

Abstract
We examine the mechanistic basis and wider implications of adopting a developmental perspective on human ageing. Previous models of ageing have concentrated on its genetic basis, or the detrimental effects of accumulated damage, but also have raised issues about whether ageing can be viewed as adaptive itself, or is a consequence of other adaptive processes, for example if maintenance and repair processes in the period up to reproduction are traded off against later decline in function. A life course model places ageing in the context of the attainment of peak capacity for a body system, starting in early development when plasticity permits changes in structure and function induced by a range of environmental stimuli, followed by a period of decline, the rate of which depends on the peak attained as well as the later life conditions. Such path dependency in the rate of ageing may offer new insights into its modification. Focusing on musculoskeletal and cardiovascular function, we discuss this model and the possible underlying mechanisms, including endothelial function, oxidative stress, stem cells and nutritional factors such as vitamin D status. Epigenetic changes induced during developmental plasticity, and immune function may provide a common mechanistic process underlying a life course model of ageing. The life course trajectory differs in high and low resource settings. New insights into the developmental components of the life course model of ageing may lead to the design of biomarkers of later chronic disease risk and to new interventions to promote healthy ageing, with important implications for public health.
Abbreviations
- CVD
cardiovascular disease
- DOHaD
developmental origins of health and disease
- FMD
flow‐mediated dilatation
- NCD
non‐communicable disease
- NO
nitric oxide
Introduction
There is increased awareness that the process of human ageing commences as early as conception with the inheritance of a specific genome, and it does not cease until death. Epidemiological studies have demonstrated that environmental influences during intrauterine and early postnatal life are associated with alterations in form and function across a range of systems, which establish predisposition to age‐related system decline (Sayer et al. 1998). The biological process through which these changes in gene expression are established is known as ‘developmental plasticity’. This process is ubiquitous throughout the animal world, and in some plants, and it provides the mechanism whereby a given genotype can develop into a range of phenotypes which are best adapted to the environment that they are likely to meet once development is completed, a concept formalized as a predictive adaptive response (Gluckman & Hanson, 2004). Such adaptive responses are usually thought to operate on Darwinian fitness, i.e. survival to the time of successful reproduction, rather than on ageing per se. New concepts challenge this view, and this paper reviews these using musculoskeletal and cardiovascular ageing as examples, and discusses some common underlying mechanisms. It concludes by noting the potential for translational public health strategies to promote healthy ageing by adopting a developmental perspective.
Theories of ageing can be grouped using several criteria. One distinction views ageing as either genetically predetermined (‘the biological clock’ concept) or as a response to accumulated detrimental but otherwise random events over time (Medvedev, 1990). Another contrasts ageing which evolved as an adaptive process or as a non‐adaptive by‐product of other processes. These groups are not mutually exclusive. This paper starts from the premise that ageing is one aspect of a life course strategy initiated in early life in each individual as part of a cassette of evolved adaptive responses (Gluckman et al. 2007). In this sense, ageing can be considered as a normal process: this in turn raises the question of whether abnormal ageing can occur. Clearly it can in unusual conditions such as progeria (Gabr et al. 1960). However, in the population as a whole there are wide variations in the rate of ageing in terms of declining functional capacity, which may be regarded in part either as normal responses to common challenges or as pathological processes. Distinguishing between these processes may have important consequences for the identification of individuals or groups at risk of accelerated ageing, and for interventions. In this paper we discuss these issues with specific reference to musculoskeletal and cardiovascular disorders, using a life course model of functional capacity.
Models of ageing
Accumulated damage
One model of ageing treats it as the result of a series of random events, and the role of chance in ageing is widely recognized (Bjorksten, 1958). The underlying mechanisms may be conceived in terms of molecular changes such as DNA damage (Varizi et al. 1994), or alteration in gene expression associated with errors in transcription (Kirkwood & Austad 2000). Human somatic cells are believed to possess the ability to undergo a finite number of possible mitoses (the Hayflick limit), although this may not be the basis for ageing (Williams, 1957). In cell lines, accumulation of errors in protein transcription (Miller, 1999), and of cross‐links in macromolecules, may cause structural and functional changes associated with ageing. A number of theories have focused on the role of exposure to metabolic or waste products, for example reducing sugars or free radicals, as the cause of such deteriorative changes (Rattan, 2008). At a more holistic level, homeostatic systems such as the hypothalamic–pituitary axis or the immune system may play a fundamental role (see below) and a link has been proposed between psychosocial factors such as stress and ageing (Nilsson, 1996).
As proposed by López‐Otín and colleagues (López‐Otín et al. 2013), biomarkers (or hallmarks) of ageing can be grouped into primary (gene and protein stability, telomere length, epigenetic processes), secondary (regulation of nutrient sensing, mitochondrial function, cellular senescence) and integrative (stem cell provision and intercellular communication) mechanisms. The primary markers are believed to represent the underlying causes of ageing, while the secondary markers represent potential compensatory responses. The integrative markers are believed to contribute directly to ageing‐related phenotypes of health and disease (López‐Otín et al. 2013). López‐Otín and co‐workers provide an elegant model of temporal events, which may contribute either directly or indirectly to physiological disturbances related to ageing. Such temporal events interact at multiple levels of biological organization, which may involve several feed‐forward and feedback mechanisms. Disturbances in homeostatic mechanisms which regulate these markers of ageing are likely to play an important role in the development of ageing‐related disorders.
Disposable soma
The adaptive pleiotropy or disposable soma theory proposes that some genes expressed in early life up to the time of reproduction promote Darwinian fitness but that they may have detrimental effects in the post‐reproductive period (Kirkwood & Austad 2000). Many candidate ageing genes have been proposed but while their individual effects may be small collectively they may be important. In terms of the disposable soma theory, ageing may have evolved as part of the life course of a species because of the necessity for a trade‐off between survival to achieve reproduction versus somatic processes such as maintenance and repair which place large demands on available energy and other biological resources. The theory suggests that the optimal level of investment in growth, maintenance and repair of the soma provides adequate protection against endogenous and exogenous damage for survival in the short term, but that this is less than that required for prolonged survival in the post‐reproductive period. Life course theory suggests that developing in a high‐risk environment will accelerate the acquisition of reproductive capacity at the expense of tissue maintenance/repair; a low‐risk environment will delay reproductive function. Recently, there have been some excellent examples of this in wild animal populations (Dantzer et al. 2013), and some human observations support the concept (Sloboda et al. 2007). However, examination of the timing of fertility and mortality across a wide range of species (Jones et al. 2014) demonstrates considerable variations in the pattern across the life course, which questions the assumption that the disposable soma strategy has formed a fundamental part of evolution.
Life course perspective
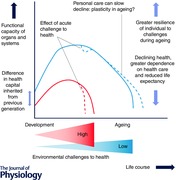
The focus of most previous research on ageing is on influences operating in later life. However, there is considerable unexplained variation in age‐related disease between individuals of a similar age and so predictive potential markers of ageing, which may be linked to early life processes, may be helpful (Menni et al. 2013). The life course approach to ageing suggests that the rate of decline in function for a particular organ or system is not only dependent on contemporary influences but on the level of peak function attained earlier in life, which in turn depends partly on developmental processes and early environmental influences (Dodds et al. 2014) (Abstract figure). The aged phenotype therefore results from intrinsic and extrinsic exposures across the life course and the corresponding response in terms of maintenance, regeneration and repair (López‐Otín et al. 2013). This life course concept helps to explain the wide variation in rates of ageing between individuals in a population at any time. The complexity may necessitate a systems biology approach (Kirkwood, 2011). The life course model of ageing (Abstract figure) also indicates that differences in both exposure and response underlie the variation in rates of ageing between systems in the same individual and between individuals, seen for example in high versus low resource settings. It also shows how the differences between high and low–middle income countries affect population ageing.
The life course model of ageing is also relevant to clinical practice as it may provide an opportunity to identify individuals at risk of accelerated ageing early in the life course, using early biomarkers of such risk (Martin‐Ruiz et al. 2011). It also suggests that the window for instituting beneficial interventions should be widened to include early life, when adverse environments may have long‐term effects on ageing (Drury et al. 2012). Furthermore, knowledge of underlying mechanisms may lead to the development of new interventions to regulate the rate of ageing or to minimize its detrimental effects.
Developmental plasticity
Plasticity is defined as the effect of a challenge to alter the structure or function of a system, often permanently. It should be distinguished from flexibility, in which a structure or function ‘bends’ when faced with a challenge, to return to its previous state when the challenge ceases, and from resilience, where the challenge is met with an opposing force (Hanson & Gluckman, 2014). Plasticity confers advantage in that it may diminish the effect of a similar challenge later, unlike flexibility, and it is less likely to lead to damage to the system, unlike resilience. There is growing recognition that the rate and manner in which individuals age can be significantly influenced by extrinsic factors at multiple points throughout life (Kuh, 2007). In animals endocrine, nutritional and other influences which affect development during critical periods of early life permanently alter the structure and function of the body's tissues and systems (Bertram & Hanson 2001; McMillen & Robinson, 2005; Gluckman et al. 2005). The effects rely on the principle of developmental plasticity, by which one genotype can give rise to a range of different physiological or morphological phenotypes in response to different prevailing environmental conditions during development (Gluckman et al. 2009; Bateson & Gluckman, 2011).
Organ systems are most susceptible to such effects during periods in which they are growing rapidly. During the embryonic period (the first 2 months of gestation in the human), progenitor cells undergo extensive differentiation without rapid cell replication. The highest growth rates are observed during the subsequent fetal period, and thus some organs are most susceptible to environmental cues at this point (Gluckman & Hanson, 2004). Relative growth slows in late gestation and continues to slow in childhood. This plasticity is lost later in life when the organism has a relatively fixed functional capacity, although there is a limited capacity for renewal through stem cells in some tissues and it is possible that developmental processes can influence the degree to which stem cell lineages can provide such renewal later on (Obernier et al. 2014). Developmental plasticity provides a fitness benefit through the ability to adapt to environmental changes over a relatively short time span, i.e. one generation (Gluckman & Hanson, 2004). In some circumstances, usually following potent disruptive exposures such as environmental chemicals, this benefit can be passed to several subsequent generations without the need for re‐exposure to the stimulus (Skinner et al. 2010), although this is not necessarily true of milder, nutritional stimuli.
The concept of path dependency permits a trajectory of intrinsic capacity or health capital to be drawn for an individual (Abstract figure). This trajectory will be a composite of that for individual organs and systems, which have different sensitive periods when early life influences the rate of acquisition and the magnitude of the maximal functional capacity attained. The rate of decline in function or in overall health partly depends on the height of this peak, examples being the attainment prenatally and the later decline of renal nephrons and cardiomyocytes, of bone and muscle mass (see Fig. 1) in late adolescence, and of frontal cortical executive functions in early adulthood (Blakemore & Choudhury, 2006). In addition, the settings of some homeostatic control systems influencing metabolism or appetite are partly established in early life (Gahagan, 2012).
Figure 1. Factors acting across the life course .
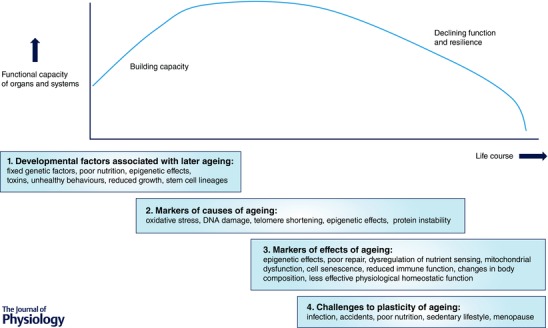
(1) Developmental factors which are associated with later ageing. (2) Markers of the causes of ageing. (3) Markers of the effects of ageing. (4) Factors which challenge the plasticity of ageing. Some of these factors give insights into underlying mechanisms and some may serve as useful biomarkers of risk or biological age.
The life course trajectory can also be used to show when an individual's health declines to a critical stage of dependence on medical care or, conversely how great their resilience to challenges such as infection or injury will be. It also relates to their likely longevity, and animal studies show that developmental influences such as nutrition in prenatal and early postnatal life can have dramatic effects on lifespan (Jennings et al. 1999). This concept can be extrapolated to different socio‐economic or resource settings (see Abstract figure) showing not only how potential longevity is influenced but also how resilience is affected. Intervention at early stages in the life course can have a dramatic effect on the entire trajectory, but some plasticity is manifest even in later life.
Evidence supporting a developmental contribution to ageing
Epidemiological studies showing the relationship between small size at birth and an increased risk of age‐related disease led to the concept of the developmental origins of health and disease (DOHaD). Forsdahl (1977) showed that the prevalence of arteriosclerotic heart disease correlated with past infant mortality in Norway and suggested that a poor standard of living in childhood and adolescence was a risk factor for later heart disease. Subsequently Barker (1995) showed that rates of death from coronary heart disease in different parts of England and Wales were correlated with previous rates of infant mortality. This led Barker and colleagues (Barker, 1998) to suggest that adverse environmental influences in utero and during infancy permanently change the body's structure, physiology and metabolism, increasing susceptibility to disease in later life.
As noted above, a range of animal studies were undertaken in parallel to investigate the mechanisms of developmental plasticity underlying DOHaD (e.g. Bertram & Hanson, 2001). The most commonly used models involve a prenatal nutrient imbalance induced by a reduction in overall maternal food intake, by protein restriction in an isocaloric diet or feeding a high fat diet (Langley‐Evans et al. 2005; Brawley et al. 2004; Armitage et al. 2005). Other models include non‐nutritional interventions such as glucocorticoid exposure or placental restriction to impact on early growth and development (Drake et al. 2005; Wadley et al. 2008). In the rat, maternal dietary restriction resulted in offspring with permanent stunting of growth, anaemia and reduced resistance to hypothermia in later life. Kahn (1968) demonstrated an earlier age‐related decline in haemoglobin in the offspring of restricted mothers. Roeder & Chow (1972) suggested that dietary restriction in early life may be followed by accelerated ageing, in contrast with the slowing in ageing seen with this intervention in adulthood. Further work showed that reduced prenatal and early postnatal nutrition led to increased levels of age‐associated enzymes in the liver and kidney and to a permanent reduction in muscle mass (Roeder, 1973). Prenatal dietary restriction is associated with shorter lifespan in female rats (Sayer et al. 2001), possibly via increased telomere shortening (Jennings et al. 1999). Effects on longevity were confirmed in a mouse model where dams were fed either a control or a low protein diet from conception until the end of pregnancy (Ozanne & Hales, 2005). The average lifespan of the female offspring exposed to a low‐protein diet in utero was reduced, especially if they showed catch‐up growth postnatally (as suggested by the ‘mismatch’ model: Gluckman & Hanson, 2006).
The relevance of these findings to humans in terms of structural and functional ageing in different body systems was explored in the Hertfordshire, UK, ageing study (Sayer et al. 1998). For men and women born in Hertfordshire between 1920 and 1930, and for whom archived birth records documented weight at birth and at 1 year of age, markers of ageing in the eye, ear, skin and muscle were measured. Low birth weight was associated with reduced bone mass and muscle strength, but poor infant growth with a greater range of markers of ageing, including increased lens opacity, worse hearing, thinner skin, and reduced muscle strength.
Cardiovascular and metabolic disease
Ageing is an independent risk factor for cardiovascular diseases (CVD) including atherosclerosis, coronary heart disease, hypertension and stroke. The life course approach to ageing is supported by the trajectory of blood pressure with age (Wills et al. 2011). Age‐related changes in the functional phenotype of both large and small arteries and in the microvasculature are thought to occur in the absence of conventional risk factors and to accelerate the development of CVD (Donato et al. 2015). Support for a developmental contribution to increased risk of CVD came originally from epidemiological studies that demonstrated an association between low birth weight and markers of adult cardio‐metabolic disease, including hypertension, coronary heart disease and stroke, insulin resistance and type 2 diabetes (Barker, 1998). The relationship is not limited to the extremes but is graded and linear, operating across the range of normal birth weights (Hanson & Gluckman, 2014), and growth in infancy and childhood also affects long‐term cardiovascular health (Eriksson et al. 2001). Although the association between low birth weight and higher adult blood pressure is only about 2 mmHg (kg birth weight)−1, the association with clinical hypertension is stronger, and this has been attributed in part to the reduced number of nephrons or the amount of elastin formed by vascular smooth muscle in individuals born small (Luyckx & Brenner, 2015). There is also evidence that the shape and size of the placenta influence subsequent risk of CVD in the offspring (Barker et al. 2010). These effects may depend on the mother's nutritional state, for example if the placental surface expands to compensate for fetal undernutrition but affects the load on the developing circulation. They may also reflect effects of the mother's body size on the transport of nutrients across the placenta (Lewis et al. 2010, 2012), which can affect a range of fetal organs.
Cardiovascular risk factors in childhood such as high blood pressure and increased body mass index (BMI) are associated with arterial wall thickening in adults, which in turn predicts the formation of arterial plaques and later CVD (Norman & Bonamy, 2005; Norman, 2008). In very low birth weight infants, arterial stiffness has been reported as early as the first week after birth attributed to altered deposition and/or organization of vascular elastin (Ligi et al. 2010; Simeoni et al. 2011). Impaired endothelium‐dependent and ‐independent vasodilatation is evident at birth, at 3 months of age, in later childhood and in early adult life in individuals born preterm, with low birth weight or with poor fetal nutrition. For example, a significant, graded, positive association between birth weight and flow‐mediated dilatation (FMD) assessed in the brachial artery has been reported in British children aged 9–11 years (Leeson et al. 1997). The association is not affected by adjustment for childhood body build, cardiovascular risk factors, parity, social class, or ethnicity. Recently, arterial stiffness in 9‐year‐old children was shown to be inversely related to mother's consumption of oily fish in late pregnancy but not to child's oily fish consumption (Bryant et al. 2015).
Several studies show that such early changes in vascular structure and function persist into adulthood. In the Cardiovascular Risk in Young Finns study, impaired fetal growth was associated with impaired FMD and preclinical atherosclerosis in young adults (24–45 years) born preterm or at term with impaired fetal growth (Skilton et al. 2011). Small size at birth, in terms of both thinness and low birth weight, is associated with later type 2 diabetes and impaired glucose tolerance, perhaps via the combination of low skeletal muscle mass with high intra‐abdominal fat mass, a phenotype particularly prevalent in some Asian populations (Yajnik, 2004). There is a relationship between low birth weight and plasma lipids in adulthood, affected by sex, although the effects of later BMI and lifestyle are greater in adulthood (Lawlor et al. 2006; Pereira & Power, 2012).
A U‐shaped association has been shown between birth weight and later risk, with rates increasing at both the highest and lowest birth weights (Curhan et al. 1996 a,b; Kyle & Pichard, 2006; Gamborg et al. 2007; Painter et al. 2008; Veenendaal et al. 2013). This may be the result of maternal obesity, high gestational weight gain or gestational diabetes, which have been shown to be associated with high birth weight. Excessive maternal weight gain in pregnancy may be associated with elevated offspring blood pressure and adiposity, although there appear to be subtle differences in the impact of maternal obesity and gestational weight gain as, in the Finnish cohort, young adults born large for gestational age at term did not show impaired FMD or other cardiovascular risk factors, although they did show an increase in carotid intimal medial thickness, a marker of subclinical atherosclerosis (Skilton et al. 2014). They were also more likely to be obese. However, birth weight is only a crude indicator of adverse environmental conditions such as suboptimal nutrition.
A major age‐related vascular phenotype and an early change responsible for the development of CVD is endothelial dysfunction (Ozaki et al. 2001; Khan et al. 2005; Seals et al. 2011; Donato et al. 2015). Endothelial dysfunction is manifest through a number of processes including a reduced angiogenic capacity, increased cell senescence (Hayashi et al. 2008), enhanced inflammatory state and impaired vasodilator capacity. Amongst the possible mechanisms that contribute to age‐dependent and CVD risk‐associated endothelial dysfunction is a reduced bioavailability of the endothelium‐synthesized dilating molecule nitric oxide (NO) augmented by NO scavenging due to oxidative stress. Implicated in this endothelial redox change is an increase in NADPH oxidase activity and in mitochondrial respiration (Bachschmid et al. 2013), in the absence of augmented antioxidant defences (Lesniewski et al. 2009). Circulating markers of antioxidants are either reduced or unchanged with ageing in humans (Seals et al. 2011). The transcription factor nuclear erythroid 2‐related factor 2 (Nrf2) regulates gene expression to counteract oxidative stress. In vascular models, activators of the Nrf2 pathway have been shown to restore redox homeostasis (Chapple et al. 2012). In utero exposure to increased expression of Nrf2 may also have a persistent effect on antioxidant capacity to limit oxidative stress‐induced DNA damage (Vanhees et al. 2013) and to improve endothelial function and afford cardiovascular protection in older age (Bonacasa et al. 2011).
Pregnancy‐related disruption of redox balance impacts on the germ cell, embryo and fetus with long‐term consequences on the mature organism, depending on the timing of these events (Dennery, 2010). For example, pre‐eclampsia remodelling of the uteroplacental circulation is associated with increased markers of oxidative stress (protein carbonyls and nitrotyrosine residues) and placental vascular dysfunction in humans (Redman & Sargent, 2000). The consequences to the offspring are both short‐ and long‐term and include adult hypertension, stroke, diabetes and obesity (Fernandez‐Twinn & Ozanne, 2006; Kajantie et al. 2009). Maternal obesity and overfeeding can also alter fetal oxidant status. In the rodent, maternal cafeteria feeding diminishes fetal plasma total antioxidant status and offspring exhibit a disrupted redox balance with long‐term cardiovascular effects (Bouanane et al. 2009; Torrens et al. 2012). In humans, increased levels of reactive oxygen species in the placenta may be linked to neonatal insulin resistance and obesity in response to maternal obesity or gestational diabetes (Coughlan et al. 2004; Roberts et al. 2009). Reduced levels of superoxide dismutase, catalase, glutathione and serum malondialdehyde have been observed in cord blood from babies born small for gestational age compared to infants born appropriate for gestational age (Gupta et al. 2004) and raised levels of biomarkers of oxidative stress in addition to insulin resistance persist in pre‐pubescent life (Franco et al. 2007; Chiavaroli et al. 2009).
Supplementation with l‐arginine to increase NO bioavailability or antioxidant levels during gestation and early postnatal life lowers blood pressure and improves vascular endothelial function in an animal model of hypertension (Alexander et al. 2004); however, randomized controlled trials of combined vitamin C and E supplementation during pregnancy have yet to show usefulness in preventing pre‐eclampsia (Rumbold & Crowther, 2005; Poston et al. 2006). Low levels of folate, an essential cofactor for epigenetic methylation, are strongly correlated with pre‐eclampsia in mothers and small for gestational age infants (Darnton‐Hill et al. 2015) and maternal supplementation with mixed antioxidant vitamins and essential minerals to prevent the later elevation of offspring blood pressure and vascular and renal dysfunction associated with poor maternal nutrition (Torrens et al. 2006; Franco et al. 2009).
Ageing‐associated damage to the endothelium may also result from a deterioration in endogenous endothelial progenitor cell levels and function, decreasing capacity for neovascularization and reducing re‐endothelialization following vascular lesions (Williamson et al. 2012).
Bone health
Ageing is associated with bone loss and osteoporosis, which increases the risk of age‐related fracture, typically of the hip, spine and distal forearm, which is associated with substantial morbidity and mortality (Harvey et al. 2010). Incidence of such fractures increases with age and is related to declining bone strength. These depend upon the peak attained during skeletal growth and the subsequent rate of bone loss. Bone mass is partly a heritable characteristic, although current fixed genetic factors do not explain more than a small proportion of the variance in bone mass at the population level. However, there is increasing evidence that early environmental influences modulate the attainment of peak bone mass.
Epidemiological studies show links between birth weight, weight in infancy, and adult bone mass (Baird et al. 2011). Initially associations were shown between weight at 1 year and bone mineral content, although not density, at the lumbar spine and femoral neck at age 21 years independent of adult BMI (Cooper et al. 1995). This relationship was replicated in men and women aged 60–75 years in Hertfordshire (Cooper et al. 1997), and persisted after adjustment for later environmental influences on bone loss including level of physical activity, dietary calcium intake, smoking, alcohol consumption and age at menopause. In these studies, there is evidence for effects of the early environment (Harvey et al. 2014) and for a gene–environment interaction: a polymorphism in the vitamin D receptor gene relates to bone mineral density in a direction that depends on birth weight (Dennison et al. 2001), indicating that the genetic processes influencing adult bone size and mineral density may be modified by undernutrition in utero, probably by epigenetic influences (Harvey et al. 2014).
Further observations in a range of countries have confirmed the association between growth in infancy and adult bone mass (Baird et al. 2011). Clinically, recent studies have shown links between childhood and adolescence growth and risk of later hip fracture (Javaid et al. 2011) and greater maternal height and low rate of weight gain between 7 and 15 years in the child (Cooper et al. 2001). In addition, adults suffering fracture were shorter at birth but of average height by 7 years, perhaps because accelerated childhood growth in the absence of adequate skeletal mineralization was associated with greater fracture risk later, another instance of the importance of a match between pre‐ and postnatal environments in influencing later risk of disease. These epidemiological studies, however, indicate that effects on bone mass alone are not sufficient to explain hip fracture risk. It is likely that skeletal muscle size and function (see below) contributes to frailty in individuals born light and thereby adds to the risk of fracture conferred by reduced bone mass.
Skeletal muscle
The relationship between birth weight and muscle strength in later life, first described in the Hertfordshire Ageing Study, has been replicated using a prospective national birth cohort of men and women aged 53 years (Kuh et al. 2002). This study also investigated the effect of childhood growth on adult muscle function and demonstrated that there was no interaction between birth weight and later body size. A potential underlying mechanism is that birth weight is related to an individual's muscle fibre number and types, established by birth, and that even in middle age, compensating hypertrophy may be inadequate. The relationship between birth weight and muscle strength may reflect a common genetic mechanism but this has been little explored. One study investigated the role of polymorphism in the insulin‐like growth factor 2 gene (IGF2) (Sayer et al. 2002). The product of this gene, insulin‐like growth factor II, is central to fetal growth and has a proliferative action in adult muscle. However, the study showed that birth weight and the polymorphism of the IGF2 gene had independent effects on grip strength in men suggesting that this polymorphism did not explain the observed association between early growth and grip. Adult muscle mass, as estimated by urinary creatinine excretion, is also related to early size independent of adult size (Phillips, 1995). A study of men and women aged 50 years showed that muscle mass was related to low birth weight and small head circumference, but not to thinness at birth (Phillips, 1995). A study of adult body composition in people aged 65–75 years using whole body dual X‐ray absorptiometry confirmed that low birth weight was associated with lower adult lean mass in both men and women (Sayer et al. 2004 a). From a clinical point of view, measures of muscle mass and function are not only relevant to bone health and frailty but also predict cardiovascular risk in diabetic patients (Lopes‐Jaramillo et al. 2014).
Epigenetic mechanisms as an underlying process
There is currently considerable interest in the role of epigenetic processes induced during development in later risk of non‐communicable diseases (NCDs) (e.g. Hanson & Gluckman, 2014). The mechanisms include changes in DNA methylation, histone modification and small non‐coding RNAs. A range of maternal dietary, body composition, metabolic and endocrine factors can initiate such changes in specific tissues of the developing fetus (Godfrey et al. 2007), or potentially in all tissues if the process was initiated in the early embryo (Fleming et al. 2004). The resulting epigenetic effects can affect gene transcription in the presence of the appropriate transcription factors. Thus, whilst measurable at birth, the epigenetic changes may not be manifest in the phenotype until later in the life course, affecting for example the responses to later nutritional or metabolic challenges (Hanson & Gluckman, 2014). Such changes have been shown to be reversible in fetal and early postnatal life (Lillycrop et al. 2005; Burdge et al. 2009), although this is not necessarily universally the case. In addition, because some epigenetic changes can be induced in the germ cells, they can potentially be passed across generations via the egg and sperm (Skinner et al. 2010).
Epigenetic changes are established to play a role in ageing via effects on telomere stability (Blasco, 2007), antioxidant protection, metabolic homeostasis and effects on specific genes (Fraga & Estellar, 2007). NCDs associated with ageing, such as some forms of cancer, atherosclerosis, obesity, osteoporosis and sarcopenia, involve epigenetic processes but it is not yet known whether these processes are accelerated by epigenetic changes induced during development.
Immune function as an underlying process
Immune cells are strongly affected by ageing, which results in, among other effects, an increase in the number of memory cells at the expense of immunologically naive cells, termed immunosenescence, which is associated with reduction of the T‐cell repertoire and the ‘immunological space’ (Franceschi et al. 1999; McElhaney & Effros, 2009; Freund et al. 2010). However, both in individuals who successfully age and in those who do not, an increase in pro‐inflammatory status is observed (Baggio, 1998; Mari et al. 1995, 1996; Freund et al. 2010) and inter‐individual pro‐inflammatory thresholds are hypothesized to exist, depending on the age and coping mechanisms available within the individual: beyond this, ageing‐related disorders may develop (Franceschi et al. 2000).
Both fetal and childhood conditions, such as fetal nutrition, childhood maltreatment and the socioeconomic status of the family may contribute to the pro‐inflammatory trajectory and thereby to the development of ageing‐related disorders in later life (Surtees et al. 2003; Danese et al. 2007; Pollitt et al. 2007; West et al. 2010; Amarasekera et al. 2013). The induction of inflammatory responses across the life course may also contribute to the pro‐inflammatory trajectory of an individual (Franceschi et al. 2000).
Inflammatory responses constitute secondary responses to a stressor, when primary homeostatic mechanisms, such as the endocrine axes, are unsuccessful. As a consequence, allocation of metabolic resources will be shifted to the immune system to benefit the immediate survival of the individual. However, when inflammatory responses are chronically induced, the set point for metabolic parameters can be permanently shifted contributing to, amongst other conditions, increased insulin resistance (Kotas & Medzhitov, 2015). There is therefore an interaction between infectious and auto‐immune effects on the immune system and NCDs. The possible interaction may underlie early life effects on longevity associated with season of birth reported in The Gambia (Moore et al. 1997), effects which have both nutritional and epigenetic components.
Hence, by overriding homeostatic mechanisms such as endocrine responses, and by shifting the set point for metabolic parameters, inflammatory responses might have a strong impact on the developmental plasticity of an individual, which, depending upon the life course history, the environment and the socioeconomic resources available, may predispose individuals towards chronic disorders in later life.
Wider social and global issues
Increasing longevity in many populations around the world creates problems as well as benefits. Ageing populations can be expected to experience greater prevalence of chronic disease, especially NCDs (Prince et al. 2015); the compression of morbidity may result in extended healthy lives for many individuals but this may require investment. The trade‐offs between investments in health care for younger versus older members of the population raise complex issues (Fries, 1980), although, as this review explains, health at the two ends of life may be more related than previously thought.
The balance between health over the life course and the need for health care can be viewed using the economic analogy of capital versus stock (Dasgupta & Serageldin, 2001). Whilst both individuals and populations may have to utilize stock to pay for health care or to make up for lost earnings or capabilities when they suffer acute health problems, a sustainable life course health care policy will allow health capital to be retained or even increased. In low socio‐economic status groups in high income countries (HICs), and in low‐ to middle‐income countries (LMICs) more widely, stock will soon be exhausted and capital growth will become negative.
Not all individuals commence the life course with the same resources (Hanson & Gluckman, 2011 a). Individuals or groups who have a good start to life and healthy childhood development already have some health capital and achieve a higher peak intrinsic capacity for many functions: they may for example not have stunted growth and have a more extensive coronary vasculature. As life proceeds they are more likely to retain this inherited health capital and to build on it. Part of this inheritance may be in the form of genetic predisposition, but factors such as maternal nutrition, body composition and lifestyle are more important, as are prenatal care, immunization and promotion of breast feeding (Gluckman et al. 2008). Some of these effects are mediated by epigenetic processes in the offspring, which can alter the individual response to later challenges (Godfrey et al. 2007). They may even be established before conception. Whilst this has implications for the health of current and future parents, in reality a wider set of social factors and age groups influence the developmental environment of the next generation. The life course approach emphasizes that the health of one age group should not be considered in isolation from that of others, and raises broad social and environmental, as well as medical, considerations.
Turning to potential interventions, the life course approach indicates that a shift in trajectory may need to be produced at a time when an individual or population group is fundamentally healthy and not accessing health services voluntarily (Hanson & Gluckman, 2011 b). This emphasizes the importance of a health literacy approach, for example in adolescents (Kleinert, 2007). It also raises questions of equity in access not only to health care, but also to education, family planning and other empowerment issues, especially for girls and young women. These are issues which resonate with the Sustainable Development Goals and which need support in the post‐2015 era.
Lastly, the life course health trajectory concept provides a theoretical basis for instituting and measuring the efficacy of interventions at critical points (Fig. 1). The response of an individual to an additional challenge at a particular time point can be used to predict their future trajectory and likely health needs. Biomarkers of risk trajectory can thus be highly informative, obviating the need to wait for later outcomes such as manifestation of disease and shortening the time frame of evaluation of intervention trials substantially. The discovery of such biomarkers of risk trajectory, which can be measured in early life, makes this argument even more powerful.
Additional information
Competing interests
None declared.
Funding
M.A.H. is supported by The British Heart Foundation. R.J.A.H.E. is grateful for support from the Biotechnology and Biological Sciences Research Council (Doctoral Training Partnership), and the Fundatie van de Vrijvrouwe van Renswoude and the Scholten‐Cordes scholarship foundations.
Biographies
Mark Hanson is a leader in the field of developmental origins of health and disease (DOHaD). He is British Heart Foundation Professor and Director of the Institute of Developmental Sciences at the University of Southampton. His research revealed that nutrition and lifestyle of parents can affect their offspring's development, even before conception, passing on risk of non‐communicable diseases. The underlying mechanisms involve epigenetic processes, by which environmental factors alter gene function. He was a founder of LifeLab which aims to promote science and health literacy in children. Mark's co‐authored popular science books include Mismatch and Fat, Fate and Disease.

Cyrus Cooper is Professor of Rheumatology and Director of the MRC Lifecourse Epidemiology Unit; Vice‐Dean of the Faculty of Medicine at the University of Southampton; and Professor of Epidemiology at the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford. He leads an internationally competitive programme of research into the epidemiology of musculoskeletal disorders, most notably osteoporosis.
Geraldine Clough is Professor of Vascular Physiology at the University of Southampton where her research focuses on the life course determinants of vascular dysfunction and particularly of the microcirculation. She has been a member of Council of the Physiological Society and Senior Editor of the Journal of Physiology. Geraldine Clough has also served as President of the British Microcirculation Society and is currently deputy Editor‐in‐Chief of Microcirculation. She was a Visiting Professor at Southern University China (2008–2011) and is currently a Visiting Professor at King's College London.
This review was presented at the symposium “Microvascular plasticity and developmental priming: Impact on human health”, which took place at the 10th World Congress for Microcirculation in Kyoto, Japan, 25–27 September 2015.
References
- Alexander BT, Llinas M, Kruckeberg WC & Granger JP (2004). l‐Arginine attenuates hypertension in pregnant rats with reduced uterine perfusion pressure. Hypertension 43, 832–836. [DOI] [PubMed] [Google Scholar]
- Amarasekera M, Prescott SL & Palmer DJ (2013). Nutrition in early life, immune‐programming and allergies: the role of epigenetics. Asian Pac J Allergy Immunol 31, 175–182. [PubMed] [Google Scholar]
- Armitage JA, Taylor PD & Poston L (2005). Experimental models of developmental programming: consequences of exposure to an energy rich diet during development. J Physiol 565, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D & Loo B (2013). Vascular aging: chronic oxidative stress and impairment of redox signaling – consequences for vascular homeostasis and disease. Ann Med 45, 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio G, Donazzan S, Monti D, Mari D, Martini S, Gabelli C, Dalla Vestra M, Previato L, Guido M, Pigozzo S, Cortella I, Crepaldi G & Franceschi C (1998). Lipoprotein(a) and lipoprotein profile in healthy centenarians: a reappraisal of vascular risk factors. FASEB J 12, 433–437. [DOI] [PubMed] [Google Scholar]
- Baird J, Kurshid MA, Kim M, Harvey N, Dennison E & Cooper C (2011). Does birthweight predict bone mass in adulthood? A systematic review and meta‐analysis. Osteoporos Int 22, 1323–1334. [DOI] [PubMed] [Google Scholar]
- Barker DJP (1995). Fetal origins of coronary heart disease. BMJ 311, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP (1997). Intrauterine programming of coronary heart disease and stroke. Acta Paediatr Suppl 423, 178–182. [DOI] [PubMed] [Google Scholar]
- Barker DJP (1998). Mothers, Babies and Health in Later Life, 2nd edn. Churchill Livingstone, Edinburgh. [Google Scholar]
- Barker DJ, Thornburg KL, Osmond C, Kajantie E & Eriksson JG (2010). The surface area of the placenta and hypertension in the offspring in later life. Int J Dev Biol 54, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateson PPG & Gluckman P (2011). Plasticity, Robustness, Development and Evolution. Cambridge University Press. [Google Scholar]
- Bertram CE & Hanson MA (2001). Animal models and programming of the metabolic syndrome. Type 2 diabetes. Br Med Bull 60, 103–121. [DOI] [PubMed] [Google Scholar]
- Bjorksten J (1958). A common molecular basis for the aging syndrome. J Am Geriatr Soc 6, 740–748. [DOI] [PubMed] [Google Scholar]
- Blakemore SJ & Choudhury S (2006). Development of the adolescent brain: implications for executive function and social cognition. J Child Psychol Psychiatry 47, 296–312. [DOI] [PubMed] [Google Scholar]
- Blasco MA (2007). The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8, 299–309. [DOI] [PubMed] [Google Scholar]
- Bonacasa B, Siow RC & Mann GE (2011). Impact of dietary soy isoflavones in pregnancy on fetal programming of endothelial function in offspring. Microcirculation 18, 270–285. [DOI] [PubMed] [Google Scholar]
- Bouanane S, Benkalfat NB, Baba Ahmed FZ, Merzouk H, Mokhtari NS, Merzouk SA, Gresti J, Tessier C & Narce M (2009). Time course of changes in serum oxidant/antioxidant status in overfed obese rats and their offspring. Clin Sci (Lond) 116, 669–680. [DOI] [PubMed] [Google Scholar]
- Brawley L, Torrens C, Anthony FW, Itoh S, Wheeler T, Jackson AA, Clough GF, Poston L & Hanson MA (2004). Glycine rectifies vascular dysfunction induced by dietary protein imbalance during pregnancy. J Physiol 55, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J, Hanson M, Peebles C, Davies L, Inskip H, Robinson S, Calder PC, Cooper C & Godfrey KM (2015). Higher oily fish consumption in late pregnancy is associated with reduced aortic stiffness in the child at age 9 years. Circ Res 116, 1202–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA, Phillips ES, Slater‐Jefferies JL, Jackson AA & Hanson MA (2009). Folic acid supplementation during the juvenile‐pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr 139, 1054–1060. [DOI] [PubMed] [Google Scholar]
- Chapple SJ, Siow RC & Mann GE (2012). Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol 44, 1315–1320. [DOI] [PubMed] [Google Scholar]
- Chiavaroli V, Giannini C, D'Adamo E, de Giorgis T, Chiarelli F & Mohn A (2009). Insulin resistance and oxidative stress in children born small and large for gestational age. Pediatrics 124, 695–702. [DOI] [PubMed] [Google Scholar]
- Cooper C, Cawley M, Bhalla A, Egger P, Ring F, Morton L & Barker D (1995). Childhood growth, physical activity, and peak bone mass in women. J Bone Miner Res 10, 940–947. [DOI] [PubMed] [Google Scholar]
- Cooper C, Eriksson JG, Forsén T, Osmond C, Tuomilehto J & Barker DJP (2001). Maternal height, childhood growth and risk of hip fracture in later life: a longitudinal study. Osteoporos Int 12, 623–629. [DOI] [PubMed] [Google Scholar]
- Cooper C, Fall C, Egger P, Hobbs R, Eastell R & Barker D (1997). Growth in infancy and bone mass in later life. Ann Rheum Dis 56, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan MT, Vervaart PP, Permezel M, Georgiou HM & Rice GE (2004). Altered placental oxidative stress status in gestational diabetes mellitus. Placenta 25, 78–84. [DOI] [PubMed] [Google Scholar]
- Curhan GC, Chertow GM, Willett WC, Spiegelman D, Colditz GA, Manson JE, Speizer FE & Stampfer MJ (1996. a). Birth weight and adult hypertension and obesity in women. Circulation 94, 1310–1315. [DOI] [PubMed] [Google Scholar]
- Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL & Stampfer MJ (1996. b). Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation 94, 3246–3250. [DOI] [PubMed] [Google Scholar]
- Danese A, Pariante CM, Caspi A, Taylor A & Poulton R (2007). Childhood maltreatment predicts adult inflammation in a life‐course study. Proc Nat Acad Sci USA 104, 1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer B, Newman AE, Boonstra R, Palme R, Boutin S, Humphries MM & McAdam AG (2013). Density triggers maternal hormones that increase adaptive offspring growth in a wild mammal. Science 340, 1215–1217. [DOI] [PubMed] [Google Scholar]
- Darnton‐Hill I & Mkparu UC (2015). Micronutrients in pregnancy in low‐ and middle‐income countries. Nutrients 7, 1744–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta P & Serageldin I (2001). Social Capital: A Multifaceted Perspective. World Bank Publications, Washington DC. [Google Scholar]
- Dennery PA (2010). Oxidative stress in development: nature or nurture? Free Radic Biol Med 49, 1147–1151. [DOI] [PubMed] [Google Scholar]
- Dennison EM, Arden NK, Keen RW, Syddall H, Day IN, Spector TD & Cooper C (2001). Birthweight, vitamin D receptor genotype and the programming of osteoporosis. Paediatr Perinat Epidemiol 15, 211–219. [DOI] [PubMed] [Google Scholar]
- Dodds RM, Syddall HE, Cooper R, Benzeval M, Deary IJ, Dennison EM, Der G, Gale CR, Inskip HM, Jagger C, Kirkwood TB, Lawlor DA, Robinson SM, Starr JM, Steptoe A, Tilling K, Kuh D, Cooper C & Sayer AA (2014). Grip strength across the life course: normative data from twelve British studies. PLoS One 9, e113637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Morgan RG, Walker AE & Lesniewski LA (2015). Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol; DOI: 10.1016/j.yjmcc.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake AJ, Walker BR & Seckl JR (2005). Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol 288, R34–R38. [DOI] [PubMed] [Google Scholar]
- Drury SS, Theall K, Gleason MM, Smyke AT, De Vivo I, Wong JY, Fox NA, Zeanah CH & Nelson CA (2012). Telomere length and early severe social deprivation: linking early adversity and cellular aging. Mol Psychiatry 17, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Osmond C & Barker DJ (2001). Early growth and coronary heart disease in later life: longitudinal study. BMJ 322, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Twinn DS & Ozanne SE (2006). Mechanisms by which poor early growth programs type‐2 diabetes, obesity and the metabolic syndrome. Physiol Behav 88, 234–243. [DOI] [PubMed] [Google Scholar]
- Fleming TP, Kwong WY, Porter R, Ursell E, Fesenko I, Wilkins A, Miller DJ, Watkins AJ & Eckert JJ (2004). The embryo and its future. Biol Reprod 71, 1046–1054. [DOI] [PubMed] [Google Scholar]
- Forsdahl A (1977). Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease?. Br J Prev Soc Med 31, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF & Esteller M (2007). Epigenetics and aging: the targets and the marks. Trends Genet 23, 413–418. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luc, M , Ottaviani E, De Benedictis, G (2000). Inflamm‐aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci 908, 244–254. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Valensin S, Fagnoni F, Barbi C & Bonafè M (1999). Biomarkers of immunosenescence within an evolutionary perspective: the challenge of heterogeneity and the role of antigenic load. Exp Gerontol 34, 911–921. [DOI] [PubMed] [Google Scholar]
- Franco Mdo C, Ponzio BF, Gomes GN, Gil FZ, Tostes R, Carvalho MH & Fortes ZB (2009). Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci 85, 327–333. [DOI] [PubMed] [Google Scholar]
- Franco MC, Kawamoto EM, Gorjão R, Rastelli VM, Curi R, Scavone C, Sawaya AL, Fortes ZB & Sesso R (2007). Biomarkers of oxidative stress and antioxidant status in children born small for gestational age: evidence of lipid peroxidation. Pediatr Res 62, 204–208. [DOI] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY & Campisi J (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries JF (1980). Aging, natural death, and the compression of morbidity. N Engl J Med 303, 130–135. [DOI] [PubMed] [Google Scholar]
- Gabr M, Hashem N, Hashem M, Fahmi A & Safouh M (1960). Progeria, a pathologic study. J Pediatr 57, 70–77. [DOI] [PubMed] [Google Scholar]
- Gahagan S (2012). The development of eating behavior: biology and context. J Dev Behav Pediatr 33, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamborg M, Byberg L, Rasmussen F, Andersen PK, Baker JL, Bengtsson C, Canoy D, Droyvold W, Eriksson JG, Forsen T, Gunnarsdottir I, Jarvelin MR, Koupil I, Lapidus L, Nilsen TI, Olsen SF, Schack‐Nielsen L, Thorsdottir I, Tuomainen TP & Sorensen TI (2007). Birth weight and systolic blood pressure in adolescence and adulthood: meta‐regression analysis of sex‐ and age‐specific results from 20 Nordic studies. Am J Epidemiol 166, 634–645. [DOI] [PubMed] [Google Scholar]
- Gluckman PD & Hanson MA (2004). Living with the past: evolution, development, and patterns of disease. Science 305, 1733–1736. [DOI] [PubMed] [Google Scholar]
- Gluckman PD & Hanson MA (2004). Maternal constraint of fetal growth and its consequences. Semin Fetal Neonatal Med 9, 419–425. [DOI] [PubMed] [Google Scholar]
- Gluckman P & Hanson M (2006). Mismatch: Why Our World No Longer Fits Our Bodies Oxford University Press. [Google Scholar]
- Gluckman PD, Hanson MA & Beedle AS (2007). Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol 19, 1–19. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C & Thornburg KL (2008). Effect of in utero and early‐life conditions on adult health and disease. N Engl J Med 359, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman P, Hanson M, Spencer HG & Bateson P (2005). Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proc Biol Sci 272, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Bateson P, Beedle AS, Law CM, Bhutta ZA, Anokhin KV, Bougneres P, Chandak GR, Dasgupta P, Smith GD, Ellison PT, Forrester TE, Gilbert SF, Jablonka E, Kaplan H, Prentice AM, Simpson SJ, Uauy R & West‐Eberhard MJ (2009). Towards a new developmental synthesis: adaptive developmental plasticity and human disease. Lancet 373, 1654–1657. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD & Hanson MA (2007). Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res 61, 5R–10R. [DOI] [PubMed] [Google Scholar]
- Gupta P, Narang M, Banerjee BD & Basu S (2004). Oxidative stress in term small for gestational age neonates born to undernourished mothers: a case control study. BMC Pediatr 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA & Gluckman PD (2011. a). Developmental origins of noncommunicable disease: population and public health implications. Am J Clin Nutr 94 (6 Suppl.), 1754S–1758S. [DOI] [PubMed] [Google Scholar]
- Hanson MA & Gluckman PD (2011. b). Developmental origins of health and disease: moving from biological concepts to interventions and policy. Int J Gynaecol Obstet 115, Suppl. 1, S3–S5. [DOI] [PubMed] [Google Scholar]
- Hanson MA & Gluckman PD (2014). Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94, 1027–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey NC, Javaid MK, Arden NK, Poole JR, Crozier SR, Robinson SM, Inskip HM, Godfrey KM, Dennison EM, Cooper C; SWS Study Team (2010). Maternal predictors of neonatal bone size and geometry: the Southampton Women's Survey. J Dev Orig Health Dis 1, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey NC, Lillycrop KA, Garratt E, Sheppard A, McLean C, Burdge G, Slater‐Jefferies J, Rodford J, Crozier S, Inskip H, Emerald BS, Gale CR, Hanson M, Gluckman P, Godfrey K & Cooper C (2012). Evaluation of methylation status of the eNOS promoter at birth in relation to childhood bone mineral content. Calcif Tissue Int 90, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey NC, Sheppard A, Godfrey KM, McLean C, Garratt E, Ntani G, Davies L, Murray R, Inskip HM, Gluckman PD, Hanson MA, Lillycrop KA & Cooper C (2014). Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J Bone Miner Res 29, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Yano K, Matsui‐Hirai H, Yokoo H, Hattori Y & Iguchi A (2008). Nitric oxide and endothelial cellular senescence. Pharmacol Ther 120, 333–339. [DOI] [PubMed] [Google Scholar]
- Javaid MK, Prieto‐Alhambra D, Lui LY, Cawthon P, Arden NK, Lang T, Lane NE, Orwoll E, Barrett‐Conner E, Nevitt MC, Cooper C, Cummings SR; Osteoporotic Fractures in Men (MrOS) Research Group (2011). Self‐reported weight at birth predicts measures of femoral size but not volumetric BMD in eldery men: MrOS. J Bone Miner Res 26, 1802–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings BJ, Ozanne SE, Dorling MW & Hales CN (1999). Early growth determines longevity in male rats and may be related to telomere shortening in the kidney. FEBS Lett 448, 4–8. [DOI] [PubMed] [Google Scholar]
- Jones OR, Scheuerlein A, Salguero‐Gómez R, Camarda CG, Schaible R, Casper BB, Dahlgren JP, Ehrlén J, García MB, Menges ES, Quintana‐Ascencio PF, Caswell H, Baudisch A & Vaupel JW (2014). Diversity of ageing across the tree of life. Nature 505, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn AJ (1968). Embryogenic effect on post‐natal changes in hemoglobin concentration with time. Growth 32, 13–22. [PubMed] [Google Scholar]
- Kajantie E, Eriksson J, Osmond C, Thornburg K & Barker DJ (2009). Pre‐eclampsia is associated with increased risk of stroke in the adult offspring. The Helsinki birth cohort study. Stroke 40, 1176–1180. [DOI] [PubMed] [Google Scholar]
- Khan IY, Dekou V, Douglas G, Jensen R, Hanson MA, Poston L & Taylor PD (2005). A high‐fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Physiol Regul Integr Comp Physiol 288, R127–R133. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB (2011). Systems biology of ageing and longevity. Philos Trans R Soc Lond B Biol Sci 366, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TBL & Austad SN (2000). Why do we age? Nature 408, 233–238. [DOI] [PubMed] [Google Scholar]
- Kleinert S (2007). Adolescent health: an opportunity not to be missed. Lancet 369, 1057–1058. [DOI] [PubMed] [Google Scholar]
- Kotas ME & Medzhitov R (2015). Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuh D, Bassey J, Hardy R, Sayer AA, Wadsworth M & Cooper C (2002). Birth weight, childhood size, and muscle strength in adult life: evidence from a birth cohort study. Am J Epidemiol 156, 627–633. [DOI] [PubMed] [Google Scholar]
- Kuh D; New Dynamics of Ageing (NDA) Preparatory Network (2007). A life course approach to healthy aging, frailty, and capability. J Gerontol A Biol Sci Med Sci 62, 717–721. [DOI] [PubMed] [Google Scholar]
- Kyle UG & Pichard C (2006). The Dutch Famine of 1944–1945: a pathophysiological model of long‐term consequences of wasting disease. Curr Opin Clin Nutr Metab Care 9, 388–394. [DOI] [PubMed] [Google Scholar]
- Langley‐Evans SC, Phillips GJ, Benediktsson R, Gardner DS, Edwards CR, Jackson AA & Seckl JR (1996). Protein intake in pregnancy, placental glucocorticoid metabolism and the programming of hypertension in the rat. Placenta 17, 169–172. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Owen CG, Davies AA, Whincup PH, Ebrahim S, Cook D & Smith GD (2006). Sex differences in the association between birth weight and total cholesterol. A meta‐analysis. Ann Epidemiol 16, 19–25. [DOI] [PubMed] [Google Scholar]
- Leeson CP, Whincup PH, Cook DG, Donald AE, Papacosta O, Lucas A & Deanfield JE (1997). Flow‐mediated dilation in 9‐ to 11‐year‐old children: the influence of intrauterine and childhood factors. Circulation 96, 2233–2238. [DOI] [PubMed] [Google Scholar]
- Lesniewski LA, Connell ML, Durrant JR, Folian BJ, Anderson MC, Donato AJ & Seals DR (2009). B6D2F1 mice are a suitable model of oxidative stress‐mediated impaired endothelium‐dependent dilation with aging. J Gerontol A Biol Sci Med Sci 64, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RM, Cleal JK, Ntani G, Crozier SR, Mahon PA, Robinson SM, Harvey NC, Cooper C, Inskip HM, Godfrey KM, Hanson MA; Southampton Women's Survey Study Group , John RM (2012). Relationship between placental expression of the imprinted PHLDA2 gene, intrauterine skeletal growth and childhood bone mass. Bone 50, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RM, Greenwood SL, Cleal JK, Crozier SR, Verrall L, Inskip HM, Cameron IT, Cooper C, Sibley CP, Hanson MA & Godfrey KM (2010). Maternal muscle mass may influence system A activity in human placenta. Placenta 31, 418–422. [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA & Burdge GC (2005). Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr 35, 1382–1386. [DOI] [PubMed] [Google Scholar]
- Ligi I, Grandvuillemin I, Andres V, Dignat‐George F & Simeoni U (2010). Low birth weight infants and the developmental programming of hypertension: a focus on vascular factors. Semin Perinatol 34, 188–192. [DOI] [PubMed] [Google Scholar]
- Lopez‐Jaramillo P, Cohen DD, Gómez‐Arbeláez D, Bosch J, Dyal L, Yusuf S & Gerstein HC (2014). Association of handgrip strength to cardiovascular mortality in pre‐diabetic and diabetic patients: a subanalysis of the ORIGIN trial. Int J Cardiol 174, 458–461. [DOI] [PubMed] [Google Scholar]
- López‐Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyckx VA & Brenner BM (2015). Birth weight, malnutrition and kidney‐associated outcomes—a global concern. Nat Rev Nephrol 11, 135–149. [DOI] [PubMed] [Google Scholar]
- McElhaney JE & Effros RB (2009). Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol 21, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen IC & Robinson JS (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85, 571–633. [DOI] [PubMed] [Google Scholar]
- Mari D, Mannucci PM, Coppola R, Bottasso B, Bauer KA & Rosenberg RD (1995). Hypercoagulability in centenarians: the paradox of successful aging. Blood 85, 3144–3149. [PubMed] [Google Scholar]
- Mari D, Mannucci PM, Duca F, Bertolini S & Franceschi C (1996). Mutant factor V (Arg506Gln) in healthy centenarians. Lancet 347, 1044. [DOI] [PubMed] [Google Scholar]
- Martin‐Ruiz C, Jagger C, Kingston A, Collerton J, Catt M, Davies K, Dunn M, Hilkens C, Keavney B, Pearce SH, den Elzen WP, Talbot D, Wiley L, Bond J, Mathers JC, Eccles MP, Robinson L, James O, Kirkwood TB, von Zglinicki T (2011). Assessment of a large panel of candidate biomarkers of ageing in the Newcastle 85+ study. Mech Ageing Dev 132, 496–502. [DOI] [PubMed] [Google Scholar]
- Medvedev ZA (1990). An attempt at a rational classification of theories of ageing. Biol Rev 65, 375–398. [DOI] [PubMed] [Google Scholar]
- Menni C, Kastenmüller G, Petersen AK, Bell JT, Psatha M, Tsai PC, Gieger C, Schulz H, Erte I, John S, Brosnan MJ, Wilson SG, Tsaprouni L, Lim EM, Stuckey B, Deloukas P, Mohney R, Suhre K, Spector TD & Valdes AM (2013). Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol 42, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA (1999). Kleemeier Award Lecture: Are there genes for ageing? J Gerontal A Biol Sci Med Sci 54, B297–B307. [DOI] [PubMed] [Google Scholar]
- Moore SE, Cole TJ, Poskitt EM, Sonko BJ, Whitehead RG, McGregor IA & Prentice AM (1997). Season of birth predicts mortality in rural Gambia. Nature 388, 434–434. [DOI] [PubMed] [Google Scholar]
- Nilsson PM (1996). Premature ageing: the link between psychosocial risk factors and disease. Med Hypotheses 47, 39–42. [DOI] [PubMed] [Google Scholar]
- Norman M (2008). Low birth weight and the developing vascular tree: a systematic review. Acta Paediatr 97, 1165–1172. [DOI] [PubMed] [Google Scholar]
- Norman M & Bonamy AK (2005). Aortic wall thickening in utero. Lancet 365, 1444–1446. [DOI] [PubMed] [Google Scholar]
- Obernier K, Tong CK & Alvarez‐Buylla A (2014). Restricted nature of adult neural stem cells: re‐evaluation of their potential for brain repair. Front Neurosci 8, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Nishina H, Hanson MA & Poston L (2001). Dietary restriction in pregnant rats causes gender‐related hypertension and vascular dysfunction in offspring. J Physiol 530, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanne SE & Hales NC (2005). Poor fetal growth followed by rapid postnatal catch‐up growth leads to premature death. Mech Ageing Dev 126, 852–854. [DOI] [PubMed] [Google Scholar]
- Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI & Roseboom TJ (2008). Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 115, 1243–1249. [DOI] [PubMed] [Google Scholar]
- Pereira SMP & Power C (2012). Life course body mass index, birthweight and lipid levels in mid‐adulthood: a nationwide birth cohort study. Eur Heart J; DOI: 10.1093/eurheartj/ehs333. [DOI] [PubMed] [Google Scholar]
- Phillips DI (1995). Relation of fetal growth to adult muscle mass and glucose tolerance. Diabet Med 12, 686–690. [DOI] [PubMed] [Google Scholar]
- Pollitt RA, Kaufman JS, Rose KM, Diez‐Roux AV, Zeng D & Heiss G (2007). Early‐life and adult socioeconomic status and inflammatory risk markers in adulthood. Eur J Epidemiol 22, 55–66. [DOI] [PubMed] [Google Scholar]
- Poston L, Briley AL, Seed PT, Kelly FJ & Shennan AH; Vitamins in Pre‐eclampsia (VIP) Trial Consortium (2006). Vitamin C and vitamin E in pregnant women at risk for pre‐eclampsia (VIP trial): randomised placebo‐controlled trial. Lancet 367, 1145–1154. [DOI] [PubMed] [Google Scholar]
- Prince MJ, Wu F, Guo Y, Robledo LMG, O'Donnell M, Sullivan R & Yusuf S (2015). The burden of disease in older people and implications for health policy and practice. Lancet 385, 549–562. [DOI] [PubMed] [Google Scholar]
- Rattan SI (2008). Increased molecular damage and heterogeneity as the basis of aging. Biol Chem 389, 267–272. [DOI] [PubMed] [Google Scholar]
- Redman CWG & Sargent IL (2000). Placental debris, oxidative stress and pre‐eclampsia. Placenta 21, 597–602. [DOI] [PubMed] [Google Scholar]
- Roberts VH, Smith J, McLea SA, Heizer AB, Richardson JL & Myatt L (2009). Effect of increasing maternal body mass index on oxidative and nitrative stress in the human placenta. Placenta 30, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder LM (1973). Effect of the level of nutrition on rates of cell proliferation and of RNA and protein synthesis in the rat. Nutr Rep Int 7, 271–288. [Google Scholar]
- Roeder LM & Chow BF (1972). Maternal undernutrition and its longterm effects on the offspring. Am J Clin Nutr 25, 812–821. [DOI] [PubMed] [Google Scholar]
- Rumbold A & Crowther CA (2005). Vitamin C supplementation in pregnancy. Cochrane Database Syst Rev CD004072. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Cooper C, Evans JR, Rauf A, Wormald RP, Osmond C & Barker DJ (1998). Are rates of ageing determined in utero ? Age Ageing 27, 579–583. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Dunn R, Langley‐Evans S & Cooper C (2001). Prenatal exposure to a maternal low protein diet shortens life span in rats. Gerontology 47, 9–14. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Syddall HE, Dennison EM, Gilbody HJ, Duggleby SL, Cooper C, Barker DJ & Phillips DI (2004. a). Birth weight, weight at 1 year of age and body composition in older men: findings from the Hertfordshire Cohort Study. Am J Clin Nutr 80, 199–203. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Syddall HE, Gilbody HJ, Dennison EM & Cooper C (2004. b). Does sarcopenia originate in early life? Findings from the Hertfordshire cohort study. J Gerontol A Biol Sci Med Sci 59, M930–M934. [DOI] [PubMed] [Google Scholar]
- Sayer AA, Syddall H, O'Dell SD, Chen XH, Briggs PJ, Briggs, R & Cooper C (2002). Polymorphism of the IGF2 gene, birth weight and grip strength in adult men. Age Ageing 31, 468–470. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL & Donato AJ (2011). Aging and vascular endothelial function in humans. Clin Sci (Lond) 120, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeoni U, Ligi I, Buffat C & Boubred F (2011). Adverse consequences of accelerated neonatal growth: cardiovascular and renal issues. Pediatr Nephrol 26, 493–508. [DOI] [PubMed] [Google Scholar]
- Skilton MR, Siitonen N, Wurtz P, Viikari JS, Juonala M, Seppala I, Laitinen T, Lehtimaki T, Taittonen L, Kahonen M, Celermajer DS & Raitakari OT (2014). High birth weight is associated with obesity and increased carotid wall thickness in young adults: The Cardiovascular Risk in Young Finns study. Arterioscler Thromb Vasc Biol 34, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Skilton MR, Viikari JS, Juonala M, Laitinen T, Lehtimaki T, Taittonen L, Kahonen M, Celermajer DS & Raitakari OT (2011). Fetal growth and preterm birth influence cardiovascular risk factors and arterial health in young adults: the Cardiovascular Risk in Young Finns study. Arterioscler Thromb Vasc Biol 31, 2975–2981. [DOI] [PubMed] [Google Scholar]
- Skinner MK, Manikkam M & Guerrero‐Bosagna C (2010). Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab 21, 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloboda DM, Hart R, Doherty DA, Pennell CE & Hickey M (2007). Age at menarche: influences of prenatal and postnatal growth. J Clin Endocrinol Metab 92, 46–50. [DOI] [PubMed] [Google Scholar]
- Surtees P, Wainwright N, Day N, Brayne C, Luben R & Khaw KT (2003). Adverse experience in childhood as a developmental risk factor for altered immune status in adulthood. Int J Behav Med 10, 251–268. [DOI] [PubMed] [Google Scholar]
- Torrens C, Brawley L, Anthony FW, Dance CS, Dunn R, Jackson AA, Poston L & Hanson MA (2006). Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension 47, 982–987. [DOI] [PubMed] [Google Scholar]
- Torrens C, Ethirajan P, Bruce KD, Cagampang FR, Siow RC, Hanson MA, Byrne CD, Mann GE & Clough GF (2012). Interaction between maternal and offspring diet to impair vascular function and oxidative balance in high fat fed male mice. PLoS One 7, e50671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhees K, van Schooten FJ, van Waalwijk van Doorn‐Khosrovani SB, van Helden S, Munnia A, Peluso M, Briedé JJ, Haenen GR & Godschalk RW (2013). Intrauterine exposure to flavonoids modifies antioxidant status at adulthood and decreases oxidative stress‐induced DNA damage. Free Radic Biol Med 57, 154–161. [DOI] [PubMed] [Google Scholar]
- Varizi H, Dragowska W, Allsopp RC, Thomas TE, Harley CB & Lansdorp PM (1994). Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci USA 91, 9857–9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenendaal MV, Painter RC, de Rooij SR, Bossuyt PM, van der Post JA, Gluckman PD, Hanson MA & Roseboom TJ (2013). Transgenerational effects of prenatal exposure to the 1944–45 Dutch famine. BJOG 120, 548–553. [DOI] [PubMed] [Google Scholar]
- Wadley GD, Siebel AL, Cooney GJ, McConell GK, Wlodek ME & Owens JA (2008). Uteroplacental insufficiency and reducing litter size alters skeletal muscle mitochondrial biogenesis in a sex‐specific manner in the adult rat. Am J Physiol Endocrinol Metab 294, E861–E869. [DOI] [PubMed] [Google Scholar]
- West CE, Videky DJ & Prescott SL (2010). Role of diet in the development of immune tolerance in the context of allergic disease. Curr Opin Pediatr 22, 635–641. [DOI] [PubMed] [Google Scholar]
- Williams G (1957). Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. [Google Scholar]
- Williamson K, Stringer SE & Alexander MY (2012). Endothelial progenitor cells enter the aging arena. Front Physiol 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills AK, Lawlor DA, Matthews FE, Sayer AA, Bakra E, Ben‐Shlomo Y & Hardy R (2011). Life course trajectories of systolic blood pressure using longitudinal data from eight UK cohorts. PLoS Med 8, e1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajnik CS (2004). Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. J Nutr 134, 205–210. [DOI] [PubMed] [Google Scholar]