

Abstract
Atherosclerosis is a disease of ageing in that its incidence and prevalence increase with age. However, atherosclerosis is also associated with biological ageing, manifest by a number of typical hallmarks of ageing in the atherosclerotic plaque. Thus, accelerated biological ageing may be superimposed on the effects of chronological ageing in atherosclerosis. Tissue ageing is seen in all cells that comprise the plaque, but particularly in vascular smooth muscle cells (VSMCs). Hallmarks of ageing include evidence of cell senescence, DNA damage (including telomere attrition), mitochondrial dysfunction, a pro‐inflammatory secretory phenotype, defects in proteostasis, epigenetic changes, deregulated nutrient sensing, and exhaustion of progenitor cells. In this model, initial damage to DNA (genomic, telomeric, mitochondrial and epigenetic changes) results in a number of cellular responses (cellular senescence, deregulated nutrient sensing and defects in proteostasis). Ultimately, ongoing damage and attempts at repair by continued proliferation overwhelm reparative capacity, causing loss of specialised cell functions, cell death and inflammation. This review summarises the evidence for accelerated biological ageing in atherosclerosis, the functional consequences of cell ageing on cells comprising the plaque, and the causal role that VSMC senescence plays in atherogenesis.
Abbreviations
- ATM
ataxia telangiectasia mutated
- BMC
bone marrow‐derived cell
- CAD
coronary artery disease
- DDR
DNA damage response
- EC
endothelial cell
- EPC
endothelial progenitor cell
- γ‐H2AX
phosphorylated form of histone 2A protein X
- IGF1
insulin‐like growth factor‐1
- iNOS
inducible nitric oxide synthase
- MtDNA
mitochondrial DNA
- ROS
reactive oxygen species
- SASP
senescence‐associated secretory phenotype
- SIPS
stress‐induced premature senescence
- SOD
superoxide dismutase
- UPR
unfolded protein response
- VSMC
vascular smooth muscle cell
Introduction
Atherosclerosis is the commonest cause of death in the UK, and is projected to be the commonest cause of death in the world. The risk factors for atherosclerosis are well known, but one of the most important is increasing age. Ageing is associated with increased incidence, prevalence and mortality associated with atherosclerosis. Atherosclerosis is a slowly progressive disease, with histological evidence of plaque formation often as early as the second or third decade of life. Plaques develop over decades, growing both by gradual expansion and by episodes of rupture and repair that lead to rapid plaque growth. The association between chronological ageing and atherosclerosis has been explained by this gradual evolution of plaques through histologically defined stages that ultimately result in advanced lesions that are associated with clinical events. However, more recent evidence suggests that plaques also manifest accelerated biological ageing that is out of proportion to the chronological age. These ‘hallmarks of ageing’ are seen in advanced lesions, but their earliest features are also present as plaques develop.
Tissue hallmarks of ageing
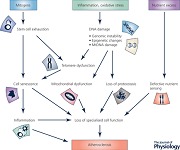
Although the functional consequences of ageing vary according to the tissue involved, there are a number of common markers or features of ageing that are seen to a variable extent across all tissues. These ‘hallmarks of ageing’ have been summarised and illustrated in an excellent recent review (Lopez‐Otin et al. (2013) and Fig. 1). These features include damage to both nuclear and mitochondrial DNA including attrition of telomeres and epigenetic changes, resulting in cellular senescence and exhaustion of stem/progenitor cells. Other consequences of cell senescence include defects in protein processing (proteostasis) and in sensing of nutrients, and also altered intercellular communication, for example by alterations in immune surveillance and release of pro‐inflammatory cytokines. We will review the evidence for these ‘hallmarks of ageing’ in atherosclerosis, with a particular focus on vascular smooth muscle cells (VSMCs).
Figure 1. Hallmarks of tissue ageing .
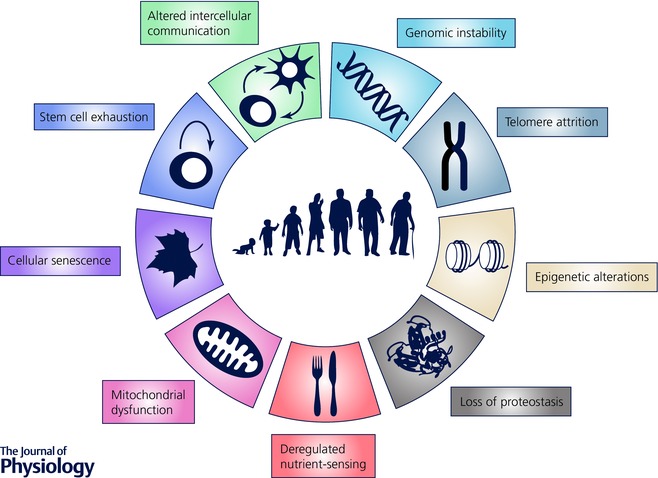
A full explanation is provided in the text. Adapted from Lopez‐Otin et al. (2013).
Genomic instability
DNA damage is evident in cells of human atherosclerotic plaques (particularly VSMCs and macrophages), including strand breaks, telomere shortening and base oxidation, particularly of guanosine residues (8‐oxo‐G). Both damage and markers of DNA repair increase with disease severity (Martinet et al. 2002; Matthews et al. 2006; Mahmoudi et al. 2008) suggesting that ongoing damage or defective DNA repair is an important feature of atherogenesis. DNA strand breaks and chromosomal damage are present in both plaque VSMCs (Gray et al. 2015) and circulating cells of patients with atherosclerosis, suggesting the interplay of both local and systemic factors that promote ageing in parallel with atherosclerosis. For example, DNA damage in circulating cells is associated with disease severity and correlates with a higher micronucleus index (a marker of genetic instability) compared with healthy controls (Botto et al. 2001).
Markers of DNA damage develop and may persist during atherosclerosis in animal models of atherosclerosis, and also in culture. For example, 8‐oxo‐G and DNA strand breaks develop in fat‐fed rabbits, associated with widespread expression of DNA repair enzymes; these changes revert on transfer to a normal diet, although 8‐oxo‐G may persist (Martinet et al. 2001). Similarly, increased DNA damage is seen in plaque‐derived VSMCs compared with normal VSMCs in culture, evidenced by increased expression of multiple DNA damage markers, including the phosphorylated forms of the ataxia telangiectasia mutated (ATM) and histone 2A protein X proteins (γ‐H2AX), and a longer tail length on Comet assay, a marker of DNA strand breaks (Mahmoudi et al. 2008; Gray et al. 2015). Thus, DNA damage (and by inference its consequences) can persist in vascular disease despite correction of the local extracellular environment.
Telomere attrition
Telomeres are high order structures composed of tandem repeats of the sequence TTAGGG that end in a single‐stranded 3′‐overhang of 50–300 nucleotides (Aubert & Lansdorp, 2008). This structure folds back to form a ‘T‐loop’ that caps the end of each chromosome (Griffith et al. 1999), and whose integrity depends on specific proteins that form the ‘shelterin complex’. This protein complex comprises telomeric repeat‐binding factor (TRF) 1, TRF2, protection of telomere protein 1 (POT1), TRF1‐interacting nuclear factor 2 (TIN2), tripeptidyl‐peptidase 1 (TPP1) and telomeric repeat‐binding factor 2‐interacting protein 1 (RAP1). Telomere protection depends particularly upon TRF2 and POT1 (van Steensel et al. 1998; Karlseder et al. 1999; Wu et al. 2006), which allow cells to distinguish between random DNA breaks and the natural end of the chromosome (Aubert & Lansdorp, 2008). Repeated cell division (d'Adda di Fagagna et al. 2003) or loss of key telomere binding proteins (Karlseder et al. 1999) destabilizes telomere integrity, which induces a DNA damage response (DDR) and leads to cellular senescence.
The telomeres of both VSMCs (Matthews et al. 2006) and ECs (Ogami et al. 2004) are shorter in atherosclerotic plaques relative to the normal vessel wall. Shorter telomeres are also seen in circulating endothelial progenitor cells (EPCs) (Carracedo et al. 2011), and in leukocytes in patients with atherosclerosis compared to control subjects (Brouilette et al. 2003, 2007), where they are inversely correlated to cardiovascular disease risks in patients with subclinical disease (Panayiotou et al. 2010; Willeit et al. 2010). Although it is hard to demonstrate critically short telomeres in these studies, ectopic activity of telomerase (the enzyme that maintains telomere length) can dramatically increase lifespan of both plaque and normal VSMCs (Matthews et al. 2006), suggesting that short telomeres and low levels of telomerase expression/activity are functionally important in VSMC senescence. In addition, whilst telomere length may reflect previous replication, arterial segments resistant to atherosclerosis, such as internal mammary artery or ascending aorta, have longer telomeres than the aortic regions prone to the disease (Nzietchueng et al. 2011; Chang & Harley, 1995). This difference is age‐independent, suggesting the existence of intrinsic genetic or developmental variations in telomere regulation may underlie location‐specific predisposition in atherogenesis.
Epigenetic modifications
Epigenetics defines genetic changes that are not related to differences in the coding sequences. For example, changes in DNA methylation and histone modification of vascular genes and growth factors are observed in the development and progression of atherosclerosis (Schiano et al. 2015). DNA hypomethylation occurs in monocytes, VSMCs and plaques of patients with atherosclerosis (Pogribny & Beland, 2009), while additional studies using ApoE−/− mice have shown that DNA hypomethylation represents a significant risk factor associated with susceptibility to atherosclerosis (Lund et al. 2004). The global DNA hypomethylation that predominates in human atherosclerosis causes activation of a number of gene clusters (Wang et al. 2014; Aavik et al. 2015).
Epigenetic changes are most likely due to changes exerted by DNA damaging agents. For example, DNA damage in vascular cells can result from exposure to oxidative stress, and under normal circumstances there are a variety of antioxidant enzymes that remove these damaging reactive oxygen species from the environment. Recent studies have shown that inflammatory cytokines can alter the expression of mediators of oxidative stress such as inducible nitric oxide synthase (iNOS) by causing changes to the chromatin structure of the promoter (Chan et al. 2005), and DNA hypomethylation of the gene for the antioxidant enzyme superoxide dismutase (SOD) results in reduced expression (Laukkanen et al. 1999). These gene expression changes are likely to force the cellular redox balance towards that of a highly oxidising environment that may potentially increase cellular DNA damage and enhance the progression of atherosclerotic lesions.
Loss of proteostasis
Proteostasis describes the biological pathways within cells that control the biogenesis, folding, trafficking and degradation of proteins present within and outside the cell. Protein storage defects, misfolding or increases in protein secretion can cause endoplasmic reticulum (ER) stress, which in turn induces activation of a series of ER‐resident proteins, including activating transcription factor‐6 (ATF‐6), inositol requiring protein‐1 (IRP‐1), and protein kinase RNA‐like ER kinase (PERK). These proteins direct signalling pathways that relieve ER stress in a process known as the unfolded protein response (UPR). Overall protein ubiquitination in atherosclerotic plaques is significantly increased, with an increase in proteasome activity in early atherosclerosis, but a decrease in advanced atherosclerosis (Wang et al. 2014). Proteasome inhibition can both reduce atherogenesis in mice, and protect vascular cells from oxidative stress (Dreger et al. 2009; Wilck et al. 2012).
UPR activation occurs in atherosclerosis, and in particular can underlie cell death in macrophages, VSMCs and endothelial cells (reviewed in Scull & Tabas, 2011). Defects in proteostasis also have major influences on other signalling pathways that underlie ageing; for example, NADP oxidases are integral signalling elements of the UPR during ER stress, with Nox4 and Nox2 regulating apoptosis after ER stress (reviewed in Laurindo et al. 2014). In addition, H2O2 is generated by the UPR in response to specific stressors, indicating that the ER surface provides a platform to spatially organize specific oxidative signalling events (Wu et al. 2010).
Deregulated nutrient sensing
Dietary restriction results in activation of a number of protective pathways, including those mediated through insulin‐like growth factor‐1 (IGF1)/Akt, 5’‐AMP‐activated protein kinase (AMPK) and the sirtuin family of histone deacetylases. The effectors of these pathways include forkhead box protein O (FOXO) proteins, mammalian target of rapamycin (mTOR) and the master regulator of metabolism, PGC1α (Fig. 2). In contrast, atherosclerosis is associated with extensive deposition and storage of both intracellular and extracellular lipid, resulting in signalling a state of nutrient ‘excess’.
Figure 2. Signalling pathways associated with nutrient excess .
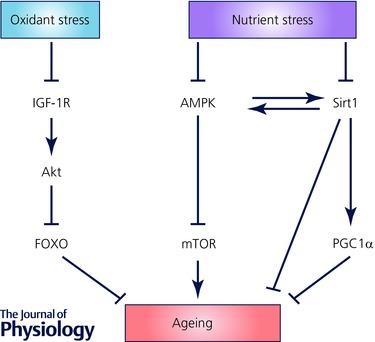
A full explanation is provided in the text.
Although there are multiple and conflicting signals, atherosclerosis is associated with down‐regulation of the IGF1 receptor, and subsequent reduction in Akt signalling and consequent activation of FOXO proteins (Patel et al. 2001; Allard et al. 2008). Loss of Akt1 results in increased atherosclerosis with multiples features of vulnerable plaques (Fernandez‐Hernando et al. 2007, 2009), a situation that is reversed by overexpression of Akt1 (Tucka et al. 2014). Atherosclerosis is also associated with down‐regulation of sirtuin 1 (Gorenne et al. 2013), which results in increased atherosclerosis, but also a more unstable plaque phenotype and aneurysm formation. In contrast, overexpression of sirtuin 1 results in reduced atherosclerosis, in part via effects on plasma lipids (Miranda et al. 2015). Oxidised LDL inhibits mTOR, which increases autophagy, whilst mTOR activation inhibits plaque EC death and atherosclerosis in ApoE−/− mice (Peng et al. 2014). Finally, knockout of PGC1α is associated with increased oxidative stress, mitochondrial abnormalities, reduced telomerase activity, reduced sirtuin 1 and the antioxidant catalase, and results in increased senescence‐associated‐beta‐galactosidase (SAβG) staining in mouse arteries and VSMC senescence in culture (Xiong et al. 2013).
Mitochondrial dysfunction
DNA damage occurs in both genomic and mitochondrial DNA (MtDNA); the latter is particularly susceptible to damage partly owing to the absence of protective histones within the DNA duplex, but also as a result of the close proximity to the ROS‐producing inner mitochondrial membrane. MtDNA damage occurs in both cells in atherosclerotic plaque and circulating cells in patients with severe coronary artery disease (CAD) (Botto et al. 2005; Yu et al. 2013). Similarly, MtDNA damage is an early event in atherosclerosis in experimental animals, and has been detected in arteries, circulating cells and other organs during atherogenesis (Ballinger et al. 2002; Yu et al. 2013).
The ‘free‐radical theory of ageing’ has been one of the major concepts behind both chronological and biological ageing, where ageing is due to an accumulation of oxidative DNA damage, predominantly driven by ROS. Mitochondrial DNA damage and dysfunction can increase ROS generation, and one of the difficulties has been separating the effects of mitochondrial ROS from loss of other mitochondrial functions. Increased vessel wall ROS and/or reduced ROS scavenger function promote atherosclerosis, and affect multiple aspects of VSMC biology, including cell proliferation, migration and release of pro‐inflammatory cytokines, controlled by a number of redox‐regulated pathways that show abnormal activity in ageing (see Li, 2010 for review). However, recent studies show that MtDNA damage is sufficient to cause mitochondrial dysfunction during atherogenesis without increased ROS, suggesting that MtDNA damage may have a causal role in atherosclerosis. For example, PolG −/− mice deficient in the mitochondrial polymerase‐γ proof‐reading enzyme (which accumulate extensive mtDNA damage and defects in oxidative phosphorylation) exhibited increased atherosclerosis associated with increased apoptosis and VSMC senescence with no change in ROS. PolG −/− bone marrow‐derived cells altered plaque characteristics, with increased necrotic core and reduction in the relative fibrous cap area, features associated with plaque vulnerability (Yu et al. 2013). PolG −/− monocytes also showed increased expression of a number of pro‐inflammatory cytokines, suggesting that mitochondrial DNA damage may promote atherosclerosis in part through inflammation (Yu et al. 2013).
Cellular senescence
DNA damage checkpoints delay the cell cycle, providing sufficient time for cells to repair lesions. However, ineffective DNA damage repair can lead to irreversible growth arrest, termed cellular senescence. In addition, the normal inability of eukaryotic cells to maintain the telomere length results in progressive telomere shortening with each division. The telomere is sensed as a strand break at a critical telomere length or structure, resulting in DDR activation and replicative senescence. Growth inhibition usually occurs in G1, and is regulated by pathways mediated by p16INK4a/retinoblastoma protein (pRB) and p53/p21 (Campisi, 2005). In addition, a range of extrinsic and intrinsic stimuli, particularly those causing intracellular oxidative stress, can induce DNA damage and senescence acutely (Herbert et al. 2008), termed ‘stress‐induced premature senescence’ (SIPS) (Toussaint et al. 2000). Both SIPS and replicative senescence induce the same phenotype of cells, and are mediated by some of the same pathways, although telomere length may be unaffected in SIPS. Aged vessels and atherosclerotic lesions show SAβG‐positive VSMCs, ECs and monocyte/macrophages, which are rarely seen in their respective young and normal counterparts (Minamino et al. 2002; Matthews et al. 2006), reinforcing the idea that atherosclerosis is associated with premature cellular senescence.
Similarly, human VSMCs derived from both aged vessels and advanced atherosclerotic plaques show markers of senescence in culture, including reduced proliferation, prolonged population doubling times (O'Brien et al. 1993; Bennett et al. 1995), and decreased S‐phase and increased G1 percentages consistent with a G1 growth arrest (Bennett et al. 1995). Growth arrest is associated with increased expression of the cyclin‐dependent kinase inhibitors p16INK4a and p21 (Matthews et al. 2006), decreased cyclin D and cyclin E (O'Sullivan et al. 2003), and pRB hypophosphorylation (Bennett et al. 1998; O'Sullivan et al. 2003; Matthews et al. 2006), similar to that seen in normal human VSMCs undergoing replicative senescence. Importantly, these cell cycle regulators become potential markers of vascular cell senescence.
Stem cell exhaustion
A number of studies suggest that some cells expressing VSMC or EC markers in the atherosclerotic plaque and normal vessel may be derived from bone marrow‐derived cells (BMCs) or endothelial progenitor cells (EPCs) that migrate and integrate into the vessel wall. Although the proportion of ECs and VSMCs from these sources appears small (Bentzon et al. 2006; Hagensen et al. 2010, 2012), dysfunctional EPCs (Heiss et al. 2005) and impaired BMC migration and adhesion (Heiss et al. 2005; Xu et al. 2011) are seen in both aged and atherosclerotic mouse models. ECs from aged animals show reduced expression of cell surface markers and cytokines for chemotaxis such as CXCR4 (Shao et al. 2011; Xu et al. 2011) and decreased HIF‐1α (Bosch‐Marce et al. 2007; Chang et al. 2007), as well as increased oxidative stress and inflammation (Zhang et al. 2009; Carracedo et al. 2011).
There is similar controversy over the presence of stem or progenitor smooth muscle cells in the vessel wall. VSMCs are not post‐mitotic, and can differentiate and adopt the phenotype of many different mesenchymal cells, including bone, cartilage and adipose tissue. Furthermore, populations of cells in the adventitia that demonstrate stem cell markers have been shown to contribute to neointima formation after vein grafting in mice, although their contribution to primary atherosclerosis appears low (Hu et al. 2004; Tsai et al. 2012). More recently, a population of multipotent vascular stem cells has been suggested that can also give rise to more specialised tissue including neurons and Schwann cells in addition to VSMCs (Tang et al. 2012). Studies such as these suggest that there is a vascular stem cell niche that may become exhausted through multiple divisions and/or damage (reviewed in Psaltis & Simari, 2015).
Altered intercellular communication
Cells undergoing senescence in response to a persistent DDR induce a ‘senescence associated secretory phenotype’ (SASP), which involves secretion of pro‐inflammatory cytokines, chemokines, growth factors and proteases (Passos et al. 2010; van Deursen, 2014). Evolutionarily the SASP may have a beneficial effect on the local environment, such that increased cell proliferation, migration or differentiation might promote tissue repair. However, immune cells can be mobilised by the SASP, which might promote atherosclerosis (Fyhrquist et al. 2013). In addition, senescent cells reinforce their phenotype to non‐senescent neighbouring cells via autocrine and paracrine effects (Acosta et al. 2008, 2013; van Deursen, 2014), which may promote atherosclerosis. For example, the matrix metalloproteinases including MMP‐1, MMP‐3 and MMP‐10 are SASP cytokines (Davalos et al. 2010) which contribute to extracellular matrix degradation and weakening of the vessel wall. Although, recent studies show that a persistent DDR stimulates interleukin‐6 and ‐8 secretion, the mechanism through which DNA damage induces the SASP is unclear (Rodier et al. 2009).
Functional consequences of cell ageing
Ageing‐associated changes are observed in multiple cell types and are conserved across species, from rodents to primates. As well as loss of normal cellular function, these changes result in specific consequences in each cell type, which may be directly pro‐atherogenic.
Endothelial cells
Aged ECs become flatter and enlarged and have an increasingly polypoid nucleus, all features associated with cellular senescence. These changes are accompanied by modulation in cytoskeleton integrity, proliferation, angiogenesis and cell migration. For example, senescent ECs show attenuated endothelial NO production (Sato et al. 1993) and increased endothelin‐1 release (Donato et al. 2009); late passage ECs also show increased expression of the adhesion molecules VCAM‐1 and ICAM‐1, increased activation of NF‐κB, and increased susceptibility to apoptosis (Khaidakov et al. 2011). Thus, EC senescence is associated with loss of EC function and a shift towards a pro‐inflammatory and pro‐apoptotic state, predicted to enhance monocyte migration into the vessel wall.
Vascular smooth muscle cells
VSMCs in human plaques or derived from plaques show reduced proliferation, early senescence and increased susceptibility to apoptosis (Bennett et al. 1995). These properties would reduce the ability to repair plaques that undergo rupture. Aged rodent aortas also show increased levels of interleukin‐6 (IL‐6) and aged aortic VSMCs have a higher basal secretion of IL‐6 than young VSMCs as part of the ‘senescence‐associated secretory phenotype’ (SASP). Moreover, aged VSMCs exhibit upregulation of chemokines (CCL2), adhesion molecules (e.g. ICAM‐1), and innate immune receptors (e.g. Toll‐like receptor 4) (Song et al. 2012). These properties generate a pro‐inflammatory environment, further promoting migration of inflammatory cells. VSMC senescence thus promotes atherosclerosis progression and inhibits plaque repair.
Inflammatory cells
As mentioned above, leukocyte telomere length is inversely associated with atherosclerosis in population studies, suggesting that leukocyte ageing has direct functional consequences that might promote atherosclerosis. Monocytes from patients with atherosclerosis demonstrate an increased burst of free radicals on activation and increased secretion of a number of cytokines, including MCP‐1, IL‐6, IL‐1β and TNF‐α (Calvert et al. 2011). Importantly, many of these differences are also observed in aged versus young monocytes and can be recapitulated by agents that disrupt telomeres (Calvert et al. 2011). Thus, there is direct evidence that ageing promotes pro‐inflammatory changes in monocytes/macrophages that are relevant to atherosclerosis (Calvert et al. 2011). Coupled with altered adhesion molecules on aged ECs, ageing would be predicted to promote both migration and activation of macrophages within plaques.
Conclusions
There is extensive evidence of accelerated biological ageing in atherosclerosis, with the large majority of the ‘hallmarks of ageing’ being present in advanced plaques. However, cellular ageing is not just a marker of disease, but contributes directly to both atherogenesis and the clinical sequelae of atherosclerosis, including plaque rupture that leads to myocardial infarction and stroke. The causal role of cellular ageing in atherosclerosis indicates that treatments aimed at both prevention of cellular ageing and reducing its consequences are viable therapeutic targets. Indeed, some of the current standard therapies for atherosclerosis may work in part by ameliorating the local arterial ageing process. For example, statins upregulate TRF2 (Spyridopoulos et al. 2004), decrease DNA damage by accelerating DNA damage repair (Pernice et al. 2006; Mahmoudi et al. 2008), and suppress oxidative stress in part by increasing antioxidant defences. Statins can also delay VSMC replicative senescence and reduce DNA damage in vivo in atherosclerosis (Mahmoudi et al. 2008). Similarly, angiotensin converting enzyme inhibitors reduce oxidative stress and subsequent DNA damage. The next step is to determine whether compounds directly targeted at ageing can safely slow the development of atherosclerosis.
Additional information
Competing interests
None.
Funding
This work was funded by British Heart Foundation Grants RG/08/009/25841, PG/11/112/29272 and PG/09/071, the NIHR Cambridge Biomedical Research Centre, and the BHF Centre for Research Excellence.
Biographies
Anna Uryga is a post‐doctoral research associate in Martin Bennett's laboratory. She undertook a MSc in Applied Biotechnology in Krakow, followed by a PhD studying the role of sirtuins in endothelial cell senescence and cytoprotection with Professor Jorge Erusalimsky in Cardiff. Her major interest is the role and regulation of senescence in vascular disease.
Martin Bennett trained in Cardiology in Birmingham and Cambridge. He was awarded one of the very first BHF 7‐year Clinician Scientist Fellowships in 1990 and undertook research training at the Imperial Cancer Research Fund Laboratories, London, studying vascular smooth muscle cell (VSMC) division and cell death in atherosclerosis and arterial injury. This was followed by a post‐doctoral position in Seattle, USA, and later award of a BHF Senior Fellowship in 1997. He currently holds the British Heart Foundation Chair of Cardiovascular Sciences at the University of Cambridge, with Honorary Consultant Cardiologist positions at Addenbrooke's and Papworth Hospitals, and heads the Division of Cardiovascular Medicine in Cambridge. He directs the Cambridge component of the Oxbridge Centre for Cardiovascular Regenerative Medicine, and co‐directs the Cambridge Cardiovascular Strategic Research Initiative and the Cambridge PhD programme in Cardiovascular Research. His major research interest is the vascular biology of atherosclerosis and artery development, and he has identified the mechanisms and consequences of VSMC apoptosis and cell senescence in atherosclerosis.

References
- Aavik E, Lumivuori H, Leppanen O, Wirth T, Hakkinen SK, Brasen JH, Beschorner U, Zeller T, Braspenning M, van Criekinge W, Makinen K & Yla‐Herttuala S (2015). Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J 36, 993–1000. [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L & Gil J (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E & Gil J (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. [DOI] [PubMed] [Google Scholar]
- Allard D, Figg N, Bennett MR & Littlewood TD (2008). Akt regulates the survival of vascular smooth muscle cells via Inhibition of FoxO3a and GSK3. J Biol Chem 283, 19739–19747. [DOI] [PubMed] [Google Scholar]
- Aubert G & Lansdorp PM (2008). Telomeres and aging. Physiol Rev 88, 557–579. [DOI] [PubMed] [Google Scholar]
- Ballinger SW, Patterson C, Knight‐Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K & Runge MS (2002). Mitochondrial integrity and function in atherogenesis. Circulation 106, 544–549. [DOI] [PubMed] [Google Scholar]
- Bennett M, Evan G & Schwartz S (1995). Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest 95, 2266–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Macdonald K, Chan SW, Boyle JJ & Weissberg PL (1998). Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 82, 704–712. [DOI] [PubMed] [Google Scholar]
- Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M & Falk E (2006). Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol 26, 2696–2702. [DOI] [PubMed] [Google Scholar]
- Bosch‐Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, Vandiver S, Rabi A, Shaked Y, Kerbel R, Lavallee T & Semenza GL (2007). Effects of aging and hypoxia‐inducible factor‐1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res 101, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Botto N, Berti S, Manfredi S, Al‐Jabri A, Federici C, Clerico A, Ciofini E, Biagini A & Andreassi MG (2005). Detection of mtDNA with 4977 bp deletion in blood cells and atherosclerotic lesions of patients with coronary artery disease. Mutat Res 570, 81–88. [DOI] [PubMed] [Google Scholar]
- Botto N, Rizza A, Colombo M, Mazzone A, Manfredi S, Masetti S, Clerico A, Biagini A & Andreassi M (2001). Evidence for DNA damage in patients with coronary artery disease. Mutat Res 493, 23–30. [DOI] [PubMed] [Google Scholar]
- Brouilette S, Singh RK, Thompson JR, Goodall AH & Samani NJ (2003). White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol 23, 842–846. [DOI] [PubMed] [Google Scholar]
- Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, Packard CJ & Samani NJ (2007). Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case‐control study. Lancet 369, 107–114. [DOI] [PubMed] [Google Scholar]
- Calvert PA, Liew TV, Gorenne I, Clarke M, Costopoulos C, Obaid DR, O'Sullivan M, Shapiro LM, McNab DC, Densem CG, Schofield PM, Braganza D, Clarke SC, Ray KK, West NE & Bennett MR (2011). Leukocyte telomere length is associated with high‐risk plaques on virtual histology intravascular ultrasound and increased proinflammatory activity. Arterioscler Thromb Vasc Biol 31, 2157–2164. [DOI] [PubMed] [Google Scholar]
- Campisi J (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. [DOI] [PubMed] [Google Scholar]
- Carracedo J, Merino A, Briceno C, Soriano S, Buendia P, Calleros L, Rodriguez M, Martin‐Malo A, Aljama P & Ramirez R (2011). Carbamylated low‐density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J 25, 1314–1322. [DOI] [PubMed] [Google Scholar]
- Chan GC, Fish JE, Mawji IA, Leung DD, Rachlis AC & Marsden PA (2005). Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J Immunol 175, 3846–3861. [DOI] [PubMed] [Google Scholar]
- Chang E & Harley CB (1995). Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci USA 92, 11190–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EI, Loh SA, Ceradini DJ, Chang EI, Lin SE, Bastidas N, Aarabi S, Chan DA, Freedman ML, Giaccia AJ & Gurtner GC (2007). Age decreases endothelial progenitor cell recruitment through decreases in hypoxia‐inducible factor 1α stabilization during ischemia. Circulation 116, 2818–2829. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP & Jackson SP (2003). A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426, 194–198. [DOI] [PubMed] [Google Scholar]
- Davalos AR, Coppe JP, Campisi J & Desprez PY (2010). Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 29, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K & Seals DR (2009). Vascular endothelial dysfunction with aging: endothelin‐1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 297, H425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreger H, Westphal K, Wilck N, Baumann G, Stangl V, Stangl K & Meiners S (2009). Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc Res 85, 395–403. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ & Sessa WC (2007). Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab 6, 446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Hernando C, Jozsef L, Jenkins D, Di Lorenzo A & Sessa WC (2009). Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol 29, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyhrquist F, Saijonmaa O & Strandberg T (2013). The roles of senescence and telomere shortening in cardiovascular disease. Nat Rev Cardiol 10, 274–283. [DOI] [PubMed] [Google Scholar]
- Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J & Bennett M (2013). Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation 127, 386–396. [DOI] [PubMed] [Google Scholar]
- Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J, Littlewood T & Bennett M (2015). Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res 116, 816–826. [DOI] [PubMed] [Google Scholar]
- Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H & de Lange T (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503–514. [DOI] [PubMed] [Google Scholar]
- Hagensen MK, Raarup MK, Mortensen MB, Thim T, Nyengaard JR, Falk E & Bentzon JF (2012). Circulating endothelial progenitor cells do not contribute to regeneration of endothelium after murine arterial injury. Cardiovasc Res 93, 223–231. [DOI] [PubMed] [Google Scholar]
- Hagensen MK, Shim J, Thim T, Falk E & Bentzon JF (2010). Circulating endothelial progenitor cells do not contribute to plaque endothelium in murine atherosclerosis. Circulation 121, 898–905. [DOI] [PubMed] [Google Scholar]
- Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M & Kalka C (2005). Impaired progenitor cell activity in age‐related endothelial dysfunction. J Am Coll Cardiol 45, 1441–1448. [DOI] [PubMed] [Google Scholar]
- Herbert KE, Mistry Y, Hastings R, Poolman T, Niklason L & Williams B (2008). Angiotensin II‐mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere‐dependent and independent pathways. Circ Res 102, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B & Xu Q (2004). Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE‐deficient mice. J Clin Invest 113, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S & de Lange T (1999). p53‐ and ATM‐dependent apoptosis induced by telomeres lacking TRF2. Science 283, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Khaidakov M, Wang X & Mehta JL (2011). Potential involvement of LOX‐1 in functional consequences of endothelial senescence. PLoS One 6, e20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukkanen MO, Mannermaa S, Hiltunen MO, Aittomaki S, Airenne K, Janne J & Yla‐Herttuala S (1999). Local hypomethylation in atherosclerosis found in rabbit EC‐SOD gene. Arterioscler Thromb Vasc Biol 19, 2171–2178. [DOI] [PubMed] [Google Scholar]
- Laurindo FR, Araujo TL & Abrahao TB (2014). Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal 20, 2755–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fukagawa NK (2010). Age‐related changes in redox signaling and VSMC function. Antioxid Redox Signal 12, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar‐Garea A, Ballestar E, Esteller M & Zaina S (2004). DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 279, 29147–29154. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T & Bennett M (2008). Statins use a novel Nijmegen breakage syndrome‐1‐dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res 103, 717–725. [DOI] [PubMed] [Google Scholar]
- Martinet W, Knaapen MW, De Meyer GR, Herman AG & Kockx MM (2001). Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 88, 733–739. [DOI] [PubMed] [Google Scholar]
- Martinet W, Knaapen MW, De Meyer GR, Herman AG & Kockx MM (2002). Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106, 927–932. [DOI] [PubMed] [Google Scholar]
- Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M & Bennett M (2006). Vascular smooth muscle cells undergo telomere‐based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res 99, 156–164. [DOI] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H & Komuro I (2002). Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105, 1541–1544. [DOI] [PubMed] [Google Scholar]
- Miranda MX, van Tits LJ, Lohmann C, Arsiwala T, Winnik S, Tailleux A, Stein S, Gomes AP, Suri V, Ellis JL, Lutz TA, Hottiger MO, Sinclair DA, Auwerx J, Schoonjans K, Staels B, Luscher TF & Matter CM (2015). The Sirt1 activator SRT3025 provides atheroprotection in Apoe‐/‐ mice by reducing hepatic PCSK9 secretion and enhancing LDLR expression. Eur Heart J 36, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nzietchueng R, Elfarra M, Nloga J, Labat C, Carteaux JP, Maureira P, Lacolley P, Villemot JP & Benetos A (2011). Telomere length in vascular tissues from patients with atherosclerotic disease. J Nutr Health Aging 15, 153–156. [DOI] [PubMed] [Google Scholar]
- O'Brien ER, Alpers CE, Stewart DK, Ferguson M, Tran N, Gordon D, Benditt EP, Hinohara T, Simpson JB & Schwartz SM (1993). Proliferation in primary and restenotic coronary atherectomy tissue. Implications for antiproliferative therapy. Circ Res 73, 223–231. [DOI] [PubMed] [Google Scholar]
- O'Sullivan M, Scott SD, McCarthy N, Figg N, Shapiro LM, Kirkpatrick P & Bennett MR (2003). Differential cyclin E expression in human in‐stent stenosis smooth muscle cells identifies targets for selective anti‐restenosis therapy. Cardiovasc Res 60, 673–683. [DOI] [PubMed] [Google Scholar]
- Ogami M, Ikura Y, Ohsawa M, Matsuo T, Kayo S, Yoshimi N, Hai E, Shirai N, Ehara S, Komatsu R, Naruko T & Ueda M (2004). Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol 24, 546–550. [DOI] [PubMed] [Google Scholar]
- Panayiotou AG, Nicolaides AN, Griffin M, Tyllis T, Georgiou N, Bond D, Martin RM, Hoppensteadt D, Fareed J & Humphries SE (2010). Leukocyte telomere length is associated with measures of subclinical atherosclerosis. Atherosclerosis 211, 176–181. [DOI] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB & von Zglinicki T (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VA, Zhang QJ, Siddle K, Soos MA, Goddard M, Weissberg PL & Bennett MR (2001). Defect in insulin‐like growth factor‐1 survival mechanism in atherosclerotic plaque‐derived vascular smooth muscle cells is mediated by reduced surface binding and signaling. Circ Res 88, 895–902. [DOI] [PubMed] [Google Scholar]
- Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B & Miao J (2014). An activator of mTOR inhibits oxLDL‐induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E‐/‐ mice. Sci Rep 4, 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernice F, Floccari F, Caccamo C, Belghity N, Mantuano S, Pacile ME, Romeo A, Nostro L, Barilla A, Crasci E, Frisina N & Buemi M (2006). Chromosomal damage and atherosclerosis. A protective effect from simvastatin. Eur J Pharmacol 532, 223–229. [DOI] [PubMed] [Google Scholar]
- Pogribny IP & Beland FA (2009). DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci 66, 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaltis PJ & Simari RD (2015). Vascular wall progenitor cells in health and disease. Circ Res 116, 1392–1412. [DOI] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR & Campisi J (2009). Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol 11, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato I, Morita I, Kaji K, Ikeda M, Nagao M & Murota S (1993). Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 195, 1070–1076. [DOI] [PubMed] [Google Scholar]
- Schiano C, Vietri MT, Grimaldi V, Picascia A, De Pascale MR & Napoli C (2015). Epigenetic‐related therapeutic challenges in cardiovascular disease. Trends Pharmacol Sci 36, 226–235. [DOI] [PubMed] [Google Scholar]
- Scull CM & Tabas I (2011). Mechanisms of ER stress‐induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol 31, 2792–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Xu Q, Wu Q, Ma Q, Salgueiro L, Wang J, Eton D, Webster KA & Yu H (2011). Defective CXCR4 expression in aged bone marrow cells impairs vascular regeneration. J Cell Mol Med 15, 2046–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shen H, Schenten D, Shan P, Lee PJ & Goldstein DR (2012). Aging enhances the basal production of IL‐6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 32, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyridopoulos I, Haendeler J, Urbich C, Brummendorf T, Hidemasa O, Schneider M, Zeiher A & Dimmeler S (2004). Statins enhance migratory capacity by upregulation of the telomere repeat binding factor TRF2 in endothelial progenitor cells. Circulation 110, 3136–3142. [DOI] [PubMed] [Google Scholar]
- Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, Helms JA & Li S (2012). Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun 3, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint O, Dumont P, Dierick JF, Pascal T, Frippiat C, Chainiaux F, Sluse F, Eliaers F & Remacle J (2000). Stress‐induced premature senescence. Essence of life, evolution, stress, and aging. Ann N Y Acad Sci 908, 85–98. [DOI] [PubMed] [Google Scholar]
- Tsai TN, Kirton JP, Campagnolo P, Zhang L, Xiao Q, Zhang Z, Wang W, Hu Y & Xu Q (2012). Contribution of stem cells to neointimal formation of decellularized vessel grafts in a novel mouse model. Am J Pathol 181, 362–373. [DOI] [PubMed] [Google Scholar]
- Tucka J, Yu H, Gray K, Figg N, Maguire J, Lam B, Bennett M & Littlewood T (2014). Akt1 regulates vascular smooth muscle cell apoptosis through FoxO3a and Apaf1 and protects against arterial remodeling and atherosclerosis. Arterioscler Thromb Vasc Biol 11, 2421–2428 [DOI] [PubMed] [Google Scholar]
- van Deursen J (2014). The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A & de Lange T (1998). TRF2 protects human telomeres from end‐to‐end fusions. Cell 92, 401–413. [DOI] [PubMed] [Google Scholar]
- Wang Z, Guo D, Yang B, Wang J, Wang R, Wang X & Zhang Q (2014). Integrated analysis of microarray data of atherosclerotic plaques: modulation of the ubiquitin‐proteasome system. PLoS One 9, e110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilck N, Fechner M, Dreger H, Hewing B, Arias A, Meiners S, Baumann G, Stangl V, Stangl K & Ludwig A (2012). Attenuation of early atherogenesis in low‐density lipoprotein receptor‐deficient mice by proteasome inhibition. Arterioscler Thromb Vasc Biol 32, 1418–1426. [DOI] [PubMed] [Google Scholar]
- Willeit P, Willeit J, Brandstatter A, Ehrlenbach S, Mayr A, Gasperi A, Weger S, Oberhollenzer F, Reindl M, Kronenberg F & Kiechl S (2010). Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler Thromb Vasc Biol 30, 1649–1656. [DOI] [PubMed] [Google Scholar]
- Wu L, Multani AS, He H, Cosme‐Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, Deng Y, Behringer RR & Chang S (2006). Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126, 49–62. [DOI] [PubMed] [Google Scholar]
- Wu RF, Ma Z, Liu Z & Terada LS (2010). Nox4‐derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 30, 3553–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong S, Salazar G, Patrushev N, Ma M, Forouzandeh F, Hilenski L & Alexander RW (2013). Peroxisome proliferator‐activated receptor γ coactivator‐1α is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol 33, 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang J, He J, Zhou M, Adi J, Webster KA & Yu H (2011). Impaired CXCR4 expression and cell engraftment of bone marrow‐derived cells from aged atherogenic mice. Atherosclerosis 219, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, Logan A, West NE, Clarke MC, Vidal‐Puig A, Murphy MP & Bennett MR (2013). Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher‐risk plaques in humans. Circulation 128, 702–712. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M & Rehman J (2009). Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor‐α via the p38 mitogen‐activated protein kinase pathway. FASEB J 23, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]