.0376. *The moyfibroblast secretome generating AngII as seen in cells harvested from an infarct scar. [DOI] [PubMed] [Google Scholar]
- 43.Ou R, Sun Y, Ganjam VK, Weber KT. In situ production of angiotensin II by fibrosed rat pericardium. J Mol Cell Cardiol. 1996;28:1319–1327. doi: 10.1006/jmcc.1996.0122. [DOI] [PubMed] [Google Scholar]
- 44.Willems IEMG, Havenith MG, De Mey JGR, Daemen MJAP. The α-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 45.Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002;8:132–140. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- 46.Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014;2:267–272. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Philip JL, Razzaque MA, Han M, et al. Regulation of mitochondrial oxidative stress by β-arrestins in cardiac fibroblasts. Dis Model Mech. 2015 doi: 10.1242/dmm.019968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316:2390–2401. doi: 10.1016/j.yexcr.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 49.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-β1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–1958. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 50.Wang Q, Usinger W, Nichols B, et al. Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4:4. doi: 10.1186/1755-1536-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roche PL, Filomeno KL, Bagchi RA, Czubryt MP. Intracellular signaling of cardiac fibroblasts. Compr Physiol. 2015;5:721–760. doi: 10.1002/cphy.c140044. [DOI] [PubMed] [Google Scholar]
- 53.Zeglinski MR, Roche P, Hnatowich M, et al. TGF β1 regulates scleraxis expression in primary cardiac myofibroblasts by a Smad-independent mechanism. Am J Physiol Heart Circ Physiol. 2015 doi: 10.1152/ajpheart.00584.2015. ajpheart 00584 02015. [DOI] [PubMed] [Google Scholar]
- 54.Ye H, Cai PC, Zhou Q, Ma WL. Transforming growth factor-β suppresses the up-regulation of matrix metalloproteinase-2 by lung fibroblasts in response to tumor necrosis factor-α. Wound Repair Regen. 2011;19:392–399. doi: 10.1111/j.1524-475X.2011.00680.x. [DOI] [PubMed] [Google Scholar]
- 55.Swynghedauw B. Molecular Cardiology for the Cardiologist. Boston: Kluwer; 1995. [Google Scholar]
- 56.Campbell SE, Rakusan K, Gerdes AM. Change in cardiac myocyte size distribution in aortic-constricted neonatal rats. Basic Res Cardiol. 1989;84:247–258. doi: 10.1007/BF01907972. [DOI] [PubMed] [Google Scholar]
- 57.Jalil JE, Janicki JS, Pick R, Abrahams C, Weber KT. Fibrosis-induced reduction of endomyocardium in the rat after isoproterenol treatment. Circ Res. 1989;65:258–264. doi: 10.1161/01.res.65.2.258. [DOI] [PubMed] [Google Scholar]
- 58.Touyz RM, Sventek P, Lariviére R, et al. Cytosolic calcium changes induced by angiotensin II in neonatal rat atrial and ventricular cardiomyocytes are mediated via angiotensin II subtype 1 receptors. Hypertension. 1996;27:1090–1096. doi: 10.1161/01.hyp.27.5.1090. [DOI] [PubMed] [Google Scholar]
- 59.Pandya K, Kim HS, Smithies O. Fibrosis, not cell size, delineates β-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci U S A. 2006;103:16864–16869. doi: 10.1073/pnas.0607700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.López JE, Myagmar BE, Swigart PM, et al. β-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res. 2011;109:629–638. doi: 10.1161/CIRCRESAHA.111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sukhanov S, Semprun-Prieto L, Yoshida T, et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. 2011;342:143–147. doi: 10.1097/MAJ.0b013e318222e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber KT, Jalil JE, Janicki JS, Pick R. Myocardial collagen remodeling in pressure overload hypertrophy: a case for interstitial heart disease. Am J Hypertens. 1989;2:931–940. doi: 10.1093/ajh/2.12.931. [DOI] [PubMed] [Google Scholar]
- 63.Collier P, Ledwidge M, McDonald K. Diagnostics and therapeutic interventions in myocardial interstitial disease, a previously neglected pathology. QJM. 2012;105:721–724. doi: 10.1093/qjmed/hcs072. [DOI] [PubMed] [Google Scholar]
- 64.Griffiths EJ. Mitochondria and heart disease. Adv Exp Med Biol. 2012;942:249–267. doi: 10.1007/978-94-007-2869-1_11. [DOI] [PubMed] [Google Scholar]
- 65.Dai DF, Chen T, Szeto H, et al. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Asemu G, O'Connell KA, Cox JW, et al. Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: effects of aging and long-term aldosterone infusion. Am J Physiol Heart Circ Physiol. 2013;304:H514–528. doi: 10.1152/ajpheart.00674.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shahbaz AU, Kamalov G, Zhao W, et al. Mitochondria-targeted cardioprotection in aldosteronism. J Cardiovasc Pharmacol. 2011;57:37–43. doi: 10.1097/FJC.0b013e3181fe1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheema Y, Sherrod JN, Zhao W, et al. Mitochondriocentric pathway to cardiomyocyte necrosis in aldosteronism: cardioprotective responses to carvedilol and nebivolol. J Cardiovasc Pharmacol. 2011;58:80–86. doi: 10.1097/FJC.0b013e31821cd83c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshida A, Asanuma H, Sasaki H, et al. H2 mediates cardioprotection via involvements of KATP channels and permeability transition pores of mitochondria in dogs. Cardiovasc Drugs Ther. 2012;26:217–226. doi: 10.1007/s10557-012-6381-5. [DOI] [PubMed] [Google Scholar]
- 70.Aurora AB, Mahmoud AI, Luo X, et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest. 2012;122:1222–1232. doi: 10.1172/JCI59327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bauersachs J. Regulation of myocardial fibrosis by MicroRNAs. J Cardiovasc Pharmacol. 2010;56:454–459. doi: 10.1097/FJC.0b013e3181ee81df. [DOI] [PubMed] [Google Scholar]
- 72.Thum T, Lorenzen JM. Cardiac fibrosis revisited by microRNA therapeutics. Circulation. 2012;126:800–802. doi: 10.1161/CIRCULATIONAHA.112.125013. [DOI] [PubMed] [Google Scholar]
- 73.Gieling RG, Burt AD, Mann DA. Fibrosis and cirrhosis reversibility - molecular mechanisms. Clin Liver Dis. 2008;12:915–937. doi: 10.1016/j.cld.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 74.Robinson CM, Watson CJ, Baugh JA. Epigenetics within the matrix: a neo-regulator of fibrotic disease. Epigenetics. 2012;7:987–993. doi: 10.4161/epi.21567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watson CJ, Horgan S, Neary R, et al. Epigenetic therapy for the treatment of hypertension-induced cardiac hypertrophy and fibrosis. J Cardiovasc Pharmacol Ther. 2015 doi: 10.1177/1074248415591698. [DOI] [PubMed] [Google Scholar]
- 76.Smits JFM, van Krimpen C, Schoemaker RG, Cleutjens JPM, Daemen MJAP. Angiotensin II receptor blockade after myocardial infarction in rats: effects on hemodynamics, myocardial DNA synthesis, and interstitial collagen content. J Cardiovasc Pharmacol. 1992;20:772–778. [PubMed] [Google Scholar]
- 77.Tsutsui H, Matsushima S, Kinugawa S, et al. Angiotensin II type 1 receptor blocker attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Hypertens Res. 2007;30:439–449. doi: 10.1291/hypres.30.439. [DOI] [PubMed] [Google Scholar]
- 78.Matsusaka H, Kinugawa S, Ide T, et al. Angiotensin II type 1 receptor blocker attenuates exacerbated left ventricular remodeling and failure in diabetes-associated myocardial infarction. J Cardiovasc Pharmacol. 2006;48:95–102. doi: 10.1097/01.fjc.0000245405.41317.60. [DOI] [PubMed] [Google Scholar]
- 79.Iwamoto M, Hirohata S, Ogawa H, et al. Connective tissue growth factor induction in a pressure-overloaded heart ameliorated by the angiotensin II type 1 receptor blocker olmesartan. Hypertens Res. 2010;33:1305–1311. doi: 10.1038/hr.2010.189. [DOI] [PubMed] [Google Scholar]
- 80.Touyz RM. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp Physiol. 2005;90:449–455. doi: 10.1113/expphysiol.2005.030080. [DOI] [PubMed] [Google Scholar]
- 81.Fujiwara Y, Shiraya S, Miyake T, et al. Inhibition of experimental abdominal aortic aneurysm in a rat model by the angiotensin receptor blocker valsartan. Int J Mol Med. 2008;22:703–708. [PubMed] [Google Scholar]
- 82.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keidar S, Kaplan M, Gamliel-Lazarovich A. ACE2 of the heart: From angiotensin I to angiotensin (1-7) Cardiovasc Res. 2007;73:463–469. doi: 10.1016/j.cardiores.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 84.Zisman LS, Keller RS, Weaver B, et al. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108:1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 85.Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26:369–375. doi: 10.1093/eurheartj/ehi114. discussion 322-364. [DOI] [PubMed] [Google Scholar]
- 86.Takeda Y, Zhu A, Yoneda T, et al. Effects of aldosterone and angiotensin II receptor blockade on cardiac angiotensinogen and angiotensin-converting enzyme 2 expression in Dahl salt-sensitive hypertensive rats. Am J Hypertens. 2007;20:1119–1124. doi: 10.1016/j.amjhyper.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 87.Sukumaran V, Veeraveedu PT, Gurusamy N, et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci. 2011;7:1077–1092. doi: 10.7150/ijbs.7.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kassiri Z, Zhong J, Guo D, et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446–455. doi: 10.1161/CIRCHEARTFAILURE.108.840124. [DOI] [PubMed] [Google Scholar]
- 89.Zhao YX, Yin HQ, Yu QT, et al. ACE2 overexpression ameliorates left ventricular remodeling and dysfunction in a rat model of myocardial infarction. Hum Gene Ther. 2010;21:1545–1554. doi: 10.1089/hum.2009.160. [DOI] [PubMed] [Google Scholar]
- 90.Dong B, Yu QT, Dai HY, et al. Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol. 2012;59:739–747. doi: 10.1016/j.jacc.2011.09.071. [DOI] [PubMed] [Google Scholar]
- 91.Trask AJ, Groban L, Westwood BM, et al. Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am J Hypertens. 2010;23:687–693. doi: 10.1038/ajh.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-β-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 93.Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guntaka RV, Varma BR, Weber KT. Triplex-forming oligonucleotides as modulators of gene expression. Int J Biochem Cell Biol. 2003;35:22–31. doi: 10.1016/s1357-2725(02)00165-6. [DOI] [PubMed] [Google Scholar]
- 95.Koilan S, Hamilton D, Baburyan N, et al. Prevention of liver fibrosis by triple helix-forming oligodeoxyribonucleotides targeted to the promoter region of type I collagen gene. Oligonucleotides. 2010;20:231–237. doi: 10.1089/oli.2010.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brilla CG, Janicki JS, Weber KT. Cardioreparative effects of lisinopril in rats with genetic hypertension and left ventricular hypertrophy. Circulation. 1991;83:1771–1779. doi: 10.1161/01.cir.83.5.1771. [DOI] [PubMed] [Google Scholar]
- 97.Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. doi: 10.1161/01.cir.102.12.1388. [DOI] [PubMed] [Google Scholar]
- 98.Varo N, Iraburu MJ, Varela M, et al. Chronic AT1 blockade stimulates extracellular collagen type I degradation and reverses myocardial fibrosis in spontaneously hypertensive rats. Hypertension. 2000;35:1197–1202. doi: 10.1161/01.hyp.35.6.1197. [DOI] [PubMed] [Google Scholar]
- 99.Díez J, Querejeta R, López B, et al. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–2517. doi: 10.1161/01.cir.0000017264.66561.3d. *Role of AT1 receptor antagonism in regressing cardiac fibrosis. [DOI] [PubMed] [Google Scholar]