

Abstract
This study investigated the effect of systemic administration of isosorbide-dinitrate (ISDN) on oxidative stress and brain monoamines in a toxic model of brain demyelination evoked by intracerebral injection (i.c.i) of ethidium bromide (10 µl of 0.1 %). Rats received saline (control) or ISDN at 5 or 10 mg/kg for 10 days prior to injection of ethidium bromide. Rats were euthanized one day later, and then the levels of reduced glutathione (GSH), lipid peroxidation (malondialdehyde; MDA), nitric oxide (nitrite/nitrate), acetylcholinesterase (AChE) activity, paraoxonase activity as well as monoamine levels (serotonin, dopamine and noradrenaline) were assessed in the brain cortex in different treatment groups. The i.c.i of ethidium bromide resulted in increased oxidative stress in the cortex one day after its injection; (i) MDA increased by 36.9 %; (ii) GSH decreased by 20.8 %, while (iii) nitric oxide increased by 60.3 %; (iv) AChE and paraoxonase activities in cortex decreased by 35.9 % and 29.4 %, respectively; (v) serotonin was significantly increased. In ethidium bromide-treated rats, pretreatment with ISDN at 10 mg/kg decreased cortical MDA by 23.9 %. Reduced glutathione was increased by 25.1 % ISDN at 10 mg/kg, while nitric oxide showed a 32.8 and 41.7 % decrease after 5 and 10 mg/kg of ISDN, respectively. Acetylcholinesterase activity increased by 24.3 % by 10 mg/kg of ISDN. Paraoxonase activity showed further decrease by 72.2 and 83.8 % after treatment with 5 and 10 mg/kg of ISDN, respectively. The administration of ISDN decreased the level of serotonin and noradrenaline compared with the ethidium bromide only treated group. Overall, the present findings suggest neuroprotective effect of ISDN against oxidative stress in this model of chemical demyelination.
Keywords: toxic demyelination, ethidium bromide, isosorbide dinitrate, rat brain
Introduction
Demyelinating diseases of the central nervous system are a heterogeneous group of chronic inflammatory disorders, the hallmark of which is loss of myelin sheath and nerve conduction deficits leading to motor and/or sensory dysfunction and are the leading cause of nontraumatic neurological disability in young adults (Hu and Lucchinetti, 2009[26]). The spectrum of demyelinating disorders includes 'autoimmune' inflammatory demyelinating diseases, the inflammatory demyelinating diseases of infectious aetiology, and the demyelinating or dysmyelinating diseases of genetic/hereditary background. In addition, primary demyelination is present in other conditions, such as brain ischaemia and intoxication (Lassmann, 2001[44]). Multiple sclerosis is by far the most common inflammatory demyelinating disease leading to focal plaques of primary demyelination with a variable degree of axonal and neuronal degeneration (Love, 2006[50]; Lassmann et al., 2007[45]).
Oxidative stress has been implicated in both normal aging and in various neurodegenerative disorders. In the brain, the high content of polyunsaturated fatty acids, the high utilization of oxygen account for the susceptibility to free radical damage. The mechanisms of tissue injury in demyelinating diseases of the central nervous system are poorly understood but increasing evidence support a role for oxidative stress due to an imbalance between free radicals generation and endogenous antioxidant mechanisms. Reactive oxygen species, nitric oxide, and proinflammatory cytokines released by monocyte-derived macrophages contribute to neuroinflammation, demyelination and axonal damage and disease progression in multiple sclerosis (Mirshafiey and Mohsenzadegan, 2009[58]; Smith, 2011[81]; de Vries et al., 2011[12]). Multiple sclerosis patients showed increased generation of superoxide free radicals in blood (Glabinski et al., 1993[18]), elevated levels of thiobarbituric acid reactive substances and reduced protein sulfhydryl groups in cerebrospinal fluid and serum (Mitosek-Szewczyk et al., 2010[59]), suggesting increased free radical production and lipid peroxidation. Oxidized lipids and DNA were highly enriched in active multiple sclerosis plaques (Haider et al., 2011[23]). Evidence also implicates increased nitric oxide generated by the inducible form of nitric oxide synthase (iNOS) in the inflammation and demyelination in multiple sclerosis. Increased iNOS activity has been demonstrated in monocytes/macrophages and/or astrocytes in demyelinating lesions of postmortem tissues in multiple sclerosis (Bagasra et al., 1995[4]; Oleszak et al., 1998[66]; Liu et al., 2001[48]). Nitric oxide is increased in serum of patients with multiple sclerosis (Ibragic et al., 2012[29]). Nitric oxide is likely to be involved in axonal and neuronal injury in demyelinating conditions (Kapoor et al., 2000[33]; Garthwaite et al., 2002[16]).
Changes in neurotransmitter concentrations in multiple sclerosis and the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis are recognized to underlie many neurological symptoms associated with the disease, and there is accumulating evidence demonstrating that immune function is directly regulated by the activity of certain neurotransmitters (Bhat et al., 2010[6]; Lee et al., 2011[46]; Vollmar et al., 2009[89]). It has been recently observed that a mouse model of EAE is associated with chronic deficits in spinal cord concentrations of noradrenaline (NE), 5-hydroxytryptamine (5-HT/ serotonin) and γ-aminobutyric acid (GABA) (Musgrave et al., 2011[63]). Furthermore, recent studies have shown that therapeutic agents that increase GABAergic and monoaminergic signaling can lessen the severity of EAE (Bhat et al., 2010[6]; Simonini et al., 2010[80]; Taler et al., 2010[84]). Nitric oxide may play a role in physiological neuronal functions such as long-term potentiation as a retrograde messenger (Shuman and Madison, 1994[79]; Medina and Izquierdo, 1995[57]) and in the regulation of gene expression (Yun et al., 1997[97]). Furthermore, it can act as a potent vasodilator and an inhibitor of platelet aggregation (Iadecola, 1997[28]; Szabo, 1996[83]) and, as has been reported, in the S-nitrosylation of proteins (Arnelle and Stamler,1995[3]; Rauhala et al., 1998[72]). Earlier studies have shown that nitric oxide exerts a regulatory influence on behavioral and physiological parameters in normal and stressed rats (Gulati and Chakraborti, 2007[21]; Masood et al, 2003[55]). The results of Hummel et al. (2006[27]) described an antioxidant effect for nitric oxide.
Isosorbide dinitrate (ISDN) (an orally active form of nitrates) is a drug widely used for the management of coronary ischaemia by virtue of its vasodilatory properties. ISDN is capable of releasing nitric oxide in a concentration- and pH-dependent manner (Jiang et al., 2001[32]). Thus, the present study was designed to investigate the effect of the nitric oxide donor ISDN on oxidative stress and brain monoamines in a model of toxic demyelination evoked by intracerebral injection of ethidium bromide in the rat. Ethidium bromide is a DNA chelating agent that is commonly used to evoke transient central nervous system demyelination in experimental animals, which can be used to study the pathogenetic mechanisms and the possible therapeutic interventions (Yajima and Suzuki, 1979[95]; Jeffery and Blakemore, 1997[31]; Mazzanti et al., 2006[56]).
Materials and Methods
Animals
Twenty five adult male Sprague Dawley rats weighing (130 ± 10 g) (age: 10-11 weeks) were used in this study. The animals were obtained from the Animal House Colony of the National Research Centre (Cairo, Egypt). They were housed in stainless steel wire meshed suspended rodent cages under environmentally controlled conditions. The ambient temperature was 25 ± 2 °C and the light/dark cycle was 12/12 hours. The animals had free access to water and standard rodent chow diet (NRC rodent chow). All animals received human care in compliance with guidelines of the Ethical Committee of National Research Centre, Egypt Centre and followed the recommendations of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985). Equal groups of 5 rats each were used in all experiments.
Drugs and chemicals
Ethidium bromide (Sigma, St Louis, MO, USA) and isosorbide dinitrate (Amrya Pharm. Ind., Cairo, Egypt) was used and dissolved in isotonic (0.9 % NaCl) saline solution immediately before use. The doses of isosorbide dinitrate in the study were based upon the human dose after conversion to that of rat according to Paget and Barnes (1964[67]) conversion tables.
Surgical procedures
Rats were anaesthetized with sodium pentobarbital (40 mg/kg, i.p.) and after shaving the hair from the fronto-occipital area antisepsis was performed with 2 % iodine solution. A hole of 0.5 Cm was made using orthodontic roof motor and number 2 drill to the right of the bregma until the dura matter was exposed. With the use of a Hamilton syringe fitted with a 30-gauge needle the solution of ethidium bromide (10 µl of 0.1 %) was injected in the cisterna pontis (basal), an enlargement of the subarachnoid space on the ventral surface of the pons. A group of rats (n=5) was undergone to the same surgical procedure but injected with saline (0.9 %) and served as negative control. The dura matter left open and the skin together with remainder of the subcutaneous tissue was sutured with a nylon thread 4.0.
Experimental design
Rats randomly assigned into 4 groups (n=5 each) received saline (control) or ISDN at 5 or 10 mg/kg orally for 10 days weeks prior to injection of ethidium bromide. Next day after ethidium bromide injection, the animals were euthanized by decapitation in deep ether anesthesia. Brains were then removed, washed with ice-cold saline solution (0.9 % NaCl), and sectioned into cortex, weighed and stored at -80 °C for further determination of biochemical parameters. The brain was homogenized with 0.1 M phosphate buffer saline at pH 7.4, to give a final concentration of 10 % w/v for the biochemical assays. For the determination of monoamine neurotransmitters, frozen samples were homogenized in cold 0.1 N-perchloric acid.
Biochemical studies
Determination of brain lipid peroxidation
Lipid peroxidation was assayed by measuring the level of malondialdehyde (MDA) in the brain tissues. Malondialdehyde was determined by measuring thiobarbituric reactive species using the method of Ruiz-Larrea et al. (1994[76]) in which the thiobarbituric acid reactive substances react with thiobarbituric acid to produce a red colored complex having peak absorbance at 532 nm.
Determination of brain reduced glutathione content
Reduced glutathione (GSH) was determined in brain tissue by Ellman's method (1959[14]). The procedure is based on the reduction of Ellman´s reagent by -SH groups of GSH to form 2-nitro-s-mercaptobenzoic acid, the nitromercaptobenzoic acid anion has an intense yellow color which can be determined spectrophotometrically. A mixture was directly prepared in a cuvette: 2.25 ml of 0.1 M K-phosphate buffer, pH 8.0; 0.2 ml of the sample; 25 μl of Ellman's reagent (10 mM 5,5′-dithio-bis-2-nitrobenzoic acid in methanol). After 1 min the assay absorbance was measured at 412 nm and the GSH concentration was calculated by comparison with a standard curve.
Determination of brain acetylcholinesterase activity
The procedure used for the determination of acetylcholinesterase activity in the cortex was a modification of the method of Ellman et al. (1961[15]) as described by Gorun et al. (1978[19]). The principle of the method is the measurement of the thiocholine produced as acetylthiocholine is hydrolyzed. The colour was read immediately at 412 nm.
Determination of brain nitric oxide
Nitric oxide measured as nitrite was determined by using Griess reagent, according to the method of Moshage et al. (1995[61]). Where nitrite, stable end product of nitric oxide radical, is mostly used as indicator for the production of nitric oxide.
Determination of brain paraoxonase activity
Arylesterase activity of paraoxonase was measured spectrophotometrically in supernatants using phenylacetate as a substrate (Higashino et al. 1972[24]; Watson et al., 1995[92]).
Determination of brain monoamines
Determination of brain serotonin, noradrenaline and dopamine was carried out using high performance liquid chromatography (HPLC) system, Agilent technologies 1100 series, equipped with a quaternary pump (Quat pump, G131A model). Separation was achieved on ODS reversed phase column (C18, 25 x 0.46 cm i.d. 5 µm). The mobile phase consisted of potassium phosphate buffer/methanol 97/3 (v/v) and was delivered at a flow rate of 1 ml/min. UV detection was performed at 270 nm and the injection volume was 20 µl. The concentration of both catecholamines and serotonin were determined by external standard method using peak areas. Serial dilutions of standards were injected and their peak areas were determined. A linear standard curve was constructed by plotting peak areas versus the corresponding concentrations. The concentration in samples was obtained from the curve.
Statistical analysis
Data are expressed as mean ± SE. Data were analyzed by one-way analysis of variance, followed by Duncan's multiple range test for post hoc comparison of group means. Effects with a probability of p < 0.05 were considered to be significant.
Results
Oxidative stress
In saline treated rats, i.c. ethidium bromide injection resulted in a significant increase in the level of MDA by 36.9 % (48.6 4.1 vs 35.5 3.0 nmol/g, p < 0.05) (Figure 1(Fig. 1)). Reduced glutathione decreased by 20.8 % (5.10 0.28 vs 6.44 0.33 µmol/g, p < 0.05) (Figure 2(Fig. 2)), while nitric oxide increased by 60.3 % (15.45 0.83 vs. 9.64 0.51 µmol/g, p < 0.05) after ethidium bromide injection compared with the saline control group (Figure 3(Fig. 3)).
Figure 1. Effect of isosorbide dinitrate (ISDN) treatment on the concentration of malondialdehyde (MDA) in the cortex of rats subjected to intracerebral injection of ethidium bromide. Data are means ± SEM. *: p < 0.05 vs the saline control group. +: p < 0.05 vs the ethidium bromide control group.
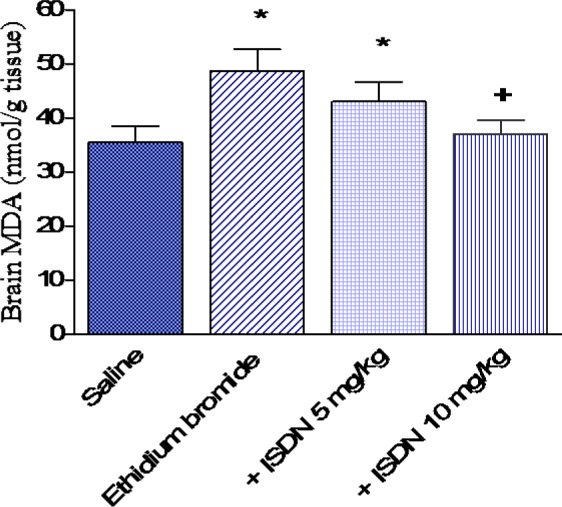
Figure 2. Effect of isosorbide dinitrate (ISDN) treatment on reduced glutathione (GSH) in the rat cortex after intracerebral administration of the demyelinating agent ethidium bromide. Data are means ± SEM. *: p < 0.05 vs the saline control group and between different groups as indicated. +: p < 0.05 vs the ethidium bromide control group.
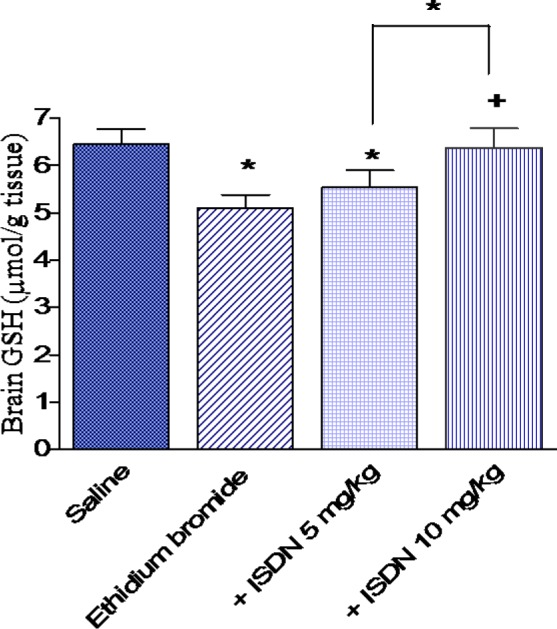
Figure 3. Effect of isosorbide dinitrate (ISDN) treatment on nitric oxide concentration in the rat cortex after intracerebral administration of the demyelinating agent ethidium bromide. Data are means ± SEM. *: p < 0.05 vs the saline control group. +: p < 0.05 vs the ethidium bromide control group.
Pretreatment with ISDN at 5 mg/kg for 10 days prior to ethidium bromide injection had no significant effect on cortical MDA (43.0 3.6 vs 48.6 4.1 nmol/g, p > 0.05). However, ISDN administered at 10 mg/kg resulted in a significant decrease in MDA in cortex by 23.9 % compared with the ethidium bromide control group (37.0 2.6 vs 48.6 4.1 nmol/g, p < 0.05) (Figure 1(Fig. 1)). Reduced glutathione was not significantly altered by ISDN treatment at 5 mg/kg, but increased by 25.1 % after treatment with the higher dose of ISDN (6.38 0.41 vs 5.1 0.28 mol/g) (Figure 2(Fig. 2)). Meanwhile, nitric oxide decreased by 32.8 and 41.7 % following ISDN administration at 5 and 10 mg/kg, respectively, compared with the ethidium bromide control group (10.38 0.64 and 9.0 0.71 vs 15.45 0.83 mol/g, p < 0.05 (Figure 3(Fig. 3)).
Acetylcholinesterase activity
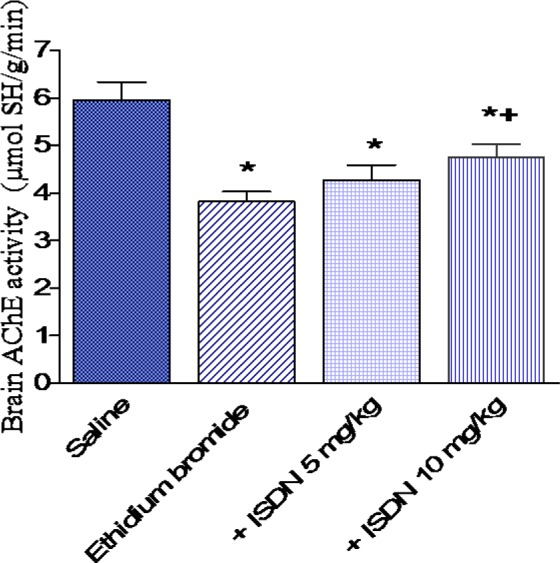
In saline treated rats, AChE activity decreased by 35.9 % after i.c. ethidium bromide injection (3.82 ± 0.21 vs 5.96 ± 0.38 µmol SH/g/min). AChE activity was unaltered in rats treated with ISDN at 5 mg/kg. The higher dose of ISDN, however, increased AChE activity by 24.3 % compared with the ethidium bromide control group (4.75 0.28 vs 3.82 0.21 µmol SH/g/min, p < 0.05 (Figure 4(Fig. 4)).
Figure 4. Effect of isosorbide dinitrate (ISDN) treatment on nitric oxide concentration in the rat cortex after intracerebral injection of the demyelinating agent ethidium bromide. Data are means ± SEM. *: p < 0.05 vs the saline control group. +: p < 0.05 vs the ethidium bromide control group.
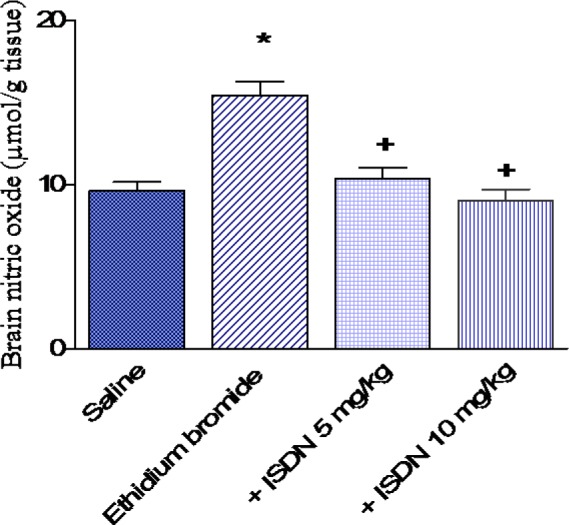
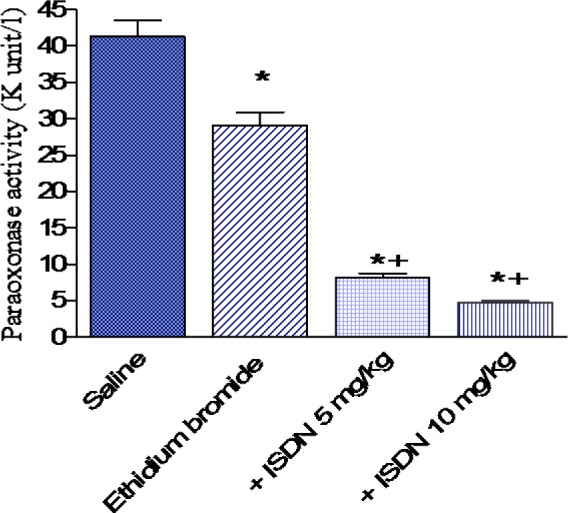
Paraoxonase activity
In saline treated rats, paraoxonase activity decreased by 29.4 % after i.c. ethidium bromide injection (29.1 ± 1.8 vs 41.22 ± 2.3 kU/l). Paraoxonase activity showed a further decrease by 72.2 and 83.8 % after treatment with ISDN at 5 or 10 mg/kg, respectively (8.1 0.62 and 4.7 0.38 vs 29.1 1.8 kU/l, p < 0.05 (Figure 5(Fig. 5)).
Figure 5. Effect of isosorbide dinitrate (ISDN) treatment on paraoxonase activity in the rat cortex after intracerebral injection of the demyelinating agent ethidium bromide. Data are means ± SEM. *: p < 0.05 vs the saline control group. +: p < 0.05 vs the ethidium bromide control group.
Brain monoamines
The levels of dopamine and noradrenaline were not significantly altered by ethidium bromide injection, whereas serotonin was increased compared with the saline control group. ISDN given at 5 or 10 mg/kg resulted in 41.6, 70.6 % decrease in serotonin and 28.6, 31.9 % decrease in noradrenaline, respectively when compared with the ethidium bromide control group (Table 1(Tab. 1)).
Table 1. Effect of ISDN on serotonin, dopamine and noradrenaline in mice cortex after ethidium bromide injection.

Results are mean ± S.E. Six mice were used per each group. Data were analyzed by one way ANOVA and means of different groups were compared by Duncan's multiple range test. P < 0.05 was considered statistically significant. *: p < 0.05 vs saline control group. +: p < 0.05 vs the ethidium control group.
Discussion
The present study provides evidence that the administration of the vasodilator and nitric oxide releasing agent ISDN in a model of toxic demyelination resulted in amelioration of oxidative stress markers. The intracerebral or intraspinal administration of the DNA chelating agent ethidium bromide has been widely utilized to evoke toxic demyelination in rodents, which can be used to study the pathogenetic mechanisms involved in the destruction of myelin as well as to evaluate possible therapeutic interventions (Yajima and Suzuki, 1979[95]; Honmou et al., 1996[25]; Jeffery and Blakemore, 1997[31]; Graça et al., 2001[20]; Mazzanti et al., 2006[56]). In the present study, the local injection of ethidium bromide into the rat brain resulted in elevated MDA, an index of lipid peroxidation (Gutteridge, 1995[22]), which indicates increased free radical production in cerebral cortex. There was also a significant decrease in the level of GSH, the major thiol present in brain tissue, and the most important redox buffer in cells, which has an important role in the protection against oxidative injury due to reactive oxygen species (Wang and Ballatori, 1998[90]). This suggests consumption of GSH by the increased free radical production following ethidium bromide injection. Nitric oxide was markedly increased after ethidium bromide. These findings suggest increased oxidative stress by ethidium bromide in the cerebral cortex and are in line with other studies indicating increased oxidative stress in different brain areas by the toxin (Abdel-Salam el al., 2011[1]). The increase in oxidative stress following ethidium bromide injection was decreased by prior treatment with the nitric oxide donor ISDN, which decreased MDA and increased GSH in the cortex. These findings have important clinical implications in view of the evidence that oxidative stress is involved in demyelination disorders. Multiple sclerosis patients were found to have elevated lipid peroxidation and decreased levels endogenous antioxidants, suggesting consumption of the scavenger molecules by free radical excess (Karg et al., 1999[34]; Mitosek-Szewczyk et al., 2010[59]). In addition, GSH levels measured in the brain with magnetic resonance spectroscopy were lower in patients with multiple sclerosis compared with control (Srinivsan et al., 2010[82]; Choi et al., 2011[9]). Oxidized lipids and DNA were highly enriched in active multiple sclerosis plaques and oxidative injury of oligodendrocytes and neurons were associated with demyelination and axonal or neuronal injury (Haider et al., 2011[23]). Studies have also indicated increased oxidative and nitrosative stress in experimental models eg., the ethidium bromide-induced damage (Abdel-Salam et al., 2011[1]) and in autoimmune encephalomyelitis (Ljubisavljevic et al., 2011[49]; Vana et al., 2011[87]).
Nitric oxide is an important molecule involved in synaptic transmission and regulation of vascular tone. Nitric oxide is produced within the central nervous system from L-arginine by a constitutive (neuronal) form of nitric oxide synthase (nNOS), an endothelial form in vascular endothelium (eNOS) or an inducible form (iNOS) localized to glia, and requires activation by endotoxin and cytokines (Moncada and Bolaños, 2006[60]). The production of nitric oxide is increased in brain and serum of multiple sclerosis patients (De Groot et al., 1997[11]; Liu et al., 2001[48]; Koch et al., 2008[38]). Nitric oxide is a free radical and can react with many other free radicals e.g., superoxide radical generating peroxynitrite radical, capable of causing oxidative changes to macromolecules e.g., proteins, lipids and DNA (Moncada and Bolaños, 2006[60]).
Elevated nitric oxide concentrations which occur in neuroinflammatory states can thus result in neurodegeneration. Increased levels of nitric oxide causes axonal degeneration (Kapoor et al., 2000[33]; Garthwaite et al., 2002[16]) and activation of nNOS in oligodendrocytes leads to oligodendrocyte injury resulting in demyelination (Yao et al., 2010[96]). Evidence also implicates iNOS in the inflammation and demyelination of optic neuritis, where localized loss of myelin proteins, myelin breakdown, and the presence of iNOS and nitrotyrosine were associated with inflammatory infiltrates on the edges of the nerve and reactive astrocytes (Tsoi et al., 2006[85]). In experimental allergic encephalomyelitis (EAE), nitrotyrosine, an indicator of peroxynitrite formation is increased in the spinal cord white matter, which correlated with loss of mature oligodendrocytes (Li et al., 2011[47]).
Given that nitric oxide is likely to be involved in axonal and neuronal injury in demyelinating conditions (Kapoor et al., 2000[33]; Garthwaite et al., 2002[16]), in the present study, the nitric oxide donor ISDN was used to evaluate a possible modulating effect. ISDN is capable of releasing nitric oxide in a concentration- and pH-dependent manner. ISDN increased nNOS and eNOS activities in the presence oxyhemoglobin under hypoxia due to the increase in molecular oxygen concentration (Jiang et al., 2001[32]). Interestingly, pretreatment with ISDN decreased MDA, whilst elevating the level of reduced glutathione in cerebral cortex. Moreover, ISDN resulted in marked decrease in the level of nitric oxide in cortex. These results suggest that nitric oxide donors are likely to exert beneficial effects on the demyelination process. The results are also unexpected in view of the evidence that implicates nitric oxide in demyelinating diseases of the central nervous system. Studies thus have shown that the production of nitric oxide is increased in brain and serum of multiple sclerosis patients (De Groot et al., 1997[11]; Liu et al., 2001[48]; Koch et al., 2008[38]). Increased levels of nitric oxide causes axonal degeneration (Kapoor et al., 2000[33]; Garthwaite et al., 2002[16]) and activation of nNOS in oligodendrocytes which leads to oligodendrocyte injury resulting in demyelination (Yao et al., 2010[96]). Evidence also implicates iNOS in the inflammation and demyelination of optic neuritis, where localized loss of myelin proteins, myelin breakdown, and the presence of iNOS and nitrotyrosine were associated with inflammatory infiltrates on the edges of the nerve and reactive astrocytes (Tsoi et al. 2006[85]). In experimental allergic encephalomyelitis, nitrotyrosine, an indicator of peroxynitrite formation is increased in the spinal cord white matter, which correlated with loss of mature oligodendrocytes (Li et al., 2011[47]). Nitric oxide is a free radical and can react with many other free radicals e.g., superoxide radical generating peroxynitrite radical, capable of causing oxidative changes to macromolecules e.g., proteins, lipids and DNA. Increased nitric oxide production by microglia which occurs in neuroinflammatory states can thus result in neurodegeneration. While nitric oxide normally functions as a physiological neuronal mediator, excess production of nitric oxide mediates cellular toxicity by damaging critical metabolic enzymes and by reacting with superoxide to form an even more potent oxidant, peroxynitrite (Bredt, 1999[7]).
On the other hand, the effect of nitric oxide donors in neurodegenerative and demyelinating conditions is not clear. Nitric oxide donors exerted cytotoxic effects on dopaminergic neurons (Nunes et al., 2008[64]; Di Matteo et al., 2009[13]; Kurauchi et al., 2009[40]) via mechanisms that include mitochondrial dysfunction (Nunes et al., 2008[64]). Nitric oxide donors cause reversible conduction block in both normal and demyelinated axons of the central and peripheral nervous systems. Notably, conduction in demyelinated and early remyelinated axons is particularly sensitive to block by nitric oxide (Redford et al., 1997[73]). In cultured hippocampal neurons, ISDN as well as another newly developed nitric oxide-releasing agent rapidly and significantly reduced axonal transport in anterograde and retrograde directions (Kiriyama et al., 2002[37]). Nitric oxide donors exert metabolic effects e.g., nitroglycerin, ISDN, molsidomine, and sodium nitroprusside induced stimulation of glycolysis and shortened adenosine triphosphate (ATP)-turnover time in rat erythrocytes (Maletic et al., 2000[53]). In rat reticulocytes, ISDN, stimulated glycolysis and decreased ATP production via oxidative phosphorylation (Maletic et al., 1999[52]). The NO donor "spermine NONOate" decreased stimulated release of ATP from rabbit erythrocytes (Olearczyk et al., 2004[65]). In hippocampal synaptosomes of rats, sodium nitroprusside (but not other nitric oxide donors such as S-nitroso-N-acetyl-penicillamine and ISDN) inhibited adenosine triphosphate diphosphohydrolase and 5'-nucleotidase involved in an enzymatic chain for the hydrolysis of ATP to adenosine in the synaptic cleft (Kirchner et al., 2001[36]). Thus, whilst nitric oxide plays a physiological role in neuronal cell signaling, its over-production may cause neuronal energy compromise leading to neurodegeneration. Other researchers provided data suggesting that the administration of the exogenous nitric oxide donor molsidomine, a drug used for the treatment of coronary artery disease, limits the development of autoimmune encephalomyelitis and other T helper 1 (Th1) cell-mediated inflammatory diseases (Kwak et al., 2003[41]). Studies also suggested that enhanced nitric oxide production by the nitric oxide donor SIN-1 (3-morpholinosydnonimine hydrochloride) during the priming phase of autoimmune encephalomyelitis promotes apoptosis, down-regulates disease-promoting immune reactivities, and ameliorates clinical EAE, without depending on NOS (Xu et al., 2001[94]). Moreover, the increase in iNOS, nNOS and nitrotyrosine induced in the cerebral cortex of rats subjected to ischemia was prevented by the nitric oxide donor LA 419 (Serrano et al., 2007[78]). It has been suggested that nitric oxide may be a double-edged sword, mediating tissue damage on the one hand and on the other hand modulating complex immunological functions which may be protective (Giovannoni et al., 1998[17]).
Isosorbide dinitrate has been reported to preserve cell viability in the hippocampus after focal ischemia (Ramos-Zúñiga et al., 1998[71]). In addition, intravenous administration of nitric oxide donors reduces the infarct size after transient focal cerebral ischemia in rats (Salom et al., 2000[77]). Although there are several potential mechanisms for nitric oxide neuroprotective effects during brain ischemia (Verrecchia et al., 1995[88]), a rationale for the use of nitric oxide promoting strategies lies on the ability of nitric oxide to increase brain perfusion in areas of compromised perfusion around the ischemic core. NO has many additional roles outside the cardiovascular system. It appears to promote or prevents cellular inflammation and death. Evidence shows that nitric oxide can be anti-inflammatory through several activities; inhibition of maturation of cytokines, such as IL-18 and IL 1β (Kim et al., 1997[35]); blocking the effect of INF-γ (Murphy, 2000[62]) and preventing the expression of cellular expression molecules via effects on NF-κB (Laroux et al, 2001[43]; Brüne et al., 1998[8]). Alternatively, nitric oxide can enhance neuronal survival by attenuation of Ca++ influx via antagonism of the NMDA glutamate receptors. The reasons for the dual effects of nitric oxide are unclear, although it may be that where there are high local concentrations of nitric oxide or where it is derived from a particular source (such as iNOS or nNOS) the toxic effects predominate (Willmot and Bath, 2003[93]).
An important new observation in the present study was the decrease in paraoxonase activity in cortex by ethidium bromide. Paraoxonase is a calcium-dependent serum esterase that is synthesized by the liver and is released into the circulation, where it associates mainly with high density lipoproteins and protects LDL and cellular membranes against lipid peroxidation (La Du, 1992[42]; Primo-Parmo et al., 1996[69]). The paraoxonase gene family in humans includes three members: PON1, PON2 and PON3. PON1 possesses organophosphatase, arylesterase and lactonase activity and it hydrolyzes many different substrates (Rajkovic et al., 2011[70]). Serum PON1 and PON3 are inactivated under oxidative stress (Marsillach et al., 2004[54]). The enzyme PON1 is largely thought to have a role in protection against oxidative stress (Watson et al., 1995[92]; Mackness et al., 2006[51]; Amengual-Cladera et al., 2011[2]). It has been proposed that this enzyme might have a function related to the inactivation of oxidative stress by-products (either at a cellular level or blood-vessel wall) and other environmental chemicals (Rodrigo et al., 2001[74]). Lead exposed workers (Permpongpaiboon et al., 2011[68]) and patients with coronary heart disease (Kotur-Stevuljevic et al., 2008[39]) showed increased lipid peroxidation and decreased PON1 activity. In multiple sclerosis patients, PON1 activity does not change in the course of stable and progressive type of multiple sclerosis. However, PON1 activity in relapse was significantly lower in comparison to the other multiple sclerosis groups (Jamroz-Wisniewska et al., 2009[30]). In the present study, paraoxonase activity was markedly decreased in the cortex of ethidium bromide treated rats and showed a further decrease following ISDN treatment. This occurred despite a decrease of in lipid peroxidation (MDA) and increased GSH by the nitric oxide donor. One intriguing possibility is that PON1 represents an early defense mechanism against oxidative stress, resulting in an initial sparing of GSH. With higher levels of oxidative stress, depletion of the antioxidant glutathione will ensue. Studies have also shown that incubation of myelin suspensions with the peroxynitrite donor 3-morpholinosydnonimine (SIN-1) (but not nitric oxide or superoxide alone) resulted in the formation of the lipid peroxidation product, MDA, indicating that peroxynitrite formation is required for myelin-lipid peroxidation. Nitric oxide actually inhibited lipid peroxidation in myelin, as demonstrated using simple nitric oxide donors (van der Veen and Roberts, 1999[86]).
In the present study, ethidium bromide injection resulted in increased serotonin concentration in cortex. The increase in serotonin was partially restored by ISDN treatment which also decreased noradrenaline, compared with the ethidium bromide only treated group. Patients with multiple sclerosis were found to have increased cerebrospinal fluid noradrenaline and excitatory amino acid (glutamate and aspartate) levels (Barkhatova et al., 1998[5]). It has been shown that a mouse model of EAE is associated with chronic deficits in spinal cord concentrations of noradrenaline, serotonin and -aminobutyric acid (GABA) (Musgrave et al., 2011[63]) and that therapeutic agents that increase GABAergic and monoaminergic signaling can lessen the severity of EAE (Wang et al., 2008[91]; Simonini et al., 2010[80]). The findings of the current study also indicated that AChE activity decreased in the cortex early after ethidium bromide injection, suggesting alterations in cholinergic neurotransmission induced by the toxic agent (Taler et al., 2010[84]). Moreover, AChE activity is increased by the higher dose of ISDN. Thus the administration of ISDN appears to correct the neurochemical alterations induced by the toxic agent in the cortex. Changes in neurotransmitter levels and AChE activity has been demonstrated in patients with multiple sclerosis and in experimental models of demyelination. AChE decreased in the cerebrospinal fluid of subjects with multiple sclerosis (and in Huntington's chorea patients). This suggested that cerebrospinal fluid AChE activity may globally reflect brain AChE, but pathology-induced changes may not be directly reflected (Ruberg et al., 1987[75]). Alterations in butyrylcholinesterase activity, another enzyme capable of hydrolysing acetylcholine, were observed in multiple sclerosis white matter lesions diminished enzyme activity associated with myelin and an increased activity in cells with microglial morphology (Darvesh et al., 2010[10]). In ethidium bromide-treated rats, AChE activity was found to vary in all the brain structures in accordance with the day studied (Mazzanti et al., 2006[56]; Abdel-Salam et al., 2011[1]).
In summary, the present study indicated that the administration of the nitric oxide donor ISDN in a model of toxic demyelination in rats resulted in decreased lipid peroxidation, increased reduced glutathione. ISDN also lessened the elevation in nitric oxide and partially prevented the alterations in AChE activity induced by the toxic agent in the cortex. These findings suggest that nitric oxide donors might demonstrable therapeutic benefits in demyelinating conditions.
References
- 1.Abdel-Salam OME, Khadrawy YA, Salem NA, Sleem AA. Oxidative stress in a model of toxic demyelination in rat brain: the effect of piracetam and vinpocetine. Neurochem Res. 2011;36:1062–1072. doi: 10.1007/s11064-011-0450-1. [DOI] [PubMed] [Google Scholar]
- 2.Amengual-Cladera E, Nadal-Casellas A, Gómez-Pérez Y, Gomila I, Prieto RM, Proenza AM, Lladó I. Phytotherapy in a rat model of hyperoxaluria: the antioxidant effects of quercetin involve serum paraoxonase 1 activation. Exp Biol Med. 2011;236:1133–1138. doi: 10.1258/ebm.2011.011090. [DOI] [PubMed] [Google Scholar]
- 3.Arnelle DR, Stamler JS. NO1, NO, NO2 donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–85. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 4.Bagasra O, Michaels FH, Zheng YM, Bobroski LE, Spitsin SV, Fu ZF, et al. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc Natl Acad Sci USA. 1995;92:12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barkhatova VP, Zavalishin IA, Askarova LSh, Shavratskii VKh, Demina EG. Changes in neurotransmitters in multiple sclerosis. Neurosci Behav Physiol. 1998;28:341–344. doi: 10.1007/BF02464784. [DOI] [PubMed] [Google Scholar]
- 6.Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci USA. 2010;107:2580–5. doi: 10.1073/pnas.0915139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 8.Brüne B, von Knethen A, Sandau KB. Nitric oxide and its role in apoptosis. Eur J Pharmacol. 1998;351:261–272. doi: 10.1016/s0014-2999(98)00274-x. [DOI] [PubMed] [Google Scholar]
- 9.Choi IY, Lee SP, Denney DR, Lynch SG. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult Scler. 2011;17:289–296. doi: 10.1177/1352458510384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darvesh S, Leblanc AM, Macdonald IR, Reid GA, Bhan V, Macaulay RJ, et al. Butyrylcholinesterase activity in multiple sclerosis neuropathology. Chem Biol Interact. 2010;187:425–431. doi: 10.1016/j.cbi.2010.01.037. [DOI] [PubMed] [Google Scholar]
- 11.de Groot CJ, Ruuls SR, Theeuwes JW, Dijkstra CD, van der Valk P. Immunocytochemical characterization of the expression of inducible and constitutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions. J Neuropathol Exp Neurol. 1997;56:10–20. doi: 10.1097/00005072-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 12.de Vries HE, Schreibelt G, van Horssen G. Oxidative stress in multiple sclerosis pathology and therapeutic potential of Nrf2 activation. In: Armstrong D, editor. Oxidative stress in applied basic research and clinical practice. New York: Springer; 2011. pp. 65–77. [Google Scholar]
- 13.di Matteo V, Pierucci M, Benigno A, Crescimanno G, Esposito E, Di Giovanni G. Involvement of nitric oxide in nigrostriatal dopaminergic system degeneration: a neurochemical study. Ann N Y Acad Sci. 2009;1155:309–315. doi: 10.1111/j.1749-6632.2008.03678.x. [DOI] [PubMed] [Google Scholar]
- 14.Ellman GL. Tissue sulfhydryl groups. Arch Biochem. 1959;82:70–7. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 15.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharm. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 16.Garthwaite G, Goodwin DA, Batchelor AM, Leeming K, Garthwaite J. Nitric oxide toxicity in CNS white matter: an in vitro study using rat optic nerve. Neuroscience. 2002;109:145–155. doi: 10.1016/s0306-4522(01)00447-x. [DOI] [PubMed] [Google Scholar]
- 17.Giovannoni G, Heales SJ, Land JM, Thompson EJ. The potential role of nitric oxide in multiple sclerosis. Mult Scler. 1998;4:212–216. doi: 10.1177/135245859800400323. [DOI] [PubMed] [Google Scholar]
- 18.Glabinski A, Tawsek NS, Bartosz G. Increased generation of superoxide radicals in the blood of MS patients. Acta Neurol Scand. 1993;88:174–7. doi: 10.1111/j.1600-0404.1993.tb04212.x. [DOI] [PubMed] [Google Scholar]
- 19.Gorun V, Proinov I, Baltescu V, Balaban G, Barzu O. Modified Ellman procedure for assay of cholinesterases in crude enzymatic preparation. Anal Biochem. 1978;86:324–326. doi: 10.1016/0003-2697(78)90350-0. [DOI] [PubMed] [Google Scholar]
- 20.Graça DL, Bondan EF, Pereira LA, Fernandes CG, Maiorka PC. Behaviour of oligodendrocytes and Schwann cells in an experimental model of toxic demyelination of the central nervous system. Arq Neuropsiquiatr. 2001;59(2-B):358–361. doi: 10.1590/s0004-282x2001000300009. [DOI] [PubMed] [Google Scholar]
- 21.Gulati K, Chakraborti A, Ray A. Modulation of stress-induced neurobehavioral changes and brain oxidative injury by nitric oxide (NO) mimetics in rats. Behav Brain Res. 2007;183:226–230. doi: 10.1016/j.bbr.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 22.Gutteridge JMC. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem. 1995;41:1819–1828. [PubMed] [Google Scholar]
- 23.Haider L, Fischer MT, Frischer JM, Bauer J, Höftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashino K, Takahashi Y, Yamamura Y. Release of phenyl acetate esterase from liver microsomes by carbon tetrachloride. Clin Chim Acta. 1972;41:313–20. doi: 10.1016/0009-8981(72)90526-8. [DOI] [PubMed] [Google Scholar]
- 25.Honmou O, Felts PA, Waxman SG, Kocsis JD. Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci. 1996;16:3199–3208. doi: 10.1523/JNEUROSCI.16-10-03199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu W, Lucchinetti CF. The pathological spectrum of CNS inflammatory demyelinating diseases. Semin Immunopathol. 2009;31:439–453. doi: 10.1007/s00281-009-0178-z. [DOI] [PubMed] [Google Scholar]
- 27.Hummel SG, Fischer AJ, Martin SM, Schafer FQ, Buetner GR. Nitric oxide as a cellular antioxidant: a little goes a long way. Free Rad Biol Med. 2006;40:501–6. doi: 10.1016/j.freeradbiomed.2005.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–9. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 29.Ibragic S, Sofic E, Suljic E, Avdagic N, Bajraktarevic A, Tahirovic I. Serum nitric oxide concentrations in patients with multiple sclerosis and patients with epilepsy. J Neural Transm. 2012;119:7–11. doi: 10.1007/s00702-011-0686-6. [DOI] [PubMed] [Google Scholar]
- 30.Jamroz-Wisniewska A, Beltowski J, Stelmasiak Z, Bartosik-Psujek H. Paraoxonase 1 activity in different types of multiple sclerosis. Mult Scler. 2009;15:399–402. doi: 10.1177/1352458508098371. [DOI] [PubMed] [Google Scholar]
- 31.Jeffery ND, Blakemore WF. Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain. 1997;120:27–37. doi: 10.1093/brain/120.1.27. [DOI] [PubMed] [Google Scholar]
- 32.Jiang HB, Yoneyama H, Furukawa A, Hamamoto T, Takahara J, Ichikawa Y. Effect of isosorbide dinitrate on nitric oxide synthase under hypoxia. Pharmacology. 2001;62:10–16. doi: 10.1159/000056066. [DOI] [PubMed] [Google Scholar]
- 33.Kapoor R, Blaker PA, Hall SM, Davies M, Smith KJ. Protection of axons from degeneration resulting from exposure to nitric oxide. Rev Neurol (Paris) 2000;156(Suppl. 3):S67. [Google Scholar]
- 34.Karg E, Klivényi P, Németh I, Bencsik K, Pintér S, Vécsei L. Nonenzymatic antioxidants of blood in multiple sclerosis. J Neurol. 1999;246:533–539. doi: 10.1007/s004150050399. [DOI] [PubMed] [Google Scholar]
- 35.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 36.Kirchner SM, Bonan CD, Battastini AM, Sarkis JJ. Effect of nitric oxide donors on extracellular ATP, ADP, and AMP catabolism in rat hippocampal synaptosomes. Brain Res Bull. 2001;55:469–473. doi: 10.1016/s0361-9230(01)00541-x. [DOI] [PubMed] [Google Scholar]
- 37.Kiriyama Y, Hiruma H, Kobayashi H, Tomita T, Kawakami T. Inhibitory effects of nitric oxide (NO) on fast axonal transport in cultured rat hippocampal neurons. Kitasato Med. 2002;32:203–209. [Google Scholar]
- 38.Koch M, Mostert J, Arutjunyan A, Stepanov M, Teelken A, Heersema D, et al. Peripheral blood leukocyte NO production and oxidative stress in multiple sclerosis. Mult Scler. 2008;14:159–165. doi: 10.1177/1352458507082075. [DOI] [PubMed] [Google Scholar]
- 39.Kotur-Stevuljevic J, Spasic S, Jelic-Ivanovic Z, Spasojevic-Kalimanovska V, Stefanovic A, Vujovic A, et al. PON1 status is influenced by oxidative stress and inflammation in coronary heart disease patients. Clin Biochem. 2008;41:1067–1073. doi: 10.1016/j.clinbiochem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 40.Kurauchi Y, Hisatsune A, Isohama Y, Katsuki H. Nitric oxide-cyclic GMP signaling pathway limits inflammatory degeneration of midbrain dopaminergic neurons: cell type-specific regulation of heme oxygenase-1 expression. Neuroscience. 2009;158:856–866. doi: 10.1016/j.neuroscience.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 41.Kwak HJ, Pae HO, Oh GS, Choi BM, Jang SI, Jung S, et al. Molsidomine ameliorates experimental allergic encephalomyelitis in Lewis rats. Immunopharmacol Immunotoxicol. 2003;25:41–52. doi: 10.1081/iph-120018282. [DOI] [PubMed] [Google Scholar]
- 42.La Du BN. Human serum paraoxonase/arylesterase. In: Kalow W, editor. Pharmacogenetics of drug metabolism. New York: Pergamon Elmford; 1992. pp. 51–91. [Google Scholar]
- 43.Laroux FS, Pavlick KP, Hines IN, Kawachi S, Harada H, Bharwani S, et al. Role of nitric oxide in inflammation. Acta Physiol Scand. 2001;173:113–118. doi: 10.1046/j.1365-201X.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- 44.Lassmann H. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr Opin Neurol. 2001;14:253–258. doi: 10.1097/00019052-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 45.Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17:210–218. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–65. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 47.Li S, Vana AC, Ribeiro R, Zhang Y. Distinct role of nitric oxide and peroxynitrite in mediating oligodendrocyte toxicity in culture and in experimental autoimmune encephalomyelitis. Neuroscience. 2011;184:107–119. doi: 10.1016/j.neuroscience.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 48.Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ljubisavljevic S, Stojanovic I, Pavlovic D, Sokolovic D, Stevanovic I. Aminoguanidine and N-acetyl-cysteine supress oxidative and nitrosative stress in EAE rat brains. Redox Rep. 2011;16:166–172. doi: 10.1179/1351000211Y.0000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love S. Demyelinating diseases. J Clin Pathol. 2006;59:1151–9. doi: 10.1136/jcp.2005.031195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackness B, Quarck R, Verreth W, Mackness M, Holvoet P. Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:1545–1550. doi: 10.1161/01.ATV.0000222924.62641.aa. [DOI] [PubMed] [Google Scholar]
- 52.Maletić SD, Dragicević LM, Zikić RV, Stajn AS, Kostić MM. Effects of nitric oxide donor, isosorbide dinitrate, on energy metabolism of rat reticulocytes. Physiol Res. 1999;48:417–427. [PubMed] [Google Scholar]
- 53.Maletic SD, Dragicevic-Djokovic LM, Zikic RV, Stajn AS, Milenkovic P, Kostic MM. Effects of nitric oxide donors on energy metabolism of rat erythrocytes. J Environ Pathol Toxicol Oncol. 2000;19:383–390. [PubMed] [Google Scholar]
- 54.Marsillach J, Camps J, Ferré N, Beltran R, Rull A, Mackness B, et al. Paraoxonase-1 is related to inflammation, fibrosis and PPAR delta in experimental liver disease. BMC Gastroenterol. 2009;9:3. doi: 10.1186/1471-230X-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masood A, Banerjee BD, Vijayan VK, Ray A. Modulation of stress induced neurobehavioral changes by nitric oxide in rats. Eur J Pharmacol. 2003;458:138–9. doi: 10.1016/s0014-2999(02)02688-2. [DOI] [PubMed] [Google Scholar]
- 56.Mazzanti CM, Spanevello RM, Pereira LB, Gonçalves JF, Kaizer R, Corrêa M, et al. Acetylcholinesterase activity in rats experimentally demyelinated with ethidium bromide and treated with interferon beta. Neurochem Res. 2006;31:1027–1034. doi: 10.1007/s11064-006-9112-0. [DOI] [PubMed] [Google Scholar]
- 57.Medina JH, Izquierdo I. Retrograde messengers, long-term potentiation and memory. Brain Res Rev. 1995;21:185–94. doi: 10.1016/0165-0173(95)00013-5. [DOI] [PubMed] [Google Scholar]
- 58.Mirshafiey A, Mohsenzadegan M. Antioxidant therapy in multiple sclerosis. Immunopharmacol Immunotoxicol. 2009;31:13–29. doi: 10.1080/08923970802331943. [DOI] [PubMed] [Google Scholar]
- 59.Mitosek-Szewczyk K, Gordon-Krajcer W, Walendzik P, Stelmasiak Z. Free radical peroxidation products in cerebrospinal fluid and serum of patients with multiple sclerosis after glucocorticoid therapy. Fol Neuropathol. 2010;48:116–122. [PubMed] [Google Scholar]
- 60.Moncada S, Bolaños JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- 61.Moshage H, Kok B, Huizenga JR. Nitrite and nitrate determination in plasma: a critical evaluation. Clin Chem. 1995;41:892–896. [PubMed] [Google Scholar]
- 62.Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 63.Musgrave TM, Tenorio G, Rauw G, Baker GB, Kerr BJ. Tissue concentration changes of amino acids and biogenic amines in the central nervous system of mice with experimental autoimmune encephalomyelitis (EAE) Neurochem Int. 2011;59:28–38. doi: 10.1016/j.neuint.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 64.Nunes C, Almeida L, Laranjinha J. 3,4-Dihydroxyphenylacetic acid (DOPAC) modulates the toxicity induced by nitric oxide in PC-12 cells via mitochondrial dysfunctioning and ATP depletion. Neurotoxicology. 2008;29:998–1007. doi: 10.1016/j.neuro.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Olearczyk JJ, Ellsworth ML, Stephenson AH, Lonigro AJ, Sprague RS. Nitric oxide inhibits ATP release from erythrocytes. J Pharmacol Exp Ther. 2004;309:1079–1084. doi: 10.1124/jpet.103.064709. [DOI] [PubMed] [Google Scholar]
- 66.Oleszak EL, Zaczynska E, Bhattacharjee M, Butunoi C, Legido A, Katsetos CD. Inducible nitric oxide synthase and nitrotyrosine are found in monocytes/macrophages and/or astrocytes in acute, but not in chronic, multiple sclerosis. Clin Diagn Lab Immunol. 1998;5:438–445. doi: 10.1128/cdli.5.4.438-445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paget GE, Barnes JM. Toxicity tests. In: Laurence DR, Bacharach AL, editors. Evaluation of drug activities. Pharmacometrics. London: Academic Press; p. 161. [Google Scholar]
- 68.Permpongpaiboon T, Nagila A, Pidetcha P, Tuangmungsakulchai K, Tantrarongroj S, Porntadavity S. Decreased paraoxonase 1 activity and increased oxidative stress in low lead−exposed workers. Hum Exp Toxicol. 2011;30:1196–1203. doi: 10.1177/0960327110388536. [DOI] [PubMed] [Google Scholar]
- 69.Primo-Parmo SL, Sorenson RC, Teiber J, La Du BN. The human serum paraoxonase/arylesterase gene (PON1) is one member of a multigene family. Genomics. 1996;33:498–507. doi: 10.1006/geno.1996.0225. [DOI] [PubMed] [Google Scholar]
- 70.Rajkovic MG, Rumora L, Barisic K. The paraoxonase 1, 2 and 3 in humans. Biochem Med (Zagreb) 2011;21:122–130. doi: 10.11613/bm.2011.020. [DOI] [PubMed] [Google Scholar]
- 71.Ramos-Zúñiga R, Velázquez-Santana H, Mercado-Pimentel R, Cerda-Camacho F. Neuroprotection in selective focal ischemia in rats by nitrates, an alternative redox manipulation of nitric oxide: experimental model. Minim Invasive Neurosurg. 1998;41:152–60. doi: 10.1055/s-2008-1052033. [DOI] [PubMed] [Google Scholar]
- 72.Rauhala P, Lin AM, Chiueh CC. Neuroprotection by S-nitrosoglutathione of brain dopamine neurons from oxidative stress. FASEB J. 1998;12:165–73. doi: 10.1096/fasebj.12.2.165. [DOI] [PubMed] [Google Scholar]
- 73.Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120:2149–2157. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- 74.Rodrigo L, Hernández AF, López-Caballero JJ, Gil F, Pla A. Immunohistochemical evidence for the expression and induction of paraoxonase in rat liver, kidney, lung and brain tissue: implications for its physiological role. Chem Biol Interact. 2001;137:123–137. doi: 10.1016/s0009-2797(01)00225-3. [DOI] [PubMed] [Google Scholar]
- 75.Ruberg M, Villageois A, Bonnet AM, Pillon B, Rieger F, Agid Y. Acetylcholinesterase and butyrylcholinesterase activity in the cerebrospinal fluid of patients with neurodegenerative diseases involving cholinergic systems. J Neurol Neurosurg Psych. 1987;50:538–543. doi: 10.1136/jnnp.50.5.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruiz-Larrea MB, Leal AM, Liza M, Lacort M, de Groot H. Antioxidant effects of estradiol and 2-hydroxyestradiol on iron-induced lipid peroxidation of rat liver microsomes. Steroids. 1994;59:383–388. doi: 10.1016/0039-128x(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 77.Salom JB, Ortí M, Centeno JM, Torregrosa G, Alborch E. Reduction of infarct size by the NO donors sodium nitroprusside and spermine/NO after transient focal cerebral ischemia in rats. Brain Res. 2000;865:149–156. doi: 10.1016/s0006-8993(00)02095-3. [DOI] [PubMed] [Google Scholar]
- 78.Serrano J, Fernández AP, Martínez-Murillo R, Alonso D, Rodrigo J, Salas E, et al. The nitric oxide donor LA 419 decreases ischemic brain damage. Int J Mol Med. 2007;19:229–236. [PubMed] [Google Scholar]
- 79.Shuman EM, Madison DV. Nitric oxide and synaptic function. Annu Rev Neurosci. 1994;17:153–83. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- 80.Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL. Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharmacol. 2010;5:252–9. doi: 10.1007/s11481-009-9182-2. [DOI] [PubMed] [Google Scholar]
- 81.Smith KJ. Newly lesioned tissue in multiple sclerosis-a role for oxidative damage? Brain. 2011;134:1877–1881. doi: 10.1093/brain/awr144. [DOI] [PubMed] [Google Scholar]
- 82.Srinivasan R, Ratiney H, Hammond-Rosenbluth KE, Pelletier D, Nelson SJ. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn Reson Imaging. 2010;28:163–170. doi: 10.1016/j.mri.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 83.Szabo C. Physiological and pathophysiological roles of nitric oxide in the central nervous system. Brain Res Bull. 1996;41:131–41. doi: 10.1016/0361-9230(96)00159-1. [DOI] [PubMed] [Google Scholar]
- 84.Taler M, Gil-Ad I, Korob I, Weizman A. The immunomodulatory effect of the antidepressant sertraline in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Neuroimmunomodulation. 2010;18:117–22. doi: 10.1159/000321634. [DOI] [PubMed] [Google Scholar]
- 85.Tsoi VL, Hill KE, Carlson NG, Warner JE, Rose JW. Immunohistochemical evidence of inducible nitric oxide synthase and nitrotyrosine in a case of clinically isolated optic neuritis. J Neuroophthalmol. 2006;26:87–94. doi: 10.1097/01.wno.0000223266.48447.1b. [DOI] [PubMed] [Google Scholar]
- 86.van der Veen RC, Roberts LJ. Contrasting roles for nitric oxide and peroxynitrite in the peroxidation of myelin lipids. J Neuroimmunol. 1999;95:1–7. doi: 10.1016/s0165-5728(98)00239-2. [DOI] [PubMed] [Google Scholar]
- 87.Vana AC, Li S, Ribeiro R, Tchantchou F, Zhang Y. Arachidonyl trifluoromethyl ketone ameliorates experimental autoimmune encephalomyelitis via blocking peroxynitrite formation in mouse spinal cord white matter. Exp Neurol. 2011;231:45–55. doi: 10.1016/j.expneurol.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 88.Verrecchia C, Boulu RG, Plotkine M. Neuroprotective and deleterious effects of nitric oxide on focal cerebral ischemia-induced neurone death. Adv Neuroimmunol. 1995;5:359–78. doi: 10.1016/0960-5428(95)00023-2. [DOI] [PubMed] [Google Scholar]
- 89.Vollmar P, Nessler S, Kalluri SR, Hartung HP, Hemmer B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int J Neuropsychopharmacol. 2009;12:525–36. doi: 10.1017/S1461145708009425. [DOI] [PubMed] [Google Scholar]
- 90.Wang W, Ballatori N. Endogenous glutathione conjugates: occurrence and biological functions. Pharmacol Rev. 1998;50:335–356. [PubMed] [Google Scholar]
- 91.Wang Y, Feng D, Liu G, Luo Q, Xu Y, Lin S, et al. Gamma-aminobutyric acid transporter 1 negatively regulates T cell-mediated immune responses and ameliorates autoimmune inflammation in the CNS. J Immunol. 2008;181:8226–36. doi: 10.4049/jimmunol.181.12.8226. [DOI] [PubMed] [Google Scholar]
- 92.Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, et al. Protective effect of high density lipoprotein associated paraoxonase: inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96:2882–2891. doi: 10.1172/JCI118359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Willmot MR, Bath PMW. The potential of nitric oxide therapeutics in stroke. Expert Opin Investig Drugs. 2003;12:455–470. doi: 10.1517/13543784.12.3.455. [DOI] [PubMed] [Google Scholar]
- 94.Xu LY, Yang JS, Link H, Xiao BG. SIN-1, a nitric oxide donor, ameliorates experimental allergic encephalomyelitis in Lewis rats in the incipient phase: the importance of the time window. J Immunol. 2001;166:5810–5816. doi: 10.4049/jimmunol.166.9.5810. [DOI] [PubMed] [Google Scholar]
- 95.Yajima K, Suzuki K. Demyelination and remyelination in the rat central nervous system following ethidium bromide injection. Lab Invest. 1979;41:385–92. [PubMed] [Google Scholar]
- 96.Yao SY, Ljunggren-Rose A, Chandramohan N, Whetsell WO, Jr, Sriram S. In vitro and in vivo induction and activation of nNOS by LPS in oligodendrocytes. J Neuroimmunol. 2010;229:146–156. doi: 10.1016/j.jneuroim.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yun H-Y, Dawson VL, Dawson TM. Nitric oxide in health and disease of the nervous system. Mol Psychiatry. 1997;2:300–10. doi: 10.1038/sj.mp.4000272. [DOI] [PubMed] [Google Scholar]