

Abstract
Recent studies suggest that normobaric hyperoxia (HO) protects the rat brain from ischemia reperfusion (IR) injury. Protein kinase C (PKC) is a key signaling molecule involved in protection against IR injury but its role in protective effect of HO in brain injury in unknown. In this study we attempted to see if PKC is involved in the effect of HO. Rats were divided into four main experimental groups. The first two were exposed to 95 % oxygen (HO) in a chamber 4 h/day for 6 consecutive days. Each of these groups had a control group exposed to 21 % oxygen. To investigate the role of PKC during HO, chelerythrin chloride (CHEL, 1 mg/kg/day), a PKC inhibitor, or its vehicle was given to animals for 6 days. After 24 h, the rats were subjected to 60 min of right middle cerebral artery occlusion (MCAO). After 24 h reperfusion neurological deficit scores, infarct volume, brain edema and blood-brain Barrier (BBB) permeability were assessed. HO decreased the infarct volume and brain edema in comparison with controls. PKC inhibition was associated with a significant increase in infarct size in both HO and control animals. PKC inhibition was unable to change brain edema in the experimental groups. Both HO and PKC inhibition reduced the BBB permeability within 24 h post occlusion of middle cerebral artery. Although both HO and PKC inhibition were associated with inhibition of BBB permeability during ischemic brain injury in rats, the neuroprotective effect of HO was independent of PKC in the MCAO model.
Keywords: normobaric hyperoxia, ischemic preconditioning, stroke, neuroprotection, PKC, chelerythrin chloride
Introduction
Ischemic stroke is the major cause of morbidity and mortality in modern society (Murray and Lopez, 1997[19]). Ischemic preconditioning (IPC) is an endogenous phenomenon that whereby brief periods of ischemia, causes a tissue more resistant to subsequent sustained loss of blood flow and can induce ischemic tolerance (IT) in a variety of organ systems such as the heart and brain (Kitagawa et al., 1990[15]). Ischemic tolerance involves mediating mechanisms such as the synthesis of new protective proteins, anti-oxidant enzymes, growth factors, or anti-apoptotic gene products (Chen and Simon, 1997[5]; Kato et al., 1992[12]; Perez-Pinzon et al., 1996[23]; Kirino, 2002[14]; Ohtsuki et al., 1992[20]). Recent studies suggest that normobaric hyperoxia (HO) protects the rat brain tissue from ischemia reperfusion (IR) injury (Bigdeli et al., 2007[2]). The mechanism of this effect is not well understood but it appears that enhancement of endogenous defense mechanisms are involved.
Protein kinase C (PKC) is a ubiquitous histone protein kinase activated by phospholipids, diacylglycerol (DAG) and Ca2+ (Hoshida et al., 1993[11]), this family of enzymes has been implicated as an important signal transduction pathway for many cellular and physiological events such as enhancement of neurotransmitter release, desensitization of receptors and modulation of ion channels (Tahepold et al., 2003[26]). PKC is a key signaling molecule involved in protection against ischemia reperfusion (IR) injury. PKC activation might be a common signal transduction factor in both heart and brain which initiates different signaling pathways during IPC-induced tolerance (Perez-Pinzon et al., 2005[22]). PKC is abundant in neural cells; the heterogeneous topographical distribution of PKC is formed within particular brain regions and is referred to special activities (Thornton et al., 1990[27]). When PKC is activated with phorbol esters, transcription factor activator protein-1 (AP-1) is bounded to TRE which is its BNA binding site. It was demonstrated that this binding of AP-1 to DNA and DNA activation is sensitive to changes in its oxidation-reduction status (Hoshida et al., 1993[11]), and that PKC-specific activity is increased in response to reactive oxygen species (Thornton et al. 1990[27]). When cells were exposed to 95 % O2 for 5 days, a 2-fold increase PKC specific activity has been observed (Perez-Pinzon et al., 2005[22]).
In spite of above information, the role of PKC in the induction of the ischemic tolerance by HO is still not well understood. Activation of PKC by oxidants, prompted us to investigate the potential role of PKC under HO in rat brain stroke model.
Material and Methods
Adult male Wistar rats (250-350 g) were housed under conditions of controlled temperature (22 ± 2 °C) and constant humidity, with 12 h light/dark cycle (light on 07:00-19:00) for all experiments. All experimental animal procedures were conducted with the approval of the Ethics Committee of the Tarbiat Modares University. Every effort was made to minimize the number of animals used and their suffering.
Chelerythrin chloride (CHEL, 1.0 mg/ kg/day) was dissolved in sterile saline and was subcutaneously injected to the rats for 6 days, 20 minutes before housing the animals in the normobaric chamber.
Rats were divided randomly into 4 main groups (n=18 animals in each group). Two of these groups were placed in an environmental chamber and exposed to a hyperoxic atmosphere (95 % oxygen: normobaric hyperoxic groups, or HO), for 4 continuous hours for six consecutive days. The first group received daily injection of CHEL 20 min prior to HO and the other one was given daily injection of vehicle (saline). The two other groups were similarly placed in the environmental chamber and exposed to room-air (RA) equivalent (21 % oxygen: normobaric normoxic groups) for similar time periods. Each group was subdivided for evaluation of infarct volume, brain water content measurement and blood-brain barrier (BBB) permeability. In addition of the main groups, we included 2 sham operated groups (n=12 animals in each group). One of these groups was used for evaluation of brain water content and another for measurement of BBB permeability. HO treatment was initiated in the chamber (650×350×450 mm3). Oxygen concentration inside the container was continuously monitored using an oxygen sensor (Lutron-Do5510, Taiwan), and soda lime; a carbon dioxide absorber was used (BDH Limited, Poole, UK) at the sides of the chamber. The two other groups were similarly placed in the environmental chamber and exposed to RA (21 % oxygen). Oxygen concentration was maintained at 95 % or 21 % according to the experimental protocol.
At the beginning of day 7 after induction of HO, animals were anesthetized with chloral hydrate (400 mg/kg) and were subjected to middle cerebral artery occlusion (MCAO) as described (Longa et al., 1989[18]). In brief, the right common carotid artery (CCA) was carefully separated from the vagus nerve and surrounding tissues. Using microscopic surgery, a 3-0 silicone coated nylon suture was introduced through the external carotid artery stump. The occluder was advanced into the internal carotid artery (ICA) 20-22 mm beyond the carotid bifurcation until mild resistance indicated that the tip was lodged in the anterior cerebral artery and blocked the blood flow to the middle cerebral artery (MCA). Rectal temperature was monitored (Citizen-513w) and maintained at 37 °C by surface heating and cooling during surgery. The body temperature, blood gases, heart rate were maintained within the physiologic range throughout the operation. Continuous Laser-Doppler flowmetry (LDF; Moor Instrument, UK) was used to monitor cerebral perfusion to ensure adequacy of MCAO through the CCA until the Laser-Doppler signal showed a steep decrease (more than 75 % reduction of cerebral blood flow is necessary for successful induction of ischemia). Reperfusion was started by removing the suture after 1 hour of ischemia. The sham operated rats were subjected to the same surgical procedures except the filament insertion.
After the operation, the rats were returned to their separate cages. 24 h after surgery, the rats were assessed neurologically by an observer who was blind to the animal groups. The neurobehavioral scoring was performed using a six-point scale (Longa et al., 1989[18]): normal motor function=0; failure to extend left forepaw fully=1; circling to the contralateral side but have normal posture at rest=2; falling to the left, a severe focal deficit=3; rats that did not walk spontaneously and had a depressed level of consciousness=4. Death was considered as score 5 only when a large infarct volume was present in the absence of subarachnoid hemorrhage. If the rats died due to subarachnoid hemorrhage or pulmonary insufficiency and asphyxia, they were eliminated from the study.
After scarification of rats with chloral hydrate (800 mg/kg), the animals were decapitated and the brains were rapidly removed and cooled in 4 °C saline for 15 minutes. Eight 2-mm-thick coronal sections were cut using a rat brain matrix through the brain, beginning at the olfactory bulb. The slices were immersed in 2 % 2,3,5-triphenyltetrazolium chloride solution (Merck, Germany), and kept at 37 °C in a water bath for 15 min. The slices were then photographed by a digital camera (Canon SX210IS, Japan) connected to a computer. Unstained areas were defined as infarct, and were measured using image analysis software (Image Tools, NIH, USA). The infarct volume was calculated by measuring the unstained and stained area in each hemisphere slice, and multiplied by slice thickness (2 mm), and then summating all of the eight slices according to the method of Swanson (Swanson et al., 1990[25]): corrected infarct volume=left hemisphere volume− (right hemisphere volume − infarct volume).
After decapitation, the brain was removed. The cerebellum, pons, and olfactory bulb were separated and their wet weights (WW) measured (Bigdeli et al., 2007[2]). Dry weights (DW) were assessed after 24 h at 120 °C. Brain water content (BWC) was calculated as [(WW−DW)/WW] ×100.
The integrity of BBB was evaluated by using Evans Blue (EB, Sigma Chemicals, USA) dye extravasations (Kaya et al., 2003[13]). Briefly, the rats received 4 ml/kg of 2 % EB solution in saline by tail vein injection 30 min after MCAO. 24 h after reperfusion, the thoracic cavity was opened under anesthesia. The rats were perfused with 250 ml saline transcardially to wash out intravascular EB until colorless perfusion fluid was obtained from the atrium. After decapitation, the brains were removed and the hemispheres separated and weighed. The right and left hemispheres were separately homogenized in 2.5 ml phosphate-buffered saline to extract the EB, and to precipitate protein 2.5 ml of 60 % trichloroacetic acid was added and mixed by vortex for 3 min. The samples were then placed at 4 °C for 30 min and centrifuged for 30 min at 1000×g. The amount of EB in the supernatants was measured at 610 nm using spectrophotometer (Genova, Jenway). EB levels were expressed as μg/g of brain tissue against a standard curve (Bigdeli et al., 2007[2]).
Data were expressed as means ± SEM. Infarct volumes and EB extravasations, brain water content and arterial blood gases (ABG) were compared using two-way ANOVA test. The neurologic deficit scores (NDS) were analyzed using the Bonferoni post-hoc test. P<0.05 was considered significant.
Results
Figure 1(Fig. 1) shows oxygen content (%) in the container in HO and normoxia conditions. Arterial blood gas analysis confirmed clinical HO in the treated groups (Table 1(Tab. 1)).
Figure 1. Oxygen concentration in the hyperoxia-inducing chamber during hyperoxia (HO) in comparison with room air (RA).
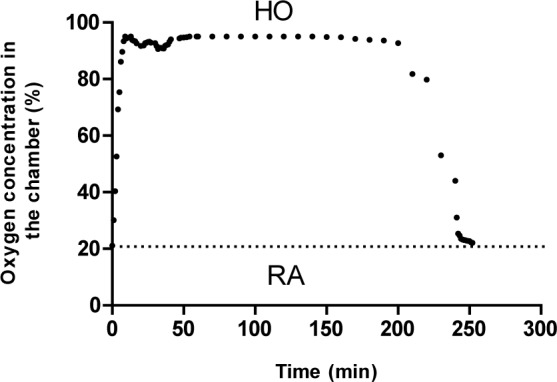
Table 1. ABG tests at the end of pretreatment.

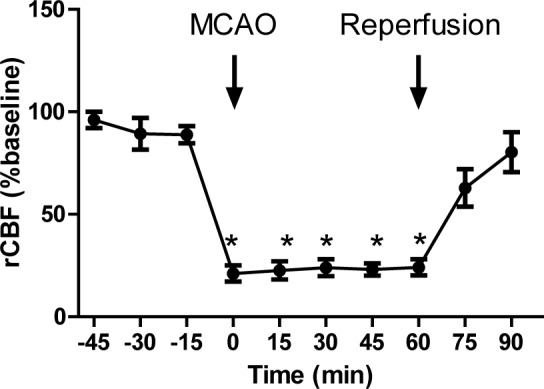
As shown in Figure 2(Fig. 2), MCAO was able to reduce regional cerebral blood flow to less than 25 % of that at baseline.
Figure 2. Regional cerebral blood flow (rCBF) before and during middle cerebral artery occlusion (MCAO), and after reperfusion (n=6).
CHEL increased the neurologic deficit scores (NDS) in groups of normoxia and HO. In the group of normoxia this increase was greater. The median of NDS was 2.12 (range: 0-4), 0.83 (range: 0-2), 3.08 (range: 1-4) and 1.95 (range: 0-4) in the HO, HO-CHEL, RA-CHEL and RA groups, respectively (Table 2(Tab. 2)). The harmful effects of CHEL were confirmed by an increase in infarct volume (Figure 2(Fig. 2)).
Table 2. The distribution of neurologic deficit score in each groups.

The putative beneficial effects of HO were confirmed by a reduction in infarct volume (Figure 3(Fig. 3)). Pretreatment with CHEL resulted in an increase of infarct volume.
Figure 3. Photographs showing the neuroprotective effect of hyperoxia on focal cerebral ischemia. All sections of the brain were stained with TTC at 24h after 60 min of ischemia. Each column presents the series of rat brain coronal sections: (A) Brain sections of control rats, (B) Brain sections of rats with HO, (C) Brain sections of rats with HO + CHEL and (D) Brain sections of CHEL in stroke groups.
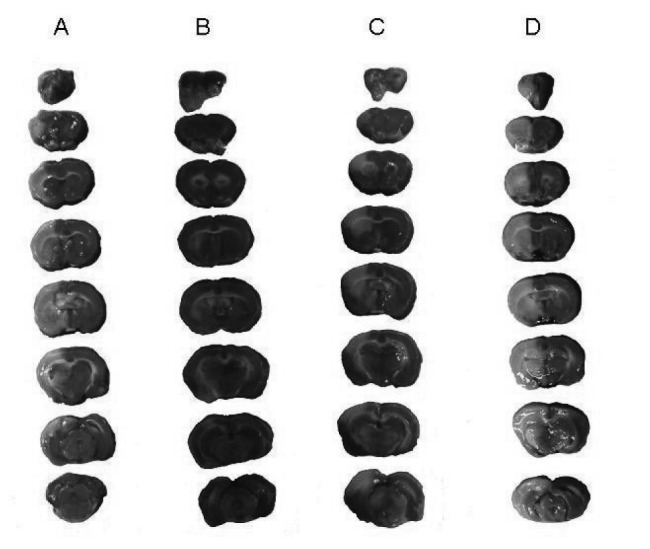
Figure 4A(Fig. 4) shows the effect of HO on infarct volume in whole brain. Statistical analysis using two-way ANOVA showed that HO induced a significant reduction in infarct volume in comparison with RA groups (FHO vs RA=14.19, P<0.05). CHEL significantly increased infarct volume in both groups (FCHEL vs vehicle=9.896, P<0.05). The effect of HO on infarct volume did not show a significant interaction with the PKC inhibition (Finteraction=0.157, P=0.695). Figure 4B(Fig. 4) shows the effect of HO on infarct volume in the cortical layer. Likewise, HO induced a significant reduction in infarct volume in comparison with RA group (FHO vs RA=19.44, P<0.001). CHEL significantly increased infarct volume in the cortical layer in both groups (FCHEL vs vehicle=10.88, P<0.01). However, the effect of HO on infarct volume did not show a significant interaction with CHEL (Finteraction=0.592, P=0.4506). In the subcortical area, as shown in Figure 4C(Fig. 4), there was no significant difference between the groups (FHO vs RA=3.015, P=0.098, FCHEL vs vehicle=1.114, P=0.307, Finteraction=0.073, P=0.789).
Figure 4. Figure 4: The effect of hyperoxia on infarct volume in different experimental groups. Results were confirmed by infarct volumetry: whole brain (A), cortex (B), and subcortex (C) area of brain (n=6).
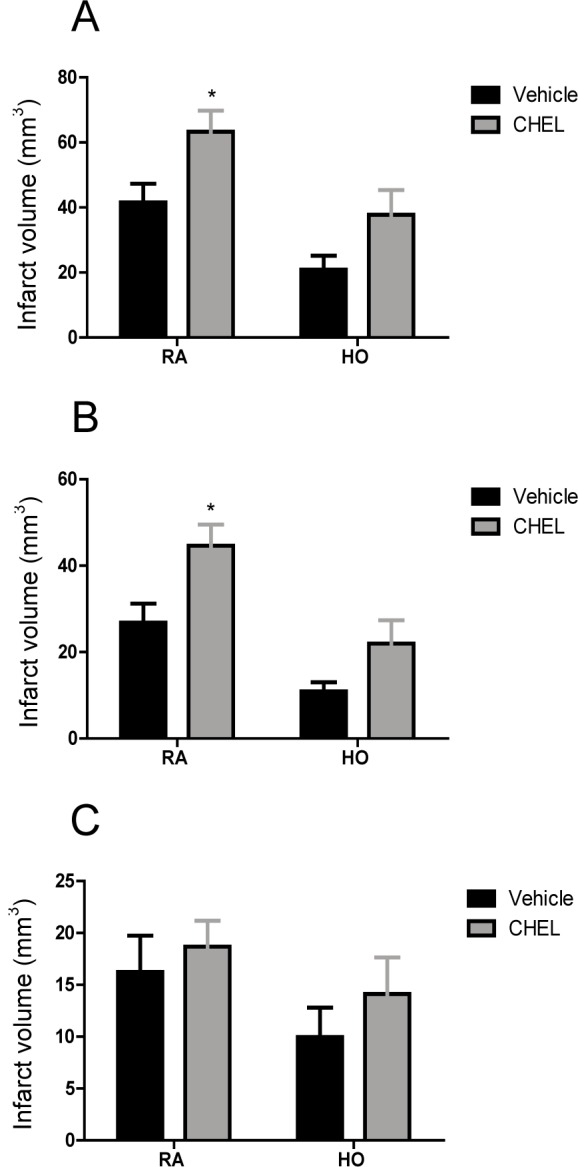
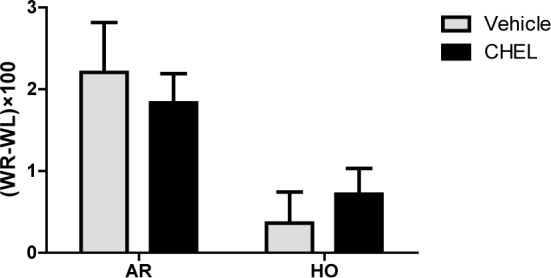
Figure 5(Fig. 5) shows the effect of HO and CHEL on brain water content which is an indicator of brain edema. Data are expressed as the difference between the left and right hemispheres. HO significantly decreased brain edema in comparison with RA animals (FHO vs RA=11.79, P<0.01). The effect of CHEL on brain edema was not statistically significant in the experimental groups (FCHEL vs vehicle=0.0005, P=0.982). And there was no significant interaction between HO and CHEL on brain edema induced by MCAO (Finteraction=0.711, P=0.409).
Figure 5. Difference between right and left hemisphere water content (WR-WL) in rats given room air (RA), hyperoxia (HO) in CHEL or vehicle treated groups (n=6).
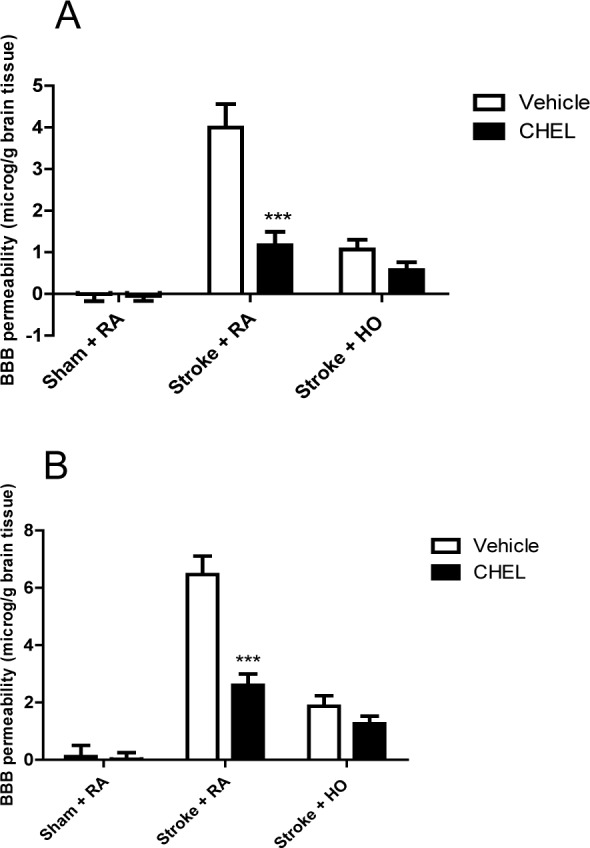
The effect of HO and CHEL on BBB permeability is shown in Figure 6(Fig. 6). MCAO was able to increase the BBB permeability in the experimental groups in both cortical and subcortical areas (Figure 6A and 6B(Fig. 6)). In cortical areas, HO was able to significantly decrease BBB permeability in comparison with RA groups (FHO vs RA=37.6, P<0.0001). Likewise, PKC inhibition was associated with a significant reduction in BBB permeability in both groups (FCHEL vs vehicle=20.11, P<0.0001). Moreover, the effect of HO on BBB protection showed a significant interaction with CHEL (Finteraction=11.87, P=0.0001). The effect of HO and CHEL was similar in the subcortical and cortical areas (FHO vs RA=62.48, P<0.0001; FCHEL vs vehicle=20.97, P<0.0001; Finteraction=12.58, P=0.0001).
Figure 6. Blood brain barrier permeability in various experimental groups. EBR: Evans blue extravasation right; EBL: Evans blue extravasations left. Cortex (A), subcortex (B) (n = 6).
Discussion
We have previously shown that intermittent and prolonged HO induces ischemic tolerance, as demonstrated by decreases in infarct volume, neurological deficit scores, brain edema, and BBB permeability (Bigdeli et al., 2007[2]). Ischemic tolerance also decreases excitotoxicity via reduction of extracellular glutamate in rat brain (Grabb et al., 2002[10]). We have also previously demonstrated that preconditioning with prolonged and intermittent normobaric HO up-regulates glutamate transporters (EAAT1, EAAT2, EAAT3, and TACE) and serum TNFα level as well as phospho-IkB level in rat brain (Bigdeli et al., 2009[3]). The MCAO 'suture' model used in this study is a reliable and reproducible model (Longa et al., 1989[18]) which induces less subarachnoid hemorrhage and hypothalamically-induced hyperthermia than the intraluminal filament model (Dittmar et al., 2003[7]). To further study the mechanism of HO-induced ischemic tolerance, we used a rat model of HO and investigated the role of PKC in HO-induced ischemic tolerance. Our data suggest that PKC inhibitor in the cortical areas significantly increase infarct volume; indicating the importance of PKC in brain protection during stroke. HO significantly and without interaction with PKC reduced infarct size, this suggests the neuroprotective effect of HO on infarct size is independent of PKC in the MCAO model. In the subcortical areas HO or CHEL didn't have significant effect on infarct volume. The lack of protective effect of HO in the subcortical area could be due to the more severe damage in the subcortical areas than the cortical areas (Garcia et al., 1997[9]).
Previous studies have shown that NF-κB inhibitors abrogate the infarct-limiting effect, which has been shown in both early and delayed phase of hyperoxic preconditioning. The cellular mechanism by which IPC activates NF-κB is known to involve formation of reactive oxygen species (ROS) and activation of PKC and tyrosine kinase dependent pathways (Xuan et al., 1999[28]).
Cerebral ischemia/reperfusion injury typically causes the disruption of water homeostasis, and an excess of water accumulates in the brain parenchyma, causing brain edema, the serious sequelae of brain edema includes deficits in neurological function as well as increased intracranial pressure. Aquaporins are homologous water-transporting channel proteins. They provide the major means of water permeability across plasma membranes. Among the family of aquaporin proteins, aquaporin 4 (AQP4) is the major water-selective channel in the central nervous system (Kobayashi et al., 2004[16]). It is extensively expressed in specialized cells that form the blood-brain and brain-cerebral spinal fluid interfaces, including astrocyte end-feet that surround cerebral blood vessels, ependymal cells, and microvascular endothelial cells (Rash et al., 1998[24]). Further work has shown that PKC is implicated in the regulation of AQP4 expression. For example in cultured astrocytes, the phorbol ester, 12-O-tetradecanoylphorbol 13-acetate (TPA), a PKC activator, reduced AQP4 mRNA and inhibited its biological activity (Yamamoto et al., 2001[29]). In support of previous work we investigated the effect of a PKC inhibitor on brain edema in a rat model of stroke. Our results showed that inhibition of PKC didn't have a significant effect on brain water in the MCAO model. However, HO was able to decrease brain edema in rat, although this effect was independent of PKC.
Our experiments in support of previous work showed that HO improves BBB function. The means through which HO improves BBB integrity are not clear, but may hinge upon the production of ROS and TNF-α in the brain tissue (Currie and Tanguay, 1991[6]). HO can lead to increase in free radical oxygen, the increase of free radical oxygen and superoxide dismutase is associated with decreased expression of hypoxia induced factor-1α, leading to improved blood brain barrier function via decreased vascular growth factor (Ostrowski et al., 2005[21]). PKC inhibitor significantly decreased BBB permeability. This finding corroborates with the recent reports which showed that activation of the various PKC isozymes have different effect on the integrity of BBB. PKC has 11 isozymes, some of them (α, βІ, βΙІ) require Ca2+ for activation and other isozymes (μ, ε, η, δ, θ, ζ, λ) are independent of Ca2+. The Ca2+-induced of signaling pathways such as phosphorylation of actin-associated proteins and occluding is in agreement with changes in BBB function (Brown and Davis, 2002[4]). When ischemia occurs α, βІ, βΙІ isozymes are active which increase permeability of the cerebral endothelial cells, and PKC inhibitor reduces this effect of PKC in brain areas, whereas through HO; μ, ε, η, δ, θ, ζ, λ are active, and protect the BBB integrity (Fleegal et al., 2005[8]).
Another hypothesis is that PKC may indirectly alter the tight junction proteins by activating a downstream intracellular signaling pathway that in turn directly affects the tight junction proteins (Kumar et al., 2009[17]). Previous studies have demonstrated that changes in nitric oxide and intracellular Ca2+ may contribute to increases in paracellular permeability (Brown and Davis, 2002[4]; Fleegal et al., 2005[8]). Ca2+ increases in the expression of the Ca2+-dependent isozymes PKC-βΙІ and PKC-γ along with PKC-ζ suggest that PKC may interact with this signaling molecule to contribute to BBB permeability alterations (Banan et al., 2003[1]).
In summary, this is the first report showing the inhibition of PKC after normobaric HO in the rat brain, and its neuroprotective role in ischemic tolerance. These data indicate the importance of PKC activation in response to HO which is important in brain integrity; whereas effect of HO on brain edema and infarct volume is independent of PKC in the MCAO model. And finally elucidation of other mechanisms that regulate the acquisition of brain ischemia tolerance may help in the development of effective methods of HO preconditioning to protect the brain or reduce ischemic injury.
Acknowledgements
The authors are grateful to Dr Ali R. Mani for his scientific comments. We would also like to express our thanks to Miss Fatemeh Mohagheghi and Miss Azam Asgari for their cooperation. This study was supported by a grant from Dean of Research, Tarbiat Modares University.
References
- 1.Banan A, Farhadi A, Fields J, Zhang L, Shaikh M, Keshavarzian A. The delta-isoform of protein kinase C causes inducible nitricoxide synthase and nitric oxide up-regulation: key mechanism for oxidant induced carbonylation, nitration, and disassembly of the microtubule cytoskeleton and hyperpermeability of barrier of intestinal epithelia. J Pharmacol Exp Ther. 2003;305:482–494. doi: 10.1124/jpet.102.047308. [DOI] [PubMed] [Google Scholar]
- 2.Bigdeli MR, Hajizadeha S, Froozandeh M, Rasulianc B, Heidarianpour A, Khoshbaten A. Prolonged and intermittent normobaric hyperoxia induce different degrees of ischemic tolerance in rat brain tissue. Brain Res. 2007;1152:228–233. doi: 10.1016/j.brainres.2007.03.068. [DOI] [PubMed] [Google Scholar]
- 3.Bigdeli MR, Rasoulian B, Meratan AA. In vivo normobaric hyperoxia preconditioning induces different degrees of antioxidant enzymes activities in rat brain tissue. Neuropharmacol Analg. 2009;611:22–9. doi: 10.1016/j.ejphar.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 4.Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: a potential mechanism for blood–brain barrier disruption after stroke. Stroke. 2002;33:1706–1711. doi: 10.1161/01.str.0000016405.06729.83. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Simon R. Ischemic tolerance in the brain. Neurology. 1997;48:306–311. doi: 10.1212/wnl.48.2.306. [DOI] [PubMed] [Google Scholar]
- 6.Currie R, Tanguay R. Analysis of RNA for transcripts for catalase and HSP71 in rat hearts after in vivo hyperthermia. Biochem Cell Biol. 1991;69:375–382. doi: 10.1139/o91-057. [DOI] [PubMed] [Google Scholar]
- 7.Dittmar M, Spruss T, Schuierer G, Horn M. External carotid artery territory ischemia impairs outcome in the endovascular filament model of middle cerebral artery occlusion in rats. Stroke. 2003;34:2252–7. doi: 10.1161/01.STR.0000083625.54851.9A. [DOI] [PubMed] [Google Scholar]
- 8.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Physiol Heart Circ Physiol. 2005;289:2012–2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 9.Garcia JH, Liu KF, Ye ZR, Gutierrez JA. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2303–9. doi: 10.1161/01.str.28.11.2303. [DOI] [PubMed] [Google Scholar]
- 10.Grabb M, Lobner D, Turetsky M, Choi D. Preconditioned resistance to oxygen-glucose deprivation-induced cortical neuronal death: alterations in vesicular GABA and glutamate release. Neuroscience. 2002;115:173–83. doi: 10.1016/s0306-4522(02)00370-6. [DOI] [PubMed] [Google Scholar]
- 11.Hoshida S, Kuzuya T, Fuji H, Yamashita N, Oe H, Hori M, et al. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am J Physiol. 1993;264:33–39. doi: 10.1152/ajpheart.1993.264.1.H33. [DOI] [PubMed] [Google Scholar]
- 12.Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- 13.Kaya M, Kalayci R, Kucuk M, Arican N, Elmas I, Kudat H, et al. Effect of losartan on the blood–brain permeability in diabetic hypertensive rats. Life Sci. 2003;73:3235–44. doi: 10.1016/j.lfs.2003.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Kirino T. Ischemic tolerance. Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- 15.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. Ischemic tolerance phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi H, Yanagita T, Yokoo H, Wada A. Molecular mechanisms and drug development in aquaporin water channel diseases: aquaporins in the brain. J Pharmacol Sci. 2004;96:264–270. doi: 10.1254/jphs.fmj04004x5. [DOI] [PubMed] [Google Scholar]
- 17.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;30:11–15. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 19.Murray C, Lopez A. Mortality by cause for eight regions of the world Global Burden of Disease Study. Lancet. 1997;349:1269–76. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 20.Ohtsuki T, Matsumoto M, Kuwabara K, Kitagawa K, Suzuki K, Taniguchi N, et al. Influence of oxidative stress on induced tolerance to ischemia in gerbil hippocampal neurons. Brain Res. 1992;599:246–252. doi: 10.1016/0006-8993(92)90398-s. [DOI] [PubMed] [Google Scholar]
- 21.Ostrowski R, Colohan A, Zhang J. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Pinzon MA, Dave KR, Raval AP. Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxid Redox Signal. 2005;7:1150–7. doi: 10.1089/ars.2005.7.1150. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Pinzon MA, Mumford PL, Rosenthal M, Sick TJ. Anoxic preconditioning in hippocampal slices: role of adenosine. Neuroscience. 1996;75:687–694. doi: 10.1016/0306-4522(96)00311-9. [DOI] [PubMed] [Google Scholar]
- 24.Rash J, Yasumura T, Hudson C, Aqre P, Nielsen S. Direct immunogold labeling of Aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci USA. 1998;95:11981–11986. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp F. A semiautomated method for measuring brain infarct volume. Cereb Blood Flow Metab. 1990;10:290–3. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 26.Tahepold P, Vaage J, Starkopf J, Valen G. Hyperoxia elicits myocardial protection through a nuclear factor kappaB–dependent mechanism in the rat heart. J Thorac Cardiovasc Surg. 2003;125:650–660. doi: 10.1067/mtc.2003.36. [DOI] [PubMed] [Google Scholar]
- 27.Thornton J, Striplin S, Liu G, Swafford A, Stanley A, van Winkle D, et al. Inhibition of protein synthesis does not block myocardial protection afforded by preconditioning. Am J Physiol. 1990;259:1822–5. doi: 10.1152/ajpheart.1990.259.6.H1822. [DOI] [PubMed] [Google Scholar]
- 28.Xuan Y, Tang X, Banerjee S, Takano H, Li R, Han H, et al. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto N, Sobue K, Miyachi T, Inagaki M, Miura Y, Katsuya H, et al. Differential regulation of aquaporin expression in astrocytes by protein kinase C. Brain Res Mol Brain Res. 2001;95:110–116. doi: 10.1016/s0169-328x(01)00254-6. [DOI] [PubMed] [Google Scholar]