

A V. cholerae MATE transporter was crystallized using the lipidic cubic phase (LCP) method. X-ray diffraction data sets were collected from single crystals obtained in a sandwich plate and a sitting-drop plate to resolutions of 2.5 and 2.2 Å, respectively.
Keywords: MATE transporter, multidrug resistance, lipidic cubic phase, LCP
Abstract
Multidrug and toxic compound extrusion (MATE) transporters, one of the multidrug exporter families, efflux xenobiotics towards the extracellular side of the membrane. Since MATE transporters expressed in bacterial pathogens contribute to multidrug resistance, they are important therapeutic targets. Here, a MATE-transporter homologue from Vibrio cholerae, VcmN, was overexpressed in Escherichia coli, purified and crystallized in lipidic cubic phase (LCP). X-ray diffraction data were collected to 2.5 Å resolution from a single crystal obtained in a sandwich plate. The crystal belonged to space group P212121, with unit-cell parameters a = 52.3, b = 93.7, c = 100.2 Å. As a result of further LCP crystallization trials, crystals of larger size were obtained using sitting-drop plates. X-ray diffraction data were collected to 2.2 Å resolution from a single crystal obtained in a sitting-drop plate. The crystal belonged to space group P212121, with unit-cell parameters a = 61.9, b = 91.8, c = 100.9 Å. The present work provides valuable insights into the atomic resolution structure determination of membrane transporters.
1. Introduction
The increase in multidrug-resistant pathogens that are difficult to treat using conventional antibiotics has become a global public health problem (Reimer et al., 2011 ▸; Miwanda et al., 2015 ▸). One of the multidrug-resistance mechanisms is to actively export the invading xenobiotics using multidrug-resistance (MDR) transporters (Blair et al., 2014 ▸). MDR transporters are classified into at least five families, including the ATP-binding cassette (ABC) superfamily, the resistance–nodulation–division (RND) family, the major facilitator superfamily (MFS), the small multidrug-resistance (SMR) family and the multidrug and toxic compound extrusion (MATE) family (Du et al., 2015 ▸). MATE transporters are secondary active transporters that use the electrochemical energy of an Na+ or H+ gradient across the membrane (Morita et al., 2000 ▸; He et al., 2004 ▸; Jin et al., 2014 ▸) and are conserved among bacteria, archaea and eukarya (Omote et al., 2006 ▸). They efflux chemically and structurally distinct organic cations, such as fluoroquinolones (e.g. norfloxacin) and fluorescent dyes (e.g. Hoechst 33342). Since MATE transporters from bacterial pathogens contribute to their multidrug resistance (Kaatz et al., 2005 ▸; McAleese et al., 2005 ▸), they are potential therapeutic targets. To date, the crystal structures of five prokaryotic MATE-transporter homologues have been reported (He et al., 2010 ▸; Lu, Symersky et al., 2013 ▸; Tanaka et al., 2013 ▸; Lu, Radchenko et al., 2013 ▸; Radchenko et al., 2015 ▸; Mousa et al., 2016 ▸). The structures have provided insight into substrate recognition and transport by MATE transporters. MATE transporters are composed of 12 transmembrane (TM) helices, which are divided into the N-lobe (TM1–TM6) and the C-lobe (TM7–TM12). A cleft is formed between the N- and C-lobes, and is divided into the N- and C-lobe cavities. This central cleft is opened towards the extracellular side in all of the reported structures. The structures of complexes with substrates demonstrated that the substrates bind to the N-lobe cavity (Tanaka et al., 2013 ▸; Lu, Radchenko et al., 2013 ▸). Furthermore, the inhibitor-bound structures revealed that inhibitors partially occupy the substrate-binding site, resulting in inhibition of the substrate-transport activity by MATE transporters (Tanaka et al., 2013 ▸; Radchenko et al., 2015 ▸). However, the binding sites of substrates and inhibitors are diverse among the MATE-transporter homologues, and the amino-acid residues involved in the recognition of substrates and inhibitors are not conserved. For the development of effective inhibitors against MATE transporters, structural analyses of MATE-transporter homologues from clinically relevant multidrug-resistant pathogens, such as Staphylococcus aureus and Vibrio cholerae, are needed. To date, six MATE-transporter homologues from V. cholerae have been identified (Brown et al., 1999 ▸; Huda et al., 2001 ▸, 2003 ▸; Begum et al., 2005 ▸); however, of these homologues, only medium-resolution structures of NorM-VC (∼3.65 Å) have been reported (He et al., 2010 ▸). Here, we performed a structural analysis of one of the V. cholerae MATE-transporter homologues, VcmN, which shares approximately 17% sequence identity with NorM-VC. We report the expression, purification, lipidic cubic phase (LCP) crystallization and X-ray diffraction analysis of VcmN. Moreover, we describe its co-crystallization with macrocyclic peptides and a substrate-soaking experiment with the aim of determinating complex structures. Since there is little literature on the practical LCP crystallization method for membrane transporters, our study provides valuable insights into crystallization techniques for the growth of crystals of membrane transporters that diffract to high resolution.
2. Materials and methods
2.1. Macromolecule production
The gene encoding full-length VcmN (UniProt ID C3LWQ2) was subcloned into a modified pET-28a vector. This modified pET-28a vector contains a C-terminal Tobacco etch virus (TEV) protease cleavage site (ENLYFQG) followed by a His6 tag. The ten C-terminal residues of full-length VcmN and the two vector-derived residues were truncated by a PCR-based method using PrimeSTAR Max DNA polymerase (Takara Bio). The resulting construct was designated VcmNΔC. The plasmid was introduced into Escherichia coli C41(DE3) Rosetta cells harbouring pRARE, which encodes the tRNAs for codons rarely used in E. coli. The typical purification method is described below. The transformed cells were grown in 2.5 l Luria–Bertani (LB) medium containing 30 µg ml−1 kanamycin at 310 K. When the absorbance at 600 nm (A 600) reached 0.5–0.8, expression was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the cells were grown for about 20 h at 293 K. The cells were centrifuged at 5000g for 10 min and disrupted by 2–3 passes at 103 MPa using a Microfluidizer (Microfluidics). After centrifugation to remove debris at 28 000g for 30 min, the supernatant was ultracentrifuged at 125 000g for 1 h to collect the membrane fraction. The membrane fraction was resuspended in 20 mM Tris–HCl pH 8.0, 300 mM NaCl and stored at 193 K until use. The membrane fraction was solubilized in 20 mM Tris–HCl pH 8.0, 300 mM NaCl, 20 mM imidazole, 1.5% n-dodecyl-β-d-maltoside (DDM) for 1 h at 277 K. After the removal of debris by ultracentrifugation at 125 000g for 30 min, the supernatant was mixed with 5 ml Ni–NTA resin (Qiagen) equilibrated with buffer A (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 0.1% DDM) containing 20 mM imidazole for about 1 h at 277 K. The mixture was loaded into an Econo-column (Bio-Rad) and the flowthrough fraction was discarded. The resin was washed with ten column volumes of buffer A containing 30 mM imidazole and the protein sample was eluted with buffer A containing 300 mM imidazole. To cleave the His6 tag, His-tagged TEV protease (produced in-house) was added to the eluted fraction in a 10:1(w:w) protein:TEV protease ratio. During the overnight cleavage reaction at 277 K, the solution was dialyzed against 20 mM Tris–HCl pH 8.0, 300 mM NaCl, 0.02% DDM. The solution was mixed with Ni–NTA resin again for 1 h at 277 K to remove the TEV protease. The flowthrough fraction was concentrated using an Amicon Ultra centrifugal filter (50 kDa molecular-weight cutoff, Millipore) and applied onto a Superdex 200 Increase (GE Healthcare) column equilibrated with 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.1% DDM. The peak fractions were concentrated to approximately 10 mg ml−1 using an Amicon Ultra filter (50 kDa molecular-weight cutoff, Millipore). To remove the Na+ ions, a portion of the sample was dialyzed against 20 mM Tris–HCl pH 8.0, 0.02% DDM. The purity of the protein sample was assessed by SDS–PAGE. The macromolecule-production information is summarized in Table 1 ▸.
Table 1. Macromolecule-production information.
Cloning artifacts are underlined. The truncated residues are in parentheses.
| Source organism | V. cholerae |
| DNA source | UniProt ID C3LWQ2 |
| Expression vector | Modified pET-28a |
| Expression host | E. coli strain C41(DE3) Rosetta |
| Complete amino-acid sequence of the construct produced | MAMQTSTSSLAKQLFQMTWPMLFGVLSLMSFQLVDSAFIGQLGVLPLAAQGFTMPIQMVIIGIQVGLGIATTAVISRAIGAGKTEYAKQLGGLVIVIGGIGVALIALVLYLLRQPLLGLLGAPETVFAIIDHYWLWWLASAWTGAMLYFYYSVCRANGNTLLPGTLMMVTSVLNLILDPIFIFTFDLGIDGAAIATIIAFGVGIAIVAPKVAQRQWTSYQWQDLNISQSLTALGHIMGPAMLSQLLPPLSSMFATKLLASFGTAAVAAWALGSRFEFFALVAVLAMTMSLPPMIGRMLGAKEITHIRQLVRIACQFVLGFQLLIALVTYVFATPLAELMTSETEVSQILNLHLVIVPISLGALGICMLMVSVANALGKSYVALTISALRLFAFYLPCLWLGAHFYGIEGLFIGALVGNIIAGWAAWLAYQKALR(QLEGAHHTSAPN)SENLYFQGQVDKLAAALEHHHHHH |
2.2. Crystallization
Previously, co-crystallization with macrocyclic peptides improved the quality of crystals of selenomethionine-labelled Pyrococcus furiosus MATE (PfMATE), thereby facilitating its structure determination. Moreover, the macrocyclic peptides showed inhibitory activity against PfMATE (Tanaka et al., 2013 ▸; Hipolito et al., 2013 ▸). Thus, to improve the quality of the VcmN crystals, an in vitro selection of macrocyclic peptides that bind to VcmN was also performed using the random nonstandard peptide integrated discovery (RaPID) system (Hipolito & Suga, 2012 ▸). Crystallization was performed using the LCP method (Caffrey & Cherezov, 2009 ▸), as the structure of PfMATE was also determined at high resolution using this crystallization method (Tanaka et al., 2013 ▸). The protein solution containing Na+ was mixed with 8 mM macrocyclic peptides dissolved in dimethyl sulfoxide in a 10:1(v:v) ratio, and the mixture was incubated for 1 h at 277 K. The sample was mixed with monoolein (Nu-Chek Prep) in a 2:3(w:w) protein:lipid ratio using coupled syringes. Drops of the mixture (50 nl) were dispensed onto a 96-well plastic sandwich plate (SWISSCI) and were overlaid with 600 nl reservoir solution using a Mosquito LCP crystallization robot (TTP Labtech). Initial crystallization screening was performed at 293 K using the MemMeso crystallization kit (Molecular Dimensions) and in-house-produced grid-screening crystallization kits as reservoir solutions. For crystallization optimization, StockOptions Salt and Additive Screen (Hampton Research) were added to the reservoir solutions, in addition to optimization of the pH and the concentrations of precipitants and salts. Furthermore, 40 nl portions of a mixture of protein samples without Na+ and monoolein in a 2:3(w:w) ratio were spotted on a 96-well sitting-drop plate (AS ONE) and overlaid with 800 nl reservoir solution. The obtained crystals were overlaid with 800 nl reservoir solution containing 10 mM Hoechst 33342 as a substrate (Begum et al., 2005 ▸) and were incubated at 293 K. The crystals were picked up using MicroMounts (MiTeGen) or LithoLoops (Protein Wave) and were flash-cooled in liquid nitrogen. Crystallization information is provided in Table 2 ▸.
Table 2. Crystallization.
| Form A crystal | Form B crystal | |
|---|---|---|
| Method | Lipidic cubic phase | Lipidic cubic phase |
| Plate type | 96-well plastic sandwich plate | 96-well sitting-drop plate |
| Temperature (K) | 293 | 293 |
| Protein concentration (mg ml−1) | 10 | 10 |
| Buffer composition of protein solution | 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.1% DDM | 20 mM Tris–HCl pH 8.0, 0.02% DDM |
| Co-crystallized compound (final concentration in mixture with protein solution) | 0.8 mM macrocyclic peptide | — |
| Composition of reservoir solution | 30% PEG 300, 100 mM sodium citrate pH 5.0, 100 mM ammonium fluoride | 28–33% PEG 500 DME, 100 mM Tris–HCl pH 7.5–8.0, 50–100 mM magnesium formate |
| Volume of LCP drop (nl) | 50 | 40 |
| Volume of reservoir (nl) | 600 | 800 |
| Soaked compound (final concentration in crystallization drop) | — | 5 mM Hoechst 33342 |
2.3. Data collection and processing
All X-ray diffraction data sets were collected by the helical data-collection method using a micro-focus beam at SPring-8 beamline BL32XU (Hirata et al., 2013 ▸). Data processing was performed using XDS (Kabsch, 2010 ▸), DIALS (Waterman et al., 2013 ▸) and AIMLESS (Evans & Murshudov, 2013 ▸). Molecular replacement was performed with Phaser (McCoy et al., 2007 ▸) using the structure of PfMATE (PDB entry 3vvn; Tanaka et al., 2013 ▸) as the search model. Model building and refinement were performed with Coot (Emsley & Cowtan, 2004 ▸; Emsley et al., 2010 ▸) and phenix.refine (Adams et al., 2010 ▸; Afonine et al., 2012 ▸), respectively. The data-collection statistics are summarized in Table 3 ▸.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Form A crystal | Form B crystal | |
|---|---|---|
| Diffraction source | BL32XU, SPring-8 | BL32XU, SPring-8 |
| Wavelength (Å) | 1.0 | 1.0 |
| Temperature (K) | 100 | 100 |
| Detector | Rayonix MX225HE | Rayonix MX225HS |
| Crystal-to-detector distance (mm) | 240 | 240 |
| Rotation range per image (°) | 1.0 | 1.0 |
| Total rotation range (°) | 180 | 178 |
| Exposure time per image (s) | 1.0 | 1.0 |
| Space group | P212121 | P212121 |
| a, b, c (Å) | 52.3, 93.7, 100.2 | 61.9, 91.8, 100.9 |
| Resolution range (Å) | 93.74–2.50 (2.60–2.50) | 45.91–2.20 (2.26–2.20) |
| Total No. of reflections | 118705 | 210722 |
| No. of unique reflections | 17737 | 29974 |
| Completeness (%) | 99.8 (98.5) | 99.8 (97.2) |
| Multiplicity | 6.7 (5.9) | 7.0 (5.0) |
| 〈I/σ(I)〉 | 5.9 (1.9) | 7.8 (1.2) |
| R meas | 0.252 (0.995) | 0.208 (1.611) |
| CC1/2 | 0.989 (0.635) | 0.995 (0.477) |
| Overall B factor from Wilson plot (Å2) | 31.7 | 24.3 |
3. Results and discussion
In general, the expression level and stability of the protein sample are important factors for structural analysis. We first overexpressed and purified full-length VcmN. However, the full-length VcmN partially precipitated during the concentration step. In addition, the size-exclusion chromatography (SEC) chromatogram peak was not monodisperse, thus indicating instability of full-length VcmN. To improve the stability, the C-terminal 12 residues of full-length VcmN were truncated as the C-terminal residues of VcmN were predicted to be disordered based on a DISOPRED2 analysis (Ward et al., 2004 ▸). During the purification of VcmNΔC, the loss of sample by aggregation was suppressed compared with that of the full-length protein, and the SEC chromatogram peak showed monodispersity. The final yield increased from 1 to 5 mg per 2.5 l culture on truncating the C-terminal 12 residues. The schematic diagram of the constructs, the SEC chromatograms and the Coomassie Brilliant Blue-stained gel are shown in Fig. 1 ▸.
Figure 1.

Protein preparation. (a) Schematic diagram of the VcmN crystallization construct. (b) Chromatograms of full-length VcmN and the VcmNΔC construct. (c) SDS–PAGE analysis with Coomassie Brilliant Blue staining. Lane 1, VcmNΔC; lane 2, molecular-weight markers (labelled in kDa).
We performed an LCP crystallization screening of VcmNΔC. Initially, tiny crystals of a mixture of VcmNΔC and the macrocyclic peptide (Fig. 2 ▸ a) were obtained under several conditions containing precipitants such as polyethylene glycol (PEG) 300 and PEG 400 using sandwich plates. As a result of optimization efforts, including the screening of additives using the StockOptions Salt and Additive Screen kits, rectangular prism-shaped crystals (form A) were obtained with a reservoir solution composed of 30% PEG 300, 100 mM sodium citrate pH 5.0, 100 mM ammonium fluoride (Fig. 2 ▸ b). Crystals appeared in a day and grew to approximate dimensions of 10 × 10 × 25 µm in a week. We also attempted crystallization of VcmNΔC without the macrocyclic peptides. In contrast to the crystallization of PfMATE, we unfortunately found that the peptides did not affect the quality of the VcmNΔC crystals. Next, crystallization screening was performed using sitting-drop plates. In contrast to crystallization in sandwich plates, crystallization in sitting-drop plates is suitable for substrate-soaking experiments to determine the structures of complexes with substrates. Rod-shaped crystals (form B) were obtained in a reservoir composed of 30% PEG 500 dimethyl ether (DME), 100 mM Tris–HCl pH 8.0, 100 mM magnesium formate. The approximate dimensions of the form B crystals were 10 × 10 × 50 µm. The sizes of the crystals obtained in the sitting-drop plates were usually larger than those produced in the sandwich plates. Finally, as a result of optimization of the salt concentration and the pH of the condition, crystals with approximate dimensions of 10 × 10 × 100 µm were obtained in reservoir solutions composed of 28–33% PEG 500 DME, 100 mM Tris–HCl pH 7.5, 50 mM magnesium formate (Fig. 2 ▸ c). For the substrate-soaking experiments, an 800 nl portion of reservoir solution containing 10 mM Hoechst 33342 was added to drops containing crystals. The protein crystals were incubated for about one month at 293 K and then cooled in liquid nitrogen.
Figure 2.
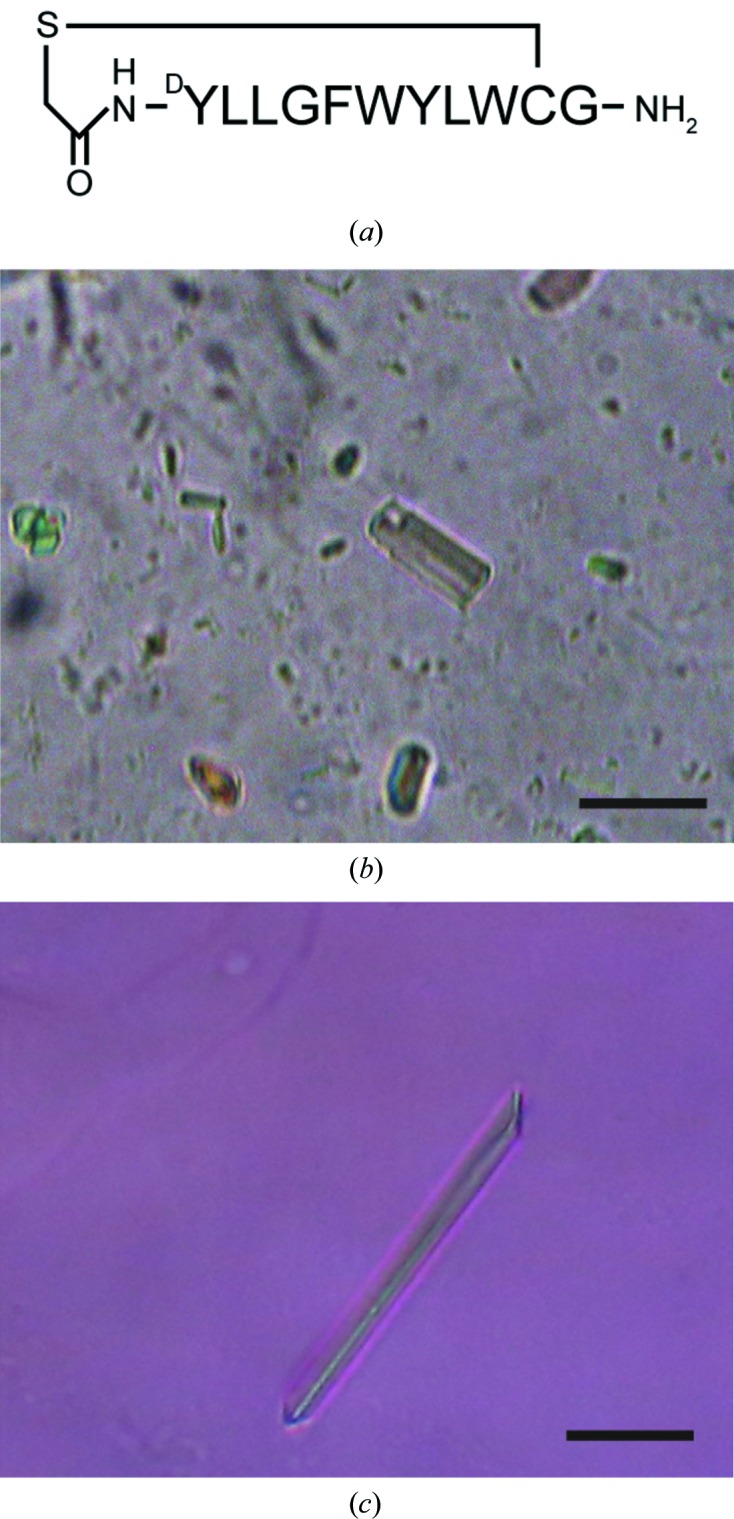
Crystals of VcmNΔC. (a) Schematic representation of the macrocyclic peptide. (b) Form A crystal. (c) Form B crystal. The scale bars represent 30 µm.
A form A single crystal diffracted X-rays to 2.5 Å resolution (Fig. 3 ▸ a) and belonged to space group P212121, with unit-cell parameters a = 52.3, b = 93.7, c = 100.2 Å. The calculated Matthews coefficient is 2.58 Å3 Da−1 with 52.4% solvent content, assuming the presence of one molecule in the asymmetric unit. The form B single crystal diffracted X-rays to 2.2 Å resolution (Fig. 3 ▸ b) and belonged to space group P212121, with unit-cell parameters a = 61.9, b = 91.8, c = 100.9 Å. The calculated Matthews coefficient is 3.01 Å3 Da−1 with 59.2% solvent content, assuming the presence of one molecule in the asymmetric unit. Molecular replacement was performed using the structure of PfMATE as the search model and an interpretable electron-density map was obtained. However, we have not yet observed an electron-density peak corresponding to either the macrocyclic peptide or Hoechst 33342. Model building and structure refinement are under way.
Figure 3.

X-ray diffraction images of the form A (a) and form B (b) crystals. The rings indicate 2.5 Å (a) and 2.2 Å (b) resolution, respectively.
Acknowledgments
We thank T. Nishizawa, H. Nishimasu, T. Nakane and M. Fukuda for useful suggestions and the beamline staff members at BL32XU of SPring-8 for technical support during data collection. The X-ray diffraction experiments were performed at BL32XU of SPring-8 (proposal Nos. 2012B1146, 2013A1128, 2013A1168, 2014A1061 and 2014A1116) with the approval of RIKEN. This work was supported by the Platform for Drug Discovery, Informatics and Structural Life Science, funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), by a Grant-in-Aid for Scientific Research (S) (24227004) and a Grant-in-Aid for Scientific Research (B) (25291011) from the Japan Society for the Promotion of Science (JSPS) to ON and RI, respectively, and by a Grant-in-Aid for JSPS Fellows.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Begum, A., Rahman, M. M., Ogawa, W., Mizushima, T., Kuroda, T. & Tsuchiya, T. (2005). Microbiol. Immunol. 49, 949–957. [DOI] [PubMed]
- Blair, J. M. A., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. V. (2014). Nature Rev. Microbiol. 13, 42–51. [DOI] [PubMed]
- Brown, M. H., Paulsen, I. T. & Skurray, R. A. (1999). Mol. Microbiol. 31, 394–395. [DOI] [PubMed]
- Caffrey, M. & Cherezov, V. (2009). Nature Protoc. 4, 706–731. [DOI] [PMC free article] [PubMed]
- Du, D., van Veen, H. W., Murakami, S., Pos, K. M. & Luisi, B. F. (2015). Curr. Opin. Struct. Biol. 33, 76–91. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- He, G., Kuroda, T., Mima, T., Morita, Y., Mizushima, T. & Tsuchiya, T. (2004). J. Bacteriol. 186, 262–265. [DOI] [PMC free article] [PubMed]
- He, X., Szewczyk, P., Karyakin, A., Evin, M., Hong, W.-X., Zhang, Q. & Chang, G. (2010). Nature (London), 467, 991–994. [DOI] [PMC free article] [PubMed]
- Hipolito, C. J. & Suga, H. (2012). Curr. Opin. Chem. Biol. 16, 196–203. [DOI] [PubMed]
- Hipolito, C. J., Tanaka, Y., Katoh, T., Nureki, O. & Suga, H. (2013). Molecules, 18, 10514–10530. [DOI] [PMC free article] [PubMed]
- Hirata, K., Kawano, Y., Ueno, G., Hashimoto, K., Murakami, H., Hasegawa, K., Hikima, T., Kumasaka, T. & Yamamoto, M. (2013). J. Phys. Conf. Ser. 425, 012002.
- Huda, M. N., Chen, J., Morita, Y., Kuroda, T., Mizushima, T. & Tsuchiya, T. (2003). Microbiol. Immunol. 47, 419–427. [DOI] [PubMed]
- Huda, M. N., Morita, Y., Kuroda, T., Mizushima, T. & Tsuchiya, T. (2001). FEMS Microbiol. Lett. 203, 235–239. [DOI] [PubMed]
- Jin, Y., Nair, A. & van Veen, H. W. (2014). J. Biol. Chem. 289, 14624–14632. [DOI] [PMC free article] [PubMed]
- Kaatz, G. W., McAleese, F. & Seo, S. M. (2005). Antimicrob. Agents Chemother. 49, 1857–1864. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Lu, M., Radchenko, M., Symersky, J., Nie, R. & Guo, Y. (2013). Nature Struct. Mol. Biol. 20, 1310–1317. [DOI] [PMC free article] [PubMed]
- Lu, M., Symersky, J., Radchenko, M., Koide, A., Guo, Y., Nie, R. & Koide, S. (2013). Proc. Natl Acad. Sci. USA, 110, 2099–2104. [DOI] [PMC free article] [PubMed]
- McAleese, F., Petersen, P., Ruzin, A., Dunman, P. M., Murphy, E., Projan, S. J. & Bradford, P. A. (2005). Antimicrob. Agents Chemother. 49, 1865–1871. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miwanda, B., Moore, S., Muyembe, J., Nguefack-Tsague, G., Kabangwa, I. K., Ndjakani, D. Y., Mutreja, A., Thomson, N., Thefenne, H., Garnotel, E., Tshapenda, G., Kakongo, D. K., Kalambayi, G. & Piarroux, R. (2015). Emerg. Infect. Dis. 21, 847–851. [DOI] [PMC free article] [PubMed]
- Morita, Y., Kataoka, A., Shiota, S., Mizushima, T. & Tsuchiya, T. (2000). J. Bacteriol. 182, 6694–6697. [DOI] [PMC free article] [PubMed]
- Mousa, J. J., Yang, Y., Tomkovich, S., Shima, A., Newsome, R. C., Tripathi, P., Oswald, E., Bruner, S. D. & Jobin, C. (2016). Nature Microbiol. 1, 15009. [DOI] [PMC free article] [PubMed]
- Omote, H., Hiasa, M., Matsumoto, T., Otsuka, M. & Moriyama, Y. (2006). Trends Pharmacol. Sci. 27, 587–593. [DOI] [PubMed]
- Radchenko, M., Symersky, J., Nie, R. & Lu, M. (2015). Nature Commun. 6, 7995. [DOI] [PMC free article] [PubMed]
- Reimer, A. et al. (2011). Emerg. Infect. Dis. 17, 2151–2154. [DOI] [PMC free article] [PubMed]
- Tanaka, Y., Hipolito, C. J., Maturana, A. D., Ito, K., Kuroda, T., Higuchi, T., Katoh, T., Kato, H. E., Hattori, M., Kumazaki, K., Tsukazaki, T., Ishitani, R., Suga, H. & Nureki, O. (2013). Nature (London), 496, 247–251. [DOI] [PubMed]
- Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F. & Jones, D. T. (2004). J. Mol. Biol. 337, 635–645. [DOI] [PubMed]
- Waterman, D. G., Winter, G., Parkhurst, J. M., Fuentes-Montero, L., Hattne, J., Brewster, A., Sauter, N. K. & Evans, G. (2013). CCP4 Newsl. Protein Crystallogr. 49, 16–19.