

Abstract
In this study, hydrophilic and hydrolytically degradable poly (ethylene glycol) (PEG) hydrogels were formed via Michael-type addition and employed for sustained delivery of a monoclonal antibody against the protective antigen of anthrax. Taking advantage of the PEG-induced precipitation of the antibody, burst release from the matrix was avoided. These hydrogels were able to release active antibodies in a controlled manner from 14 days to as long as 56 days in vitro by varying the polymer architectures and molecular weights of the precursors. Analysis of the secondary and tertiary structure and the in vitro activity of the released antibody showed that the encapsulation and release did not affect the protein conformation or functionality. The results suggest the promise for developing PEG-based carriers for sustained release of therapeutic antibodies against toxins in various applications.
Keywords: PEG hydrogel, controlled release, anthrax, monoclonal antibody, protective antigen
INTRODUCTION
Anthrax is caused by Bacillus anthracis, a Gram-positive, rod-shaped, spore-forming bacterium that primarily affects livestock but can spread to humans.1,2 Due to the ability of the pathogen to form endospores that can be easily concealed, transported, and released, B. anthracis poses a great threat as a bioterrorism agent, highlighted by the anthrax postal attack in 2001.3,4 The pathogenesis of B. anthracis is mediated by a tripartite toxin. This exotoxin consists of protective antigen (PA) and two enzymatically active proteins: lethal factor (LF) and edema factor (EF). PA functions as a cell-binding receptor for LF and EF to form lethal toxin (LeTx) and edema toxin, respectively, making it an ideal target for vaccine and countermeasure development. The development of biotechnology and genetic engineering methodologies has enabled monoclonal antibody (mAb) therapy to be developed as an effective countermeasure for protection against anthrax.5,6 The utilization of mAbs that target specific cells or proteins permits anthrax toxin neutralization by a variety of mechanisms, including neutralizing pathogen growth, limiting its spread from infected to adjacent cells, or by inhibiting the toxin’s biological activity.7 During the past 10 years, several human antibodies against anthrax PA have been demonstrated to provide passive protection in variety of animal models including rats, rabbits, guinea pigs and non-human primates.8–10 One such mAb was developed by Fraunhofer USA Center for Molecular Biotechnology (FhCMB) and shown to provide full protection against an inhalation anthrax spore challenge in non-human primates.11,12 FhCMB engineered this mAb in their plant-based production platform to be a non-glycosylated (NG) version of a mAb against PA, termed PANG. This NG variant was shown to have superior half-life and protective efficacy compared to a glycosylated counterpart. Therefore, PANG was selected as the mAb of interest for the work described below.
Similar to most protein therapeutics, antibodies can suffer from poor stability due to chemical degradation as well as physical aggregation.13 Also, repetitive dosing may be required to achieve a therapeutic effect, which compromises patient’s comfort, convenience, and compliance.14–16 Water-swollen polymeric hydrogels have been extensively investigated as vehicles for the delivery of a variety of small and large molecules, including proteins.17–21 By encapsulation in the network, proteins can be protected against degradation and released from the hydrogel matrix in a controlled manner over an extended period of time, either in blood circulation or in the surrounding tissues.22–24 Degradable hydrogels are desirable for protein delivery, since the release rate of the therapeutic proteins can be manipulated by the degradation of the matrix, and clearance of the device from the body can be achieved when the release is completed.25–28 Recently, several hydrogels based on synthetic polymers, natural polymers, and peptides have been formulated to offer local and sustained release of antibodies including immunoglobulin (IgG), Herceptin (a breast cancer antibody), and Bevacizumab (an anti-VEGF antibody), with enhanced therapeutic efficacy that reduces the number of injections and lowers the administered dose.29–34
In this study, we present hydrolytically degradable poly (ethylene glycol) (PEG) hydrogels as a reservoir system for the controlled delivery of PANG, an anthrax LeTx neutralizing antibody. Degradable PEG hydrogels were formed via Michael-type addition using multi-arm PEG thiols (-SH) and linear PEG acrylates (-Ac). These hydrogels were rendered hydrolytically degradable via the acrylate ester linkages (see polymer structures in Scheme 1). We characterized the swelling properties of these hydrogels and demonstrated that the release rate of PANG can be adjusted by varying the molecular structures of the hydrogel precursors. Post-release and in-gel characterizations including polyacrylamide gel electrophoresis (SDS-PAGE), size-exclusion chromatography (SEC), circular dichroism (CD), and fluorescence indicated that PANG remained stable when encapsulated and released from the gel. A toxin neutralization assay (TNA) showed that the released PANG remained biologically active and exhibited toxin-neutralizing activity in a concentration-dependent manner.
SCHEME 1.
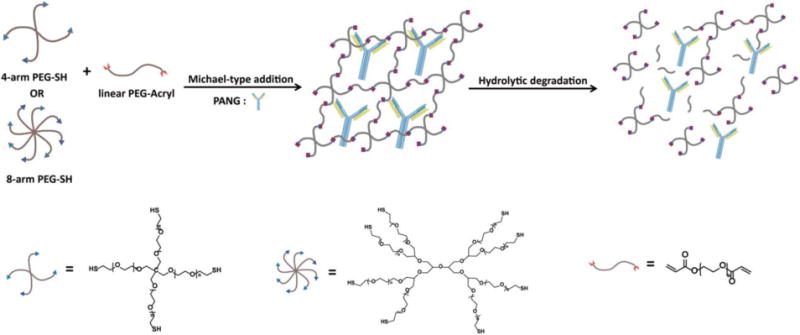
Formation and degradation of PANG-loaded PEG hydrogels.
MATERIALS AND METHODS
Materials
Four-arm, thiol-functionalized PEG (PEG-4SH, Mn = 5000; 10,000; and 20,000 g/mol), eight-arm, thiol-functionalized PEG (PEG-8SH, Mn = 10,000 and 20,000 g/mol) and linear diacrylated PEG (PEG-2Ac, Mn = 2000, 3500, 5000, and 7500 g/mol) were purchased from JenKem Technology USA Inc. (Allen, TX). Low molecular weight, diacrylated PEG (PEG-2Ac, Mn = 700) was purchased from Sigma-Aldrich (St. Louis, MO). The non-glycosylated mAb (PANG) was produced by Fraunhofer USA (Newark, DE). All other reagents and materials were purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise noted.
Purification of plant-produced mAb PANG
Engineering, expression, and purification of PANG followed methods previously described with minor modifications.12 Briefly, Agrobacterium tumefaciens transformed with the plasmid pGR-D4, carrying either the heavy chain or light chain, were used for agroinfiltration of Nicotiana benthamiana. Infiltrated plant material was blended in 1 volume of Tris based extraction buffer and centrifuged at 16,000 × g for 15 min at 4°C. The supernatant was collected and the pH adjusted to 5.3, mixed for 15 min at room temperature, and centrifuged a second time. The pH of the supernatant was then raised to 7.5, followed by filtration through a 0.45/0.2 μm Sartopore filter and loading onto a 10 mL HiTrap MAb Select resin (GE Healthcare, Piscataway, NJ). The column was washed with 15 column volumes of 50 mM Tris, pH7.5 buffer before elution with 100 mM citrate buffer (pH 3.0). Eluted PANG was buffered to neutral with pH 9 Tris buffer. The protein was concentrated using a Centricon Plus–70 (3000 MWCO) and dialyzed into PBS.
Formation of degradable PEG hydrogels
The hydrogels were formed by a Michael-type addition between multi-arm PEG-SH and hydrolytically degradable crosslinkers of linear PEG-diacrylate. The molar ratio of thiols/acrylates was 1:1 for all hydrogels. Each polymer precursor was dissolved in a phosphate-buffered saline (PBS) solution (pH = 7.4). The identities of the polymer precursors for various hydrogel formulations are summarized in Table I. The convention for naming the hydrogel samples is based on the number of arms and molecular weight of the multi-functional PEG and the molecular weight of the linear PEG “crosslinker.” For example, the notation “8-arm 10/2K” indicates a hydrogel comprising a 10 kDa, 8-arm PEG-SH mixed with a 2 kDa linear PEG-diacrylate. Immediately after mixing, the solution was quickly vortexed, and then transferred to a scintillation vial and allowed to gel at 37°C. Gelation occurred in several minutes, but the hydrogels were left in the incubator overnight to achieve maximum crosslinking. For protein encapsulation, a solution of PANG (15.7 mg/mL) was used to dissolve the polymer precursor to achieve a final protein concentration of 5 mg/mL or 2.5 mg/mL.
TABLE I.
Formulations of Different PEG Hydrogels
| Sample | Polymers employed | Polymer concentration (wt %) | PANG loading concentration (mg/mL) |
|---|---|---|---|
| 4-arm 10/5K | 10K PEG-4SH/5K PEG-2Ac | 10, 20, 30 | 5, 2.5 |
| 4-arm 5/2K | 5K PEG-4SH/2K PEG-2Ac | 20 | 2.5 |
| 4-arm 5/3.5K | 5K PEG-4SH/3.5K PEG-2Ac | 20 | 2.5 |
| 8-arm 20/5K | 20K PEG-8SH/5K PEG-2Ac | 20 | 2.5 |
| 8-arm 10/2K | 10K PEG-8SH/2K PEG-2Ac | 20 | 2.5 |
| 8-arm 10K/700 | 10K PEG-8SH/700 PEG-2Ac | 20 | 2.5 |
Rheological characterization of PEG hydrogels
Oscillatory rheology experiments conducted on a stress-controlled AR-G2 rheometer (TA Instruments, New Castle, DE) were used to characterize the gelation time of the PEG hydrogels. Experiments were conducted at 37°C using a 20 mm-diameter, cone and plate geometry with a 25 lm gap distance, with oscillatory time, frequency, and strain sweeps performed. Strain sweeps were performed on samples from 0.1% to a maximum strain of 1000% to determine the limit of the linear viscoelastic region. Dynamic oscillatory time sweeps were performed to monitor the in situ gelation of different PEG hydrogel solutions at angular frequencies of 6 rad/s and 1% strain chosen from the linear viscoelastic region. The PEG-SH and PEG-Ac precursors were dissolved in PBS separately with a final polymer concentration of 20 wt %. The precursors were then combined, vortexed, and loaded onto the rheometer stage. The temporal evolution of G′ (storage modulus) and G″ (loss modulus) was recorded throughout the crosslinking experiments.
Swelling experiments
Hydrogels (100 μL) were incubated at 37°C in water. Hydrogel samples were collected at regular intervals of swelling, excess water removed by blotting, and their mass after swelling was measured. The hydrogels were then dried under vacuum overnight, and their dry mass was measured. The equilibrium mass swelling ratio q was experimentally obtained by the following equation:
where Wswollen and Wdry are the weights of the polymer at swollen and dried states, respectively.
In vitro protein release experiments
The protein release experiments were performed at 37°C in a glass vial with PBS buffer. Hydrogels (400 μL) were placed on the bottom of the vial and 5 mL of PBS was added over the gel. One mL of PBS was sampled from the vial at predetermined time points and replaced with 1 mL of fresh PBS. The final time point of the release experiments was defined as the time point when the buffer solutions above the gels became too viscous for sampling (which was prior to 100% hydrogel degradation). The resulting release samples were distributed into several aliquots and stored at −80°C to avoid potential damage to the PANG before characterization. Samples were reserved for determination of concentration and determination of cumulative release. (Samples were also reserved for characterization of protein conformation and activity; see below.) Protein samples were concentrated by centrifugation using Amicon Ultra-0.5 mL Centrifugal Filters (3 kDa, Billerica, MA), and protein concentration was characterized via UV–Vis spectrometry with detection at 280 nm. Protein stability was confirmed by SDS-PAGE electrophoresis.
The cumulative protein release (given as a percentage of encapsulated protein, (Pr)) was calculated using the following equation:
Where mPANG represents the amount of protein encapsulated in the hydrogel, V0 is the whole volume of the release media (5 mL), Ve is the volume of each sample that is being taken out at each time point (1 mL), and Ci represents the concentration of the PANG protein measured by UV–Vis in the ith sample (Cn represents the nth sample).
In-gel and post-release characterization
Size-exclusion chromatography
Size-exclusion chromatography (SEC) studies were performed on an Agilent 1260 HPLC with PANG resolved over a ZENIX SEC-300 7.8 × 300 mm column (Sepax Technologies, Newark, DE, USA) with PBS as the mobile phase. All samples were analyzed at a flow rate of 1.0 mL per minute, with UV detection at 280 nm.
Circular dichroism spectroscopy
Circular dichroic spectra were recorded on a Jasco J-810 spectropolarimeter (Jasco Inc, Easton, MD) equipped with a Jasco PTC-424S temperature controller. Background scans of PBS were recorded and subtracted automatically from the sample scans. For in-gel experiments, the samples (50 μL) were loaded into a 0.20 mm path length demountable cuvette. For solution-phase experiments, the samples were loaded into a 1 mm path length quartz cuvette. Data points for the wavelength-dependent CD spectra were recorded at every nanometer with a 1 nm bandwidth and an averaging time of 10s for each data point from 190 to 250 nm, at room temperature. The mean residue ellipticity [θ]MRE (degrees cm2 dmol−1), was calculated via use of the concentration, molecular weight of the samples, number of residues, and cell path length.
Fluorescence emission spectroscopy
Fluorescence emission of released PANG and PANG control was measured by using a HORIBA Jobin Yvon SPEX FluoroMax-4 spectrofluorometer (HORIBA Scientific, Edison, NJ) at room temperature with quartz cuvettes of 1 cm path length. Emission spectra were recorded between 310 and 400 nm after excitation at 300 nm. In-gel experiments were carried out using a QuantaMaster spectrofluorometer (Photon Technologies International, Edison, NJ) at room temperature with quartz cuvettes of 1.0 mm path length. Emission spectra were recorded between 310 nm and 400 nm on excitation at 295 nm. The excitation and emission slit widths were set at 5.0 and 2.5 nm, respectively. All spectra were corrected for the background emission of PBS.
Anthrax lethal toxin neutralization assay
Samples collected after release were characterized for toxin-binding activity via an anthrax LeTx neutralization assay that was performed using decreasing amounts of the released samples. Specifically, the biological activity of PANG was measured using a LeTx neutralization assay as described previously with modifications.35 J774A.1 (ATCC TIB-67, Manassas, VA) cells were plated in 96-well flat-bottomed tissue culture plates at 2.5 × 104 cells/well in 50 μL and incubated for 16–19 h at 37°C with 5% CO2. Test samples were diluted and titrated on the plated cells in the presence or absence of LeTx (List Biological Laboratories, Campbell, CA). After a 4 h incubation at 37°C with 5% CO2, cell viability was assessed by the addition of WST-1 (Roche Applied Sciences, Indianapolis, IN), a proliferation reagent, followed by a spectrophotometric measurement at 450 nm. A four-parameter logistic-log regression model was used to analyze data plotted as OD versus the reciprocal of the sample dilution. The inflection point for each curve from this model is reported as the dilution of the sample that provides 50% inhibition of LeTx; termed effective dilution 50 (ED50).
RESULTS
Hydrogel compositions and formation
The Michael-type addition reaction has long been used as a versatile crosslinking strategy for the development of injectable biomaterials, owing to its rapid kinetics and mild reaction conditions.36 Hydrogels were formed in situ from multi-arm PEG-SH precursors with linear PEG diacrylate cross-linkers via Michael-type addition (Scheme 1) in PBS buffer. Gelation occurred within 1–3.5 min depending on the molecular structures of the precursors, as characterized by oscillatory rheology time sweeps (Fig. 1 and Supporting Information Fig. S1). Figure 1 shows the time sweep profile of the evolution of G′ and G″ for the 20 wt % 8-arm 10/2K hydrogel. (The convention 8-arm 10/2K indicates a hydrogel comprising a 10 kDa, 8-arm PEG-SH mixed with a 2 kDa linear PEG-diacrylate; this convention is used throughout the manuscript to indicate the compositions of the various hydrogels studied.) A rapid increase of G′ was observed after mixing, with a crossover point (where G′ becomes larger than G″) at approximately 2 min.37 Although the apparent gelation times estimated by a tube inversion method were greater (ca. 2–5 min) compared to those observed in the rheological profiles, they were proportionate for all PEG hydrogels studied (i.e., longer inversion times correlated with longer rheological crossover times). The composition of hydrogels was varied by using polymers with different functionalities, molecular weights, and concentrations (Table I), to tune the rate of release of the antibody.
FIGURE 1.
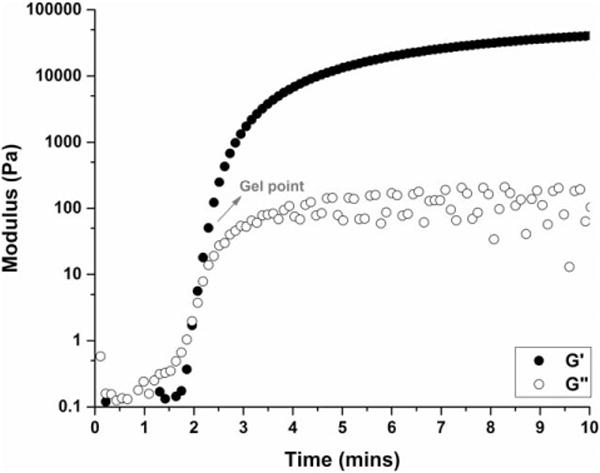
In situ gelation of 20 wt % 8-arm 10/2K hydrogel, as characterized by oscillatory rheology. The evolution of G′ (storage modulus) and G″ (loss modulus) was monitored as a function of time.
Swelling experiments
Swelling is a critical parameter for hydrogels employed in biomedical and pharmaceutical applications as the equilibrium swelling ratio is inversely correlated with the hydrogel crosslinking density, which in turn influences solute diffusion in and release from the hydrogel.38 In Figure 2, the relationship between network composition and hydrogel crosslinking density was explored by evaluating the equilibrium swelling ratio. The data in Figure 2(a) show the equilibrium swelling ratios (q) of 4-arm 10/5K gels at various polymer concentrations. It was observed that the q value of 4-arm 10/5K hydrogels decreased from 13.8 ± 0.5 to 11.8 ± 0.7 and then further to 8.8 ± 0.2 as the precursor concentration increased from 10 wt % to 20 wt % to 30 wt %, which can be attributed to the lower degrees of intramolecular reactions at higher monomer concentrations in step-growth polymerizations, resulting in an increased overall crosslinking density.39–42
FIGURE 2.
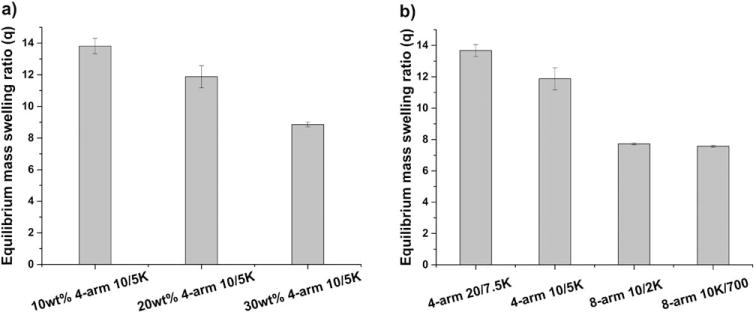
Equilibrium swelling properties of various PEG hydrogels. (a) 4-arm 10/5K gels at different concentration; (b) 20 wt % hydrogels prepared from polymer precursors with different functionalities and molecular weights.
The equilibrium swelling ratios (q) of hydrogels synthesized from precursors with different functionalities and molecular weights were also investigated. The data in Figure 2(b) demonstrate that hydrogels polymerized from 4-arm PEG-SH precursors have significantly higher swelling ratios than those of the hydrogels polymerized from 8-arm PEG-SH precursors (ca. 12 vs. 7.5), as expected given the increased crosslinking density of the 8-arm precursors. The data in Figure 2(b) also confirm that the equilibrium swelling ratio of the 4-arm 20/7.5K hydrogel is greater than that of the 4-arm 10/5K hydrogel (13.6 vs. 11.8), consistent with the increased molecular weight between crosslinks (MC) for the 4-arm 20/7.5K hydrogels and as would be expected according to Flory-Rehner theory.41,43 The changes in the hydrogels formed from 8-arm precursors are not as significant as those for the hydrogels formed from 4-arm precursors, as the extent of crosslinking for the 8-arm hydrogels is already high. Comparable swelling ratio data from PEG hydrogels with similar architectures were reported by Metters and Hubbell,39 in which such dependence of swelling ratio on precursor concentration, functionality, and molecular weight was also observed. These results confirm that the network density and physical swelling of these hydrogels can be easily tuned, which provides insights in the design of hydrogels that meet the performance criteria demanded by a specific biomedical application.
In vitro studies of PANG release from PEG-based hydrogels
The PEG hydrogels formed by Michael-type addition were employed as the delivery vehicles for PANG, a plant-produced non-glycosylated mAb against the PA of anthrax, which is able to neutralize anthrax LeTx activity in vivo and in vitro.12 The release of PANG from various 4-arm 10/5K gels (Supporting Information Fig. S2) was measured every 24 h for the first 10 days and then monitored every week until the buffer solutions above the gels became too viscous for sampling (as a result of matrix degradation). The 20 wt % hydrogel formulation was chosen for expanded studies of PANG release, given the steady release kinetics of PANG and the injection-friendly viscosity of precursor solutions. Interestingly, precipitation of the antibody was observed within most of the hydrogel formulations, consistent with previously observed PEG-induced protein precipitation44 that is generally attributed to depletion effects. The exclusion of PEG chains from the solvent space between protein molecules (as a result of entropic considerations) results in osmotically driven attractive forces between proteins.45 Therefore, a lower initial loading concentration of PANG (2.5 mg/mL) was adopted in these studies in order to minimize the influence of protein precipitation on the release properties of different hydrogel compositions.
A series of 20 wt % PEG hydrogels prepared from 4-arm and 8-arm PEGs with various linear PEG crosslinkers were targeted to manipulate antibody delivery rates; results are presented in Figure 3. Controlled release of the antibody without initial burst was observed from most of these hydrogels. The timespan of the release of the antibody varied from 14 days up to 56 days depending on the polymer architectures and molecular weights of the precursors, which is consistent with results reported in similar systems for protein release.22,40,46 For 4-arm 10/5K hydrogels, the antibody was released in a sustained fashion for approximately two weeks, with approximately 60% of PANG released at day 14. For 8-arm 20/5K hydrogels, approximately 90% of the antibody was released with zero-order kinetics over 28 days, with release of 3–4% of the antibody each day. For 8-arm 10/2K and 10K/700 hydrogels, following a slow initial release of approximately 10% of the antibody for the first 14 days, an increased rate of antibody release was observed, achieving a release of approximately 95% at day 35 and 85% at day 56, respectively.
FIGURE 3.
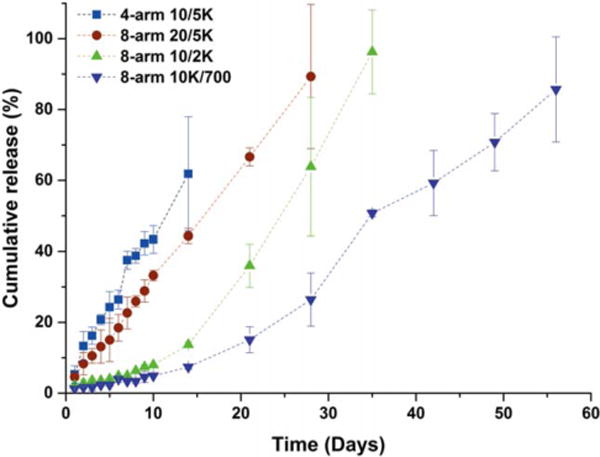
In vitro cumulative release of PANG from various PEG hydrogels at a precursor concentration of 20 wt %, with an initial loading concentration of 2.5 mg/mL, as a function of time. The release of protein was monitored by measuring the absorbance, at 280 nm, of the buffer solution above the gel at the indicated time intervals.
In agreement with previous swelling experiments, release of PANG from 8-arm hydrogels was slower than that from 4-arm hydrogels due to the increase in crosslinking density. While approximately 60% of PANG was released on day 14 from the 4-arm 10/5K hydrogels, the amount of PANG released at the same time from 8-arm hydrogels was only 44%, 13% and 7% for the 8-arm 20/5K, 10/2K and 10K/700 hydrogels, respectively, and, the release of PANG could be extended from 28 days to 56 days. These observed trends are in accordance with theory47 as well as with other existing reports.39,43,48. However, the release kinetics of PANG were unexpectedly similar for almost all of the 4-arm hydrogels, despite the variation in the molecular weight of the precursors (Supporting Information Fig. S3). During the first week of the experiment, the cumulative release curves of 4-arm 5/2K, 5/3.5K and 10/5K almost overlap, with approximately 43%, 38% and 37% of PANG released at day 7, respectively. This discrepancy might be explained by the effect of the PEG molecular weight on the precipitation of PANG. PEGs with lower molecular weights produce smaller depletion interactions between protein molecules, owing to the reduction in entropic driving force for the smaller macromolecules.49,50 The solubility of PANG thus would be expected to increase as the molecular weight of the linear PEG decreases, which would facilitate the release of PANG from the 4-arm 5/2K and 5/3.5K hydrogels and might subsequently offset the expected reduction in release rate from the gels with decreased MC.
In-gel and post-release characterization of PANG
To characterize PANG after its release from the hydrogels, SEC and SDS-PAGE were used to examine aggregation and possible degradation of the released protein. To obtain further insight into the conformational properties of PANG during encapsulation and after release, CD and fluorescence spectroscopy in both hydrogel and solution were used to examine the secondary and tertiary structures of the protein, respectively.
For characterization of protein stability, samples of PANG released from 8-arm 10/2K and 10K/700 hydrogels were selected as a more stringent test owing to the longer duration of retention of PANG in these networks. PANG samples collected after 28 days of release were examined by SEC, as shown in Figure 4. Peaks of monomeric PANG, from all the samples, were observed at an elution volume of 7.8 mL. A soluble PANG control incubated at 37°C (without hydrogel) showed a similar elution profile to that observed for the PANG solution stored at −80°C, indicating that PANG was stable over the test period. Quantitative analysis of the amount of monomeric PANG in these samples is also shown in Table II. The PANG released from both type of hydrogels contained approximately 90–92% monomer, which is very close to that of a control solution of PANG dissolved directly (ca. 92–97% monomer), indicating that PANG released from both types of hydrogels was mainly in a monomeric state and showed no significant aggregation. In a similar report,40 Leach and coworkers also confirmed that bovine serum albumin (BSA) was mostly maintained as monomer (>90%) after its release from PEG hydrogels. The small peak at about 9.5 mL in the elution profile of the PANG released from the 10/2K hydrogel is possibly due to the presence of PEG fragments caused by hydrogel degradation [visible in the UV owing to the ester linkage at the termini (Supporting Information Fig. S4)]. The lack of these fragments from the 10/700 hydrogel is likely a result of the slower rate of degradation of the 10/700 hydrogel.
FIGURE 4.
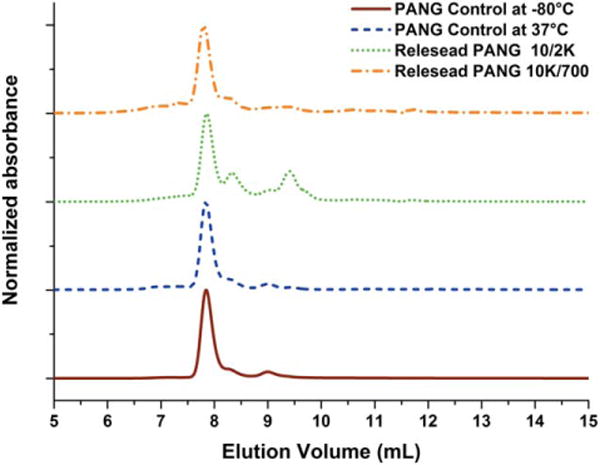
Normalized SEC trace of PANG controls and PANG released from the hydrogels after 28 days in PBS. Elution was monitored by UV absorbance at 280 nm.
TABLE II.
SEC Analysis of the Physical State of Released PANG
| Sample type | Time (day) | % monomer |
|---|---|---|
| PANG control at −80°C | 97.7% | |
| PANG control at 37°C | 28 | 91.5% |
| PANG released from 10/2K gel | 28 | 92.0% |
| PANG released from 10K/700 gel | 28 | 89.3% |
Protein samples collected at later time points, when almost all of the PANG had been released (day 35 and day 56, respectively), were also characterized via SDS-PAGE (Fig. 5). Similar to most antibodies, PANG comprises two heavy chains (50 kDa) and two light chains (25 kDa) connected by disulfide bonds. Therefore, electrophoresis under reducing (with DTT) and non-reducing (without DTT) conditions was used to examine the structural integrity of the antibody. Generally, separate heavy chains and light chains are observed in SDS-PAGE under reducing conditions while the intact antibody is observed under non-reducing condition. PANG released from 8-arm 10/2K and 10K/700 hydrogels was loaded in Lane 1 and Lane 4, respectively. PANG controls stored at either 37°C or −80°C were loaded in Lane 2–3 and Lane 5–6, respectively, for parallel comparison with the release samples. In Figure 5(a), two bands representing the heavy chains (50 kDa) and light chains (25 kDa) of the antibody were observed as expected in each lane. In Figure 5(b), bands indicative of the intact antibody were observed at approximately 220 kDa, significantly greater than the expected molecular weight (~145 kDa), likely due to incomplete linearization and unfolding of the antibody under these conditions. Some fragmentation products were observed for the higher concentration PANG samples (Lane 1–3) under non-reducing conditions, which might be a result of disulfide-bond scrambling catalyzed by free sulfhydryl groups in the antibody itself.51–53 Nevertheless, these data demonstrate that the majority of PANG released from the hydrogels, even after extended periods, showed an electrophoretic profile comparable to the PANG controls.
FIGURE 5.
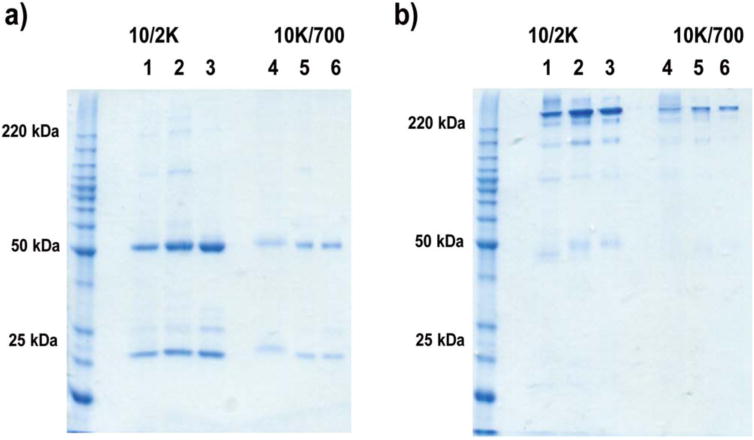
SDS-PAGE electrophoresis depicting protein release under (a) reducing and (b) non-reducing conditions. Lane 1: D35 sample from 10/2K; Lane 2: D35 PANG Control at 37°C; Lane 3: PANG Standard at −80°C; Lane 4: D56 sample from 10K/700; Lane 5: D56 PANG Control at 37°C; Lane 6: PANG Standard at −80°C.
The conformational properties of PANG after its release from the matrices were investigated by CD and fluorescence emission spectroscopy respectively, as shown in Figure 6. Figure 6(a) illustrates CD confirmation of the secondary structure of PANG after its release. Negative peaks were observed at 218 nm for both of the release samples as well as for the antibody control, which is consistent with the presence of β-sheet conformations in the protein.54 More importantly, the CD spectra of the released PANG closely resembled that of the PANG control. The low signal-to-noise ratio observed in some spectra, especially in the PANG solution collected from the 10K/700 hydrogel after 56 days, may be attributed to the relatively low concentration (even after concentration via centrifugal filtration) of released PANG in these samples, along with the possible presence of detached PEG fragments from the hydrogel matrix.55 Figure 6(b) shows the data from fluorescence emission spectroscopy, which was used to detect changes in the tertiary structure of the PANG after its release from the hydrogel, as the emission spectrum is sensitive to the tryptophan micro-environment within the 3D structure of the protein. The data in Figure 6(b) show an emission maximum of tryptophan at 340 nm; the emission spectra of the released PANG were similar to those of the PANG control with respect to both the emission maximum and fluorescence intensity normalized for concentration. These results suggest that PANG maintained its structural conformation after undergoing the precipitation and re-dissolution process, as well as release from the PEG hydrogels.
FIGURE 6.
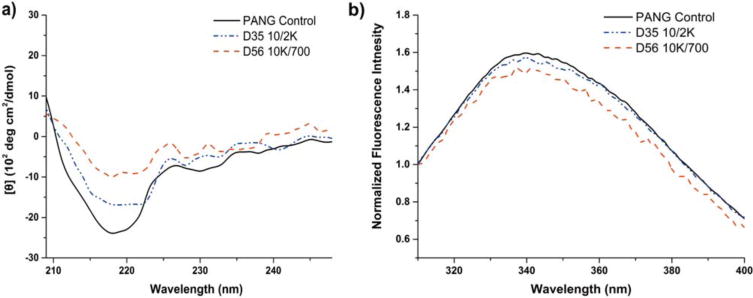
Conformational characterization of PANG control solutions and of PANG released from the hydrogels. (a) CD spectra of solutions of PANG control and PANG released from hydrogels; (b) Normalized fluorescence emission spectra of PANG control and PANG released from hydrogels.
Although hydrogel strategies have been widely used to protect proteins from degradation and denaturation, the conformational properties of proteins during in situ gelation and encapsulation have rarely been studied. In order to understand the conformation of PANG when incorporated within the hydrogel, PANG-loaded PEG hydrogels were also studied via CD and fluorescence spectroscopy (Fig. 7). In this case, a relatively low polymer concentration (10 wt % hydrogel) was employed to alleviate the opacity caused by PEG-induced precipitation. Similar to the data in Figure 6, the characteristic negative peak at approximately 218 nm of β-sheet conformations were observed [Fig. 7(a)] while the expected fluorescence emission maximum of PANG was observed at 331–334 nm [Fig. 7(b)]. The data in Figure 7 confirm that soluble PANG encapsulated in the polymer network exhibited similar secondary and tertiary structures as those of the antibody control. The slight red shift of the emission maximum of the in-gel PANG (334 nm) compared to that of the PANG control (331 nm) in Figure 7(b) might be attributed to a slightly more hydrophilic local environment within the hydrogel.56 These data indicate that the processes involved during the in situ gelation in the presence of PANG did not adversely affect the antibody conformation.
FIGURE 7.
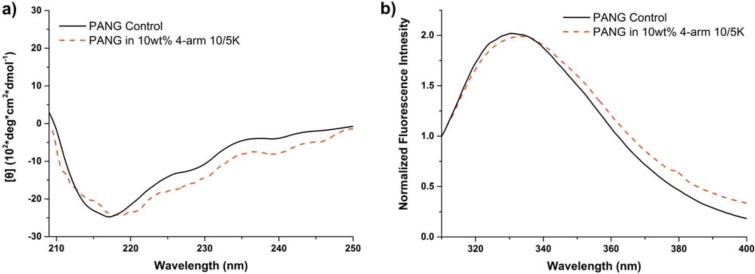
Conformational characterizations of PANG (5 mg/mL) when encapsulated within the hydrogels. (a) CD spectra of PANG control and encapsulated PANG; (b) Normalized fluorescence emission spectra of PANG control and encapsulated PANG.
Biological activity of released PANG
To examine the biological activity of the antibody released from the hydrogels, an anthrax lethal TNA was employed to assess the ability of PANG to neutralize anthrax LeTx. Test samples were applied to cells in the presence of LeTx, and the dilution of the sample at which 50% of the cells are protected from LeTx was determined to be the effective dilution 50 (ED50).12 ED50 values establish a quantitative criterion to a dose response curve that allows for the comparison of the biological activity of PANG in each test sample. Higher ED50 values indicate that a higher sample dilution yields 50% protection, and thus indicate a sample with greater neutralizing activity (which is related to the concentration of functional PANG in the sample). As shown in Figure 8(a), the ED50 values of these release samples gradually increased over time, after an initial lag period. For 8-arm 20/5K hydrogels, little toxin-neutralizing activity was observed for the first 5 days. After that, the release samples exhibited a significant increase in activity, reaching an ED50 value of 1550 at day 28. Similarly, following an initial delay in biological activity over the first 10 days, samples from 8-arm 10/2K and 10K/700 hydrogels showed increased activity, achieving an ED50 value of 1890 at day 35 and 670 at day 56, respectively. The PANG released from the hydrogel matrix clearly retained its ability to neutralize anthrax LeTx in vitro up to 56 days. The initial delay in toxin-neutralization might be attributed to the slow initial release of PANG from the hydrogels. Furthermore, the toxin-neutralizing profile of the released proteins closely mirrored the concentration profile determined via UV–Vis [Fig. 8(b)], further confirming that the functionality of the antibody was not affected by its extended encapsulation in the PEG hydrogels.
FIGURE 8.
In vitro characterization of PANG released from PEG hydrogels. (a) Toxin-neutralizing activity of released PANG on J774A.1 cells incubated with LeTx; (b) Concentration profile of the in vitro released protein samples as characterized by UV–Vis at 280 nm.
DISCUSSION
In contrast to many other existing antibody release vehicles, PANG release from these PEG hydrogels exhibited zero-order kinetics without any significant burst effect during the initial release stage, likely a result of the PEG-induced precipitation of the antibodies. At an early stage of the experiment, PANG release from the hydrogel is hindered due to the fact that only a minor, non-precipitated fraction of the incorporated antibody is available for diffusion out of the matrix, resulting in a lack of burst release. (Opacity associated with the precipitation of PANG was observed within the hydrogel for most formulations until approximately 40% of the antibody was released.) In most hydrogel delivery systems, swelling of the polymer network and release of protein result in a decreasing protein concentration gradient, leading to a decrease in release rate.57,58 The consistent release rate observed for our experiments, in contrast, may be a result of a balance between hydrogel degradation and dissolution of the antibody precipitate throughout the course of the experiment. As the gels swell/degrade, the soluble antibody concentration within the network decreases (decreased concentration gradient), and the dissolution of antibody precipitate increases (increased concentration gradient). These two limiting factors of antibody release counteract each other, which might yield a more consistent concentration gradient and lead to an almost constant release rate of PANG over prolonged periods of time.22 This proposed mechanism, which still requires hydrogel degradation for the release of antibody from the hydrogels, is further evidenced by the release profiles of the 8-arm 10/2K and 10K/700 hydrogels with high network density. The swelling and degradation of these hydrogels was observed to be very limited during initial time points, which would be expected to retard protein release. Consistent with this expectation, only minimal protein release (ca. 10%) was observed over the first 10 days of the experiment, and zero-order release was observed only once hydrogel swelling and degradation became significant, leading to increased dissolution and subsequent release of the precipitated antibody.
The observation of these release profiles for PANG, in conjunction with the precipitation of the protein, is consistent with results reported by others. For example, Hubbell and coworkers took advantage of the precipitation of human growth hormone (hGH) by linear PEG (or complexation of zinc) to protect the protein during in situ gelation and dramatically decrease the rate of protein release from PEG hydrogels.48 Hennink and coworkers also confirmed the effect of protein precipitation by NaCl or concentrated dextran on prolonging the release of hGH from dextran hydrogel microspheres.59 In addition, Herrmann et al. demonstrated that PEG-containing lipidic implants significantly reduced the burst release of rh-interferon α-2a (IFN-α), due to the PEG-induced, reversible precipitation of protein.60 Similar to these reports, our results suggest that the precipitation and subsequent dissolution of PANG have contributed significantly to its prolonged release from the hydrogel network.
Altogether, our studies illustrate the facile use of hydrogel based methods to control the delivery of an antibody that is active for the neutralization of the LeTx of anthrax. The data presented here for PANG suggest more general applications for the controlled release of therapeutic proteins/antibodies by PEG-induced precipitation methods. Although the ability of PEG to precipitate proteins has been extensively studied and widely used in the field of protein separation and purification,44,45 only few studies have explored the precipitation of protein within PEG hydrogel-based controlled release systems and its potential benefit in controlling delivery profiles.48,59–61 The in situ, reversible precipitation of the protein induced by PEG in this report may not only provide protein stabilization during the gelation process without the addition of any exogenous precipitants, but also offer a useful strategy to prevent burst release and prolong the release of protein over an extended period of time. In addition, the in-gel characterization of the protein provides insight about the conformational properties of proteins during in situ gelation and encapsulation, potentially relevant to the clinical translation and approval of such protein delivery systems.
CONCLUSIONS
In this study, hydrolytically degradable PEG hydrogels have been synthesized and used as injectable local delivery materials for sustained release of the antibody PANG. The delivery properties of the hydrogels can be easily adjusted by varying the chemical composition of the precursors. PANG was released from the hydrogel matrix in a controlled manner ranging from 14 to 56 days in vitro. Burst release of the antibody was prevented by PEG-induced protein precipitation. The secondary and tertiary structures of the hydrogel released antibodies as well as their biological activities were not affected by encapsulation and release from the hydrogel. These results suggest that the injectable PEG hydrogels, combined with PEG-induced protein precipitation, hold significant potential in the sustained and long-term delivery of therapeutic antibodies.
Supplementary Material
Acknowledgments
Contract grant sponsor: Fraunhofer USA Center for Molecular Biotechnology under the project “Efficacy and Stability Testing of Biologicals in Novel, Biologically Inspired Matrices”
Contract grant sponsor: University of Delaware
Contract grant sponsor: National Institute of General Medical Sciences – NIGMS; contract grant number: 1 P30 GM110758-01
Contract grant sponsor: National Institutes of Health
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ross JM. The pathogenesis of anthrax following the administration of spores by the respiratory route. J Pathol Bacteriol. 1957;73:485–494. [Google Scholar]
- 2.Vanness GB. Ecology of anthrax. Science. 1971;172:1303–1307. doi: 10.1126/science.172.3990.1303. [DOI] [PubMed] [Google Scholar]
- 3.Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Hauer J, McDade J, Osterholm MT, O’Toole T, et al. Anthrax as a biological weapon—Medical and public health management. JAMA. 1999;281:1735–1745. doi: 10.1001/jama.281.18.1735. [DOI] [PubMed] [Google Scholar]
- 4.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, Tapper M, Fisk TL, Zaki S, Popovic T, et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen ZC, Moayeri M, Purcell R. Monoclonal antibody therapies against anthrax. Toxins. 2011;3:1004–1019. doi: 10.3390/toxins3081004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding GP, Chen XM, Zhu J, Duesbery NS, Cheng XJ, Cao BA. A human/murine chimeric Fab antibody neutralizes anthrax lethal toxin in vitro. Clin Dev Immunol. 2013;2013:475809. doi: 10.1155/2013/475809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JW, Smolen J. Monoclonal antibody therapy. Adv Protein Chem. 2001;56:369–421. doi: 10.1016/s0065-3233(01)56010-6. [DOI] [PubMed] [Google Scholar]
- 8.Mohamed N, Clagett M, Li J, Jones S, Pincus S, D’Alia G, Nardone L, Babin M, Spitalny G, Casey L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect Immunity. 2005;73:795–802. doi: 10.1128/IAI.73.2.795-802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson JW, Comer JE, Noffsinger DM, Wenglikowski A, Walberg KG, Chatuev BM, Chopra AK, Stanberry LR, Kang AS, Scholz WW, et al. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect Immunity. 2006;74:1016–1024. doi: 10.1128/IAI.74.2.1016-1024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson JW, Comer JE, Baze WB, Noffsinger DM, Wenglikowski A, Walberg KG, Hardcastle J, Pawlik J, Bush K, Taormina J, et al. Human monoclonal antibody AVP-21D9 to protective antigen reduces dissemination of the Bacillus anthracis ames strain from the lungs in a rabbit model. Infect Immunity. 2007;75:3414–3424. doi: 10.1128/IAI.00352-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chichester JA, Manceva SD, Rhee A, Coffin MV, Musiychuk K, Mett V, Shamloul M, Norikane J, Streatfield SJ, Yusibov V. A plant-produced protective antigen vaccine confers protection in rabbits against a lethal aerosolized challenge with Bacillus anthracis Ames spores. Hum Vaccin Immunother. 2013;9:544–552. doi: 10.4161/hv.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mett V, Chichester JA, Stewart ML, Musiychuk K, Bi H, Reifsnyder CJ, Hull AK, Albrecht MT, Goldman S, Baillie LWJ, et al. A non-glycosylated, plant-produced human monoclonal antibody against anthrax protective antigen protects mice and non-human primates from B. anthracis spore challenge. Hum Vaccin. 2011;7:183–190. doi: 10.4161/hv.7.0.14586. [DOI] [PubMed] [Google Scholar]
- 13.Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58:686–706. doi: 10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Szlachcic A, Zakrzewska M, Otlewski J. Longer action means better drug: Tuning up protein therapeutics. Biotechnol Adv. 2011;29:436–441. doi: 10.1016/j.biotechadv.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 16.Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J Pharm Sci. 2004;93:2184–2204. doi: 10.1002/jps.20125. [DOI] [PubMed] [Google Scholar]
- 17.Li YL, Rodrigues J, Tomas H. Injectable and biodegradable hydrogels: Gelation, biodegradation and biomedical applications. Chem Soc Rev. 2012;41:2193–2221. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 18.Lin C-C, Anseth K. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2009;26:631–643. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853–2888. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 20.Censi R, Di Martino P, Vermonden T, Hennink WE. Hydrogels for protein delivery in tissue engineering. J Control Release. 2012;161:680–692. doi: 10.1016/j.jconrel.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2010;62:83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 22.Elbert DL, Pratt AB, Lutolf MP, Halstenberg S, Hubbell JA. Protein delivery from materials formed by self-selective conjugate addition reactions. J Control Release. 2001;76:11–25. doi: 10.1016/s0168-3659(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi N, Zhang L, Chae BS, Palla CS, Furst EM, Kiick KL. Growth factor mediated assembly of cell receptor-responsive hydrogels. J Am Chem Soc. 2007;129:3040–3041. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freudenberg U, Hermann A, Welzel PB, Stirl K, Schwarz SC, Grimmer M, Zieris A, Panyanuwat W, Zschoche S, Meinhold D, et al. A star-PEG-heparin hydrogel platform to aid cell replacement therapies for neurodegenerative diseases. Biomaterials. 2009;30:5049–5060. doi: 10.1016/j.biomaterials.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Kraehenbuehl TP, Ferreira LS, Zammaretti P, Hubbell JA, Langer R. Cell-responsive hydrogel for encapsulation of vascular cells. Biomaterials. 2009;30:4318–4324. doi: 10.1016/j.biomaterials.2009.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharkar PM, Kloxin AM, Kiick KL. Dually degradable click hydrogels for controlled degradation and protein release. J Mater Chem B. 2014;2:5511–5521. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahoo S, Chung C, Khetan S, Burdick JA. Hydrolytically degradable hyaluronic acid hydrogels with controlled temporal structures. Biomacromolecules. 2008;9:1088–1092. doi: 10.1021/bm800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffin DR, Schlosser JL, Lam SF, Nguyen TH, Maynard HD, Kasko AM. Synthesis of photodegradable macromers for conjugation and release of bioactive molecules. Biomacromolecules. 2013;14:1199–1207. doi: 10.1021/bm400169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Licciardi M, Grassi M, Di Stefano M, Feruglio L, Giuliani G, Valenti S, Cappelli A, Giammona G. PEG-benzofulvene copolymer hydrogels for antibody delivery. Int J Pharm. 2010;390:183–190. doi: 10.1016/j.ijpharm.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Lee ALZ, Ng VWL, Gao SJ, Hedrick JL, Yang YY. Injectable hydrogels from triblock copolymers of vitamin E-functionalized polycarbonate and poly (ethylene glycol) for subcutaneous delivery of antibodies for cancer therapy. Adv Funct Mater. 2014;24:1538–1550. [Google Scholar]
- 31.Hu CC, Chaw JR, Chen CF, Liu HW. Controlled release bevacizumab in thermoresponsive hydrogel found to inhibit angiogenesis. Bio-Med Mater Eng. 2014;24:1941–1950. doi: 10.3233/BME-141003. [DOI] [PubMed] [Google Scholar]
- 32.Tian WM, Zhang CL, Hou SP, Yu X, Cui FZ, Xu QY, Sheng SL, Cui H, Li HD. Hyaluronic acid hydrogel as Nogo-66 receptor antibody delivery system for the repairing of injured rat brain: In vitro. J Control Release. 2005;102:13–22. doi: 10.1016/j.jconrel.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 33.Guziewicz N, Best A, Perez-Ramirez B, Kaplan DL. Lyophilized silk fibroin hydrogels for the sustained local delivery of therapeutic monoclonal antibodies. Biomaterials. 2011;32:2642–2650. doi: 10.1016/j.biomaterials.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koutsopoulos S, Zhang SG. Two-layered injectable self-assembling peptide scaffold hydrogels for long-term sustained release of human antibodies. J Control Release. 2012;160:451–458. doi: 10.1016/j.jconrel.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 35.Hull AK, Criscuolo CJ, Mett V, Groen H, Steeman W, Westra H, Chapman G, Legutki B, Baillie L, Yusibov V. Human-derived, plant-produced monoclonal antibody for the treatment of anthrax. Vaccine. 2005;23:2082–2086. doi: 10.1016/j.vaccine.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 36.Mather BD, Viswanathan K, Miller KM, Long TE. Michael addition reactions in macromolecular design for emerging technologies. Prog Polym Sci. 2006;31:487–531. [Google Scholar]
- 37.Ghosh K, Shu XZ, Mou R, Lombardi J, Prestwich GD, Rafailovich MH, Clark RAF. Rheological characterization of in situ cross-linkable hyaluronan hydrogels. Biomacromolecules. 2005;6:2857–2865. doi: 10.1021/bm050361c. [DOI] [PubMed] [Google Scholar]
- 38.Weber LM, Lopez CG, Anseth KS. Effects of PEG hydrogel cross-linking density on protein diffusion and encapsulated islet survival and function. J Biomed Mater Res Part A. 2009;90A:720–729. doi: 10.1002/jbm.a.32134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metters A, Hubbell J. Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules. 2004;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 40.Zustiak SP, Leach JB. Characterization of protein release from hydrolytically degradable poly(ethylene glycol) hydrogels. Biotechnol Bioeng. 2011;108:197–206. doi: 10.1002/bit.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih H, Lin C-C. Cross-linking and degradation of step-growth hydrogels formed by thiol–ene photoclick chemistry. Biomacromolecules. 2012;13:2003–2012. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elliott JE, Anseth JW, Bowman CN. Kinetic modeling of the effect of solvent concentration on primary cyclization during polymerization of multifunctional monomers. Chem Eng Sci. 2001;56:3173–3184. [Google Scholar]
- 43.Zustiak SP, Leach JB. Hydrolytically degradable poly(ethylene glycol) hydrogel scaffolds with tunable degradation and mechanical properties. Biomacromolecules. 2010;11:1348–1357. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annunziata O, Asherie N, Lomakin A, Pande J, Ogun O, Benedek GB. Effect of polyethylene glycol on the liquid-liquid phase transition in aqueous protein solutions. Proc Natl Acad Sci U S A. 2002;99:14165–14170. doi: 10.1073/pnas.212507199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Lomakin A, Latypov RF, Laubach JP, Hideshima T, Richardson PG, Munshi NC, Anderson KC, Benedek GB. Phase transitions in human IgG solutions. J Chem Phys. 2013;139 doi: 10.1063/1.4811345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buwalda SJ, Dijkstra PJ, Feijen J. In situ forming poly(ethylene glycol)-poly(L-lactide) hydrogels via Michael addition: mechanical properties, degradation, and protein release. Macromol Chem Phys. 2012;213:766–775. [Google Scholar]
- 47.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Hydrogels in regenerative medicine. Adv Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Wetering P, Metters AT, Schoenmakers RG, Hubbell JA. Poly(ethylene glycol) hydrogels formed by conjugate addition with controllable swelling, degradation, and release of pharmaceutically active proteins. J Control Release. 2005;102:619–627. doi: 10.1016/j.jconrel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 49.Bhat R, Timasheff SN. Steric Exclusion Is the Principal Source of the Preferential Hydration of Proteins in the Presence of Polyethylene Glycols. Protein Sci. 1992;1:1133–1143. doi: 10.1002/pro.5560010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Latypov RF, Lomakin A, Meyer JA, Kerwin BA, Vunnum S, Benedek GB. Quantitative evaluation of colloidal stability of antibody solutions using PEG-induced liquid-liquid phase separation. Mol Pharm. 2014;11:1391–1402. doi: 10.1021/mp400521b. [DOI] [PubMed] [Google Scholar]
- 51.Liu H, Gaza-Bulseco G, Chumsae C, Newby-Kew A. Characterization of lower molecular weight artifact bands of recombinant monoclonal IgG1 antibodies on non-reducing SDS-PAGE. Biotechnol Lett. 2007;29:1611–1622. doi: 10.1007/s10529-007-9449-8. [DOI] [PubMed] [Google Scholar]
- 52.Taylor FR, Prentice HL, Garber EA, Fajardo HA, Vasilyeva E, Blake Pepinsky R. Suppression of sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample preparation artifacts for analysis of IgG4 half-antibody. Anal Biochem. 2006;353:204–208. doi: 10.1016/j.ab.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 53.Zhu ZC, Chen Y, Ackerman MS, Wang B, Wu W, Li B, Obenauer-Kutner L, Zhao R, Tao L, Ihnat PM, et al. Investigation of monoclonal antibody fragmentation artifacts in non-reducing SDS-PAGE. J Pharm Biomed Anal. 2013;83:89–95. doi: 10.1016/j.jpba.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 54.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protocol. 2007;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koutsopoulos S, Unsworth LD, Nagaia Y, Zhang SG. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc Natl Acad Sci U S A. 2009;106:4623–4628. doi: 10.1073/pnas.0807506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varlan A, Hillebrand M. Bovine and human serum albumin interactions with 3-carboxyphenoxathiin studied by fluorescence and circular dichroism spectroscopy. Molecules. 2010;15:3905–3919. doi: 10.3390/molecules15063905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Dijk-Wolthuis WNE, Hoogeboom JAM, van Steenbergen MJ, Tsang SKY, Hennink WE. Degradation and release behavior of dextran-based hydrogels. Macromolecules. 1997;30:4639–4645. [Google Scholar]
- 58.Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release. 2001;73:121–136. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 59.Vlugt-Wensink KDF, Meijer YJ, van Steenbergen MJ, Verrijk R, Jiskoot W, Crommelin DJA, Hennink WE. Effect of excipeients on the encapsulation efficiency and release of human growth hormone from dextran microspheres. Eur J Pharm Biopharm. 2007;67:589–596. doi: 10.1016/j.ejpb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 60.Herrmann S, Mohl S, Siepmann F, Siepmann J, Winter G. New insight into the role of polyethylene glycol acting as protein release modifier in lipidic implants. Pharm Res. 2007;24:1527–1537. doi: 10.1007/s11095-007-9271-y. [DOI] [PubMed] [Google Scholar]
- 61.Johnson OL, Cleland JL, Lee HJ, Charnis M, Duenas E, Jaworowicz W, Shepard D, Shahzamani A, Jones AJS, Putney SD. A month-long effect from a single injection of microencapsulated human growth hormone. Nat Med. 1996;2:795–799. doi: 10.1038/nm0796-795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.