

Abstract
The ageing endothelium progressively loses its remarkable and crucial ability to maintain homeostasis of the vasculature, as it acquires a proinflammatory phenotype. Cellular and structural changes gradually accumulate in the blood vessels, and markedly in artery walls. Most changes in aged arteries are comparable to those occurring during the atherogenic process, the latter being more marked: pro‐oxidant and proinflammatory molecules, mainly deriving from or triggered by oxidized low density lipoproteins (oxLDLs), are undoubtedly a major driving force of this process. Oxysterols, quantitatively relevant components of oxLDLs, are likely candidate molecules in the pathogenesis of vascular ageing, because of their marked pro‐oxidant, proinflammatory and proapoptotic properties. An increasing bulk of experimental data point to the contribution of a variety of oxysterols of pathophysiological interest, also in the age‐related genesis of endothelium dysfunction, intimal thickening due to lipid accumulation, and smooth muscle cell migration and arterial stiffness due to increasing collagen deposition and calcification. This review provides an updated analysis of the molecular mechanisms whereby oxysterols accumulating in the wall of ageing blood vessels may ‘activate’ endothelial and monocytic cells, through expression of an inflammatory phenotype, and ‘convince’ smooth muscle cells to proliferate, migrate and, above all, to act as fibroblast‐like cells.
Abbreviations
- α‐EPOX
5α,6α‐epoxide
- β‐EPOX
5β,6β‐epoxide
- 25‐OH
25‐hydroxycholesterol
- 27‐OH
27‐hydroxycholesterol
- 7α‐OH
7α‐hydroxycholesterol
- 7β‐OH
7β‐hydroxycholesterol
- 7‐K
7‐ketocholesterol
- 24S‐OH
24S‐hydroxycholesterol
- AngII
angiotensin II
- CVD
cardiovascular disease
- EC
endothelial cell
- ECM
extracellular matrix
- EDR
endothelium‐dependent relaxation
- EGFR
epidermal growth factor receptor
- eNOS
endothelial nitric oxide synthase
- ER
endoplasmic reticulum
- ERK
extracellular signal‐regulated kinase
- ET‐1
endothelin‐1
- GSH
reduced glutathione
- HNE
4‐hydroxy‐2‐nonenal
- HUVECs
human umbilical vein ECs
- IL
interleukin
- MCP‐1
monocyte chemoattractant protein
- MMP
matrix metalloproteinase
- NO
nitric oxide
- NOX
reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase
- oxLDLs
oxidized low density lipoproteins
- PG
prostaglandin
- RAS
renin–angiotensin system
- ROS
reactive oxygen species
- SMC
smooth muscle cell
- TGF‐β1
transforming growth factor‐β1
- TIMPs
tissue inhibitors of MMPs
- TLR4
Toll‐like receptor 4
- TNF‐α
tumour necrosis factor‐α
- Triol
cholesterol‐3β,5α,6β‐triol
- VCAM‐1
vascular cell adhesion molecule
- VSMC
vascular smooth muscle cell
Introductory remarks
Ageing is a gradual and irreversible process caused by environmental, genetic, and epigenetic factors and lifestyle conditions, which together cause progressive alterations in the homeostatic mechanisms of an organism. Although the improvement of lifestyle and hygienic conditions and the great advances in medicine have lengthened life expectancy, ageing is considered to be an independent risk factor for age‐related diseases in the developed countries.
The vascular endothelium plays a crucial role in maintaining the body's health, adapting its functions and structures to different insults. In particular, the endothelium makes a major contribution to blood homeostasis, appropriate organ perfusion, immune response activation, and regulation of peripheral resistances (Busse & Fleming, 2006; Donato et al. 2015). The compensative mechanisms of the endothelium are progressively impaired during ageing, causing peculiar changes in vascular cell morphology and vessel structure.
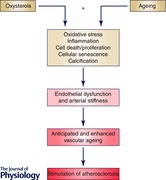
A growing bulk of evidence suggests that oxysterols, cholesterol oxidation products contained in oxidized low density lipoproteins (oxLDLs), significantly contribute to the vascular remodelling that occurs in atherosclerosis, since they are involved in various key steps of this age‐related disease. Oxysterols are also involved in endothelial dysfunction: they favour platelet aggregation and impair arterial relaxation by reducing nitric oxide (NO) bioavailability; moreover, they promote migration and proliferation of vascular smooth muscle cells (VSMCs), causing intimal thickening, and collagen deposition and calcification with consequent arterial stiffness. Oxysterols might also play a crucial role in vascular ageing thanks to their ability to induce inflammation, oxidative stress and apoptosis (Fig. 1).
Figure 1. Involvement of oxysterols in endothelial dysfunction .

LDLs pass through the activated endothelium where they are oxidized in the subendothelial space leading to oxysterol formation. These compounds induce and sustain inflammatory processes in the vascular wall by upregulating adhesion molecules on the ECs surface and by augmenting the release of MCP‐1, consequently amplifying recruitment of monocytes from the bloodstream. In the intima layer, oxysterols promote monocyte differentiation into lipid‐laden macrophages, called foam cells, which are the first sign of atherogenesis. Oxysterols alter endothelial permeability by inserting into EC membranes and modulating their fluidity and structure; further, they contribute to endothelial dysfunction by favouring platelet aggregation. In this connection, these oxysterols impair arterial relaxation by reducing NO bioavailability and promoting VSMC calcification, which leads to arterial stiffness. Finally, oxysterols are triggers of oxidative stress, one of the major processes implicated in the development of age‐related disease. ECs, endothelial cells; NO, nitric oxide; LDLs, low density lipoproteins; VSMCs, vascular smooth muscle cells.
Vascular ageing
Aged blood vessels are characterized by arterial stiffness due to increased intimal‐to‐media thickness, reduced elasticity consequent on increased collagen accumulation and decreased elastin content, and vascular calcification (Wang & Bennett, 2012). Augmented proliferation and migration of medial VSMCs and increased numbers of inflammatory cells have been observed in the arteries of aged rats in vivo, together with an elevated ratio between matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) (Li et al. 1999; Monk & George, 2014). Moreover, collagen levels in the arteries of aged rodents are reported to be higher than in young animals, while the content of elastin remains unchanged, favouring arterial stiffness (Harvey et al. 2015). During ageing, VSMCs shift their contractile phenotype into a fibroblast‐like one, thus becoming able to secrete extracellular matrix (ECM) components, and to migrate and proliferate from the tunica media to the intima of the artery (Monk & George, 2014).
Endothelial dysfunction is one of the first signs of age‐related alteration of the vasculature, and may appear well before the clinical manifestation of cardiovascular diseases (Donato et al. 2015). A healthy endothelium retains antithrombotic, vasodilatory, anti‐inflammatory and antioxidant properties, which are tightly regulated to respond to the organism's needs. During ageing, these features gradually deteriorate, and the endothelium acquires a proinflammatory, proaggregating and pro‐oxidant phenotype. One hallmark of endothelium dysfunction is the progressively impaired vasodilatory response to blood flow and vasodilating compounds, that is altered endothelium‐dependent relaxation (EDR) (Deanfield et al. 2007). The reduced vasodilation is mainly due to a diminished bioavailability of NO, which is the most important vasodilating factor synthesized by endothelial NO synthase (eNOS) having modulatory effects on several autocrine and paracrine mediators (Michel & Vanhoutte, 2010). Moreover, NO exerts vasoprotective, cardioprotective and antiatherogenic effects: it prevents vasospasm of the coronary arteries, inhibits the release of vasoconstrictor endothelin‐1 (ET‐1), and decreases endothelium permeability thus contrasting LDL infiltration; it also reduces VSMC proliferation and migration, as well as inflammation and endothelial cell (EC) apoptosis (Gradinaru et al. 2015).
Several studies support the implication of vascular oxidative stress in diminishing endothelium relaxation and NO production (Moncada et al. 1991; Cohen et al. 1997; Taddei et al. 2001; Puca et al. 2013). The bioavailability of NO is reduced by oxidative stress because of the reaction with superoxide anion (O2 · −) (Harrison, 1997; Hamilton et al. 2001) and because of a reduced eNOS activity (Gradinaru et al. 2015). Moreover, NO interaction with O2 · − generates peroxynitrite (ONOO−), a highly reactive radical molecule able to damage proteins, lipids and DNA (Bosch‐Marce et al. 2007).
Oxidative stress is known to be a major driving force in ageing and in the development of cardiovascular diseases (CVDs). Reactive oxygen species (ROS) are produced by enzyme systems, such as NOX (reduced nicotinamide adenine dinucleotide phosphate oxidases: NADPH oxidases), xanthine oxidase, cytochrome P450, as well as by mitochondria through the electron transfer chain. Several studies have demonstrated the crucial role of NOX enzymes in ageing (Krause, 2007; Brandes et al. 2014), and stressed the association between up‐regulation of NOX isoforms and endothelial dysfunction in CVDs (Wind et al. 2010; Montezano et al. 2011; Montezano & Touyz, 2012). Another relevant source of ROS is the mitochondrial dysfunction induced by biological ageing, which leads to altered electron flow in the respiratory complexes and increases oxygen consumption (Kokoszka et al. 2001). A vicious circle thus comes about, since ROS production in turn causes mitochondrial DNA and respiratory chain damage. The increased cellular ROS steady‐state due to altered NOX and mitochondria during ageing is further supported by the quite frequent dysregulation of antioxidant defence, for example through decreased levels of reduced glutathione (GSH), decreased superoxide dismutase and nuclear factor erythroid 2 p45‐related factor 2 (Nrf2) transcription factor activities (Puca et al. 2013).
It has been demonstrated that oxidative stress is one of the main triggers of cell apoptosis, via both mitochondria‐dependent and ‐independent pathways (Sinha et al. 2013). Apoptosis, or programmed cell death, is a crucial mechanism necessary to regulate cell turnover and homeostasis; this process depends on the balance between proapoptotic and antiapoptotic genes. Inflammation, oxidative stress, genomic damage, and other stimuli impair this complex equilibrium, making different cell types more sensitive to apoptosis. Among these stimuli, ageing is responsible for mitochondrial DNA damage (Szklarczyk et al. 2014), impairment of human retinal vessels (Catita et al. 2015) and alterations of cardiac function (Quarles et al. 2015), reflecting an increase in the rate of cell apoptosis.
Cell loss by programmed death, and alteration of cell repair mechanisms, are amplified by cellular senescence, a key marker of vascular ageing. Senescence is described as the loss of replicative potential, and occurs at the end of proliferative cell lifespan, or prematurely under the influence of stress factors, such as hypoxia, shearing, mechanical injury and oxidative stress. Senescent ECs are detached from the basement membrane, with consequent enhanced permeability, infiltration of inflammatory molecules and cells, and thrombotic events (Lakatta, 2013). Moreover, premature VSMC senescence has been observed in old arterial wall, probably associated with an increase of NOX4 activity and angiotensin II (AngII) signalling (Yang et al. 2007). The AngII pathway is implicated in arterial remodelling during the ageing process: its age‐related perturbation, as well as that of aldosterone/mineralocorticoid receptor and ET‐1/ET‐1 receptor A systems, represents a chronic proinflammatory stimulus for ECs and VMSCs. These pathways trigger and sustain inflammation by releasing several molecules, such as various MMPs, the chemokine monocyte chemoattractant protein 1 (MCP‐1), transforming growth factor‐β1 (TGF‐β1), NOX and ROS (Wang et al. 2014). Chronic inflammation is certainly the pivotal force in the development of the above structural and functional changes occurring in the arterial wall during ageing.
Of note, the molecular, cellular, and structural changes occurring in aged vessels, leading to endothelial dysfunction, vascular inflammation, remodelling, and increased arterial stiffness, are comparable to those occurring in the process of atherogenesis (Harvey et al. 2015). For this solid reason, ageing is considered to be a non‐modifiable and important risk factor, associated with the incidence and prevalence of CVDs; importantly, traditional risk factors of atherosclerosis, such as hypercholesterolaemia, obesity and smoking, promote the pathogenesis of atherosclerosis by accelerating the vascular ageing process (Wang & Bennett, 2012).
Atherosclerosis, oxLDLs and oxysterols
Atherosclerosis is a multifactorial, degenerative disease which affects large‐ and medium‐sized arteries, and is characterized by chronic inflammation of the vascular wall. This inflammation entails continuous monocyte recruitment and migration from the blood into the subendothelial space; it is favoured by the up‐regulation of endothelial adhesion molecules, chemokines, cytokines, and growth factors. Once in the sub‐intimal space, monocytes differentiate into macrophages and take up oxLDLs through the scavenger receptors CD36 and SR‐A. Unlike LDL receptors, scavenger receptors are not regulated by a negative feedback loop, so that macrophages avidly accumulate oxidized lipids, becoming foam cells, and meanwhile release a large variety of proinflammatory cytokines. Perpetuation of this process promotes a chronic inflammatory state, and progression of the atherosclerotic lesion.
A causative role in atherosclerosis has now been established for oxLDLs; these micelles are involved in the initiation, formation, progression and destabilization of the fibrotic plaque, as they induce inflammation, apoptosis, VSMC proliferation and migration, and endothelial activation (Lusis, 2000). It thus appears possible that oxLDLs might also significantly contribute to vascular ageing. Although the implication of oxidized lipoproteins in atherogenesis and ageing is undeniable, the different roles of the various components present in oxLDLs has not yet been fully elucidated. Several reactive molecules derive from oxidation of the LDL lipid fraction, including peroxides, hydroxides, aldehydes, oxidized phospholipids and cholesterol oxidation products (Parthasarathy et al. 2010; Leonarduzzi et al. 2012). The latter class of compounds, quantitatively relevant in oxLDLs, has drawn the attention of various research groups; oxysterols, especially, are of interest as they are thought to be closely involved in the pathogenesis of atherosclerosis. Oxysterols are 27‐atom carbon compounds that originate from cholesterol oxidation by both enzymatic and non‐enzymatic mechanisms and present one or more carbonyl, keto, hydroxyl or epoxide groups in the sterol ring and/or in the side chain (Fig. 2). They are present in both free and esterified form. Several foods of animal origin contain oxysterols, as the consequence of food preparation, storage and processing; the amount of oxysterols thus produced in foods can reach or even exceed 10% of total cholesterol (Lordan et al. 2009) (Table 1). In biological systems, oxidation of the cholesterol side chain is an enzymatic process, generally catalysed by enzymes of the cytochrome P450 family, while oxidation of the sterol ring occurs through non‐enzymatic reactions, with the exception of the formation of 7α‐hydroxycholesterol (7α‐OH), which is produced by the enzyme cholesterol 7α‐hydroxylase. From the quantitative stand point, 27‐hydroxycholesterol (27‐OH), 7‐ketocholesterol (7‐K), 5α,6α‐epoxide (α‐EPOX), 5β,6β‐epoxide (β‐EPOX) and cholesterol‐3β,5α,6β‐triol (Triol) are the most abundant oxysterols in the plasma and atherosclerotic lesions (Lordan et al. 2009; Poli et al. 2009). Of note, oxysterols are involved in many physiological processes, such as cholesterol metabolism, and the synthesis of steroid hormones and vitamin D, and are present in cholesterol‐rich domains of membranes (lipid rafts), where they contribute to modulating membrane fluidity and permeability, flux of ions, and the activity of membrane‐bound enzymes. Conversely, an excessive accumulation of these compounds in tissues and organs has been related to the progression of major chronic disease processes. Indeed, certain oxysterols exercise strong pro‐oxidant, proinflammatory and proapoptotic effects at pathological concentrations detectable in the lesions typical of atherosclerosis, neurodegenerative diseases, inflammatory bowel diseases, age‐related macular degeneration, and other pathological conditions characterized by altered cholesterol uptake and/or metabolism (Poli et al. 2013). In particular, there is no longer any doubt that oxysterols of pathophysiological relevance play a pivotal role in contributing to the various steps of atheroma formation, from endothelial dysfunction to plaque fibrosis and rupture, through macrophage infiltration and VSMC differentiation (Table 2). In this connection, an excessive amount of oxysterols has been detected in human atherosclerotic plaques (Table 3). It has been observed that the oxysterols commonly found in plasma from hypercholesterolaemic patients are recovered in atherosclerotic plaques, and a strong direct correlation between the total oxysterols and the total amount of cholesterol have been observed in the same plaques. This finding further points to cholesterol oxidation as an important or even crucial event in vascular remodelling due to atherosclerosis. Conversely, it has been observed that unoxidized cholesterol does not exert a proatherosclerotic effect, since it is less polar and reactive than oxysterols (Leonarduzzi et al. 2007, 2012; Poli et al. 2009; Zarrouk et al. 2014).
Figure 2.

Chemical structures of the main oxysterols
Table 1.
Origin of the most relevant oxysterols
| Endogenous origin | Exogenous origin | |
|---|---|---|
| Enzymatic formation | Non‐enzymatic formation | Autoxidation |
| 7α‐Hydroxycholesterol (7α‐OH) | 7α‐Hydroxycholesterol (7α‐OH) | 7α‐Hydroxycholesterol (7α‐OH) |
| 27‐Hydroxycholesterol (27‐OH) | 7β‐Hydroxycholesterol (7β‐OH) | 7β‐Hydroxycholesterol (7β‐OH) |
| 24S‐Hydroxycholesterol (24S‐OH) | 7‐Ketocholesterol (7‐K) | 7‐Ketocholesterol (7‐K) |
| 22‐Hydroxycholesterol (22‐OH) | Cholesterol‐5,6‐epoxide (EPOX) | Cholesterol‐5,6‐epoxide (EPOX) |
| 25‐Hydroxycholesterol (25‐OH) | Cholesterol‐3β,5α,6β‐triol (Triol) | Cholesterol‐3β,5α,6β‐triol (Triol) |
Table 2.
Effects of oxysterols on atherogenesis and vascular ageing
| Oxysterol | Effect | Experimental model | Reference | |
|---|---|---|---|---|
| Oxidative stress | 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX (alone/mixture) | NOX1 ↑ | CaCo‐2 | Biasi et al. 2009, 2013; Mascia et al. 2010 |
| 7‐K | NOX4 ↑ | Human aortic SMCs | Pedruzzi et al. 2004 | |
| 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX, Triol, 25‐OH, 27‐OH (mixture) | NOX2 ↑ | U937 | Gargiulo et al. 2011 | |
| Inflammation | 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX (alone/mixture) | ILs, chemokines ↑ | CaCo‐2 | Biasi et al. 2009, 2013; Mascia et al. 2010 |
| 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX, Triol, 25‐OH (alone/mixture) | TGF‐β1, MCP‐1, CD36, β1‐integrin ↑ | J774A.1, U937 | Leonarduzzi et al. 2001, 2005, 2008, 2010; Gargiulo et al. 2012 | |
| 7‐K, α‐EPOX, β‐EPOX (mixture) | TGF‐β1, MCP‐1 ↑ | Raw264.7 | Ferré et al. 2009 | |
| 7‐K, α‐EPOX (alone) | MMP‐2/9, EGFR ↑ | Rat aortic SMCs | Liao et al. 2010 | |
| 7‐K | MMP‐9, TNF‐α, IL‐6 ↑ | Human M1/M2 | Buttari et al. 2013 | |
| 27‐OH | MMP‐9, IL‐1β, IL‐8, TNF‐α ↑ | U937 | Gargiulo et al. 2015 | |
| 27‐OH | MMP‐9 ↑ | THP‐1 | Kim et al. 2015 | |
| High fat diet containing 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX, Triol | MCP‐1, MMP‐2/9 ↑ | ApoE−/− mice | Sato et al. 2012 | |
| 7‐K, 7α‐OH, 7β‐OH (alone) | IL‐1β, TNFα, adhesion molecules ↑ | HUVECs | Lemaire et al. 1998 | |
| 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX, 25‐OH, 27‐OH (alone) | IL‐1β, and/or TNFα, MCP‐1, MIP‐1β ↑ | U937, THP1 | Prunet et al. 2006 | |
| 25‐OH | VCAM‐1 ↑ | Human aortic ECs | Naito et al. 2005 | |
| 7α‐OH, 27‐OH (alone) | TNF‐α ↑ | THP‐1 | Kim et al. 2013 | |
| Cell death | 7‐K, 7α‐OH, 7β‐OH, α‐EPOX, β‐EPOX (alone/mixture) | Caspase‐3 ↑ | CaCo‐2 | Biasi et al. 2009; Mascia et al. 2010 |
| 7‐K | TNFα death pathway ↑ | Human aortic SMCs | Lee et al. 2005 | |
| 7‐K, 25‐OH (alone) | Proapoptotic mitochondrial pathway ↑ | Rat VSMCs | Appukuttan et al. 2013 | |
| 7α‐OH, 7β‐OH, α‐EPOX (alone) | Caspase‐3/7 ↑ | CaCo‐2 | Biasi et al. 2013 | |
| 7‐K | ER stress | Human aortic SMCs | Pedruzzi et al. 2004 | |
| Triol | Mitochondrial membrane potential impairment, Ca2+ ↑ | Rat VSMCs | Tang et al. 2005 | |
| 25‐OH | Cytochrome c release, caspase ↑ | CHO‐K1 | Yang & Sinensky 2000 | |
| 7β‐OH | Caspase‐3/9 ↑ | U937 | Ryan et al. 2004 | |
| 7‐K, 7β‐OH (alone) | p53 ↑ | THP‐1, J774, U937 | Li W. et al. 2012; Miah et al. 2013 | |
| 7β‐OH, 25‐OH (alone) | G2/M arrest, caspases ↑ | THP‐1 | Lim et al. 2003 | |
| 7‐K | GSH ↓, cytochrome c, pro‐caspase ↑ | U937 | Lizard et al. 1998 | |
| 7‐K | Phospholipidosis | U937 | Vejux et al. 2009 | |
| 7‐K, 7β‐OH, 24S‐OH (alone) | Oxiapoptophagy | Murine oligodendrocytes | Nury et al. 2015 | |
| 7‐K, α‐EPOX (alone) | Cytotoxicity | Rat aortic SMCs | Liao et al. 2010 | |
| Triol | ATPase ↑, cytotoxicity | ECs | Ramasamy et al. 1992 | |
| Endothelial dysfunction/cell phenotype changes | 7‐K, 7α‐OH, 27‐OH | Endothelial stiffness | Bovine aortic ECs | Shentu et al. 2012 |
| 7‐K | Na+–K+‐ATPase ↓ | Human ECs | Duran et al. 2010 | |
| 7‐K, 7α‐OH, 7β‐OH (alone/mixture) | EDR ↓ | Rat aorta rings | Wong et al. 2011 | |
| 7‐K, 7β‐OH (alone) | NO ↓ | HUVECs | Deckert et al. 1998 | |
| 7‐K, 7β‐OH (alone) | EDR ↓ | Rabbit aorta rings | Deckert et al. 1997 | |
| 7‐K, α‐EPOX, 25‐OH, Triol (alone) | PGI2 ↓ | HUVECs | Peng et al. 1993 | |
| 7‐K, 5,6‐secosterol (alone) | EDR ↓ | HUVECs, rat aorta rings | Luchetti et al. 2015 | |
| 7‐K, 7β‐OH (alone) | von Willebrand factor ↑ | HUVECs | Li et al. 2011 | |
| 7‐K, α‐EPOX (alone) | Proliferation, migration | Rat aortic SMCs | Liao et al. 2010 | |
| 25‐OH | Morphological changes, PG/eicosanoids ↑ | Rabbit pulmonary artery SMCs | Wohlfeil & Campbell, 1999 | |
| Vascular calcification | 20‐OH, 22‐OH (alone/mixture) | Osteogenic differentiation | Mouse mesenchymal and embryonic stem cells | Kha et al. 2004; Kwon et al. 2015 |
| Oxysterol mixture | Osteogenic differentiation, calcium deposition | Rat mesenchymal stem cells | Wöltje et al. 2015 | |
| 25‐OH | Mineralized nodule formation ↑ | Bovine aortic SMCs | Watson et al. 1994 | |
| 7‐K | Increased inorganic phosphate‐induced osteogenesis, apoptosis and calcium deposition | Bovine VSMCs | Saito et al. 2008 | |
| Triol | ROS/apoptosis‐mediated calcification | Rat VSMCs | Liu et al. 2004 |
Table 3.
Summary of recent studies quantifying cholesterol and oxysterols in human normal vessels and atherosclerotic plaques
| Sample | Cholesterol | 7α‐OH | 7β‐OH | 7‐K | 25‐OH | 27‐OH | α‐EPOX | β‐EPOX | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Peripheral artery: | |||||||||
| Normal vessel | n.a. | n.a. | n.a. | n.a. | 0.91 | 0.43 | n.a. | n.a. | Virginio et al. |
| (1.04) | (0.92) | 2015 | |||||||
| Atherosclerotic plaque (ng (mg tissue)−1)a | n.a. | n.a. | n.a. | n.a. | 0.50 | 9.27 | n.a. | n.a. | |
| (0.64) | (29.8) | ||||||||
| Carotid: | |||||||||
| Atherosclerotic plaque (ng (mg free cholesterol)−1)b,* | n.a. | 128 (75.6–216.8) | 607.1 | n.a. | n.a. | 110.7 | 112.2 | Helmschrodt | |
| (205.9–1407.6) | (34.4–165.4) | (95.9–334.2) | et al. 2013 | ||||||
| Carotid: | |||||||||
| Atherosclerotic plaque (pmol (mg tissue)−1) | 93,328 ± 45,009 | n.a. | 146 ± 88 | 322 ± 279 | n.a. | 432 ± 323 | n.a. | n.a. | Leonarduzzi |
| et al. 2007 | |||||||||
| Carotid: | |||||||||
| Normal vessel | 4000 ± 2400 | n.a. | 0.2 ± 0.1 | 1.9 ± 1 | n.a. | n.a. | n.a. | n.a. | Micheletta et al. 2004 |
| Atherosclerotic plaque (pmol (mg tissue)−1) | 40,600 ± 28,700 | n.a. | 9.8 ± 7.8 | 39.7 ± 14 | n.a. | n.a. | n.a. | n.a. | |
| Carotid: | |||||||||
| Normal vessel | 2200 ± 600 | n.a. | 0.17 ± 0.06 | 0.77 ± 0.7 | n.a. | n.a. | n.a. | n.a. | Iuliano et al. 2003 |
| Atherosclerotic plaque (pmol (mg tissue)−1) | 40,600 ± 28,600 | n.a. | 7.75 ± 3.9 | 35.9 ± 21.5 | n.a. | n.a. | n.a. | n.a. | |
| Carotid: | |||||||||
| Atherosclerotic plaque (pmol (mg tissue)−1) | 117,000 ± 87,730 | n.a. | 420 ± 724 | n.a. | n.a. | n.a. | n.a. | n.a. | Carpenter et al. 2003 |
| Aorta: | |||||||||
| Normal vessel | 10,600 (1200) | n.a. | n.a. | 30 (10) | n.a. | 20 (20) | n.a. | n.a. | Upston et al. 2002 |
| Atherosclerotic plaque (pmol (mg protein)−1)c | 79,900 (19,700) | n.a. | n.a. | 90 (20) | n.a. | 640 (80) | n.a. | n.a. | |
| Carotid/aorta: | |||||||||
| Normal vessel | 3710 ± 1540 | 0.91 ± 0.87 | 0.51 ± 0.7 | 0.49 ± 0.98 | n.a. | 1.6 ± 4.6 | 0.99 ± 1.6 | 0.69 ± 1.4 | Garcia‐Cruset et al. 2001 |
| Atherosclerotic plaque (ng (mg tissue)−1) | 38,570 ± 11,590 | 130 ± 110 | 57 ± 40 | 100 ± 63 | n.a. | 220 ± 225 | 60 ± 38 | 34 ± 16 | |
| Carotid: | |||||||||
| Normal vessel | 37,600 ± 6400 | n.a. | n.a. | 10 ± 10 | n.a. | 30 ± 10 | 10 ± 10 | 10 ± 10 | Vaya et al. 2001 |
| Atherosclerotic plaque (ng (mg tissue)−1) | 51,200 ± 11,200 | n.a. | 20 ± 10 | 70 ± 30 | n.a. | 250 ± 110 | 10 ± 10 | 30 ± 10 | |
| Coronary: | |||||||||
| Normal vessel | 4500 ± 1000 | n.a. | 10 ± 20 | 20 ± 10 | n.a. | 20 ± 10 | 20 ± 10 | 40 ± 30 | Vaya et al. 2001 |
| Atherosclerotic plaque (ng (mg tissue)−1) | 50,300 ± 15,600 | 40 ± 10 | 140 ± 90 | 80 ± 40 | n.a. | 250 ± 100 | 60 ± 30 | 120 ± 70 | |
Data represent the total content of sterols (free and esterified subfractions) and are expressed as mean ± SD unless noted otherwise.
Median (interquartile range), bmedian (range), cmean (SEM), *only free oxysterols. n.a., not applicable.
Further, among the aldehydic end‐products of polyunsaturated fatty acid peroxidation, 4‐hydroxy‐2‐nonenal (HNE) may contribute to the pathophysiology of vascular ageing by forming adducts on cellular proteins, leading to a progressive protein dysfunction (Leonarduzzi et al. 2012). HNE increases endothelial permeability (Usatyuk & Natarajan, 2012), modifies cytoskeletal proteins, including actin and microtubules, by regulating cell‐cell contacts and endothelial barrier function (Usatyuk et al. 2006). HNE may also stimulate the adhesion of macrophages to the vascular endothelium during atherosclerosis (Go et al. 2007). In addition, HNE covalently modifies LDLs by binding to lysine and histidine residues, leading to adduct formation: this favours LDL uptake by macrophages, thus resulting in macrophage activation and foam cell formation, a crucial event in atherosclerosis lesion development (Annangudi et al. 2008).
Aldehyde‐adducts have been detected in human aorta, and their levels were increased in aged aortas; however, it is not known whether aldeydes accumulate on ECM proteins, contributing to arterial stiffness. Although in these aged aortas the structure of elastin fibre is strongly altered, elastin was shown to be very poorly modified by HNE‐adducts (Zarkovic et al. 2015).
Pro‐oxidant effect of oxysterols
Vascular ROS mainly derive from the activity of multi‐subunit enzyme NOXs present in ECs and SMCs, which catalyse the reduction of oxygen to O2 − using NAD(P)H as electron donor. These enzymes comprise five subunits (p47phox, p67phox, p22phox, p40phox and gp91phox) which assembly into the membrane upon complex activation by different stimuli. Different NOX isoenzymes, namely NOX1, NOX4 and NOX5, are constitutively present in the vasculature, while NOX2, typically expressed in cells of the macrophage lineage, may be involved when ECs show an inflammatory phenotype. All these enzymes release O2 − which acts as signalling molecule in many pathophysiological processes, such as cell growth, migration, fibrosis, apoptosis and inflammation. Excessive activation of these enzymes, by AngII, proinflammatory cytokines and oxLDLs, occurs in age‐related diseases, including diabetes, hypertension and atherosclerosis.
Oxysterols appear to up‐regulate the activity of these NOX isoenzymes, at least that of constitutive NOX1, NOX4 and induced NOX2. With regard to NOX1, a dietary representative mixture of oxysterols has been reported to up‐regulate this enzyme in a differentiated epithelial colonic cell line (CaCo2) leading to the overexpression and synthesis of various proinflammatory molecules, more efficiently than unoxidized cholesterol (Biasi et al. 2009; Mascia et al. 2010); in the same cell model, NOX1 up‐regulation induced by 7α‐OH, 7β‐hydroxycholesterol (7β‐OH) and α‐EPOX was observed to trigger apoptosis (Biasi et al. 2013). The NOX4 content is increased in atherosclerotic plaques, and it has been demonstrated that 7‐K promotes endoplasmic reticulum (ER) stress and apoptosis by up‐regulating NOX4 in human aortic SMCs (Pedruzzi et al. 2004). ER stress occurs when stress signals, such as oxidative stress, cause the accumulation of misfolded or unfolded proteins in the organelle. Under normal conditions, the unfolded protein response is then activated to restore ER homeostasis; if this response is not sufficient to contrast the stress stimuli, ER triggers apoptosis (Chaudhari et al. 2014). As regards NOX2, in U937 promonocytic cells, an oxysterol mixture mimicking that present in atherosclerotic plaques induced the early generation of ROS, the effect being at least partly dependent on activation of this enzyme (Gargiulo et al. 2011).
More recently, in the same cell model 27‐OH was shown to markedly up‐regulate NOX2, but also induced the derangement of the mitochondrial membrane potential, thus amplifying ROS production (Vurusaner et al. 2014). In aged ECs, it appears reasonable to expect reduced mitochondrial function; oxysterols might then further interfere with these aged organelles, and either favour the onset of, or sustain, an oxidative imbalance of EC redox state.
Oxysterols and inflammation
Atherosclerosis, which resembles the accelerated ageing of certain tracts of the arterial system, is characterized by chronic inflammation. This process begins as a protective mechanism, and then becomes detrimental when it is activated in an uncontrolled and continuous manner in the arterial wall. Inflammation is a crucial trigger of the initial stage of atherosclerosis, and supports its pathogenesis at all stages, including the rupture of unstable plaques. In the atherosclerotic process, oxLDLs are the typical trigger of the expression of adhesion molecules and chemokines on ECs, with consequent recruitment of circulating monocytes; the inflammation‐dependent increase in the permeability of the endothelial layer allows inflammatory cells to transmigrate into the subendothelial space, where they differentiate into macrophages and start secreting chemokines and cytokines to attract other inflammatory cells and activate vascular cells, i.e. ECs and VSMCs. Indeed, inflammation stimulates the shift of VSMCs from the contractile phenotype to the fibroblast‐like phenotype, enabling the cells to proliferate and migrate from the tunica media into the intima, thus determining a net and irreversible alteration of the structure of the arterial wall. All these events lead to endothelium dysfunction and arterial stiffness. Inflammation also participates in vascular remodelling due to ageing, by progressively disrupting the ECM, both by stimulating fibrogenesis (the inflammatory cytokine TGF‐β1 is strongly profibrogenic) and by markedly up‐regulating MMPs. In this connection, oxysterols have been shown to induce expression and synthesis of TGF‐β1 in U937 human promonocytic cells (Leonarduzzi et al. 2001) and in Raw264.7 murine macrophages (Ferré et al. 2009). Both of these studies used a biologically compatible mixture of oxysterols, in which 7‐K was preponderant. Conversely, no effect on TGF‐β1 expression and synthesis was found with unoxidized cholesterol in U937 cells (Leonarduzzi et al. 2001).
Irregular and uncontrolled ECM degradation would clearly contribute to the age‐related disruption of vascular wall homeostasis. MMPs are the main enzymes responsible for this process, together with their specific inhibitors, TIMPs; their expression may be modulated by inflammation. In this connection, oxysterols are among the active molecules that contribute to disrupting arterial ECM homeostasis: MMP‐2 and ‐9 have been found to increase in VSMCs by 7‐K and α‐EPOX through activation of extracellular signal‐regulated kinase (ERK) (Liao et al. 2010), a signalling kinase involved in nuclear factor‐κB (NF‐κB) activation, and thus in overexpression of inflammatory genes. Interestingly, 7‐K can orientate human macrophages from peripheral blood monocytes towards a proinflammatory phenotype by inducing many proinflammatory mediators, including MMP‐9 (Buttari et al. 2013). In addition, one of the most represented oxysterols in human blood, i.e. 27‐OH, has been shown to markedly up‐regulate MMP‐9 expression and synthesis in human promonocytic U937 cells, by activating Toll‐like receptor 4 (TLR4) (Gargiulo et al. 2015) and/or by inducing CD14, a coreceptor of TLR4 (Kim et al. 2015). As far as TIMPs are concerned, 27‐OH is reported not to modulate their levels in promonocytic cells (Gargiulo et al. 2011).
Of great interest, arteries from oxysterol‐fed ApoE−/− mice showed accelerated plaque destabilization and rupture, associated with increased monocyte infiltration, and MCP‐1 and MMP activity. All these effects were prevented, in the same animal model, when oxysterol intestinal absorption was inhibited by administering ezetimibe; this points to a role of oxysterols in plaque vulnerability in high‐risk patients (Sato et al. 2012). Moreover, MCP‐1 as well as β1‐integrin were found to be up‐regulated in U937 by an oxysterol mixture mimicking that detectable in the plasma of hypercholesterolaemic individuals (Leonarduzzi et al. 2005; Gargiulo et al. 2012). The same oxysterol mixture was also found to induce foam cell formation through up‐regulation of scavenger receptor CD36 (Leonarduzzi et al. 2008, 2010). Unlike the oxysterol mixture, unoxidized cholesterol did not modulate MCP‐1 and β1‐integrin expression and synthesis as well as CD36 (Leonarduzzi et al. 2005, 2008, 2010; Gargiulo et al. 2012).
Quite a number of oxysterols appear able to trigger and sustain an inflammatory process within the vasculature, through overexpression of a variety of inflammatory cytokines, chemokines and adhesion molecules. 7β‐OH and 7‐K induce interleukin‐1β (IL‐1β) expression in human vascular ECs, as well as E‐selectin, intercellular adhesion molecule‐1 (ICAM‐1) and vascular cell adhesion molecule (VCAM‐1) (Lemaire et al. 1998). Moreover, 27‐OH was found to up‐regulate IL‐1β, IL‐8, TNF‐α and MMP‐9 in U937 cells (Gargiulo et al. 2015). In addition, 7β‐OH, 7‐K and 25‐hydroxycholesterol (25‐OH) display potent induction of MCP‐1, macrophage inflammatory protein‐1β (MIP‐1β), tumour necrosis factor‐α (TNF‐α) and IL‐8 in human macrophagic cell lines (Prunet et al. 2006). IL‐8 appears to be up‐regulated by 7β‐OH and 25‐OH in THP‐1 cells via the ERK/activator protein 1 (AP‐1) pathway (Lemaire‐Ewing et al. 2009) and by 27‐OH in U937 cells via the TLR4/NF‐κB pathway (Gargiulo et al. 2015). Again in connection with the oxysterol‐stimulated expression of adhesion molecules, 25‐OH has been shown to enhance VCAM‐1 expression in human aortic ECs, as well as augmenting monocyte adhesion to ECs (Naito et al. 2005).
Oxysterols and apoptosis
It is known that cell death and survival are important events in arterial wall degeneration and atherosclerosis progression. Macrophage apoptosis and defective clearance of these cells contribute to the formation and enlargement of the lipid‐rich necrotic core, a structural change that favours the further development of atherosclerotic lesions. In this connection, apoptosis of VSMCs eventually favours plaque instability, since the thickness of the atheroma's fibrotic cap essentially depends on the proliferation and activity of these cells: an increased number of apoptotic VSMCs has, indeed, been found in symptomatic plaques (Khatib & Vaya, 2014).
Apoptosis is genetically programmed cell death that has been conserved throughout evolution, and which is used to maintain normal cell numbers in the organism, and during growth. It is a coordinated and active process characterized by specific morphological changes: chromatin condensation and nuclear fragmentation, membrane blebbing and apoptotic body formation.
Apoptosis is triggered both through an extrinsic or death‐receptor‐mediated pathway, and through an intrinsic or mitochondrial pathway. Both pathways appear to be activated by a variety of biologically relevant oxysterols that exert strong proapoptotic effects. Some findings support the involvement of death receptors in oxysterol‐induced apoptosis. 27‐OH and 22‐hydroxycholesterol induce TNF‐α in macrophagic cells (Landis et al. 2002; Kim et al. 2013; Gargiulo et al. 2015) and the up‐regulation of TNF receptors has been observed in human aortic SMCs after challenge with 7‐K (Lee et al. 2005).
With regard to the intrinsic pathway of apoptosis, a large number of reports corroborate its activation by oxysterols. To give some examples, 25‐OH and 7‐K have been shown to regulate mitochondrial proapoptotic Bax translocation and ROS release in VSMCs via activation of soluble adenylyl cyclase/protein kinase A (Appukuttan et al. 2013). Again in VSMCs, Triol was found to markedly impair mitochondrial membrane potential (Tang et al. 2005); 25‐OH caused cytochrome c release and activation of caspase‐3 and ‐9 in CHO‐K1 (Yang & Sinensky, 2000), while 7β‐OH activated caspase‐3 and ‐9 in U937 cells (Ryan et al. 2004).
An important point in oxysterol‐induced‐apoptosis is the effect of the compounds on cell cycle regulation: oxysterols can lead to p53 activation, a key protein involved in DNA surveillance, promotion of cell cycle arrest, repair and eventually apoptosis. The role of p53 in cell death associated with atherosclerosis progression is confirmed by the finding that p53 expression is correlated with necrotic core formation in human carotid atherosclerotic plaques (Yuan et al. 2010). Challenging macrophagic cells (THP‐1 and J774 cell lines) with either 7β‐OH or 7‐K has been observed to promote p53 phosphorylation, and its translocation into the nucleus, with consequent triggering of a series of cell reactions, namely lysosomal membrane permeabilization, cathepsin release, and eventually mitochondrial membrane damage (Li et al. 2011). The activation of p53 by these two oxysterols is thought to be dependent upon an early induction of ROS and a transient up‐regulation of early growth response protein, a proinflammatory protein also present in human atheroma (Miah et al. 2013). Besides up‐regulation of p53 function, another common mechanism that interferes with cell cycle completion is the inhibition of certain cyclin‐dependent kinases, with consequent accumulation of cells in the G1, S and G2/M phases of the cycle. In this connection, treatment of THP‐1 monocyte‐like cells with 7β‐OH or with 25‐OH blocked them in the G2/M phase, through an oxysterol‐dependent inhibition of cyclin B1 and its dependent kinase cdc2. The cells then underwent apoptosis via reduction of the antiapoptotic protein Bcl‐2 and activation of caspase‐9 and ‐3 (Lim et al. 2003).
Further evidence of primary interest with regard to proapoptotic effects of some oxysterols is that the ability to induce apoptosis appears consistently to be correlated with an increased intracellular ROS steady state (Cheng et al. 2005; Sottero et al. 2009; Miah et al. 2013). The key role exerted by ROS in oxysterol‐mediated apoptosis is indirectly validated by its inhibition upon cell pre‐treatment with antioxidants. For instance, when promonocytic U937 cells were challenged with 7‐K, it provoked oxidation of GSH, a versatile molecule that also controls transmembrane mitochondrial potential; the consequence was an increased rate of cell apoptosis. However, when oxysterol‐treated cells were supplemented with GSH or N‐acetylcysteine, the number of apoptotic cells was drastically diminished (Lizard et al. 1998). Supplementation of U937 cells with α‐tocopherol counteracted 7‐K‐provoked lysosomal degradation, caspase activation and phospholipidosis (Vejux et al. 2009).
Moreover, some oxysterols, for example 7‐K, 7β‐OH and 24S‐hydroxycholesterol (24S‐OH), are reported to trigger apoptosis and, at the same time, autophagy; activation of the autophagic process was demonstrated by the presence of cytoplasmic phagosomes and by increased levels of the specific marker of autophagy, the microtubule‐associated protein light chain 3 (LC3)‐II derived from LC3‐I cleavage (Nury et al. 2015).
Oxysterols thus appear able to induce a complex type of cellular death mechanism, which includes oxidative stress, apoptosis and autophagy; this has very recently been termed ‘oxiapoptophagy’. Of interest, α‐tocopherol and docosahexaenoic acid effectively contrasted the various aspects associated with oxiapoptophagy; this was observed in murine oligodendrocytes (Nury et al. 2015).
Oxysterols and endothelial dysfunction
Alteration of the endothelium is an early sign of vascular ageing and atherosclerosis development, leading over time to altered endothelial relaxation and vascular stiffness. Quiescent ECs become senescent when exposed to vascular risk factors; for example, oxLDLs promote the senescence of human umbilical vein ECs (HUVECs) by decreasing sirtuin 1 and up‐regulating both the ROS steady‐state level and the inflammatory reaction (Tian & Li, 2014), probably through hyperactivation of NOX (Zarzuelo et al. 2013). On becoming senescent, ECs gradually lose their replicative potential, so that endothelium repair is markedly or even dramatically impaired. The latter event, together with intima stiffness, leads to the loss of endothelial integrity, which in the long run and in localized areas of medium and large arteries may also initiate the atherosclerotic process (Huynh et al. 2011).
Oxysterols may contribute to endothelial senescence by making ECs more susceptible to stress stimuli and to CVD pathogenesis: oxysterols may alter the endothelium layer in at least two ways: (i) in circulating oxLDLs, these compounds are localized on the surface of the micelles and may directly affect endothelial structure and function, thus favouring the deposition of LDLs and inflammatory cells in the subendothelial space; (ii) once they have accumulated in the subintimal spaces, and thus reached relatively high concentrations, oxysterols promote EC detachment from basement membrane, enhance inflammatory reactions, and favour thrombotic events.
In the initial stage of atherosclerosis, oxysterols are involved in the impairment of several endothelial functions, such as endothelial‐eNOS‐dependent NO generation, ROS production and cell permeability. Among the various lipid oxidation products present in oxLDLs, 7‐K, 7α‐OH and 27‐OH are reported to be the most effective at endothelial stiffening, suggesting these compounds play an important role in EDR and endothelium dysfunction. Of note, unoxidized cholesterol has no effect on endothelial stiffness. Interestingly, enriching the cells with cholesterol before oxysterol treatment protect ECs against the detrimental effect of oxysterols (Shentu et al. 2012). It has been hypothesized that oxysterols alter the endothelium by inserting themselves into the lipid bilayer and decreasing lipid packing of the ordered domains rich in cholesterol, as was experimentally observed for 7‐K (Sleer et al. 2001; Shentu et al. 2010), and that, conversely, increasing the levels of membrane cholesterol may be protective (Shentu et al. 2012). The insertion of oxysterols into biomembranes might also induce endothelial dysfunction by affecting the activity of membrane‐bound enzymes, such Na+–K+‐ATPase, as was found to occur in cells treated with Triol and 7‐K (Ramasamy et al. 1992; Duran et al. 2010). The different effects of different oxysterols on endothelial stiffness may be due to their differing ability to modify lipid membrane content and structure, depending on their chemical structure and properties. The incubation of aortic rings with 7‐K, 7α‐OH and 7β‐OH confirmed the ability of these oxysterols to affect EDR; of note, arterial relaxation was inhibited more markedly when oxysterols were used in a mixture, suggesting possible synergistic reactions in vivo. This effect on rabbit aortic rings was due to diminished NO availability following direct interaction of oxysterols with NO, and not to eNOS inhibition (Wong et al. 2011). Conversely, eNOS inhibition was observed in histamine‐stimulated HUVECs, in which 7‐K or 7β‐OH significantly decreased NO release, while cholesterol, β‐EPOX and 19‐hydroxycholesterol did not (Deckert et al. 1998). It is known that a decline in NO bioavailability may also be due to NO accelerated degradation by ROS (Harrison, 1997). Since oxysterols are well‐known NOX activators and ROS inducers, they may increase the production of O2 −, which rapidly reacts with NO, thus decreasing its bioavailability (Bosch‐Marce et al. 2007).
Again with regard to EDR, pretreatment of rabbit aortic segments with native LDLs did not affect endothelial relaxation, while oxLDLs significantly did so; remarkably, among the major components derived from LDL oxidation, e.g. lysophosphatidylcholine and the lipoperoxides, oxysterols most strongly impaired EDR (Deckert et al. 1997; Wong et al. 2011). These results strengthen the case for the oxidation of cholesterol being a crucial reaction in rendering LDLs proatherogenic molecules, and also point to cholesterol oxidation derivatives as key actors in endothelium dysfunction related to hypercholesterolaemia. Another mechanism whereby oxysterols, such as 25‐OH, 7‐K, α‐EPOX and Triol, could alter endothelial integrity is by inhibiting prostaglandin (PG) I2 synthesis and release by HUVECs, in consequence enhancing platelet adhesion to ECs. Unoxidized cholesterol, conversely, had no effect on PGI2 production or platelet adhesion (Peng et al. 1993).
A recently characterized oxysterol, 5,6‐secocholesterol, stemming from a non‐free‐radical‐mediated autoxidation process, was demonstrated to affect endothelial integrity by inducing p53‐dependent apoptosis and by strongly inhibiting EDR in in vivo and in vitro models (Luchetti et al. 2015). Further, the ability of 7‐oxysterols, which derive from cholesterol oxidation at C7, including 7β‐OH and 7‐K, to induce endothelial dysfunction was suggested by research showing that these oxysterols induced EC apoptosis by accumulating in lysosomes at an early stage, followed by lysosomal activation, oxidative stress and the mitochondrial pathway of programmed death. In particular, 7β‐OH and 7‐K induced cell release of lysosomal cathepsin and von Willebrand factor, thus further contributing to endothelial damage. Again, cell treatment with cholesterol did not show any toxic effect (Li et al. 2011).
Among the various factors involved in the regulation of endothelial function is AngII, which stems from the renin–angiotensin system (RAS). This peptide has a vasoconstrictor effect, and stimulates the release of aldosterone from the adrenal glands, regulating reabsorption of water and sodium by kidneys. This system is thus crucial to the regulation of vascular tone and systemic blood pressure levels, and its dysregulation is associated with vascular damage in hypertension and atherosclerosis (Schiffrin & Touyz, 2004). The classical source of renin are the kidneys, but it has recently been reported that angiotensin peptides may be formed in the brain, adrenal glands, reproductive tissue, pituitary glands, gastrointestinal tract, haematopoietic tissues, heart and blood vessels (Ferrario et al. 2014). The effects of AngII on vascular cells is mediated by binding to its G‐protein‐coupled receptor AT1R and receptor AT2R, and by the activation of several signalling pathways. AngII may contribute to cell proliferation, apoptosis and inflammation by activating the mitogen‐activated protein kinases ERK, p38 and c‐Jun N‐terminal kinases (JNK), Janus kinase/signal transducer and activator of transcription (JAK/STAT); further, AngII may promote arterial remodelling by activating tyrosine kinase receptors, such as epidermal growth factor receptor (EGFR), platelet‐derived growth factor receptor, and insulin‐like 1 growth factor receptor (Montezano et al. 2014).
Increasing evidence supports the synergistic effect of hyperlipidaemia and RAS in endothelial dysfunction during atherogenesis. Indeed, oxLDLs can induce the expression of angiotensin‐converting enzyme and AT1R in primary human HUVECs (Catar et al. 2007); this interaction was confirmed in ApoE−/− mice fed a high‐cholesterol diet, in which the combined administration of the statin rosuvastatin and the AT1R inhibitor candesartan significantly counterbalanced the up‐regulation of lectin‐like oxLDL receptor‐1 and p38 activation, as provoked by the dietary regimen adopted (Chen et al. 2006). Again, the combination of pravastatin and irbesartan in patients with stable coronary artery disease improved endothelial function (Morawietz et al. 2006). Blocking of AT1R abolished oxLDL‐induced EDR alteration in murine aortic vascular ring (Yamamoto et al. 2015) and foam cell formation in human macrophages (Osada‐Oka et al. 2012).
The role of oxysterols as important mediators of the oxLDL effect on RAS has been little investigated to date. It has been demonstrated that 27‐OH and 24S‐OH up‐regulate RAS, in particular angiotensin‐converting enzyme activity, and angiotensinogen levels, in mouse brain and in primary neurons and astrocytes (Mateos et al. 2011). Since AngII activates signalling pathways similar to that promoted by oxysterols, and since this peptide is involved in key events in atherosclerosis, it might be interesting to investigate the relationship between these molecules and their likely synergistic interaction in greater depth.
Oxysterols and VSMC phenotypic changes
VSMCs are crucial to plaque formation, progression and stability, and the changes observed in these cells during ageing reflect that occurring during atherogenesis. As described above, VSMCs migrate and proliferate, causing intimal thickening, and they contribute to arterial stiffness by increasing collagen deposition and calcification. These cells undergo a phenotypic change and start synthesizing ECM components (a process called SMC activation); in addition, SMCs migrate into the intimal layer of the arterial wall and they determine the fibrous cap formation.
With regard to the ability of compounds in the oxysterol family to activate VSMCs, it has been shown that low doses of 7‐K or of α‐EPOX (2.5 μm) induce proliferation and migration of rat aortic SMCs by activating the EGFR/phosphoinositide 3‐kinase (PI3K)/Akt pathway and up‐regulating MMP‐2 and MMP‐9 activities; cell treatment with the same concentration of cholesterol had no significant effect on SMC proliferation (Liao et al. 2010). The activation of survival pathways by oxLDLs has been confirmed in a study that demonstrated that oxLDLs induced MMP‐2 via the PI3K/Akt pathway in primary cultures of rat aortic VSMCs (Li HX et al. 2012). Conversely, 7‐K and α‐EPOX at high concentration (>25 μm) markedly decreased the number of viable cells by activating apoptotic death (Liao et al. 2010); this dual effect of the two oxysterols fits quite well with the view that they may first initiate impairment of arterial wall structures by favouring activation of VSMCs, and subsequently, following excessive accumulation, they could contribute to a dramatic derangement of the entire vascular wall, the latter stage being especially evident in the vascular areas affected by the atherosclerotic process. It has been hypothesized that oxysterols might activate VSMCs by a different mechanism: 25‐OH induced severe morphological changes in SMCs and ECs of rabbit pulmonary artery by up‐regulating PGG/H synthase‐2 activity thus enhancing eicosanoid production, mainly PGE2 (Wohlfeil & Campbell, 1999); PGE2 increases endothelial permeability, VSMC proliferation and migration, and vascular tone (Gomez et al. 2013). The enzyme PGG/H synthase‐2 is expressed after vascular cellular activation, often in response to inflammatory stimuli; thus, 25‐OH could induce it directly or by overexpressing proinflammatory cytokines.
The contribution of oxysterols to vascular calcification
Vascular calcification is a dynamic process similar to osteogenesis, which is often associated with arterial stiffness and atherogenic degeneration. It principally depends on the VSMCs acquiring an osteoblastic phenotype, whereupon they begin producing and secreting hydroxyapatite crystals. This specific cell shift may, for instance, be promoted by oxidative stress, in turn induced by high phosphataemia, advanced glycation end‐products, bone morphogenetic proteins, and inflammatory cytokines (McCarty & DiNicolantonio, 2014).
Several oxysterols of pathophysiological relevance have been shown to exert pro‐oxidant effects, and also to stimulate phenotypic changes in VSMCs (see above). This makes them good candidate molecules for determining accumulation of calcium in the tunica media of arterial walls. In addition, the proapoptotic effect that they exert on vascular cells would provide suitable nucleating sites for the formation of hydroxyapatite crystals (Proudfoot et al. 2000).
An increasing bulk of data now points to the property of certain oxysterols of exerting potent osteogenic activity. The first evidence of this emerged from a study in which rat bone marrow stromal cells were treated with a 1:1 combination of 22(S)‐hydroxycholesterol and 20(S)‐hydroxycholesterol (Kha et al. 2004). The same oxysterol mixture was confirmed to induce osteogenic differentiation in a murine embryonic stem cell line, in which it enhanced mitochondrial activity and triggered the Hedgehog and Wnt/β‐catenin signalling pathways (Kwon et al. 2015). Again, an oxysterol mixture enhanced calcium deposition and osteogenic protein expression, in particular alkaline phosphatase, osteopontin and osteocalcin, in rat mesenchymal stem cells (Wöltje et al. 2015). Similar effects were exerted by 25‐OH and 7‐K on bovine VSMCs (Watson et al. 1994; Saito et al. 2008) and Triol on rat VSMCs (Liu et al. 2004). At least with regard to Triol, experimental data point to the oxysterol's pro‐oxidant effect as being reponsible for cell calcification, given that incubation with antioxidants (vitamin C + vitamin E) reversed that action (Liu et al. 2004); this explanation is also compelling for oxLDLs, since they are able to induce ROS and calcification by promoting inflammation, apoptosis and the release of osteogenic factors (Farrokhi et al. 2015). A recent study provides indirect support to the hypothesis that oxysterols make a significant contribution to the abnormal calcium deposition in arterial wall; human coronary artery SMCs were challenged with oxLDLs and induced net osteopontin synthesis, and through this protein marked proliferation and migration of the cells occurred; both these effects are primary events in arterial stiffness (Liu et al. 2014).
Conclusions
The accumulating experimental evidence in favor of oxysterols as candidate molecules involved in vascular ageing mechanisms appears convincing for a number of reasons. First, the biochemical effects exerted by a large variety of these oxysterols, in particular their proinflammatory properties, closely fit the type of events occurring in ageing blood vessels. Second, oxysterols have a remarkable ability to induce vascular cells to modify their phenotypic features, and thus their function. Third, the relatively high concentrations that these compounds can reach in the vasculature, transported into the sub‐intimal space by LDLs, is noteworthy; it also probably limits the contribution of oxysterols to vascular ageing mainly in the case of the arteries and, specifically, to medium and large arteries, in which lipids do actually accumulate. However, a generalized effect on the vascular endothelium of oxysterols contained in circulating oxLDLs, favoured by the fact that they are localized on the surface of these micelles, should not be excluded a priori (see Abstract figure).
Additional information
Competing interests
None declared.
Acknowledgements
The authors thank the University of Torino for having enabled them to investigate in depth over the years the potential role of oxysterols in human pathophysiology.
Biography
Giuseppe Poli is Professor of General Pathology and Pathophysiology at the School of Medicine, University of Torino. For more than 40 years he has been involved in elucidating the role of lipid peroxidation, and then the roles of a variety of lipid oxidation products, in the pathogenesis of major chronic diseases. More recently, he focused his research interest on the pathophysiology of cholesterol oxidation products. Simona Gargiulo is a molecular biologist with expertise in the role of oxidized lipids in the pathogenesis of age‐related diseases such as atherosclerosis. In particular, she has been studying the involvement of compounds derived from cholesterol oxidation in inflammation and oxidative stress processes. She is a postdoctoral fellow at the University of Torino, School of Medicine.

References
- Annangudi SP, Deng Y, Gu X, Zhang W, Crabb JW & Salomon RG (2008). Low density lipoprotein has an enormous capacity to bind (E)‐4‐hydroxynon‐2‐enal (HNE): detection and characterization of lysyl and histidyl adducts containing multiple molecules of HNE. Chem Res Toxicol 21, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appukuttan A, Kassecker SA, Kumar S, Reusch HP & Ladilov Y (2013). Oxysterol‐induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc Res 99, 734–742. [DOI] [PubMed] [Google Scholar]
- Biasi F, Chiarpotto E, Sottero B, Maina M, Mascia C, Guina T, Gamba P, Gargiulo S, Testa G, Leonarduzzi G & Poli G (2013). Evidence of cell damage induced by major components of a diet‐compatible mixture of oxysterols in human colon cancer CaCo‐2 cell line. Biochimie 93, 632–640. [DOI] [PubMed] [Google Scholar]
- Biasi F, Mascia C, Astegiano M, Chiarpotto E, Nano M, Leonarduzzi G & Poli G (2009). Prooxidant and proapoptotic effects of cholesterol oxidation products on human colonic epithelial cells: a potential mechanism of inflammatory bowel disease progression. Free Radic Biol Med 47, 1731–1741. [DOI] [PubMed] [Google Scholar]
- Bosch‐Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, Vandiver S, Rabi A, Shaked Y, Kerbel R, Lavallee T & Semenza GL (2007). Effects of aging and hypoxia‐inducible factor‐1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res 101, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N & Schröder K (2014). Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med 76, 208–226. [DOI] [PubMed] [Google Scholar]
- Busse R & Fleming I (2006). Vascular endothelium and blood flow. Handb Exp Pharmacol 176, 43–78. [DOI] [PubMed] [Google Scholar]
- Buttari B, Segoni L, Profumo E, D'Arcangelo D, Rossi S, Facchiano F, Businaro R, Iuliano L & Riganò R (2013). 7‐Oxo‐cholesterol potentiates pro‐inflammatory signaling in human M1 and M2 macrophages. Biochem Pharmacol 86, 130–137. [DOI] [PubMed] [Google Scholar]
- Carpenter KL, Kirkpatrick PJ, Weissberg PL, Challis IR, Dennis IF, Freeman MA & Mitchinson MJ (2003). Oral α‐tocopherol supplementation inhibits lipid oxidation in established human atherosclerotic lesions. Free Radic Res 37, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Catar R, Müller G, Heidler J, Schmitz G, Bornstein SR & Morawietz H (2007). Low‐density lipoproteins induce the renin‐angiotensin system and their receptors in human endothelial cells. Horm Metab Res 39, 801–805. [DOI] [PubMed] [Google Scholar]
- Catita J, López‐Luppo M, Ramos D, Nacher V, Navarro M, Carretero A, Sánchez‐Chardi A, Mendes‐Jorge L, Rodriguez‐Baeza A & Ruberte J (2015). Imaging of cellular aging in human retinal blood vessels. Exp Eye Res 135, 14–25. [DOI] [PubMed] [Google Scholar]
- Chaudhari N, Talwar P, Parimisetty A, Lefebvre d'Hellencourt C & Ravanan P (2014). A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci 8, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li D, Schaefer R & Mehta JL (2006). Cross‐talk between dyslipidemia and renin‐angiotensin system and the role of LOX‐1 and MAPK in atherogenesis studies with the combined use of rosuvastatin and candesartan. Atherosclerosis 184, 295–301. [DOI] [PubMed] [Google Scholar]
- Cheng YW, Kang J, Shih YL, Lo YL & Wang C (2005). Cholesterol‐3‐beta, 5‐alpha, 6‐beta‐triol induced genotoxicity through reactive oxygen species formation. Food Chem Toxicol 43, 617–622. [DOI] [PubMed] [Google Scholar]
- Cohen R, Plane F, Najibi S, Huk I, Malinski T & Garland C (1997). Nitric oxide is the mediator of both endothelium‐dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc Natl Acad Sci USA 94, 4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deanfield JE, Halcox JP & Rabelink TJ (2007). Endothelial function and dysfunction: testing and clinical relevance. Circulation 115, 1285–1295. [DOI] [PubMed] [Google Scholar]
- Deckert V, Brunet A, Lantoine F, Lizard G, Millanvoye‐van Brussel E, Monier S, Lagrost L, David‐Dufilho M, Gambert P & Devynck MA (1998). Inhibition by cholesterol oxides of NO release from human vascular endothelial cells. Arterioscler Thromb Vasc Biol 18, 1054–1060. [DOI] [PubMed] [Google Scholar]
- Deckert V, Perségol L, Viens L, Lizard G, Athias A, Lallemant C, Gambert P & Lagrost L (1997). Inhibitors of arterial relaxation among components of human oxidized low‐density lipoproteins. Cholesterol derivatives oxidized in position 7 are potent inhibitors of endothelium‐dependent relaxation. Circulation 95, 723–731. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Morgan RG, Walker AE & Lesniewski LA (2015). Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol 89, 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran MJ, Pierre SV, Lesnik P, Pieroni G, Bourdeaux M, Dignat‐Georges F, Sampol J & Maixent JM (2010). 7‐Ketocholesterol inhibits Na,K‐ATPase activity by decreasing expression of its α1‐subunit and membrane fluidity in human endothelial cells. Cell Mol Biol (Noisy‐le‐grand) 56, Suppl., OL1434–1441. [PubMed] [Google Scholar]
- Farrokhi E, Samani K & Chaleshtori M (2015). Oxidized low‐density lipoprotein increases bone sialoprotein expression in vascular smooth muscle cells via runt‐related transcription factor 2. Am J Med Sci 349, 240–243. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N & Dell'italia LJ (2014). An evolving story of angiotensin‐II‐forming pathways in rodents and humans. Clin Sci (Lond) 126, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré N, Martinez‐Clemente M, Lopez‐Parra M, Gonzales‐Periz A, Horrillo R, Planagumà A, Camps J, Joven J, Tres A, Guardiola F, Bataller R, Arroyo V & Clària J (2009). Increased susceptibility to exacerbated liver injury in hypercholesterolemic Apo‐E deficient mice: potential involvement of oxysterols. Am J Physiol Gastrointest Liver Physiol 296, G553–G562. [DOI] [PubMed] [Google Scholar]
- Garcia‐Cruset S, Carpenter KL, Guardiola F, Stein BK & Mitchinson MJ (2001). Oxysterol profiles of normal human arteries, fatty streaks and advanced lesions. Free Radic Res 35, 31–41. [DOI] [PubMed] [Google Scholar]
- Gargiulo S, Gamba P, Testa G, Sottero B, Maina M, Guina T, Biasi F, Poli G & Leonarduzzi G (2012). Molecular signaling involved in oxysterol‐induced β₁‐integrin over‐expression in human macrophages. Int J Mol Sci 13, 14278–14293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo S, Gamba P, Testa G, Rossin D, Biasi F, Poli G & Leonarduzzi G (2015). Relation between TLR4/NF‐κB signaling pathway activation by 27‐hydroxycholesterol and 4‐hydroxynonenal, and atherosclerotic plaque instability. Aging Cell 14, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo S, Sottero B, Gamba P, Chiarpotto E, Poli G & Leonarduzzi G (2011). Plaque oxysterols induce unbalanced up‐regulation of matrix metalloproteinase‐9 in macrophagic cells through redox‐sensitive signaling pathways: Implications regarding the vulnerability of atherosclerotic lesions. Free Radic Biol Med 51, 844–855. [DOI] [PubMed] [Google Scholar]
- Go YM, Halvey PJ, Hansen JM, Reed M, Pohl J & Jones DP (2007). Reactive aldehyde modification of thioredoxin‐1 activates early steps of inflammation and cell adhesion. Am J Pathol 171, 1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez I, Foudi N, Longrois D & Norel X (2013). The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids 89, 55–63. [DOI] [PubMed] [Google Scholar]
- Gradinaru D, Borsa C, Ionescu C & Prada GI (2015). Oxidized LDL and NO synthesis – Biomarkers of endothelial dysfunction and ageing. Mech Ageing Dev 151, 101–113. [DOI] [PubMed] [Google Scholar]
- Hamilton CA, Brosnan MJ, McIntyre M, Graham D & Dominiczak AF (2001). Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension 37, 529–534. [DOI] [PubMed] [Google Scholar]
- Harrison DG (1997). Endothelial function and oxidant stress. Clin Cardiol 20, II‐11–17. [PubMed] [Google Scholar]
- Harvey A, Montezano A & Touyz RM (2015). Vascular biology of ageing – implications in hypertension. J Mol Cell Cardiol 83, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmschrodt C, Becker S, Schröter J, Hecht M, Aust G, Thiery J & Ceglarek U (2013). Fast LC‐MS/MS analysis of free oxysterols derived from reactive oxygen species in human plasma and carotid plaque. Clin Chim Acta 425, 3–8. [DOI] [PubMed] [Google Scholar]
- Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB & Reinhart‐King CA (2011). Age‐related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med 3, 112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuliano L, Micheletta F, Natoli S, Ginanni Corradini S, Iappelli M, Elisei W, Giovannelli L, Violi F & Diczfalusy U (2003). Measurement of oxysterols and α‐tocopherol in plasma and tissue samples as indices of oxidant stress status. Anal Biochem 312, 217–223. [DOI] [PubMed] [Google Scholar]
- Kha HT, Basseri B, Shouhed D, Richardson J, Tetradis S, Hahn TJ & Parhami F (2004). Oxysterols regulate differentiation of mesenchymal stem cells: pro‐bone and antifat. J Bone Miner Res 19, 830–840. [DOI] [PubMed] [Google Scholar]
- Khatib S & Vaya J (2014). Oxysterols and symptomatic versus asymptomatic human atherosclerotic plaque. Biochem Biophys Res Commun 446, 709–713. [DOI] [PubMed] [Google Scholar]
- Kim SM, Jang H, Son Y, Lee SA, Bae SS, Park YE, Eo SK & Kim K (2013). 27‐Hydroxycholesterol induces production of tumor necrosis factor‐alpha from macrophages. Biochem Biophys Res Commun 430, 454–459. [DOI] [PubMed] [Google Scholar]
- Kim SM, Kim BY, Eo SK, Kim CD & Kim K (2015). 27‐Hydroxycholesterol up‐regulates CD14 and predisposes monocytic cells to superproduction of CCL2 in response to lipopolysaccharide. Biochim Biophys Acta 1852, 442–450. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Coskun P, Esposito LA & Wallace DC (2001). Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age‐related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA 98, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause KH (2007). Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp Gerontol 42, 256–262. [DOI] [PubMed] [Google Scholar]
- Kwon IK, Lee SC, Hwang YS & Heo JS (2015). Mitochondrial function contributes to oxysterol‐induced osteogenic differentiation in mouse embryonic stem cells. Biochim Biophys Acta 1854, 561–572. [DOI] [PubMed] [Google Scholar]
- Lakatta EG (2013). The reality of aging viewed from the arterial wall. Artery Res 7, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis MS, Patel HV & Capone JP (2002). Oxysterol activators of liver X receptor and 9‐cis‐retinoic acid promote sequential steps in the synthesis and secretion of tumor necrosis factor‐α from human monocytes. J Biol Chem 277, 4713–4721. [DOI] [PubMed] [Google Scholar]
- Lee HS, Chang J, Baek JA, Chung MY, Lee HC, Rhim BY, Sok DE, Rho MC, Kim YK & Kim K (2005). TNF‐α activates death pathway in human aorta smooth muscle cell in the presence of 7‐ketocholesterol. Biochem Biophys Res Commun 333, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Lemaire S, Lizard G, Monier S, Miguet C, Gueldry S, Volot F, Gambert P & Néel D (1998). Different patterns of IL‐1β secretion, adhesion molecule expression and apoptosis induction in human endothelial cells treated with 7α‐, 7β‐hydroxycholesterol, or 7‐ketocholesterol. FEBS Lett 440, 434–439. [DOI] [PubMed] [Google Scholar]
- Lemaire‐Ewing S, Berthier A, Royer MC, Logette E, Corcos L, Bouchot A, Monier S, Prunet C, Raveneau M, Rébé C, Desrumaux C, Lizard G & Néel D (2009). 7β‐Hydroxycholesterol and 25‐hydroxycholesterol‐induced interleukin‐8 secretion involves a calcium‐dependent activation of c‐fos via the ERK1/2 signaling pathway in THP‐1 cells: oxysterols‐induced IL‐8 secretion is calcium‐dependent. Cell Biol Toxicol 25, 127–139. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Gamba P, Gargiulo S, Biasi F & Poli G (2012). Inflammation‐related gene expression by lipid oxidation‐derived products in the progression of atherosclerosis. Free Radic Biol Med 52, 19–34. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Gamba P, Gargiulo S, Sottero B, Kadl A, Biasi F, Chiarpotto E, Leitinger N, Vendemiale G, Serviddio G & Poli G (2008). Oxidation as a crucial reaction for cholesterol to induce tissue degeneration: CD36 overexpression in human promonocytic cells treated with a biologically relevant oxysterol mixture. Aging Cell 7, 375–382. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Gamba P, Sottero B, Kadl A, Robbesyn F, Calogero R, Biasi F, Chiarpotto E, Leitinger N, Sevanian A & Poli G (2005). Oxysterol‐induced up‐regulation of MCP‐1 expression and synthesis in macrophage cells. Free Radic Biol Med 39, 1152–1161. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Gargiulo S, Gamba P, Perrelli MG, Castellano I, Sapino A, Sottero B & Poli G (2010). Molecular signaling operated by a diet‐compatible mixture of oxysterols in up‐regulating CD36 receptor in CD68 positive cells. Mol Nutr Food Res 54, Suppl. 1, S31–41. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Poli G, Sottero B & Biasi F (2007). Activation of the mitochondrial pathway of apoptosis by oxysterols. Front Biosci 12, 791–799. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Sevanian A, Sottero B, Arkan MC, Biasi F, Chiarpotto E, Basaga H & Poli G (2001). Up‐regulation of the fibrogenic cytokine TGF‐β1 by oxysterols: a mechanistic link between cholesterol and atherosclerosis. FASEB J 15, 1619–1621. [DOI] [PubMed] [Google Scholar]
- Li HX, Kong FJ, Bai SZ, He W, Xing WJ, Xi YH, Li GW, Guo J, Li HZ, Wu LY, Wang R, Yang GD, Tian Y & Xu CQ (2012). Involvement of calcium‐sensing receptor in oxLDL‐induced MMP‐2 production in vascular smooth muscle cells via PI3K/Akt pathway. Mol Cell Biochem 362, 115–122. [DOI] [PubMed] [Google Scholar]
- Li W, Ghosh M, Eftekhari S & Yuan XM (2011). Lipid accumulation and lysosomal pathways contribute to dysfunction and apoptosis of human endothelial cells caused by 7‐oxysterols. Biochem Biophys Res Commun 409, 711–716. [DOI] [PubMed] [Google Scholar]
- Li W, Laskar A, Sultana N, Osman E, Ghosh M, Li Q & Yuan XM (2012). Cell death induced by 7‐oxysterols via lysosomal and mitochondrial pathways is p53‐dependent. Free Radic Biol Med 53, 2054–2061. [DOI] [PubMed] [Google Scholar]
- Li Z, Froehlich J, Galis Z & Lakatta EG (1999). Increased expression of matrix metalloproteinase‐2 in the thickened intima of aged rats. Hypertension 33, 116–123. [DOI] [PubMed] [Google Scholar]
- Liao P, Cheng Y, Li C, Wang YT & Kang J (2010). 7‐Ketocholesterol and cholesterol‐5α,6α‐epoxide induce smooth muscle cell migration and proliferation through the epidermal growth factor receptor/phosphoinositide 3‐kinase/Akt signaling pathways. Toxicol Lett 197, 88–96. [DOI] [PubMed] [Google Scholar]
- Lim HK, Kang HK, Yoo ES, Kim BJ, Kim YW, Cho M, Lee JH, Lee YS, Chung MH & Hyun JW (2003). Oxysterols induce apoptosis and accumulation of cell cycle at G2/M phase in the human monocytic THP‐1 cell line. Life Sci 72, 1389–1399. [DOI] [PubMed] [Google Scholar]
- Liu H, Yuan L, Xu S, Zhang T & Wang K (2004). Cholestane‐3β, 5α, 6β‐triol promotes vascular smooth muscle cells calcification. Life Sci 76, 533–543. [DOI] [PubMed] [Google Scholar]
- Liu J, Ren Y, Kang L & Zhang L (2014). Oxidized low‐density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med 33, 1341–1347. [DOI] [PubMed] [Google Scholar]
- Lizard G, Gueldry S, Sordet O, Monier S, Athias A, Miguet C, Bessede G, Lemaire S, Solary E & Gambert P (1998). Glutathione is implied in the control of 7‐ketocholesterol‐induced apoptosis, which is associated with radical oxygen species production. FASEB J 12, 1651–1663. [DOI] [PubMed] [Google Scholar]
- Lordan S, Mackrill JJ & O'Brien NM (2009). Oxysterols and mechanisms of apoptotic signaling: implications in the pathology of degenerative diseases. J Nutr Biochem 20, 321–336. [DOI] [PubMed] [Google Scholar]
- Luchetti F, Canonico B, Cesarini E, Betti M, Galluzzi L, Galli L, Tippins J, Zerbinati C, Papa S & Iuliano L (2015). 7‐Ketocholesterol and 5,6‐secosterol induce human endothelial cell dysfunction by differential mechanisms. Steroids 99, 204–211. [DOI] [PubMed] [Google Scholar]
- Lusis AJ (2000). Atherosclerosis. Nature 407, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MF & DiNicolantonio JJ (2014). The molecular biology and pathophysiology of vascular calcification. Postgrad Med 126, 54–64. [DOI] [PubMed] [Google Scholar]
- Mascia C, Maina M, Chiarpotto E, Leonarduzzi G, Poli G & Biasi F (2010). Proinflammatory effect of cholesterol oxidation products on CaCo‐2 human enterocyte‐like cells: effective protection by epigallocathechin‐3‐gallate. Free Radic Biol Med 49, 2049–2057. [DOI] [PubMed] [Google Scholar]
- Mateos L, Ismail M, Gil‐Bea F, Schüle R, Schöls L, Heverin M, Folkesson R, Björkhem I & Cedazo‐Mínguez A (2011). Side chain‐oxidized oxysterols regulate the brain renin‐angiotensin system through a liver X receptor‐dependent mechanism. J Biol Chem 286, 25574–25585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miah S, Zadeh SN, Yuan XM & Li W (2013). Expression of Egr1 and p53 in human carotid plaques and apoptosis induced by 7‐oxysterol or p53. Exp Toxicol Pathol 65, 677–682. [DOI] [PubMed] [Google Scholar]
- Michel T & Vanhoutte PM (2010). Cellular signaling and NO production. Pflugers Arch 459, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheletta F, Natoli S, Misuraca M, Sbarigia E, Diczfalusy U & Iuliano L (2004). Vitamin E supplementation in patients with carotid atherosclerosis: reversal of altered oxidative stress status in plasma but not in plaque. Arterioscler Thromb Vasc Biol 24, 136–140. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer R & Higgs E (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43, 109–142. [PubMed] [Google Scholar]
- Monk BA & George SJ (2014). The effect of ageing on vascular smooth muscle cell behaviour – a mini‐review. Gerontology 61, 416–426. [DOI] [PubMed] [Google Scholar]
- Montezano AC, Burger D, Ceravolo GS, Yusuf H, Montero M & Touyz RM (2011). Novel Nox homologues in the vasculature: focusing on Nox4 and Nox5. Clin Sci (Lond) 120, 131–141. [DOI] [PubMed] [Google Scholar]
- Montezano AC, Nguyen Dinh Cat A, Rios FJ & Touyz RM (2014). Angiotensin II and vascular injury. Curr Hypertens Rep 16, 431. [DOI] [PubMed] [Google Scholar]
- Montezano AC & Touyz RM (2012). Molecular mechanisms of hypertension – reactive oxygen species and antioxidants: a basic science update for the clinician. Can J Cardiol 28, 288–295. [DOI] [PubMed] [Google Scholar]
- Morawietz H, Erbs S, Holtz J, Schubert A, Krekler M, Goettsch W, Kuss O, Adams V, Lenk K, Mohr FW, Schuler G & Hambrecht R (2006). Endothelial protection, AT1 blockade and cholesterol‐dependent oxidative stress: the EPAS trial. Circulation 114, I296‐301. [DOI] [PubMed] [Google Scholar]
- Naito Y, Shimozawa M, Kuroda M, Nakabe N, Manabe H, Katada K, Kokura S, Ichikawa H, Yoshida N, Noguchi N & Yoshikawa T (2005). Tocotrienols reduce 25‐hydroxycholesterol‐induced monocyte‐endothelial cell interaction by inhibiting the surface expression of adhesion molecules. Atherosclerosis 180, 19–25. [DOI] [PubMed] [Google Scholar]
- Nury T, Zarrouk A, Mackrill JJ, Samadi M, Durand P, Riedinger JM, Doria M, Vejux A, Limagne E, Delmas D, Prost M, Moreau T, Hammami M, Delage‐Mourroux R, O'Brien NM & Lizard G (2015). Induction of oxiapoptophagy on 158N murine oligodendrocytes treated by 7‐ketocholesterol‐, 7β‐hydroxycholesterol‐, or 24(S)‐hydroxycholesterol: Protective effects of α‐tocopherol and docosahexaenoic acid (DHA; C22:6 n‐3). Steroids 99, 194–203. [DOI] [PubMed] [Google Scholar]
- Osada‐Oka M, Kita H, Yagi S, Sato T, Izumi Y & Iwao H (2012). Angiotensin AT1 receptor blockers suppress oxidized low‐density lipoprotein‐derived formation of foam cells. Eur J Pharmacol 679, 9–15. [DOI] [PubMed] [Google Scholar]
- Parthasarathy S, Raghavamenon A, Garelnabi MA & Santanam N (2010). Oxidized low‐density lipoprotein. Methods Mol Biol 610, 403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, O'Dowd Y, Bens M, Vandewalle A, Gougerot‐Pocidalo MA, Lizard G & Ogier‐Denis E (2004). NAD(P)H oxidase Nox‐4 mediates 7‐ketocholesterol‐induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 24, 10703–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SK, Hu B, Peng AY & Morin R (1993). Effect of cholesterol oxides on prostacyclin production and platelet adhesion. Artery 20, 122–134. [PubMed] [Google Scholar]
- Poli G, Biasi F & Leonarduzzi G (2013). Oxysterols in the pathogenesis of major chronic diseases. Redox Biol 1, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Sottero B, Gargiulo S & Leonarduzzi G (2009). Cholesterol oxidation products in the vascular remodeling due to atherosclerosis. Mol Aspects Med 30, 180–189. [DOI] [PubMed] [Google Scholar]
- Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM & Weissberg PL (2000). Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res 87, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Prunet C, Montange T, Véjux A, Laubriet A, Rohmer JF, Riedinger JM, Athias A, Lemaire‐Ewing S, Néel D, Petit JM, Steinmetz E, Brenot R, Gambert P & Lizard G (2006). Multiplexed flow cytometric analyses of pro‐ and anti‐inflammatory cytokines in the culture media of oxysterol‐treated human monocytic cells and in the sera of atherosclerotic patients. Cytometry A 69, 359–373. [DOI] [PubMed] [Google Scholar]
- Puca A, Carrizzo A, Villa F, Ferrario A, Casaburo M, Maciąg A & Vecchione C (2013). Vascular ageing: the role of oxidative stress. Int J Biochem Cell Biol 45, 556–559. [DOI] [PubMed] [Google Scholar]
- Quarles EK, Dai DF, Tocchi A, Basisty N, Gitari L & Rabinovitch PS (2015). Quality control systems in cardiac aging. Ageing Res Rev 23, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy S, Boissonneault GA & Hennig B (1992). Oxysterol‐induced endothelial cell dysfunction in culture. J Am Coll Nutr 11, 532–538. [DOI] [PubMed] [Google Scholar]
- Ryan L, O'Callaghan YC & O'Brien NM (2004). Generation of an oxidative stress precedes caspase activation during 7β‐hydroxycholesterol‐induced apoptosis in U937 cells. J Biochem Mol Toxicol 18, 50–59. [DOI] [PubMed] [Google Scholar]
- Saito E, Wachi H, Sato F & Seyama Y (2008). 7‐Ketocholesterol, a major oxysterol, promotes Pi‐induced vascular calcification in cultured smooth muscle cells. J Atheroscler Thromb 15, 130–137. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano K, Katsuki S, Matoba T, Osada K, Sawamura T, Sunagawa K & Egashira K (2012). Dietary cholesterol oxidation products accelerate plaque destabilization and rupture associated with monocyte infiltration/activation via the MCP‐1‐CCR2 pathway in mouse brachiocephalic arteries: therapeutic effects of ezetimibe. J Atheroscler Thromb 19, 986–998. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL & Touyz RM (2004). From bedside to bench to bedside: role of renin‐angiotensin‐aldosterone system in remodeling of resistance arteries in hypertension. Am J Physiol Heart Circ Physiol 287, H435–H446. [DOI] [PubMed] [Google Scholar]
- Shentu TP, Singh DK, Oh M, Sun S, Sadaat L, Makino A, Mazzone T, Subbaiah PV, Cho M & Levitan I (2012). The role of oxysterols in control of endothelial stiffness. J Lipid Res 53, 1348–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shentu TP, Titushkin I, Singh DK, Gooch KJ, Subbaiah PV, Cho M & Levitan I (2010). oxLDL‐induced decrease in lipid order of membrane domains is inversely correlated with endothelial stiffness and network formation. Am J Physiol Cell Physiol 299, C218–C229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha K, Das J, Pal PB & Sil PC (2013). Oxidative stress: the mitochondria‐dependent and mitochondria‐independent pathways of apoptosis. Arch Toxicol 87, 1157–1180. [DOI] [PubMed] [Google Scholar]
- Sleer LS, Brown AJ & Stanley KK (2001). Interaction of caveolin with 7‐ketocholesterol. Atherosclerosis 159, 49–55. [DOI] [PubMed] [Google Scholar]
- Sottero B, Gamba P, Gargiulo S, Leonarduzzi G & Poli G (2009). Cholesterol oxidation products and disease: an emerging topic of interest in medicinal chemistry. Curr Med Chem 16, 685–705. [DOI] [PubMed] [Google Scholar]
- Szklarczyk R, Nooteboom M & Osiewacz HD (2014). Control of mitochondrial integrity in ageing and disease. Philos Trans R Soc Lond B Biol Sci 369, 20130439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A & Salvetti A (2001). Age‐related reduction of NO availability and oxidative stress in human. Hypertension 38, 274–279. [DOI] [PubMed] [Google Scholar]
- Tang R, Liu H, Wang T & Huang K (2005). Mechanisms of selenium inhibition of cell apoptosis induced by oxysterols in rat vascular smooth muscle cells. Arch Biochem Biophys 441, 16–24. [DOI] [PubMed] [Google Scholar]
- Tian XL & Li Y (2014). Endothelial cell senescence and age‐related vascular diseases. J Genet Genomics 41, 485–495. [DOI] [PubMed] [Google Scholar]
- Upston JM, Niu X, Brown AJ, Mashima R, Wang H, Senthilmohan R, Kettle AJ, Dean RT & Stocker R (2002). Disease stage‐dependent accumulation of lipid and protein oxidation products in human atherosclerosis. Am J Pathol 160, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usatyuk PV, Parinandi NL & Natarajan V (2006). Redox regulation of 4‐hydroxy‐2‐nonenal‐mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281, 35554–35566. [DOI] [PubMed] [Google Scholar]
- Usatyuk PV & Natarajan V (2012). Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvasc Res 83, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaya J, Aviram M, Mahmood S, Hayek T, Grenadir E, Hoffman A & Milo S (2001). Selective distribution of oxysterols in atherosclerotic lesions and human plasma lipoproteins. Free Radic Res 34, 485–497. [DOI] [PubMed] [Google Scholar]
- Vejux A, Guyot S, Montange T, Riedinger JM, Kahn E & Lizard G (2009). Phospholipidosis and down‐regulation of the PI3‐K/PDK‐1/Akt signalling pathway are vitamin E inhibitable events associated with 7‐ketocholesterol‐induced apoptosis. J Nutr Biochem 20, 45–61. [DOI] [PubMed] [Google Scholar]
- Virginio VW, Nunes VS, Moura FA, Menezes FH, Andreollo NA, Rogerio F, Scherrer DZ, Quintão EC, Nakandakare E, Petrucci O, Nadruz‐Junior W, de Faria EC & Sposito AC (2015). Arterial tissue and plasma concentration of enzymatic‐driven oxysterols are associated with severe peripheral atherosclerotic disease and systemic inflammatory activity. Free Radic Res 49, 199–203. [DOI] [PubMed] [Google Scholar]
- Vurusaner B, Gamba P, Testa G, Gargiulo S, Biasi F, Zerbinati C, Iuliano L, Leonarduzzi G, Basaga H & Poli G (2014). Survival signaling elicited by 27‐hydroxycholesterol through the combined modulation of cellular redox state and ERK/Akt phosphorylation. Free Radic Biol Med 77, 376–385. [DOI] [PubMed] [Google Scholar]
- Wang JC & Bennett M (2012). Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 111, 245–259. [DOI] [PubMed] [Google Scholar]
- Wang M, Jiang L, Monticone RE & Lakatta EG (2014). Proinflammation: the key of arterial aging. Trends Endocrinol Metab 25, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson KE, Boström K, Ravindranath R, Lam T, Norton B & Demer L (1994). TGF‐beta 1 and 25‐hydroxycholesterol stimulate osteoblast‐like vascular cells to calcify. J Clin Invest 93, 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wind S, Beuerlein K, Armitage M, Taye A, Kumar A, Janowitz D, Neff C, Shah AM, Wingler K & Schmidt HH (2010). Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension 56, 490–497. [DOI] [PubMed] [Google Scholar]
- Wohlfeil ER & Campbel WB (1999). 25‐Hydroxycholesterol increases eicosanoids and alters morphology in cultured pulmonary artery smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol 19, 2901–2908. [DOI] [PubMed] [Google Scholar]
- Wöltje M, Böbel M, Heiland M, Beck‐Broichsitter B, Al‐Dam A, Gröbe A, Friedrich RE, Hanken H & Smeets R (2015). Purmorphamine and oxysterols accelerate and promote osteogenic differentiation of mesenchymal stem cells in vitro. In Vivo 29, 247–254. [PubMed] [Google Scholar]
- Wong W, Ng CH, Tsang SY, Huang Y & Chen ZY (2011). Relative contribution of individual oxidized components in ox‐LDL to inhibition on endothelium‐dependent relaxation in rat aorta. Nutr Metab Cardiovasc Dis 21, 157–164. [DOI] [PubMed] [Google Scholar]
- Yamamoto KK, Takeshita H, Hayashi N, Li L, Nakano A, Hanasaki‐Yamamoto H, Fujita Y, Imaizumi Y, Toyama‐Yokoyama S, Nakama C, Kawai T, Takeda M, Hongyo K, Oguro R, Maekawa Y, Itoh N, Takami Y, Onishi M, Takeya Y, Sugimoto K, Kamide K, Nakagami H, Ohishi M, Kurtz TW, Sawamura T & Rakugi H (2015). Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin‐like oxLDL receptor. FASEB J 29, 3342–3356. [DOI] [PubMed] [Google Scholar]
- Yang D, McCrann DJ, Nguyen H, St Hilaire C, DePinho RA, Jones MR & Ravid K (2007). Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell 6, 257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L & Sinensky M (2000). 25‐Hydroxycholesterol activates a cytochrome c release‐mediated caspase cascade. Biochem Biophys Res Commun 278, 557–563. [DOI] [PubMed] [Google Scholar]
- Yuan XM, Osman E, Miah S, Zadeh SN, Xu L, Forssell C & Li W (2010). p53 expression in human carotid atheroma is significantly related to plaque instability and clinical manifestations. Atherosclerosis 210, 392–399. [DOI] [PubMed] [Google Scholar]
- Zarkovic K, Larroque‐Cardoso P, Pucelle M, Salvayre R, Waeg G, Nègre‐Salvayre A & Zarkovic N (2015). Elastin aging and lipid oxidation products in human aorta. Redox Biol 4, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrouk A, Vejux A, Mackrill J, O'Callaghan Y, Hammami M, O'Brien N & Lizard G (2014). Involvement of oxysterols in age‐related diseases and ageing processes. Ageing Res Rev 18, 148–162. [DOI] [PubMed] [Google Scholar]
- Zarzuelo M, López‐Sepúlveda R, Sánchez M, Romero M, Gómez‐Guzmán M, Ungvary Z, Pérez‐Vizcaíno F, Jiménez R & Duarte J (2013). SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem Pharmacol 85, 1288–1296. [DOI] [PubMed] [Google Scholar]