

The crystal structure and preliminary structure-based mutagenesis and activity studies of aspartate transcarbamoylase from P. falciparum are reported.
Keywords: aspartate transcarbamoylase, X-ray structure, pyrimidine biosynthesis, Plasmodium falciparum, malaria, antimalarial drugs
Abstract
The de novo pyrimidine-biosynthesis pathway of Plasmodium falciparum is a promising target for antimalarial drug discovery. The parasite requires a supply of purines and pyrimidines for growth and proliferation and is unable to take up pyrimidines from the host. Direct (or indirect) inhibition of de novo pyrimidine biosynthesis via dihydroorotate dehydrogenase (PfDHODH), the fourth enzyme of the pathway, has already been shown to be lethal to the parasite. In the second step of the plasmodial pyrimidine-synthesis pathway, aspartate and carbamoyl phosphate are condensed to N-carbamoyl-l-aspartate and inorganic phosphate by aspartate transcarbamoylase (PfATC). In this paper, the 2.5 Å resolution crystal structure of PfATC is reported. The space group of the PfATC crystals was determined to be monoclinic P21, with unit-cell parameters a = 87.0, b = 103.8, c = 87.1 Å, α = 90.0, β = 117.7, γ = 90.0°. The presented PfATC model shares a high degree of homology with the catalytic domain of Escherichia coli ATC. There is as yet no evidence of the existence of a regulatory domain in PfATC. Similarly to E. coli ATC, PfATC was modelled as a homotrimer in which each of the three active sites is formed at the oligomeric interface. Each active site comprises residues from two adjacent subunits in the trimer with a high degree of evolutional conservation. Here, the activity loss owing to mutagenesis of the key active-site residues is also described.
1. Introduction
According to the World Health Organization (WHO), 3.3 billion people worldwide are at risk of infection with malaria. The disease is considered to be endemic in more than 100 countries. In 2015 the WHO estimated that there were more than 200 million infections and approximately half a million deaths from malaria, where the majority of deaths occurred amongst children under five years old in Africa (World Health Organization, 2015 ▸).
Malaria is caused by protozoan parasites of the genus Plasmodium (P. falciparum, P. vivax, P. knowlesi, P. malariae and P. ovale curtisi). P. falciparum is responsible for the most dangerous and fatal form of the disease. The complex life cycle of the parasite, as well as the spread of current drug-resistant strains, make the treatment of malaria highly challenging (Hyde, 2007 ▸). There is an urgent need for new antimalarial agents, making the identification of the new drug targets very important (Olliaro et al., 2001 ▸; Attaran, 2004a ▸,b ▸; White, 2008 ▸; Biamonte et al., 2013 ▸)
The intraerythrocytic stage of P. falciparum is associated with an extraordinary resource uptake from the host cell. Active proliferation during this stage requires a supply of purines and pyrimidines for parasite growth to support the production of DNA and parasite replication. Malaria parasites lack the de novo purine-synthesis pathway and salvage the host-cell purines for growth (de Koning et al., 2005 ▸; Hyde, 2007 ▸). Inhibition of this pathway has been shown to be lethal to P. falciparum in vitro (Kicska et al., 2002 ▸; Cassera et al., 2008 ▸). Early biochemical studies on the Plasmodium parasites P. berghei (Büngener & Nielsen, 1967 ▸, 1968 ▸; Van Dyke et al., 1970 ▸), P. knowlesi (Polet & Conrad, 1968 ▸) and P. lophurae (Walsh & Sherman, 1968 ▸; Tracy & Sherman, 1972 ▸) demonstrated the inability of Plasmodium species to metabolize pyrimidines. The parasites lack a thymidine kinase, the enzyme responsible for salvaging host thymidine, as was confirmed by the completion of the genome sequence (Gardner et al., 2002 ▸). As the parasite lacks the pyrimidine-import pathway (Reyes et al., 1982 ▸; Rathod & Reyes, 1983 ▸), the pyrimidine-biosynthesis pathway has been demonstrated to be a major target for antimalarial drug research (Downie et al., 2008 ▸; Cassera et al., 2011 ▸; Phillips et al., 2015 ▸).
Plasmodial pyrimidine biosynthesis is affected, directly or indirectly, by many current antimalarials (Phillips & Rathod, 2010 ▸). For example, the most widely used current antimalarial drug atovaquone (Mather et al., 2005 ▸) is known to inhibit cytochrome b in complex III of the respiratory chain and thus collapse the mitochondrial intramembrane potential. This inhibition causes a failure to provide oxidized ubiquinone to the fourth enzyme in the pyrimidine-biosynthetic pathway, dihydroororate dehydrogenase (PfDHODH; Painter et al., 2007 ▸; Vaidya & Mather, 2009 ▸), which has been validated as an essential enzyme for the parasite and a promising drug target (Phillips & Rathod, 2010 ▸; Booker et al., 2010 ▸; Phillips et al., 2015 ▸). The search for selective PfDHODH inhibitors that might lead to a new antimalarial therapy remains a current topic amongst antimalarial researchers (Coteron et al., 2011 ▸; Xu et al., 2013 ▸; Ross et al., 2014 ▸; Deng et al., 2015 ▸; Phillips et al., 2015 ▸).
Aspartate transcarbamoylase (ATC; EC 2.1.3.2) catalyses the second step of pyrimidine biosynthesis, the condensation of aspartate and carbamoyl phosphate to form N-carbamoyl-l-aspartate and inorganic phosphate. The ATC from Escherichia coli was fully characterized by Lipscomb & Kantrowitz (2012 ▸). E. coli ATC is known to be a highly regulated enzyme: the rate of pyrimidine biosynthesis is stimulated or inhibited in response to cellular levels of the end products of the pathway (Gerhart, 2014 ▸). In E. coli the ATC holoenzyme is composed of six catalytic and six regulatory subunits: three regulatory pairs coordinate two catalytic trimers.
ATCs are also promising targets for antiproliferative and antitumour drugs. Madani and coworkers showed that while almost undetectable in healthy human brain tissue, significant ATC levels were present in all studied tumour samples (Madani et al., 1987 ▸). In 2014, ATC from P. falciparum (PfATC) was also reported to be a potential target for gametocidal drug development (Sun et al., 2014 ▸).
PfATC is a polypeptide of 375 amino acids with a predicted molecular mass of 43.3 kDa (PlasmoDB ID PF3D7_1344800; Table 1 ▸). BLAST analysis of the PfATC sequence shows that the N-terminal region of approximately 36 amino acids does not have known structural homologues (Altschul et al., 1990 ▸). In addition, no homologues of the ATC regulatory chain have yet been identified in the plasmodial genome. Amino acids 55–372 can be aligned with the catalytic domain of P. abyssi ATC (Van Boxstael et al., 2003 ▸) with an identity of 38%. Based on the absence of structural analogues in the Protein Data Bank (PDB) and the predicted presence of an apicoplast-targeting signal (Foth et al., 2003 ▸), the N-terminal region of 36 amino acids was removed and the truncated construct PfATC-Met3 was used in this study. Attempts to obtain full-length PfATC failed owing to spontaneous proteolysis near the N-terminus (data not shown).
Table 1. Macromolecule-production information (truncated PfATC and PfATC-R109A/K138A mutant).
Cloning details for the truncated version of PfATC-Met3 and the mutant PfATC-RK. BsaI restriction sites in the primers for the wild type are underlined. Point mutations in the mutant are underlined. Additional Strep-tag residues at the C-terminus are shown in italics.
| Gene | Truncated PfATC (wild type) |
| Source organism | P. falciparum strain 3D7 |
| DNA source | pASK-IBA3-PfATC-full plasmid |
| Forward primer (BsaI) | 5′-GCGCGCGGTCTCCAATGTTTTATATCAATAGCAAG-3′ |
| Reverse primer (BsaI) | 5′-GCGCGCGGTCTCCGCGCTGCTAGTTGATGAAAAAATGAG-3′ |
| Expression vector | pASK-IBA3 |
| Expression host | E. coli |
| Complete amino-acid sequence of the construct produced | MFYINSKYKIDLDKIMTKMKNKSVINIDDVDDEELLAILYTSKQFEKILKNNEDSKYLENKVFCSVFLEPSTRTRCSFDAAILKLGSKVLNITDMNSTSFYKGETVEDAFKILSTYVDGIIYRDPSKKNVDIAVSSSSKPIINAGNGTGEHPTQSLLDFYTIHNYFPFILDRNINKKLNIAFVGDLKNGRTVHSLSKLLSRYNVSFNFVSCKSLNIPKDIVNTITYNLKKNNFYSDDSIKYFDNLEEGLEDVHIIYMTRIQKERFTDVDEYNQYKNAFILSNKTLENTRDDTKILHPLPRVNEIKVEVDSNPKSVYFTQAENGLYVRMALLYLIFSSTSSAWSHPQFEK |
| Gene | PfATC-R109A/K138A double mutant (PfATC-RK) |
| DNA source | pASK-IBA3-PfATC-Met3 plasmid |
| Forward primer (R109A) | 5′-GTTCCTTGAACCAAGTACAGCAACAAGATGTTCTTTTGATGC-3′ |
| Reverse primer (R109A) | 5′-GCATCAAAAGAACATCTTGTTGCTGTACTTGGTTCAAGGAAC-3′ |
| Forward primer (K138A) | 5′-CTGATATGAATTCAACTTCTTTTTATGCGGGAGAAACTGTTGAAGATGCC-3′ |
| Reverse primer (K138A) | 5′-GGCATCTTCAACAGTTTCTCCCGCATAAAAAGAAGTTGAATTCATATCAG-3′ |
| Expression vector | pASK-IBA3 |
| Expression host | E. coli |
| Complete amino-acid sequence of the construct produced | MFYINSKYKIDLDKIMTKMKNKSVINIDDVDDEELLAILYTSKQFEKILKNNEDSKYLENKVFCSVFLEPSTATRCSFDAAILKLGSKVLNITDMNSTSFYAGETVEDAFKILSTYVDGIIYRDPSKKNVDIAVSSSSKPIINAGNGTGEHPTQSLLDFYTIHNYFPFILDRNINKKLNIAFVGDLKNGRTVHSLSKLLSRYNVSFNFVSCKSLNIPKDIVNTITYNLKKNNFYSDDSIKYFDNLEEGLEDVHIIYMTRIQKERFTDVDEYNQYKNAFILSNKTLENTRDDTKILHPLPRVNEIKVEVDSNPKSVYFTQAENGLYVRMALLYLIFSSTSAWSHPQFEK |
The truncated PfATC (PfATC-Met3) consists of 349 amino acids, including 339 of wild-type PfATC (37–375) and the SAWSHPQFEK sequence of the Strep-tag (IBA3) at the C-terminus, with an approximate molecular mass of 40.3 kDa. Recent attempts to characterize PfATC have been published (Depamede & Menz, 2011 ▸; Banerjee et al., 2012 ▸), but a crystal structure is necessary to study the molecular basis of the catalytic mechanism, to support the validation of PfATC as a drug target and to potentially support structure-based drug design.
In this manuscript, the crystal structure and preliminary characterization of a truncated PfATC as well as its localization within the parasite are reported. Structural information was used to design a mutant of PfATC with significantly reduced catalytic activity; the activity profiles of the mutant and wild-type enzymes are reported. These data are essential in understanding the role of plasmodial aspartate metabolism and might lead to a novel antimalarial therapy.
2. Materials and methods
2.1. Cloning
A full-length gene encoding PfATC was amplified via reverse-transcriptase PCR using P. falciparum total RNA as a template. In the first step, the cDNA was synthesized from RNA using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Scientific). In the second step, the full-length PfATC gene was amplified from the cDNA template using sequence-specific sense (5′-GCGCGCGGTCTCCAATGATTGAAATATTTTGCACTGC-3′) and antisense (5′-GCGCGCGGTCTCCGCGCTGCTAGTTGATGAAAAAATGAG-3′) primers. The PCR reaction was performed using the SuperMix High Fidelity polymerase mixture (Invitrogen). The generated PCR product was digested with BsaI (restriction sites are underlined) and cloned into pASK-IBA3 vector (IBA Lifesciences) previously digested with the same enzyme. The final expression plasmid pASK-IBA3-PfATC-full encoded the full-length version of PfATC with the additional amino acids SAWSHPQFEK (Strep-tag) at the C-terminus (Supplementary Table S1).
Cloning of the truncated PfATC (PfATC-Met3) was performed via PCR amplification using pASK-IBA3-PfATC-full plasmid as a template with sequence-specific sense (5′-GCGCGCGGTCTCCAATGTTTTATATCAATAGCAAG-3′) and antisense (5′-GCGCGCGGTCTCCGCGCTGCTAGTTGATGAAAAAATGAG-3′) primers.
The PCR fragment was similarly cloned into pASK-IBA3 vector previously digested with BsaI. The final expression plasmid pASK-IBA3-PfATC-Met3 encoded the truncated version of PfATC (residues 37–375) with the additional amino acids SAWSHPQFEK (Strep-tag) at the C-terminus (Table 1 ▸). All plasmid samples were verified by sequencing.
2.2. Expression
PfATC-Met3 was recombinantly expressed using E. coli Rosetta 2 (DE3) pLysS (Nalgene) competent cells transformed with the pASK-IBA3-PfATC-Met3 expression plasmid. The optimal cell line and inductor concentration were chosen based on preliminary small-scale expression trials (data not shown). The culture was propagated in 1 l selective LB medium supplemented with 50 µg ml−1 ampicillin, 35 µg ml−1 chloramphenicol and 4 mM MgCl2 at 310 K in 2 l baffled Erlenmeyer flasks (Nalgene) and was induced with 60 ng ml−1 anhydrotetracycline (AHT) according to the expression-trial results. The temperature of the culture was decreased to 291 K after induction and the cells were harvested by centrifugation after overnight expression. Centrifugation was performed using an SLA-3000 rotor (Thermo Scientific) at 10 000g for 30 min.
2.3. Purification
Purification of recombinant PfATC-Met3 was performed via Strep-tag chromatography according to the manufacturer’s recommendations (IBA Lifesciences). The bacterial pellet from 1 l of culture was resuspended in 40 ml lysis buffer [50 mM Tris pH 8.0, 300 mM NaCl, 5%(v/v) glycerol, 3 mM β-mercaptoethanol (BME)]. The lysis was performed by sonication on ice. The supernatant containing soluble Strep-tagged PfATC-Met3 was clarified by centrifugation at 45 000g (SS-34 rotor, Thermo Scientific) and filtered using a 0.45 µm filter membrane (Whatman). The filtered supernatant was incubated with 2.0 ml Strep-tag affinity resin (Strep-Tactin Superflow, IBA Lifesciences) for 20 min at 277 K and subsequently poured onto a gravity-flow column (Bio-Rad) and washed with 100 ml lysis buffer. The protein was eluted with 8 ml elution buffer [50 mM Tris pH 8.0, 300 mM NaCl, 2.5 mM desthiobiotin, 5%(v/v) glycerol, 3 mM BME]. The eluate was pooled and concentrated at 277 K to 2 mg ml−1 using a Vivaspin 4 concentration column with a 10 kDa cutoff (Sartorius Stedim Biotech).
In order to select the correct buffer for further purification, a Thermofluor-based stability assay (Ericsson et al., 2006 ▸; Nettleship et al., 2008 ▸) was performed using a CFX96 Real-Time System (Bio-Rad). SYPRO Orange dye (5000× stock, Invitrogen) was added to the protein sample (2 mg ml−1) in a 1:500 ratio. Each experiment consisted of 5 µl of the protein/dye mixture and 45 µl of the buffer component to be screened. Water was used as a control instead of the buffer sample. Inflection points in the graphs of relative fluorescence units (RFU) against temperature were determined manually and were used as an indicator of the thermal stability of the sample in the presence of the screened buffer components. Components that exhibited a positive thermal shift in comparison to a water control sample were used to select a size-exclusion chromatography (SEC) buffer [20 mM Tris pH 8.0, 300 mM NaCl, 10 mM sodium malonate, 5%(v/v) glycerol, 2 mM BME]. The remaining protein was concentrated to a volume of 1 ml (Vivaspin 4, Sartorius Stedim Biotech) and purified via SEC using a HiLoad 16/60 Superdex 75 column (GE Healthcare) pre-equilibrated with SEC buffer using an NGC liquid-chromatography system (Bio-Rad).
The purified protein eluted as a single peak and was pooled and concentrated to 10 mg ml−1 at 277 K (Vivaspin 4, Sartorius Stedim Biotech). The final concentration was determined based on the protein theoretical absorbance at 280 nm [Abs 0.1% (1 mg ml−1) = 0.84; http://web.expasy.org/protparam]. The concentrated protein was immediately used in crystallization trials. The estimated protein purity was greater than 95% (Fig. 1 ▸ a) as assessed by Coomassie Brilliant Blue-stained SDS–PAGE (Laemmli, 1970 ▸). The final yield of purified PfATC-Met3 was 6 mg per litre of culture. Expression and purification of the mutant version of PfATC-Met3 were performed identically.
Figure 1.

(a) 10% SDS–PAGE of the purified PfATC-Met3. The sample was boiled in SDS loading buffer prior to loading and the gel was stained with Coomassie Blue. Left lane, unstained protein marker (Thermo Scientific; labelled in kDa); right lane, final PfATC-Met3 sample. (b) A diffraction-quality single PfATC-Met3 crystal mounted in a cryo-loop (Molecular Dimensions) grown in 200 mM sodium sulfate, 5 mM magnesium sulfate, 15%(w/v) PEG 3350 in 100 mM bis-tris propane pH 7.5. Dimensions as measured on the beamline are shown.
2.4. Crystallization
Screening for crystallization conditions for PfATC-Met3 was performed using a high-throughput crystallization robot (Gryphon, Art Robbins) against commercially available sparse-matrix screening kits (JCSG-plus and PACT premier; Molecular Dimensions). All experiments were performed at 293 K using the sitting-drop vapour-diffusion technique in 96-well MRC2 plates (Molecular Dimensions). Equal volumes (0.1 µl) of protein solution and crystallization reagent were equilibrated against 50 µl reservoir solution. Medium-sized single crystals appeared overnight in various conditions containing PEG 3350/4000. Further optimization was performed using the hanging-drop technique by varying the precipitant concentration, ionic strength, pH and buffer conditions. The optimized conditions consisted of equal amounts (1.5 µl) of 9 mg ml−1 PfATC-Met3 sample and 0.2 M sodium sulfate, 5 mM MgSO4, 15%(w/v) PEG 3350 in 0.1 M bis-tris propane pH 7.5 as the crystallization solution at 293 K. Rhomboid-shaped diffraction-quality crystals (Fig. 1 ▸ b) with maximum dimensions of 200 µm appeared overnight and were subsequently harvested using mounted round LithoLoops (Molecular Dimensions), incubated in the cryo-buffer (below) and flash-cooled in liquid nitrogen prior to shipment to the synchrotron. The cryo-buffer was chosen based on an estimation from Garman & Mitchell (1996 ▸) and consisted of 0.2 M sodium sulfate, 5 mM MgCl2, 15%(w/v) PEG 3350, 20% glycerol in 0.1 M bis-tris propane pH 7.5.
2.5. Data collection
Cryocooled PfATC-Met3 crystals were sent to the European Synchrotron Radiation Facility (ESRF), Grenoble using a dry-shipping cryo-container (Taylor-Wharton) and a 2.4 Å resolution data set was collected at 100 K in a nitrogen stream on the ID23-2 beamline. Initial characterization of the crystals and the optimization of data-collection parameters were performed using BEST (Popov & Bourenkov, 2003 ▸; Bourenkov & Popov, 2006 ▸). The space group of the PfATC crystals was calculated to be monoclinic P21, with unit-cell parameters a = 87.0, b = 103.8, c = 87.1 Å, α = 90.0, β = 117.7, γ = 90.0°, and the solvent content was calculated to be 59.4%. The data were processed using XDSAPP (Krug et al., 2012 ▸). The analysis also showed that the crystal was twinned. The Matthews coefficient (Matthews, 1968 ▸) predicted that the asymmetric unit is likely to contain three monomers of PfATC-Met3. The data-collection and processing statistics are reported in Table 2 ▸.
Table 2. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | ID23-2, ESRF |
| Wavelength (Å) | 0.98 |
| Temperature (K) | 100 |
| Detector | Pilatus 6M |
| Crystal-to-detector distance (mm) | 337.55 |
| Rotation range per image (°) | 0.1 |
| Total rotation range (°) | 129 |
| Exposure time per image (s) | 0.04 |
| Space group | P21 |
| a, b, c (Å) | 86.96, 103.80, 87.11 |
| α, β, γ (°) | 90.0, 117.6, 90.0 |
| Mosaicity (°) | 0.2 |
| Resolution range (Å) | 45.0–2.42 (2.56–2.42) |
| Total No. of reflections | 126651 (20324) |
| No. of unique reflections | 50423 (8051) |
| Completeness (%) | 95.9 (95.6) |
| Multiplicity | 2.51 (2.52) |
| 〈I/σ(I)〉 | 10.81 (1.98) |
| CC1/2 | 99.7 (76.6) |
| R meas † (%) | 7.6 (58.0) |
| Overall B factor from Wilson plot (Å2) | 56.64 |
R
meas is defined as 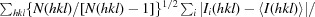
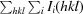 , where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average intensity from multiple observations.
, where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the average intensity from multiple observations.
2.6. Data processing and refinement
The structure of PfATC-Met3 was solved by molecular replacement using BALBES (Long et al., 2008 ▸) within the CCP4 package (Winn et al., 2011 ▸). The crystal structure of ATC from Pyrococcus abyssi (Van Boxstael et al., 2003 ▸) was used as a search model (38% identity), yielding a solution with three monomers in the asymmetric unit. The model was further optimized via manual rebuilding in Coot (Emsley et al., 2010 ▸) and was refined with REFMAC5 (Murshudov et al., 2011 ▸). The final refinement steps were carried out with global NCS restraints and TLS restraints calculated via the TLSMD web server (Painter & Merritt, 2006a ▸,b ▸). These steps also included twin refinement. The final 2.5 Å resolution model of PfATC-Met3 consisted of three molecules in the asymmetric unit, with an R factor of 0.18 and a free R factor of 0.22 (Table 3 ▸), and has been deposited in the Protein Data Bank (PDB) with accession code 5ilq. The structural figures in this manuscript were prepared with PyMOL (DeLano, 2002 ▸).
Table 3. Structure solution and refinement.
Values in parentheses are for the highest resolution shell.
| Resolution range (Å) | 20.0–2.5 |
| Completeness (%) | 95.2 |
| σ Cutoff | 1 |
| No. of reflections, working set | 42863 |
| No. of reflections, test set | 2354 |
| Final R cryst | 0.18 |
| Final R free † | 0.22 |
| No. of non-H atoms | |
| Total | 7909 |
| Protein | 7801 |
| Ion | 23 |
| Water | 88 |
| R.m.s. deviations | |
| Bonds (Å) | 0.019 |
| Angles (°) | 1.96 |
| Average B factors (Å2) | |
| Overall | 57.39 |
| Protein | 58.04 |
| Ion | 56.76 |
| Water | 51.47 |
| Ramachandran plot | |
| Most favoured (%) | 93.60 |
| Allowed (%) | 5.98 |
The free R factor was calculated using a randomly selected 5% of reflections that were omitted from refinement.
2.7. Mutagenesis
The double mutant PfATC-Met3-R109A/K138A (hereafter referred to as PfATC-RK) was created via site-directed mutagenesis using the previously described pASK-IBA3-PfATC-Met3 expression plasmid as a template. The PCR reaction was performed using Phusion Hot Start II DNA polymerase (Thermo Scientific) according to the manufacturer’s recommendations. For mutagenesis details, please refer to Table 1 ▸. The generated mutant plasmid was treated with DpnI enzyme for 2 h at 310 K to eliminate the parental template. The mutations were validated by sequencing. Similarly to the wild type, the mutant construct also encoded a C-terminal Strep-tag. Expression and purification of the PfATC-RK mutant were performed identically to those for PfATC-Met3.
2.8. Activity assays
The kinetic properties of PfATC-Met3 as well as PfATC-RK were investigated according to Müller et al. (2010 ▸), Motomizu et al. (1983 ▸) and Matoba et al. (2009 ▸) with minor modifications. Briefly, the reaction was carried out at room temperature in a total volume of 160 µl of 14 mM l-aspartate and 1 mM carbamoyl phosphate (CP) saturated substrate solution in 200 mM Tris–acetate buffer pH 8. The reaction was stopped with 80 µl 25 mM ammonium molybdate in 4.5 M H2SO4. After all of the reactions had been stopped, 160 µl 0.5 µM malachite green in 0.1%(w/v) poly(vinyl alcohol) (PVA) was added and incubated for 30 min at room temperature. Subsequently, the absorption of the samples was measured at a wavelength of 620 nm. The analyses were evaluated from at least three independent quadruplicate assays using GraphPad Prism 4 (GraphPad Software, USA).
2.9. Transfection
The open reading frame of full-length PfATC was cloned in front of the GFP gene of the expression vector pARL1a and was subsequently transfected into P. falciparum according to Müller et al. (2010 ▸). Briefly, erythrocytes were washed in Cytomix (Wu et al., 1995 ▸) and an aliquot of 450 ml was combined with approximately 100 mg of the construct resuspended in 50 ml TE buffer. The red blood cells were electroporated using a Gene-Pulser X-Cell Total System (Bio-Rad) at 0.31 kV and 900 mF. After electroporation, the cells were transferred into pre-warmed RPMI medium and inoculated with 50 ml of red blood cells infected with 10% schizonts to give a parasitaemia of 1%. 4 h post-transfection the culture medium was exchanged. Parasites were grown for 24 h without drug selection before the medium was supplemented with 5 nM WR99210. Parasites were first observed after 16–60 d of selection and live parasites were analysed by fluorescent microscopy using an Axioskop 2 plus microscope (Zeiss). In order to visualize the nucleus, parasites were incubated with Hoechst 33342 dye according to the manufacturer’s recommendation (Invitrogen). For co-localization experiments to visualize the ER, transfected Plasmodium was supplemented with 2 mM ER-Tracker Red BODIPY-TR (Invitrogen) prior to microscopy.
3. Results
A truncated version of ATC from P. falciparum was cloned, recombinantly expressed, purified and crystallized. Full-length PfATC is a protein of 375 residues with a calculated molecular weight of 43.3 kDa. BLAST (Altschul et al., 1990 ▸) analysis revealed no structural homologues of the first 36 N-terminal residues (Fig. 2 ▸). PfATC shows 38% identity (84% query cover) to the catalytic subunit of ATC from P. abyssi (PDB entry 1ml4; Van Boxstael et al., 2003 ▸) and 39% identity (84% query cover) to E. coli ATC (PDB entry 1d09; Jin et al., 1999 ▸). Nucleotide BLAST analysis also shows 37% identity (86% query cover) to the ATC domain of human CAD (carbamoyl phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase) protein. The crystal structure of this domain is not yet available, although preliminary data have been reported (Ruiz-Ramos et al., 2013 ▸). Further analysis with the PlasmoAP tool (Foth et al., 2003 ▸) showed a strong prediction for the N-terminal region to contain an apicoplast-targeting sequence, with a possible cleavage site between residues 27 and 28 (IRT–KK; Supplementary Table S1). GFP-labelling experiments in vivo showed distinct localization of PfATC compared with the ER, supporting the hypothesis of the apicomplexian nature of the enzyme (Fig. 3 ▸). Based on these data, the third methionine in the sequence (Met37) was selected as the starting point of our construct.
Figure 2.

The homology analysis of PfATC-Met3 was performed using BLAST (Altschul et al., 1990 ▸) and visualized via T-Coffee (Di Tommaso et al., 2011 ▸). Red, yellow, green and blue colours represent good, average, bad and weak alignment, respectively. Residues essential for the active sites are shown with arrows. Absolutely, strongly and slightly conserved residues are marked with ‘*’, ‘:’ and ‘.’ symbols, respectively.
Figure 3.

Fluorescent microscopy imaging of P. falciparum transfected with GFP-labelled PfATC shows distinct localization of PfATC (see §2.9).
Attempts to express and purify full-length PfATC with sufficient purity failed. SDS–PAGE analysis of the elution fraction of Strep-tag purification showed three distinct Strep-tagged (C-terminal) bands with sizes around 43 kDa, and further size-exclusion purification showed three overlapping peaks that could not be separated with sufficient purity (data not shown). Use of protease-inhibitor cocktail during the lysis and purification did not show any significant improvement. The presence of at least three versions of PfATC of different lengths in the elution fraction suggests that the full-length protein is proteolyzed near the N-terminus owing to the apicoplast-targeting nature of this region. The activity assay performed with PfATC-Met3 showed that it possesses catalytic activity, and further experiments have been performed with this truncated PfATC construct.
The recombinant PfATC-Met3 was produced with a C-terminal Strep-tag (IBA Lifesciences) and was purified by Strep-tag chromatography according to the manufacturer’s recommendations. In order to achieve higher purity and homogeneity, PfATC-Met3 was further purified via SEC and the final purity was assessed via SDS–PAGE analysis (Fig. 1 ▸ a). The yield of pure homogeneous protein was approximately 6 mg per litre of culture. In order to increase the post-purification stability of the protein sample and increase the chance of crystallization, the SEC buffer was chosen based on the results of differential scanning fluorimetry. The fresh purified protein was concentrated to 9 mg ml−1 and used in protei crystallization experiments.
Multiple crystals of PfATC-Met3 appeared overnight in various crystallization screening conditions containing PEG (polyethylene glycol) 3350/4000. Optimization of the initial conditions yielded single PfATC crystals with a maximum dimension of 0.2 mm (Fig. 1 ▸ b). The final crystallization condition was 0.1 M bis-tris propane pH 7.5, 0.2 M sodium sulfate, 5 mM magnesium sulfate, 15%(w/v) PEG 3350.
The optimized crystals diffracted X-rays to a maximum resolution of ∼2.4 Å on the ID23-2 beamline at ESRF, Grenoble, France. The space group of the PfATC-Met3 crystals was determined to be monoclinic P21, with unit-cell parameters a = 87.0, b = 103.8, c = 87.1 Å, α = 90.0, β = 117.7, γ = 90.0°. The presence of three PfATC molecules per asymmetric unit was confirmed by molecular replacement and the 2.5 Å resolution structure of PfATC-Met3 has been deposited in the PDB with accession code 5ilq.
3.1. Overall structure of PfATC-Met3
PfATC-Met3 is a homotrimer with three active sites formed at the oligomeric interfaces (Fig. 4 ▸). Superposition of the PfATC-Met3 structure and the catalytic subunit of E. coli ATC (PDB entry 1d09; Jin et al., 1999 ▸) showed a high level of sequence and secondary-structure conservation (Figs. 2 ▸ and 5 ▸). BLAST (Altschul et al., 1990 ▸) analysis against nine known homologues (structures available) showed that of 375 residues 46 (12.3%) were absolutely conserved, 43 (11.5%) were strongly conserved and 25 (6.7%) were slightly conserved. The loops formed by residues 84–91, 203–212 and 265–275 of PfATC-Met3 are slightly longer than their E. coli analogues (residues 34–36, 150–155 and 206–208, respectively). In addition, the N-terminal region of the PfATC-Met3 model (residues 37–48) is also longer and shows no secondary structure. Poor electron-density coverage suggests that this region is highly mobile. For this reason, the N-terminal residues 37–46 (chain A) and 37–43 (chains B and C) were excluded from the final model as well as the loops formed by residues 297–311 (all three chains) and the C-terminal region of chain A (residues 375–378). The presence of additional electron density near the C-terminus was modelled as the Strep-tag (SAWSHPQFEK) with the last 2–3 residues (FEK) excluded.
Figure 4.

The overall model of PfATC-Met3. (a) PfATC-Met3 is a homotrimer with three active sites (shown with stars) formed at the oligomeric interfaces. (b) Schematic view of the PfATC homotrimer.
Figure 5.

The secondary structure of PfATC-Met3 compared with E. coli ATC (a) and B. subtilis ATC (b). Coordinates of the PfATC-Met3 structure (pale green) and the catalytic subunits of E. coli ATC in the liganded R state (magenta; PDB entry 1d09; Jin et al., 1999 ▸) and of unliganded B. subtilis ATC (blue; PDB entry 3r7d; Harris et al., 2011 ▸) were superimposed using Coot (Emsley & Cowtan, 2004 ▸; Emsley et al., 2010 ▸).
In 1991, Stevens and coworkers reported the essential residues that form the active site of ATC based on the structure of E. coli ATC and confirmed by site-specific mutagenesis experiments (Stevens et al., 1991 ▸). In PfATC-Met3, all the homologous residues are conserved (BLAST) amongst the ATCs from other organisms (Figs. 2 ▸ and 6 ▸). Each subunit of the trimer hosts an active site including two residues (Ser135 and Lys138) from the adjacent chain.
Figure 6.

Superposition of key active-site residues of PfATC-Met3 (pale green) with (a) E. coli ATC in the liganded R state (magenta; PDB entry 1d09; Jin et al., 1999 ▸) and (b) unliganded B. subtilis ATC (blue, PDB code: 3r7d; Harris et al., 2011 ▸) that exhibit conservation amongst homologous species (see Fig. 2 ▸).
PISA analysis (Krissinel & Henrick, 2007 ▸) of the PfATC-Met3 structure showed that the oligomeric contact between each pair of subunits (hereafter referred to as A and B) is formed by 32 residues [buried surface area (BSA) of 899 Å2] and 27 residues (BSA of 956 Å2), respectively (Fig. 7 ▸). The interface from the subunit A consists of nine absolutely conserved, three strongly conserved and two slightly conserved residues. The adjacent interface B has eight absolutely conserved, four strongly conserved and two slightly conserved residues. Each subunit has a total surface area of approximately 14 300 Å2, of which 1855 Å2 (13%) belongs to the intra-oligomeric interfaces.
Figure 7.

PfATC-Met3 structure analysis reveals that each subunit forms two oligomeric contacts with other subunits of the trimer (a). The residues that form these contacts are shown in blue (b–f). Absolutely (red), strongly (orange) and slightly conserved (green) residues of these interfaces are shown (c, e). Active-site residues (magenta) are also shown on the surface (f).
3.2. Mutagenesis studies and activity profile of the truncated version of PfATC
Based on the mutagenic studies of E. coli ATC summarized in Lipscomb & Kantrowitz (2012 ▸) and the overall structure similarity, a mutant PfATC-Met3 version was constructed. In E. coli, point mutations of the essential active-site residues of the catalytic chain of ATC resulted in a significant loss of activity. For example, the guanidinium group of Arg54 of E. coli ATC was shown to be crucial for binding of the substrate carbamoyl phosphate (CP) and enzymatic condensation of CP and aspartate, and its removal (Arg54Ala mutation) caused a 17 000-fold loss in activity and a 13-fold reduction in the affinity for CP (Stebbins et al., 1989 ▸, 1992 ▸). The amino group of Lys84, which completes the active site of the adjacent subunit, interacts with both Arg54 and aspartate upon binding; a Lys84Asn mutation resulted in a 1200-fold activity loss for the E. coli enzyme (Jin et al., 1999 ▸). Both mutations did not affect the folding and the structure of the enzyme, as confirmed by the X-ray data (data not shown). Comparison between the crystal structures of PfATC-Met3 and E. coli ATC (PDB entry 1d09; Jin et al., 1999 ▸) showed that the Arg109 and Lys138 residues of the active site of PfATC-Met3 are homologous to Arg54 and Lys84 of E. coli ATC. The mutant version PfATC-RK (R109A/K138A) was designed and cloned using the site-specific mutagenesis technique and was recombinantly expressed according to the same protocol as PfATC-Met3 (see §2).
In order to confirm the presence of the catalytic activity of the recombinant PfATC-Met3, a phosphate-detection system based on malachite green (Motomizu et al., 1983 ▸) was established. The specific in vitro activity of PfATC-Met3 was 11.04 ± 1.04 µmol min−1 mg−1. Activity assays with the double mutant PfATC-RK were also performed and showed a significantly lower value of 0.85 ± 0.30 µmol min−1 mg−1. Additional native PAGE electrophoresis and dynamic light-scattering (DLS) experiments (data not shown) confirmed the presence of an oligomeric PfATC-Met3 assembly in solution for both the wild-type and mutant versions. These data have confirmed the initial hypothesis that the mutation of the key active-site residues (R109A and K138A) significantly reduces the catalytic activity while having no adverse effect on the oligomeric assembly.
4. Discussion
Pyrimidine biosynthesis by P. falciparum represents a highly attractive target for future antimalarial drug development. The determined crystal structure of truncated aspartate transcarbamoylase from P. falciparum, the enzyme that catalyses the second step of the pathway, shows a high degree of evolutional conservation amongst its homologues from other organisms. The active site of the enzyme shows the highest degree of conservation, making the structure-based design of specific active-site-targeting inhibitors of PfATC and further validation as a drug target highly challenging.
5. Related literature
The following reference is cited in the Supporting Information for this article: Foth et al. (2005 ▸).
Supplementary Material
PDB reference: aspartate transcarbamoylase, 5ilq
Supplementary Table S1.. DOI: 10.1107/S2053230X16008475/rr5118sup1.pdf
Acknowledgments
The authors acknowledge the staff at the ESRF (ID23-3 and MASSIF) for providing access to synchrotron radiation. The authors would like to thank the CAPES/Nuffic university network programme MALAR-ASP (053/14), the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grants 2013/10288-1 and 2013/17577-9) for financial support. Soraya S. Bosch is a recipient of an Ubbo Emmius fellowship from University of Groningen. Fernando de Assis Batista is supported by a fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) within the Science without Borders programme.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. [DOI] [PubMed]
- Attaran, A. (2004a). Lancet, 364, 1922–1923. [DOI] [PubMed]
- Attaran, A. (2004b). Nature (London), 430, 932–933. [DOI] [PubMed]
- Banerjee, A., Arora, N. & Murty, U. (2012). Med. Chem. Res. 21, 2480–2493.
- Biamonte, M. A., Wanner, J. & Le Roch, K. G. (2013). Bioorg. Med. Chem. Lett. 23, 2829–2843. [DOI] [PMC free article] [PubMed]
- Booker, M. L. et al. (2010). J. Biol. Chem. 285, 33054–33064. [DOI] [PMC free article] [PubMed]
- Bourenkov, G. P. & Popov, A. N. (2006). Acta Cryst. D62, 58–64. [DOI] [PubMed]
- Büngener, W. & Nielsen, G. (1967). Z. Tropenmed. Parasitol. 18, 456–462. [PubMed]
- Büngener, W. & Nielsen, G. (1968). Z. Tropenmed. Parasitol. 19, 185–197. [PubMed]
- Cassera, M. B., Hazleton, K. Z., Merino, E. F., Obaldia, N. III, Ho, M.-C., Murkin, A. S., DePinto, R., Gutierrez, J. A., Almo, S. C., Evans, G. B., Babu, Y. S. & Schramm, V. L. (2011). PLoS One, 6, e26916. [DOI] [PMC free article] [PubMed]
- Cassera, M. B., Hazleton, K. Z., Riegelhaupt, P. M., Merino, E. F., Luo, M., Akabas, M. H. & Schramm, V. L. (2008). J. Biol. Chem. 283, 32889–32899. [DOI] [PMC free article] [PubMed]
- Coteron, J. M. et al. (2011). J. Med. Chem. 54, 5540–5561. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL. http://www.pymol.org.
- Deng, X., Matthews, D., Rathod, P. K. & Phillips, M. A. (2015). Acta Cryst. F71, 553–559. [DOI] [PMC free article] [PubMed]
- Depamede, S. N. & Menz, I. (2011). Res. J. Microbiol. 6, 599–608.
- Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J.-M., Taly, J.-F. & Notredame, C. (2011). Nucleic Acids Res. 39, W13–W17. [DOI] [PMC free article] [PubMed]
- Downie, M. J., Kirk, K. & Mamoun, C. B. (2008). Eukaryot. Cell, 7, 1231–1237. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N. & Nordlund, P. (2006). Anal Biochem. 357, 289–298. [DOI] [PubMed]
- Foth, B. J., Ralph, S. A., Tonkin, C. J., Struck, N. S., Fraunholz, M., Roos, D. S., Cowman, A. F. & McFadden, G. I. (2003). Science, 299, 705–708. [DOI] [PubMed]
- Foth, B. J., Stimmler, L. M., Handman, E., Crabb, B. S., Hodder, A. N. & McFadden, G. I. (2005). Mol. Microbiol. 55, 39–53. [DOI] [PubMed]
- Gardner, M. J. et al. (2002). Nature (London), 419, 498–511.
- Garman, E. F. & Mitchell, E. P. (1996). J. Appl. Cryst. 29, 584–587.
- Gerhart, J. (2014). FEBS J. 281, 612–620. [DOI] [PubMed]
- Harris, K. M., Cockrell, G. M., Puleo, D. E. & Kantrowitz, E. R. (2011). J. Mol. Biol. 411, 190–200. [DOI] [PMC free article] [PubMed]
- Hyde, J. E. (2007). FEBS J. 274, 4688–4698. [DOI] [PMC free article] [PubMed]
- Jin, L., Stec, B., Lipscomb, W. N. & Kantrowitz, E. R. (1999). Proteins, 37, 729–742. [PubMed]
- Kicska, G. A., Tyler, P. C., Evans, G. B., Furneaux, R. H., Schramm, V. L. & Kim, K. (2002). J. Biol. Chem. 277, 3226–3231. [DOI] [PubMed]
- Koning, H. P. de, Bridges, D. J. & Burchmore, R. J. (2005). FEMS Microbiol. Rev. 29, 987–1020. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Krug, M., Weiss, M. S., Heinemann, U. & Mueller, U. (2012). J. Appl. Cryst. 45, 568–572.
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Lipscomb, W. N. & Kantrowitz, E. R. (2012). Acc. Chem. Res. 45, 444–453. [DOI] [PMC free article] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Madani, S., Baillon, J., Fries, J., Belhadj, O., Bettaieb, A., Ben Hamida, M. & Hervé, G. (1987). Eur. J. Cancer Clin. Oncol. 23, 1485–1490. [DOI] [PubMed]
- Mather, M. W., Darrouzet, E., Valkova-Valchanova, M., Cooley, J. W., McIntosh, M. T., Daldal, F. & Vaidya, A. B. (2005). J. Biol. Chem. 280, 27458–27465. [DOI] [PMC free article] [PubMed]
- Matoba, K., Nara, T., Aoki, T., Honma, T., Tanaka, A., Inoue, M., Matsuoka, S., Inaoka, D. K., Kita, K. & Harada, S. (2009). Acta Cryst. F65, 933–936. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Motomizu, S., Wakimoto, T. & Tôei, Y. (1983). Talanta, 30, 333–338. [DOI] [PubMed]
- Müller, I. B., Knöckel, J., Eschbach, M. L., Bergmann, B., Walter, R. D. & Wrenger, C. (2010). Cell. Microbiol. 12, 677–691. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nettleship, J. E., Brown, J., Groves, M. R. & Geerlof, A. (2008). Methods Mol. Biol. 426, 299–318. [DOI] [PubMed]
- Olliaro, P., Taylor, W. R. & Rigal, J. (2001). Trop. Med. Int. Health, 6, 922–927. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006a). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006b). J. Appl. Cryst. 39, 109–111.
- Painter, H. J., Morrisey, J. M., Mather, M. W. & Vaidya, A. B. (2007). Nature (London), 446, 88–91. [DOI] [PubMed]
- Phillips, M. A. et al. (2015). Sci. Transl. Med. 7, 296ra111. [DOI] [PMC free article] [PubMed]
- Phillips, M. A. & Rathod, P. K. (2010). Infect. Disord. Drug Targets, 10, 226–239. [DOI] [PMC free article] [PubMed]
- Polet, H. & Conrad, M. E. (1968). Proc. Soc. Exp. Biol. Med. 127, 251–253. [DOI] [PubMed]
- Popov, A. N. & Bourenkov, G. P. (2003). Acta Cryst. D59, 1145–1153. [DOI] [PubMed]
- Rathod, P. K. & Reyes, P. (1983). J. Biol. Chem. 258, 2852–2855. [PubMed]
- Reyes, P., Rathod, P. K., Sanchez, D. J., Mrema, J. E., Rieckmann, K. H. & Heidrich, H. G. (1982). Mol. Biochem. Parasitol. 5, 275–290. [DOI] [PubMed]
- Ross, L. S., Gamo, F. J., Lafuente-Monasterio, M. J., Singh, O. M., Rowland, P., Wiegand, R. C. & Wirth, D. F. (2014). J. Biol. Chem. 289, 17980–17995. [DOI] [PMC free article] [PubMed]
- Ruiz-Ramos, A., Lallous, N., Grande-García, A. & Ramón-Maiques, S. (2013). Acta Cryst. F69, 1425–1430. [DOI] [PMC free article] [PubMed]
- Stebbins, J. W., Robertson, D. E., Roberts, M. F., Stevens, R. C., Lipscomb, W. N. & Kantrowitz, E. R. (1992). Protein Sci. 1, 1435–1446. [DOI] [PMC free article] [PubMed]
- Stebbins, J. W., Xu, W. & Kantrowitz, E. R. (1989). Biochemistry, 28, 2592–2600. [DOI] [PubMed]
- Stevens, R. C., Chook, Y. M., Cho, C. Y., Lipscomb, W. N. & Kantrowitz, E. R. (1991). Protein Eng. 4, 391–408. [DOI] [PubMed]
- Sun, W., Tanaka, T. Q., Magle, C. T., Huang, W., Southall, N., Huang, R., Dehdashti, S. J., McKew, J. C., Williamson, K. C. & Zheng, W. (2014). Sci. Rep. 4, 3743. [DOI] [PMC free article] [PubMed]
- Tracy, S. M. & Sherman, I. W. (1972). J. Protozool. 19, 541–549. [DOI] [PubMed]
- Vaidya, A. B. & Mather, M. W. (2009). Annu. Rev. Microbiol. 63, 249–267. [DOI] [PubMed]
- Van Boxstael, S., Cunin, R., Khan, S. & Maes, D. (2003). J. Mol. Biol. 326, 203–216. [DOI] [PubMed]
- Van Dyke, K., Tremblay, G. C., Lantz, C. H. & Szustkiewicz, C. (1970). Am. J. Trop. Med. Hyg. 19, 202–208. [DOI] [PubMed]
- Walsh, C. J. & Sherman, I. W. (1968). J. Protozool. 15, 763–770. [DOI] [PubMed]
- White, N. J. (2008). Malar. J. 7, S8. [DOI] [PMC free article] [PubMed]
- World Health Organisation (2015). World Malaria Report 2015. Geneva: World Health Organisation. http://www.who.int/malaria/publications/world-malaria-report-2015/en/.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wu, Y., Sifri, C. D., Lei, H.-H., Su, X.-Z. & Wellems, T. E. (1995). Proc. Natl Acad. Sci. USA, 92, 973–977. [DOI] [PMC free article] [PubMed]
- Xu, M., Zhu, J., Diao, Y., Zhou, H., Ren, X., Sun, D., Huang, J., Han, D., Zhao, Z., Zhu, L., Xu, Y. & Li, H. (2013). J. Med. Chem. 56, 7911–7924. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: aspartate transcarbamoylase, 5ilq
Supplementary Table S1.. DOI: 10.1107/S2053230X16008475/rr5118sup1.pdf