

Abstract
Although it is now clear that reactive oxygen species (ROS) are not the key determinants of longevity, a number of studies have highlighted the key role that these species play in age‐related diseases and more generally in determining individual health span. Age‐related loss of skeletal muscle mass and function is a key contributor to physical frailty in older individuals and our current understanding of the key areas in which ROS contribute to age‐related deficits in muscle is through defective redox signalling and key roles in maintenance of neuromuscular integrity. This topical review will describe how ROS stimulate adaptations to contractile activity in muscle that include up‐regulation of short‐term stress responses, an increase in mitochondrial biogenesis and an increase in some catabolic processes. These adaptations occur through stimulation of redox‐regulated processes that lead to the activation of transcription factors such as NF‐κB, AP‐1 and HSF1 which mediate changes in gene expression. They are attenuated during ageing and this appears to occur through an age‐related increase in mitochondrial hydrogen peroxide production. The potential for redox‐mediated cross‐talk between motor neurons and muscle is also described to illustrate how ROS released from muscle fibres during exercise may help maintain the integrity of axons and how the degenerative changes in neuromuscular structure that occur with ageing may contribute to mitochondrial ROS generation in skeletal muscle fibres.
Introduction
The free radical theory of ageing was originally formulated in the 1950s (Harman, 1956) and has been one of the most resilient and examined of the many subsequent theories that have been proposed. It is now recognised that the free radical theory and its various derivatives cannot exclusively explain the ageing process (Romano et al. 2010; Pulliam et al. 2013) and in particular there is no invariable direct relationship between the extent of free radical, or reactive oxygen‐induced damage (i.e. oxidative damage) and the onset or rate of ageing in tissues (Muller et al. 2007 a). Nevertheless data indicate that some aspects of the ageing phenotype and age‐related disorders appear to be mediated by reactive oxygen species (ROS) (Muller et al. 2007 a; Salmon et al. 2010). In this topical review we will address two key areas relating to how ROS influence ageing of the neuromuscular system and play a role in age‐related deficits in skeletal muscle. These are the disruption of redox signalling in muscle that occurs during ageing and the role of ROS in nerve–muscle interactions that maintain muscle mass and function. The aim is to highlight current developments in these topics and to identify areas where further research is required.
Age‐related loss of skeletal muscle mass and function (sarcopenia)
The term ‘sarcopenia’ was coined over 20 years ago (Evans & Campbell, 1993), and the definition was recently revised as a ‘progressive age‐related loss of muscle mass and associated muscle weakness’ (Lynch, 2011). Between the ages of 50 and 80 years a 30–50% loss of muscle mass and decrease in strength occur that are major contributors to physical frailty which has a major negative effect on the quality of life of older people and contributes to loss of independence in older people (Young & Skelton, 1994). Despite the importance of this area limited progress has been made in understanding the mechanisms responsible for age‐associated muscle atrophy and weakness.
Analysis of post‐mortem human vastus lateralis muscles have shown a 40% reduction in total muscle area accompanied by ∼50% loss of muscle fibres between 50 and 80 years of age (Lexell et al. 1988). Old rodents also show reductions in muscle fibre number with ageing (Larkin et al. 2011). The fibre loss is associated with a loss of motor units (Campbell et al. 1973; Larson & Ansved, 1995) and the number of motor axons innervating skeletal muscles are decreased in old rodents (Larson & Ansved, 1995) and old humans (Krantic et al. 2005). Despite the strong associations between the losses of muscle fibres and motor axons, a cause–effect relationship between the loss of these two tissues has not yet been established.
The surviving motor neurons show axonal sprouting that has been proposed to rescue muscle fibres that have become temporarily denervated, resulting in an increase in average motor unit size (Brown et al. 1988). It has been proposed that the ability of motor units to increase their size is limited, and muscle fibres and motor units are eventually lost (Delbono, 2003). Ageing is associated with numerous pre‐ and postsynaptic structural abnormalities in peripheral nerve endings, including segmental demyelination (Adinolfi et al. 1991), demyelinated and remyelinated axons, and denervated Schwann cell columns (Grover‐Johnson & Spencer, 1981), synaptic detachment, partial or complete withdrawal of axons from postsynaptic sites, and fragmentation of postsynaptic motor endplates (Jang et al. 2010; Chai et al. 2011). Recent data from rodents also indicate that changes in the peripheral regions of motor units are observed prior to any loss in number of motor neuron cell bodies in the lumbar spinal cord (Chai et al. 2011), suggesting that degenerative processes in the peripheral regions of motor nerves may play an important role.
Role of oxidative damage in ageing
The effect of ageing on levels of oxidative damage in tissues of many species has been studied extensively and it is apparent that all tissues, including skeletal muscle, of old organisms contain greater oxidative damage to lipids, DNA and proteins in comparison with younger organisms (e.g. Vasilaki et al. 2006 b). In non‐mammalian models, initial interventions to reduce the ROS activities throughout life extended lifespan (Orr & Sohal, 2003), but work from Richardson and colleagues (Perez et al. 2009) and Gemms and Doonan (2009) has demonstrated a lack of any true correlation between the level of oxidative damage and lifespan in different models and argues strongly against a primary role for oxidative damage in ageing (Gems & Doonan, 2009). In mammals, few genetic manipulations to reduce ROS activities have resulted in increased lifespan (e.g. Schriner et al. 2005). It therefore seems clear that levels of ROS generation and oxidative damage are not the fundamental determinants of lifespan.
Although ROS may not be the fundamental determinant of lifespan many studies have indicated that mitochondrial ROS generation is increased in tissues, including skeletal muscle, during ageing and that this is associated with impaired mitochondrial function and oxidative damage (e.g. Vasilaki et al. 2006 b). Furthermore authors have argued that this increased ROS generation with age is important in contributing to age‐related diseases (Muller et al. 2007 a) and more generally to individual health span (Salmon et al. 2010).
Redox signalling in skeletal muscle and its dysregulation during ageing
Contractile activity increases the generation of superoxide and nitric oxide (NO) by skeletal muscle fibres with the formation of secondary reactive oxygen species (ROS) and reactive nitrogen species (Powers & Jackson, 2008). NO generation is regulated by the nitric oxide synthases, but the sites that generate superoxide during exercise have remained relatively unclear. Initial data suggested that the mitochondrial electron transport chain was the predominant source of superoxide although a number of studies have identified NADPH oxidase enzymes in the plasma membrane, T‐tubules and mitochondria (Sakellariou et al. 2014 b). Recent studies have directly compared the generation of superoxide from mitochondrial and cytosolic sources in contracting skeletal muscle (Sakellariou et al. 2013; Pearson et al. 2014) and these data indicate that NAD(P)H oxidases are the major source during a short period of contractions (Sakellariou et al. 2013; Pearson et al. 2014). This has significant implications for understanding the role of localised ROS generation in contracting muscle since the only function of NAD(P)H oxidases is to generate superoxide (or hydrogen peroxide) and hence these species are not produced by chance, or as a by‐product of metabolism.
In normal physiology ROS mediate some adaptive processes to physiological stresses through changes in gene expression (Droge, 2002). Signalling by these reactive molecules appears to be mainly achieved by targeted modifications of specific residues in proteins (Janssen‐Heininger et al. 2008). In skeletal muscle the ROS and NO generated during contractile activity appear to mediate the activation of a number of redox‐regulated transcription factors, including Nuclear Factor‐kappa B (NF‐κB), Activator Protein‐1 (AP‐1), Heat Shock Factor‐1 (HSF‐1) and nuclear factor erythroid 2 ‐related factor 2 (Nrf2) (Ji et al. 2004; Vasilaki et al. 2006 b; Ristow et al. 2009) with a subsequent increased expression of regulatory enzymes and cytoprotective proteins (McArdle et al. 2001). The full extent of the adaptive processes in muscle that are regulated through redox‐dependent systems is unclear, but appears to also include some catabolic processes and mitochondrial biogenesis (Powers & Jackson, 2008).
The finding that ROS mediate adaptations to contractile activity and other adaptive responses in tissues is based on three lines of evidence: (i) demonstration of an association of increased ROS generation with the response; (ii) the inhibitory effect of suppression or scavenging of ROS on the response; and (iii) data demonstrating that specific ROS can activate the relevant pathway. Hydrogen peroxide (H2O2) is widely viewed as the only ROS likely to play a major role in signalling and H2O2 has been shown to activate NF‐κB (Zhang et al. 2001), AP‐1 (Aggeli et al. 2006) and many other transcription factors (Marinho et al. 2014). Thus the concept has arisen that H2O2, which is generated at specific sites within muscle but is readily diffusible, can interact with activation pathways for these specific transcription factors leading to their activation. These studies have utilised H2O2 concentrations typically in the range 10−4–10−3 m and it is relevant to consider whether these concentrations have any in vivo relevance. The intracellular H2O2 concentration is in the order of 10−9–10−8 m (Sies, 2014) and we have calculated that the increase in muscle during contractions appears to be to a maximum of 10−7 m (Jackson, 2011). This is therefore a factor of 1000 below the concentrations reported to activate most transcription factors in vitro. Thus, the generally held concept of H2O2 generated from a specific enzyme system that is localised at specific sub‐cellular sites, then diffusing through the cell and encountering redox‐regulated proteins with which it reacts may be relatively naive.
An alternative explanation for the process of redox signalling has evolved to account for this recognition that most so‐called redox‐sensitive proteins are unlikely to be oxidised by H2O2 at physiological concentrations and the possibility of redox signalling through thiol oxidation by H2O2 has evolved. This potential mechanism involves the transfer of oxidative equivalents directly from a sensitive thiol peroxidase to a specific target protein through direct protein–protein contact allowing conversion of the oxidising equivalent from H2O2 into a disulphide bond that can be subsequently transmitted to other substrates through the formation of intermolecular disulphides. Thus thiol peroxidases transmit oxidising equivalents to a specific target protein to facilitate H2O2 signalling (Sobatta et al. 2015). This mechanism has been well documented in yeast (Delaunay et al. 2002; Gutscher et al. 2009), but has only recently been shown to account for activation of a transcription factor by H2O2 in animal cells (Sobatta et al. 2015). Key components of such signalling pathways are peroxiredoxins (Prx) and thioredoxins (Trx). Prx are a family of antioxidant enzymes which reduce hydroperoxides to water in the presence of electron donors. Prx are classified by the number of cysteine (Cys) residues involved in the peroxidase activity: 2‐Cys Prx and 1‐Cys Prx. The 2‐Cys Prxs form a disulphide bond by reacting with peroxides and the disulphide is reduced by thioredoxin (Trx) which is then reduced by Trx reductase and NAD(P)H (Park et al. 2014). Prx are generally considered to be important antioxidant enzymes in the cytosol (Prx1, Prx2, Prx5), mitochondria (Prx3, Prx5) and endoplasmic reticulum (Prx4). Importantly and in contrast to the relatively poor reactivity of the so‐called redox‐sensitive proteins involved in activating transcription factors discussed previously, Prx are several orders of magnitude more reactive with H2O2 (Sobatta et al. 2015) and act to scavenge H2O2 at the low concentrations found in muscle fibres.
This latter potential signalling system does not appear to have been studied in skeletal muscle, but recent studies in other cell types indicate that Prxs can function as a signal peroxidase to activate specific pathways. Prx1 has been shown to activate the transcription factor ASK1 (Jarvis et al. 2012), and Prx2 forms a redox relay with the transcription factor STAT3 such that oxidative equivalents flow from Prx2 to STAT3 generating disulphide‐linked STAT3 oligomers with modified transcriptional activity (Sobatta et al. 2015). Figure 1 shows examples of how the two potential mechanisms for redox signalling might account for the activation of a transcription factor (TF) such as NF‐κB by contraction‐induced ROS in skeletal muscle.
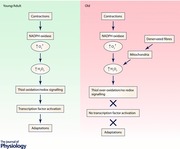
Figure 1. Schematic representation of the two potential pathways of redox signalling to account for activation of key transcription factors following contractile activity in skeletal muscle .
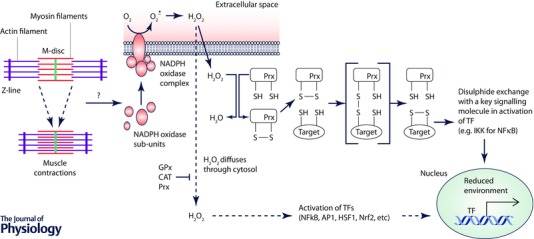
Contractions initially lead to activation of NADPH oxidase (probably Nox2) within muscle. This occurs through rapid translocation of the regulatory sub‐units of NADPH oxidase to a muscle membrane and assembly of the catalytic enzyme. It is currently unknown how contractile activity leads to activation of this enzyme. The NADPH oxidase generates superoxide that is rapidly converted to H2O2. Some evidence suggests that the major NADPH oxidases predominantly generate superoxide on the outside of the muscle fibre with some H2O2 generated rapidly diffusing into the fibre although this is not firmly established. The process by which the increased H2O2 leads to activation of transcriptional responses is the subject of debate, but the conventional view is that local concentrations of H2O2 are sufficiently high for it to diffuse through the cytoplasm and interact with redox‐sensitive components of pathways activating various transcription factors (shown as a dashed line in the scheme). Note that the cytoplasm contains various enzymes that can degrade H2O2 and compounds (e.g. glutathione) with which it can react. The alternative pathway involves the reaction of low levels of H2O2 with a highly reactive protein (e.g. Prx or Trx) that is closely associated with the NADPH oxidase with subsequent formation of disulphides and disulphide exchange with partner proteins thus transferring oxidising equivalents from H2O2 to proteins with which it would not react at low concentrations. Subsequent activation of the TF occurring through disulphide exchange with a key signalling protein. This pathway has not yet been shown to occur in skeletal muscle. Ageing appears to influence the overall scheme leading to an inability of contractile activity to further activate these transcription factors, but it is currently unknown how this occurs. GPx, glutathione peroxidase; CAT, catalase; Prx, peroxiredoxin; IKK, I kappa B kinase; TF, transcription factor.
It will be important to define which (if either) of these potential redox signalling systems plays a role in adaptations of muscle to contractions since they may provide alternative mechanisms by which aberrant ROS influence the age‐related loss of muscle mass and function. Thus for instance, ROS mediate increased expression of heat shock proteins (HSPs) and other cytoprotective proteins in muscle following contractions in adult mice (Vasilaki et al. 2006 a; Jackson & McArdle, 2011) and this response is attenuated in old mice (Vasilaki et al. 2006 a). Furthermore this attenuated response contributes to age‐related loss of muscle mass and function (Jackson & McArdle, 2011). Thus transgenic mouse studies have demonstrated that aberrant activation of adaptive responses plays a key role in age‐related muscle dysfunction since lifelong overexpression of cytosolic HSP70 or mitochondrial HSP10 normalised NF‐κB activation at rest and reduced functional deficits in muscle of old mice (Kayani et al. 2010).
We have previously proposed that aberrant hydrogen peroxide generation from mitochondria that occurs during ageing could explain this attenuation of adaptive responses leading to a failure to induce important cytoprotective and other responses (Jackson & McArdle 2011), but this has not been examined experimentally. Understanding of the processes by which the redox‐mediated adaptations to contractions occur is therefore a prerequisite to defining how they are modified by ageing.
Effect of modification of ROS on neuromuscular ageing
There are a small number of studies that indicate that a very specific manipulation of ROS activities can preserve muscle function during ageing (e.g. Schriner et al. 2005) and in collaboration with colleagues in the USA our group have undertaken studies to examine the effects of deletion of regulatory enzymes for ROS on neuromuscular ageing in mice. Despite frequent observation of increased oxidative damage in these models, no clear relationship with neuromuscular ageing was generally seen. The exception to this pattern was in mice with a whole body deletion of Cu,Zn superoxide dismutase (Sod1) which show neuromuscular changes with ageing that have been claimed to reflect an accelerated skeletal muscle ageing process (Muller et al. 2006). Adult Sod1 knockout (KO) mice show a decline in skeletal muscle mass, loss of muscle fibres and a decline in the number of motor units, loss of motor function and contractility, partial denervation and mitochondrial dysfunction by 8 months of age (Larkin et al. 2011). These are all changes that are also seen in old wild‐type (WT) mice, but not until after 22–24 months of age.
Sod1 is present in both the cytosol of cells and within the mitochondrial inter‐membrane space (Kawamata & Manfredi, 2010) and hence lack of Sod1 may influence redox homeostasis in the mitochondria and cytosol. Jang et al. (2010) showed that this model was associated with a large increase in mitochondrial H2O2 production and in our studies we examined the nature of other ROS that are generated in the cytosol of muscle from mice lacking Sod1. We concluded that increased peroxynitrite in muscle may play an important role in the phenotype of Sod1KO mice since muscle fibres from adult Sod1KO mice did not show an increase in cytosolic superoxide availability at rest, but muscles demonstrated evidence for an increase in peroxynitrite. In Sod1KO mice, this was indicated by an increased 3‐nitrotyrosine (3‐NT) content of muscle proteins and increased expression of the peroxynitrite reductase, peroxiredoxin V (Sakellariou et al. 2011).
We also showed that, in common with old WT mice, muscles of Sod1KO mice demonstrated a constitutive activation of NF‐κB with increased production of pro‐inflammatory cytokines and a constitutive increase in the content of a number of HSPs in muscle at rest and also failed to further activate cytoprotective adaptive responses to contractile activity. This results in diminished acute additional expression of HSPs and other cytoprotective proteins following contractile activity. This failed activation in response to contractile activity could potentially occur through a lack of induction of additional superoxide and/or hydrogen peroxide during contractile activity (Sakellariou et al. 2011). Other data suggest that this lack of a contraction‐induced generation of ROS in the Sod1KO mice may be due to a failure of activation of muscle NADPH oxidase activity (Sakellariou et al. 2013). Thus, a further effect of the lack of Sod1 that mimics that seen in old WT mice is a failure of redox‐mediated signalling of adaptive responses to contractile activity.
In subsequent work our group of investigators has examined whether the muscle atrophy in this model is initiated by changes within muscle fibres or motor neurons. Surprisingly, mice with skeletal muscle‐specific deletion of Sod1 (mSod1KO mice) show no evidence of neuromuscular junction degeneration (NMJ) or loss of muscle fibres and indeed showed some muscle hypertrophy (Zhang et al. 2013). Our group also examined whether the changes in ROS generation observed in Sod1KO mice were also seen in mSod1KO mice. The multiple changes seen in Sod1KO mice were not observed in the muscles of mSod1KO mice, including the increases in 3‐NT, catalase and peroxiredoxin V previously reported in muscles of Sod1KO mice (Zhang et al. 2013). To determine the role of motor neurons in the loss of muscle mass and function in Sod1KO mice, we subsequently established a transgenic Sod1KO mouse in which human SOD1 is expressed in neurons under the control of a synapsin 1 promoter (nSOD1‐Tg‐Sod1KO mice). These ‘nerve rescue’ mice expressed SOD1 in central and peripheral neurons but not other tissues. Sciatic nerve CuZnSOD content in nSOD1‐Tg‐Sod1KO mice was ∼20% of WT control mice, but they showed no loss of muscle mass or maximum isometric specific force production at 8–12 months of age, when significant reductions were seen in Sod1KO mice (Sakellariou et al. 2014 a). Thus these data implicate a lack of Sod1 specifically in motor neurons in the pathogenesis of the accelerated muscle ageing phenotype seen in the Sod1KO mice. We have also recently examined the effect of neuron‐specific Sod1 knockout in nSod1KO mice, but this model also does not recapitulate the full sarcopenia phenotype seen in Sod1KO mice and shows only minor changes in muscle mass and function (Sataranatarajan et al. 2015). The implication of this work appears to be that both neurons and muscle contribute to maintenance of neuromuscular function in this model and that deletion of Sod1 in both tissues is necessary to generate the full sarcopenic phenotype.
Thus studies of the Sod1KO model have demonstrated the importance of nerve–muscle interactions in the maintenance of neuromuscular function where ROS homeostasis is compromised during ageing. Since adult mice lacking Sod1 replicate many of the features seen in old WT mice they may indicate key mechanisms that lead to loss of muscle fibres and function that are relevant to the ageing of WT mice. Nevertheless we stress that Sod1KO mice provide a model to identify fundamental mechanisms that are highly relevant to understanding muscle ageing, but do not believe that a simple lack of Sod1 contributes to sarcopenia in WT mice or humans.
Redox cross‐talk between neurons and muscle
The situation cited above for the nerve rescue Sod1KO mice provides an example of how restoration of neuronal ROS homeostasis can restore defective function in muscle mitochondria that is associated with increased ROS generation. An analogous situation appears to occur in experimental denervation or nerve crush which has been found to lead to activation of a number of degenerative pathways in the denervated muscle, including an increased mitochondrial generation of reactive oxygen species (Muller et al. 2006) and increased generation of pro‐inflammatory cytokines (Cea et al. 2013). Muller et al. (2007 b) reported a remarkably large increase in muscle mitochondrial H2O2 generation following denervation and subsequent studies in our laboratory have shown that this increased mitochondrial H2O2 release is already apparent within 3 days of nerve transection. The reason for this rapid activation of specific degradatory pathways is unclear. It is possible that initially this may reflect an attempt to restore innervation, since products such as cytokines are released from the muscle fibre and some cytokines have been claimed to stimulate axonal sprouting, but if prolonged must inevitably lead to degradation of the denervated muscle fibres. Further work from this group also showed that other peroxides in addition to H2O2 were released from mitochondria from denervated muscle (Bhattacharya et al. 2009) and that inhibition of 12/15 lipoxygenase could ameliorate some of the muscle atrophy induced by denervation (Bhattacharya et al. 2014) . Thus together these data suggest that muscle mitochondrial ROS generation plays a role in the muscle degeneration seen following denervation.
Motor nerves and muscles are well known to play a symbiotic role in maintenance of the neuromuscular system and in particular the viability of motor neurons is recognised to be dependent upon continued exposure to neurotrophic factors generated by skeletal muscle fibres in addition to Schwann cells and neurons (Luff, 1998). Regular exercise is recognised to induce structural and functional changes in motor neurons (Gardiner et al. 2006), but in contrast to the situation with skeletal muscle there appear to be no data indicating that contractile activity or exercise training up‐regulate endogenous regulatory proteins for ROS and other cytoprotective proteins in motor neurons. Motor neurons do have the capacity to up‐regulate these cytoprotective proteins in response to exogenous reactive oxygen and nitrogen species (Bishop et al. 1999) and neurotrophic factors (e.g. Glial cell‐derived neurotrophic factor and brain‐derived neurotrophic factor) have been reported to promote neuronal survival by increasing defences against oxidative damage (Gabaizadeh et al. 1997).
In order to explain this paradox we are currently examining whether, because of proximity to their target muscle fibres, the peripheral axons of motor nerves are exposed to increased extracellular activities of ROS derived from the muscle fibres during contractile activity (Vasilaki et al. 2006 b). This must inevitably occur and preliminary data support this hypothesis. Previous studies of ROS derived from contracting skeletal muscle indicate that they cause transient oxidation in other non‐contracting tissues (Close et al. 2007) and we hypothesise that this level of oxidation is unlikely to produce substantial oxidative damage, but acts as a stimulus for the up‐regulation of cytoprotective systems.
We therefore postulate that redox cross‐talk between muscle and neurons through release of H2O2 (and potentially other ROS) plays differing roles depending on the innervation state of the muscle. In normal innervated muscle fibres contractile activity leads to generation of NADPH oxidase‐derived H2O2 in the extracellular space that interacts with adjacent neurons inducing up‐regulation of cytoprotective proteins in the axons. During ageing, studies have shown that skeletal muscle does not release equivalent amounts of ROS to the extracellular space (Vasilaki et al. 2006 b) and hence this cytoprotective cross‐talk will not occur, potentially reducing the capacity of the nerve to prevent oxidative damage. In contrast if the fibre becomes denervated (as also occurs to some extent in ageing) the fibre mitochondria release very large amounts of H2O2 (and other ROS) that can diffuse out of the fibre to neurons and other adjacent tissues. The effect of this very large and prolonged increase is unclear. It may initially represent an attempt to stimulate adaptations/axonal sprouting, but if sustained must inevitably lead to degeneration of the muscle fibre and potentially other local tissues. Figure 2 illustrates some aspects of this redox cross‐talk between muscle fibres and motor neurons that may influence neuromuscular ageing.
Figure 2. Schematic representation of putative redox cross‐talk from muscle to neurons .
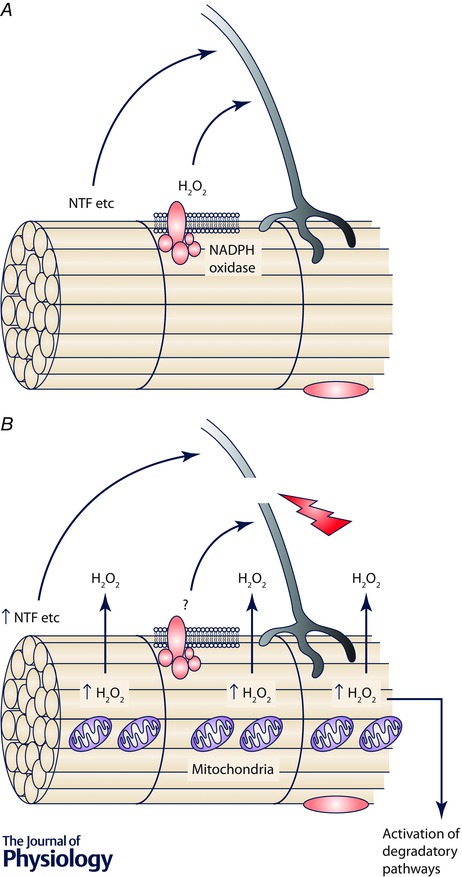
A shows the situation in innervated muscle fibres from young/adult where contractile activity leads to generation of NADPH oxidase‐derived H2O2 in the extracellular space that interacts with adjacent neurons inducing up‐regulation of cytoprotective proteins in the axons. During ageing, this process is likely to be modified since skeletal muscle from old mice does not release equivalent amounts of ROS to the extracellular space (Vasilaki et al. 2006 b) and hence this cytoprotective cross‐talk will not occur. B shows the effect of denervation in young/adult organisms and may also reflect the situation in some fibres from old organisms. Denervation of a fibre induces the fibre mitochondria to release very large amounts of H2O2 (and other ROS) that can diffuse out of the fibre to interact with neurons and other adjacent fibres or tissues. These changes may initially represent an initial attempt to stimulate adaptations/axonal sprouting, but if sustained must inevitably lead to degeneration of the muscle fibre and potentially other local tissues. NTF, neurotrophic factors.
Conclusions
Understanding how we age and ways of ameliorating the negative physical, mental and social effects of ageing is a major global challenge. Physical frailty is driven by loss of muscle mass and function and hence preventing this is key to reduction in frailty. Our current understanding of the key areas in which ROS contribute to age‐related deficits in muscle is through defective redox signalling and maintenance of neuromuscular integrity. Both are areas that still require further work to fully understand the mechanisms involved, but also appear amenable to targeted interventions that have the potential to help prevent age‐related neuromuscular decline with a consequent improvement in quality of life for older people.
Additional information
Competing interests
None declared.
Funding
The authors acknowledge generous financial support from the MRC, BBSRC, Arthritis Research UK, Research into Ageing (AgeUK) and the US National Institute on Ageing.
Acknowledgements
The authors would like to thank their many co‐workers and colleagues who have contributed to this work.
Biographies
Malcolm Jackson is Head of the Institute of Ageing and Chronic Disease at the University of Liverpool and Director of the MRC‐Arthritis Research UK Centre for Integrated Research into Musculoskeletal Ageing (CIMA). He trained in Biochemistry and undertook a PhD at University College, London with the late Professor Richard H. T. Edwards. He is a member of the BBSRC DRINC Strategy panel. His major research area is the role of free radicals and reactive oxygen species in skeletal muscle.

Anne McArdle’s PhD was from the University of Liverpool and postdoc studies included training with Dr John Faulkner in the Muscle Mechanics Laboratory at the University of Michigan. She holds a personal Chair at Liverpool and her research interests are in muscle ageing with emphasis on stress responses (particularly heat shock proteins) in muscle and the cross‐talk between muscle and inflammatory cells. She is Head of the Department of Musculoskeletal Biology and Athena SWAN lead for the Institute of Ageing and Chronic Disease.
References
- Adinolfi AM, Yamuy J, Morales FR & Chase MH (1991). Segmental demyelination in peripheral nerves of old cats. Neurobiol Aging 12, 175–179. [DOI] [PubMed] [Google Scholar]
- Aggeli IK, Gaitanaki C & Beis I (2006). Involvement of JNKs and p38‐MAPK/MSK1 pathways in H2O2‐induced upregulation of heme oxygenase‐1 mRNA in H9c2 cells. Cell Signal 18, 1801–1812. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Hamilton R, Jernigan A, Zhang Y, Sabia M, Rahman MM, Li Y, Wei R, Chaudhuri A & Van Remmen H (2014). Genetic ablation of 12/15‐lipoxygenase but not 5‐lipoxygenase protects against denervation‐induced muscle atrophy. Free Radic Biol Med 67, 30–40. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, Jang YC, Ran Q & Van Remmen H (2009). Denervation induces cytosolic phospholipase A2‐mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem 284, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop A, Marquis JC, Cashman NR & Demple B (1999). Adaptive resistance to nitric oxide in motor neurons. Free Radic Biol Med 26, 978–986. [DOI] [PubMed] [Google Scholar]
- Brown WF, Strong MJ & Snow R (1988). Methods for estimating numbers of motor units in biceps‐brachialis muscles and losses of motor units with aging. Muscle Nerve 11, 423–432. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, McComas AJ & Petito F (1973). Physiological changes in ageing muscles. J Neurol Neurosurg Psychiatry 36, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cea LA, Cisterna BA, Puebla C, Frank M, Figueroa XF, Cardozo C, Willecke K, Latorre R & Sáez JC (2013). De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc Natl Acad Sci USA 110, 16229–16234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai RJ, Vukovic J, Dunlop S, Grounds MD & Shavlakadze T (2011). Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close GL, Kayani AC, Ashton T, McArdle A & Jackson MJ (2007). Release of superoxide from skeletal muscle of adult and old mice: an experimental test of the reductive hotspot hypothesis. Aging Cell 6, 189–195. [DOI] [PubMed] [Google Scholar]
- Delaunay A, Pflieger D, Barrault MB, Vinh J & Toledano MB (2002). A thiol peroxidase is an H2O2 receptor and redox‐transducer in gene activation. Cell 111, 471–481. [DOI] [PubMed] [Google Scholar]
- Delbono O (2003). Neural control of aging skeletal muscle. Aging Cell 2, 21–29. [DOI] [PubMed] [Google Scholar]
- Dröge W (2002). Free radicals in the physiological control of cell function. Physiol Rev 82, 47–95. [DOI] [PubMed] [Google Scholar]
- Evans WJ & Campbell WW (1993). Sarcopenia and age‐related changes in body composition and functional capacity. J Nutr 123, 465–468. [DOI] [PubMed] [Google Scholar]
- Gabaizadeh R, Staecker H, Liu W & Van De Water TR (1997). BDNF protection of auditory neurons from cisplatin involves changes in intracellular levels of both reactive oxygen species and glutathione. Brain Res Mol Brain Res 50, 71–78. [DOI] [PubMed] [Google Scholar]
- Gardiner P, Dai Y & Heckman CJ (2006). Effects of exercise training on α‐motoneurons. J Appl Physiol (1985) 101, 1228–1236. [DOI] [PubMed] [Google Scholar]
- Gems D & Doonan R (2009). Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle 8, 1681–1687. [DOI] [PubMed] [Google Scholar]
- Grover‐Johnson N & Spencer PS (1981). Peripheral nerve abnormalities in aging rats. J Neuropathol Exp Neurol 40, 155–165. [DOI] [PubMed] [Google Scholar]
- Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y & Dick TP (2009). Proximity‐based protein thiol oxidation by H2O2‐scavenging peroxidases. J Biol Chem 284, 31532–31540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D (1956). Ageing: a theory based on free radical and radiation chemistry. J Gerontol 11, 298–300. [DOI] [PubMed] [Google Scholar]
- Jackson MJ (2011). Control of reactive oxygen species production in contracting skeletal muscle. Antiox Redox Signal 15, 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MJ & McArdle A (2011). Age‐related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol 589, 2139–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen‐Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG & van der Vliet A (2008). Redox‐based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis RM, Hughes SM & Ledgerwood EC (2012). Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Rad Biol Med 53, 1522–1530. [DOI] [PubMed] [Google Scholar]
- Ji LL, Gomez‐Cabrera MC, Steinhafel N & Vina J (2004). Acute exercise activates nuclear factor (NF)‐ κB signaling pathway in rat skeletal muscle. FASEB J 18, 1499–1506. [DOI] [PubMed] [Google Scholar]
- Kawamata H & Manfredi G (2010). Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid Redox Signal 13, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayani AC, Close GL, Dillmann WH, Mestril R, Jackson MJ & McArdle A (2010). Overexpression of HSP10 in skeletal muscle of transgenic mice prevents the age‐related fall in maximum tetanic force generation and muscle cross‐sectional area. Am J Physiol Regul Integr Comp Physiol 299, R268–R276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantic S, Mechawar N, Reix S & Quirion R (2005). Molecular basis of programmed cell death involved in neurodegeneration. Trends Neurosci 28, 670–676. [DOI] [PubMed] [Google Scholar]
- Larkin LM, Davis CS, Sims‐Robinson C, Kostrominova TY, Van Remmen H, Richardson A, Feldman EL & Brooks SV (2011). Skeletal muscle weakness due to deficiency of CuZn‐superoxide dismutase is associated with loss of functional innervation. Am J Physiol Regul Integr Comp Physiol 301, R1400–R1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson L & Ansved T (1995). Effects of ageing on the motor unit. Prog Neurobiol 45, 397–458. [DOI] [PubMed] [Google Scholar]
- Lexell J, Taylor CC & Sjöström M (1988). What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15‐ to 83‐year‐old men. J Neurol Sci 84, 275–294. [DOI] [PubMed] [Google Scholar]
- Luff AR (1998). Age‐associated changes in the innervation of muscle fibers and changes in the mechanical properties of motor units. Ann N Y Acad Sci 854, 92–101. [DOI] [PubMed] [Google Scholar]
- Lynch GS (ed.) (2011). Sarcopenia – Age‐Related Muscle Wasting and Weakness. Springer. [Google Scholar]
- McArdle A, Pattwell D, Vasilaki A, Griffiths RD & Jackson MJ (2001). Contractile activity‐induced oxidative stress: Cellular origin and adaptive responses. Am J Physiol Cell Physiol 280, C621–C627. [DOI] [PubMed] [Google Scholar]
- Marinho HS, Real C, Cyrne L, Soares H & Antunes F (2014). Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A & Van Remmen H (2007. a). Trends in oxidative ageing theories. Free Radic Biol Med 43, 477–503. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A & Van Remmen H (2007. b). Denervation‐induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293, R1159–R1168. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke‐Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ 2nd, Csete M, Faulkner J A & Van Remmen H (2006). Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age‐dependent skeletal muscle atrophy. Free Radic Biol Med 40, 1993–2004. [DOI] [PubMed] [Google Scholar]
- Orr WC & Sohal RS (2003). Does overexpression of Cu,Zn‐SOD extend life span in Drosophila melanogaster? Exp Gerontol 38, 227–230. [DOI] [PubMed] [Google Scholar]
- Park J, Lee S, Lee S & Kang SW (2014). 2‐Cys peroxiredoxins: emerging hubs determining redox dependency of mammalian signaling networks. Int J Cell Biol 2014, e715867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson T, Kabayo T, Ng R, Chamberlain J, McArdle A & Jackson MJ (2014). Skeletal muscle contractions induce acute changes in cytosolic superoxide, but slower responses in mitochondrial superoxide and cellular hydrogen peroxide. PLoS One 9, e96378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y & Richardson A (2009). Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam DA, Bhattacharya A & Van Remmen H (2013). Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid Redox Signal 19, 1373–1387. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR & Blüher M (2009). Antioxidants prevent health‐promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106, 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano AD, Serviddio G, de Matthaeis A, Bellanti F & Vendemiale G (2010). Oxidative stress and aging. J Nephrol 15, S29–36. [PubMed] [Google Scholar]
- Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A & Brooks SV (2014. a). Neuron‐specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD‐knockout mice. FASEB J 28, 1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Jackson MJ & Vasilaki A (2014. b). Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic Res 48, 12–29. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Pye D, Vasilaki A, Zibrik L, Palomero J, Kabayo T, McArdle F, Van Remmen H, Richardson A, Tidball JG, McArdle A & Jackson MJ (2011). Role of superoxide‐nitric oxide interactions in the accelerated age‐related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase. Aging Cell 10, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A & Jackson MJ (2013). Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18, 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon AB, Richardson A & Pérez VI (2010). Update on the oxidative stress theory of aging: does oxidative stress play a role in aging or healthy aging? Free Radic Biol Med 48, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sataranatarajan K, Qaisar R, Davis C, Sakellariou GK, Vasilaki A, Zhang Y, Liu Y, Bhaskaran S, McArdle A, Jackson M, Brooks SV, Richardson A & Van Remmen H (2015). Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype. Redox Biol 5, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC & Rabinovitch PS (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911. [DOI] [PubMed] [Google Scholar]
- Sies H (2014). Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem 289, 8735–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotta MC, Liou W, Stöcker S, Talwar D, Oehler M, Ruppert T, Scharf AN & Dick TP (2015). Peroxiredoxin‐2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11, 64–70. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, McArdle F, Iwanejko LM & McArdle A (2006. a). Adaptive responses of mouse skeletal muscle to contractile activity: The effect of age. Mech Ageing Dev 127, 830–839. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, Mansouri A, Remmen H, van der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA & Jackson MJ (2006. b). Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5, 109–117. [DOI] [PubMed] [Google Scholar]
- Young A & Skelton DA (1994). Applied physiology of strength and power in old age. Int J Sports Med 15, 149–151. [DOI] [PubMed] [Google Scholar]
- Zhang J, Johnston G, Stebler B & Keller ET (2001). Hydrogen peroxide activates NFκB and the interleukin‐6 promoter through NFκB‐inducing kinase. Antioxid Redox Signal 3, 493–504. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV & Van Remmen H (2013). CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J 27, 3536–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]