

Abstract
This review concludes that a sedentary lifestyle, obesity and ageing impair the vasodilator response of the muscle microvasculature to insulin, exercise and VEGF‐A and reduce microvascular density. Both impairments contribute to the development of insulin resistance, obesity and chronic age‐related diseases. A physically active lifestyle keeps both the vasodilator response and microvascular density high. Intravital microscopy has shown that microvascular units (MVUs) are the smallest functional elements to adjust blood flow in response to physiological signals and metabolic demands on muscle fibres. The luminal diameter of a common terminal arteriole (TA) controls blood flow through up to 20 capillaries belonging to a single MVU. Increases in plasma insulin and exercise/muscle contraction lead to recruitment of additional MVUs. Insulin also increases arteriolar vasomotion. Both mechanisms increase the endothelial surface area and therefore transendothelial transport of glucose, fatty acids (FAs) and insulin by specific transporters, present in high concentrations in the capillary endothelium. Future studies should quantify transporter concentration differences between healthy and at risk populations as they may limit nutrient supply and oxidation in muscle and impair glucose and lipid homeostasis. An important recent discovery is that VEGF‐B produced by skeletal muscle controls the expression of FA transporter proteins in the capillary endothelium and thus links endothelial FA uptake to the oxidative capacity of skeletal muscle, potentially preventing lipotoxic FA accumulation, the dominant cause of insulin resistance in muscle fibres.
Abbreviations
- Akt
protein kinase B
- bAECs
bovine aorta endothelial cells
- EC
endothelial cell
- ECL
endothelial cell layer
- eNOS
endothelial nitric oxide synthase
- FA
fatty acid
- FABP4/5
fatty acid binding protein 4/5
- FAT/CD36
fatty acid translocase/cluster of differentiation 36
- FATP3/4
fatty acid transporter protein 3/4
- GLUT1/4
glucose transporter 1/4
- IGF‐1
insulin‐like growth factor 1
- IRS1/2
insulin receptor substrate 1/2
- MVU
microvascular unit
- NO
nitric oxide
- PI3K
phosphatidylinositol 3‐kinase
- PDK1
3‐phosphoinositide dependent protein kinase‐1
- PS
permeability–surface area product
- TA
terminal arteriole
- TET
transendothelial transport
- TG
triglyceride
- TNFα
tumour necrosis factor α
- VEGF
vascular endothelial growth factor
Introduction
The aim of this symposium review is to provide an introduction and fill in the background knowledge for the subsequent reviews presented at Physiology 2014 in London in an Invited Symposium with the title: ‘Impact of physical activity, ageing, obesity and metabolic syndrome on muscle microvascular perfusion and endothelial metabolism’. Increases in plasma insulin (following meal ingestion or insulin infusion) lead to modest increases in resting skeletal muscle blood flow and/or microvascular blood volume via an endothelial nitric oxide (NO)‐dependent mechanism. During aerobic endurance exercise the blood flow in the contracting skeletal muscles increases upto 80‐fold compared to rest via a number of complementary mechanisms. In both cases more blood is present in or flowing through the muscle capillary bed, but the mechanisms by which these increases in muscle capillary blood flow occur are only partially understood. This review, therefore, will first explain the complex anatomy of the microvasculature in mammalian skeletal muscles, as a proper understanding of the anatomy is integral to an understanding of the nature and mechanisms that control muscle capillary blood flow and the supply of oxygen, fuels and hormones to the skeletal muscle fibres. This will be followed by reflections on the metabolic rationale for the very large surface area of the endothelial cell (EC) layer (ECL) in the skeletal muscle microvasculature, especially the role that it plays in the transendothelial transport of oxygen, glucose, fatty acids (FAs) and insulin from the lumen of the muscle capillaries into the interstitial fluid surrounding the skeletal muscle fibres. Finally, a summary will be given of the mechanisms by which meal‐induced increases of plasma insulin and exercise‐induced increases in blood shear stress and in the interstitial concentration of vascular endothelial growth factor‐A (VEGF‐A) activate endothelial nitric oxide synthase (eNOS), dilate terminal arterioles (TAs) and thus increase capillary blood flow. The important role of these molecular mechanisms in maintaining optimal insulin sensitivity, glucose and lipid homeostasis, skeletal muscle perfusion during exercise and the angiogenic response to regular exercise will first be explained in healthy physically active individuals and interwoven with a comprehensive summary of the most important impairments that occur in sedentary, obese and elderly individuals with and without chronic diseases.
Why are insulin‐induced increases in skeletal muscle blood perfusion important?
Research using contrast‐enhanced ultrasound has shown that meal‐induced increases in the plasma insulin concentration lead to small (+50–80%) increases in the resting blood volume of the skeletal muscle microvascular bed in lean, healthy individuals (Vincent et al. 2006). There is convincing evidence that this increase in micovascular blood volume leads to increased capillary recruitment in skeletal muscle both in rats (Rattigan et al. 1997) and humans (reviewed by Keske et al. 2015, in this issue). These increases are important, as they are part of the redistribution of the fuels that are absorbed with each of the main meals and therefore play an important role in the delivery of glucose, insulin and fatty acids to skeletal muscle as the main storage site. Following oral ingestion of a 92 g glucose load leading to physiological increases in plasma insulin in healthy lean young males, 65–70% of the ingested glucose was taken up by peripheral tissues (Katz et al. 1983). Following oral ingestion of a mixed meal (40 g fat and 40 g carbohydrate) a substantial part of the meal‐derived fat was taken up by cannulated forearm and abdominal subcutaneous adipose tissue as chylomicron‐triglyceride (CM‐TG) and very low density lipoprotein triglyceride (VLDL‐TG) (Bickerton et al. 2007). These studies collectively suggest that the peripheral tissues (primarily skeletal muscle and subcutaneous adipose tissue) make the largest contributions to the clearance of orally ingested glucose and lipid and therefore make an important contribution to postprandial glucose and lipid homeostasis. Postprandial increases in perfusion of the skeletal muscle microvascular bed, therefore, seem to be instrumental for glucose and lipid homeostasis and long‐term metabolic health. However, studies with contrast‐enhanced ultrasound have shown that physiological, meal‐induced and supraphysiological increases in plasma insulin achieved via a hyperinsulinaemic euglycaemic clamp fail to increase the blood volume that is present in the skeletal muscle microvascular bed in obese individuals (Clerk et al. 2006; Keske et al. 2009) and also in sedentary elderly individuals, type 2 diabetes patients and animal models of type 2 diabetes and insulin resistance (for references see reviews of Barrett et al. 2009 and Keske et al. 2015). This suggests that the postprandial redistribution of the blood flow to skeletal muscle and potentially subcutaneous adipose tissue is reduced or absent in obese, elderly and insulin‐resistant individuals. This impairment explains the large transient increases that are seen in plasma glucose and TG concentrations following ingestion of mixed meals in these conditions. It is the prevalence and height of the elevated postprandial glucose excursions that represent a high risk for development of type 2 diabetes (Abdul‐Ghani & deFronzo, 2009) and that impose a direct and independent risk in type 2 diabetes patients for the development of cardiovascular complications (Ceriello, 2005). Increased postprandial plasma TG concentrations also presented an independent risk for the development of cardiovascular events and insulin resistance in a large cohort of healthy women (Bansal et al. 2007).
Why are exercise‐induced increases in skeletal muscle blood perfusion important?
The microvasculature of human skeletal muscles has a complex 3‐D structure and is subject to a large number of complementary blood flow regulation mechanisms (Mortensen & Saltin, 2014). This ensures that blood supply matches the metabolic demands of the muscle fibres under resting conditions and during exercise. The energy expenditure of skeletal muscle in comparison to other tissues and organs is relatively low at rest (0.5 W (kg muscle)−1), but can increase during aerobic exercise (e.g. during running at Olympic level marathon pace) to 80 W (kg contracting muscle)−1. This is a 160‐fold increase, which trained athletes are able to maintain for more than 2 h because the density of both the mitochondrial network (ATP production capacity) and the microvascular network (oxygen and fuel supply) is much higher in endurance‐trained athletes than in sedentary individuals (Saltin et al. 1977; Saltin, 1988). All the oxygen and blood‐borne fuels (glucose, fatty acids and lipoprotein TGs) required to meet this high energy demand are delivered to the muscle via the blood (Van Loon et al. 2001). It is for this reason that leg blood flow and perfusion of the muscle microvascular bed have to increase 10‐ to 80‐fold (depending on running velocity and training status) during two‐legged running. When the active muscle mass is small and the heart (cardiac output) is not a limitation, skeletal muscle blood flow in trained individuals can even increase 100‐fold and reach perfusion rates of 300–400 ml min−1 (kg muscle)−1 (Andersen & Saltin, 1985; Richardson et al. 1993). A thorough knowledge of the anatomy of the muscle microvasculature, including the role played by the ECL and TAs in channelling blood‐borne fuels into the muscle interstitium, is essential for a basic understanding of the mechanism by which aerobic endurance exercise leads to these 10‐ to 100‐fold increases in skeletal muscle microvascular perfusion and metabolic rate.
Functional anatomy of the microvasulature of skeletal muscle
Most of the historic thinking about the supply and utilisation of oxygen and glucose in human skeletal muscle is based on spatial relationships of capillaries and skeletal muscle fibres measured in skeletal muscle cross‐sections (e.g. Krogh, 1918) (Fig. 1 A). This remains common practice today as the generation of longitudinal and 3‐D images of the microvasculature in human muscle fibres currently is an insurmountable methodological challenge. The length of human muscle fibres is between 4 cm (soleus muscle) and 40 cm (sartorius muscle) (Wickiewicz et al. 1983; Ward et al. 2009), while the diameter of capillaries is in the 6–10 μm range. The capillaries, furthermore, are densely packed between the plasma membranes of neighbouring muscle fibres, which have a diameter of 50–90 μm depending on fibre type, age and training status. The imaging problems that result from this spatial organisation seem to be the reason that today there is very little published information on the 3‐D anatomical structure of the human skeletal muscle microvasculature. We therefore depend on information from other mammalian species to build up a basic understanding, keeping in mind that there may be differences with the human skeletal muscle.
Figure 1. Structure and anatomy of skeletal muscle microvasculature .
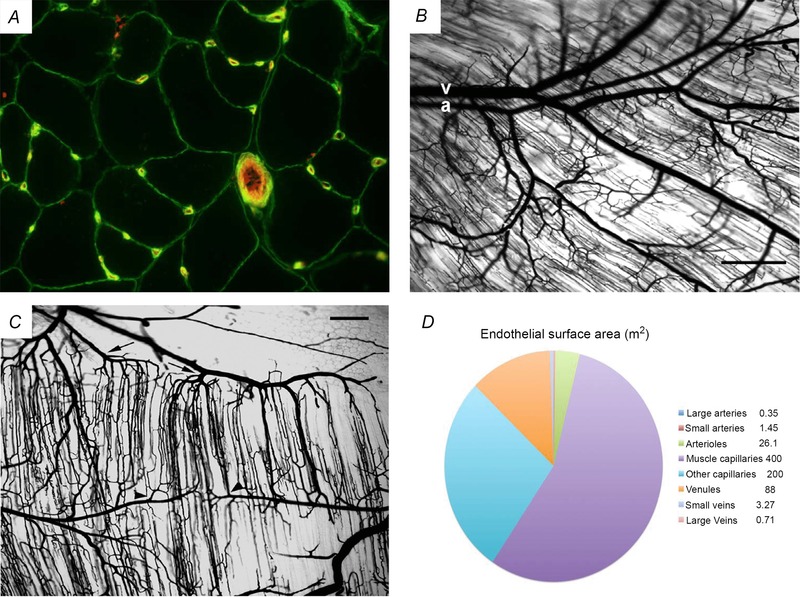
A, cross‐sectional image of muscle fibres and their microvasculature. It historically and today still is common practice to use cross‐sectional images of skeletal muscle to visualise and quantify the capillarisation of skeletal muscle fibres. This image shows in green the plasma membrane of the skeletal muscle fibres and the membrane surrounding the ECL of capillaries (visualised with an antibody stain against collagen IV, which is a main component of the basement membrane). The glycocalyx of the capillaries and of the larger 4th order arteriole (approximately 40 μm in diameter) was stained in red with a fluorescent lectin (Ulex europaeus). This unpublished image comes from the collection of Dr Chris Shaw and was obtained by Chris during his PhD with Anton Wagenmakers at the University of Birmingham, UK. B and C show microvascular casts in mouse gluteus maximus muscle. In these whole‐mount preparations, the vasculature was cast using Microfil and muscle fibres were cleared in glycerine to enhance the visibility of microvessels. B shows that the arteriolar and venular networks are paired in skeletal muscle. A 3rd order arteriole (a) and its paired venule (v) both enter at left. Note the proximity of arteriolar–venular segments through the 4th order branches, which then branch into the TAs that supply red blood cells to the capillaries. The ‘shadows’ of parallel muscle fibres and surrounding capillaries are oriented obliquely across the figure. C shows that capillaries are organised into microvascular units (MVUs). TAs (arrowheads indicate 2 of the several that are present) branch off vertically from 4th order arterioles. Each TA gives rise to a group of approximately 20 capillaries. The capillaries in the MVU run half and half in both directions parallel to muscle fibres and have a length of approximately 1 mm. The capillaries converge on collecting venules (arrows indicate 2 of the several that are present). Muscle fibres are transparent in this image. Scale bar in panels B and C = 250 μm. Panels B and C are reproduced with permission of the author and the publisher from Segal (2005), Microcirculation, © John Wiley and Sons. D is a visual representation of the relative size of the endothelial surface area, in square metres, present in arteries, the microvasculature and veins. Every blood vessel in the human vasculature is covered on the luminal side with a continuous monolayer of ECs. About 400 m2 ECL surface is present in skeletal muscle capillaries to generate the transport capacity for oxygen, fuels and hormones that is required to meet the high metabolic demands of skeletal muscle during aerobic exercise via recruitment of additional MVUs (compared to rest) and rhythmic increases in the dilatation of TAs and capillary blood flow in the recruited MVUs (compared to rest).
Figure 1 B and C are images of a plaster cast of the highly branched network of the microvasculature of the mouse gluteus maximus muscle originally published by Segal (2005). A more recent educational description, linking anatomy to blood flow control in skeletal muscle, has been published by Segal & Bearden (2012) again in mouse gluteus maximus muscle. In brief, upon leaving the heart the convection of blood through large conduit arteries such as the brachial and femoral arteries rapidly conveys blood to skeletal muscle with minimal haemodynamic resistance. The large conduit arteries then split into muscular feed arteries, which are millimeters wide in humans and are still external to skeletal muscles. These feed arteries are positioned to control the total amount of blood entering the muscle and are assumed to present close to half of the total resistance to blood flow (Segal & Bearden, 2012). Upon entering skeletal muscle, feed arteries branch into the arteriolar networks, which are assumed to provide the major resistance to blood flow (Segal & Bearden, 2012). Primary or 1st order arterioles branch into 2nd and 3rd order arterioles (Fig. 1 B). These intermediate branches distribute blood along the full length and full depth of the muscle and thereby control regional perfusion. Arising from the distributing 3rd order arterioles are the 4th order and terminal or 5th order arterioles (TAs). The TAs control the perfusion of capillaries with red blood cells.
Capillaries are organised into microvascular units
Capillaries in mouse gluteus maximus muscle (Segal, 2005; Segal & Bearden, 2012) and in hamster tibialis anterior muscle (Lund et al. 1987; Delashaw & Duling, 1988) are organised into microvascular units (MVUs). The latter represent the smallest volume of muscle to which blood flow and, therefore, oxygen, glucose and fatty acids (fuel) and insulin (hormone) delivery can be independently controlled. MVUs consist of all the capillaries arising from a common TA. TAs are oriented perpendicular to muscle fibres branching into a group of about 20 capillaries that run along the length of, and between, muscle fibres for a distance of about 1 mm. Usually blood flow in half of the capillaries is in one direction, and in the other half in the opposite direction, along the length of the same muscle fibres. The two sections of the MVU with flow in opposite directions have been named a ‘unit pair’ (Lund et al. 1987). The capillaries empty on both sides into collecting venules (Fig. 1 C). The volume of muscle tissue within an MVU with two unit pairs has been estimated at 0.2 mm3 (or 2 mg wet weight), with average dimensions being 2 mm long, 0.5 mm wide, and 0.2 mm thick. This volume contains segments of about 20 different muscle fibres, which must originate from different motor units as the muscle fibres in a motor unit are spread over a much larger cross‐sectional area.
Control of blood flow in microvascular units
In order to investigate the control of the blood flow in the capillaries of individual MVUs and potential differences between adjacent MVUs Lund et al. (1987) and Delashaw & Duling (1988) performed an impressive series of experiments in which they removed the skin and fascia from the tibialis anterior muscle in anaesthetised hamsters. This allowed these researchers to use intravital epifluorescence microscopy to visualise the flow of red blood cells from the TA through capillaries to the collecting venule in the 160–180 μm layer (one MVU deep) immediately below the surface of the muscle. When this preparation in control experiments was superfused with a Krebs–Ringer buffer that did not contain oxygen, basically all the capillaries in all MVUs were continuously perfused with blood. Exposure to a superfusate containing 10% O2 typically resulted in closure of approximately 50% of the capillaries, with the blood flow being simultaneously arrested in all capillaries of a given MVU by complete closure of the TA. An important observation in these studies was that in the MVUs that remained to be perfused, the blood flow in all capillaries showed rhythmic oscillations that were attributable to a spontaneous rhythmic change of the diameter of their respective TA. This phenomenon was called arteriolar ‘vasomotion’. During vasomotion, flow in all of the capillaries of a given MVU tended to vary in synchrony with the same phase and frequency (3–6 cycles min–1), while phase and frequency tended to vary between adjacent MVUs.
Delashaw & Duling (1988) in the same muscle preparation investigated the effects of (1) direct electrical stimulation leading to muscle contractions, (2) topical application of adenosine (vasodilator), and (3) topical application of phenylephrine (vasoconstrictor). The predominant response to these stimuli again consisted of a coordinated change in the blood flow in virtually all the capillaries of the investigated MVUs. Electrical stimulation of muscles in which blood flow was arrested in 95% of the MVUs led to the simultaneous recruitment of virtually all capillaries (of both unit pairs) in previously closed MVUs. Topical application of adenosine had a similar result, while topical application of phenylephrine resulted in the simultaneous arrest of the blood flow in all capillaries of previously perfused MVUs.
The study of Lund et al. (1987) also revealed that the capillaries of neighbouring MVUs often run in parallel and overlap and cross with each other, implying that multiple MVUs supply oxygen, fuel and insulin to the same sections of the same muscle fibres. The flow in adjacent MVUs was both concurrent and countercurrent with 25–50% of the adjacent capillaries having a countercurrent flow. Suggestions have been made that the percentage countercurrent flow is around 50% for capillaries in deeper muscle layers originating from adjacent MVUs on both sides. Such a mixed concurrent–countercurrent flow pattern allows for the diffusion of oxygen between adjacent capillaries and on theoretical grounds has been suggested to give the most homogeneous type of tissue oxygenation (Grunewald & Sowa, 1977). In more recent studies (reviewed by Pittman, 2013) convincing evidence has also been presented that muscle arterioles also exchange oxygen via diffusion to adjacent capillaries (which they often cross) and that oxygen diffusion occurs between arteriole–venule pairs which often run next to each other over distances up to several millimetres (Fig. 1 B).
Relevance of these findings for resting and contracting human skeletal muscles
Although the above studies have generated a wealth of information on MVUs in skeletal muscle of anaesthetised hamsters, a major problem that remains is how to relate these findings to intact undisturbed human skeletal muscles in the in vivo resting state and during exercise or contraction. As the same structural organisation and similar size of MVUs has also been observed in rabbit tenuissimus muscle (Lindbom, 1983) and in monkey skeletal muscle (Weibel, 1984), it is assumed in this review that this will also be the case for other mammalian species, including man. A limitation of the superfused hamster muscle preparations indicated by Lund et al. (1987) is that the of the superfusion buffer has a major impact on vasoconstriction and vasomotion of TAs and therefore on capillary blood flow in MVUs. As such it was not possible for Lund et al. (1987) to decide what the ‘normal’ state in this muscle is. Is it the continuous perfusion of all MVUs and capillaries observed with a superfusion solution containing no oxygen (condition 1), or the mixture of closed MVUs and MVUs showing cyclic vasomotion observed with a superfusion solution gassed with 10% oxygen (condition 2)? The vasodilatation observed during chronic electrical stimulation and in response to adenosine and the vasoconstriction seen in response to phenylephrine in condition 2 (Delashaw & Duling, 1988) is very similar to in vivo effects of voluntary exercise and of arterial infusion of adenosine and phenylephrine on limb and local muscle blood flow seen in humans in vivo. Cyclic vasomotion of the skeletal muscle microvasculature has also been studied in rats in vivo with laser Doppler flowmetry with insertion of the probe in the tibialis muscle parallel to the muscle fibre direction (Newman et al. 2009). This study revealed that vasomotion occurred with a frequency in the range of 6 cycles min–1, which is in the same frequency range as observed by Lund et al. (1987) in MVUs of the hamster tibialis anterior muscle using a superfusion solution gassed with 10% oxygen.
Assumptions on the regulation of capillary blood flow in human skeletal muscle currently not supported by human data
In the remainder of this review, therefore, assumptions will be made that MVUs in human skeletal muscles fibres are of similar size to those in other mammals and that part of these MVUs will be closed in the normal resting state, while others undergo cyclic vasomotion with a large variation in the perfusion rate between adjacent MVUs. Cyclic vasomotion of TAs in combination with a mixed concurrent–countercurrent flow pattern in adjacent capillaries (originating from adjacent MVUs) is assumed to play an important role in ensuring that skeletal muscles are evenly perfused by periodically redistributing blood from one MVU to adjacent ones. Vasomotion will allow switches in time both in recruitment of different MVUs and in the overall blood perfusion rate of a given MVU by step‐wise changes in the lumen of the common TA. Vasomotion as such may contribute to the local blood flow control mechanism that can lead to an even perfusion rate over the full length of the up to 40 cm‐long multinucleated muscle fibres in human skeletal muscle and may also help to ensure that the approximately 20 fibre sections that are present in a single MVU are perfused in proportion to their metabolic demand. Please note that future research is required to confirm these assumptions.
Anatomy and function of the endothelial cell layer in the microvasculature of human skeletal muscle
Every blood vessel in the human vasculature is covered on the luminal side with a continuous monolayer of endothelial cells (ECL). The total surface area of the ECL in an adult human has been estimated to cover > 700 m2 and to have a weight of about 700 g (Wolinsky, 1980). This makes the ECL into one of the largest diffuse tissues in the human body with a significant weight despite the 0.3 μm thickness. The ECL of all arteries and veins in our body together covers only about 6 m2 (Fig. 1 D), while the ECL of the microvasculature (arterioles, capillaries and venules) contributes 99% of the total surface area. Of the latter 85% (about 600 m2) is present in capillaries (Fig. 1 D). As skeletal muscle in a 70 kg lean, physically active adult has a mass between 35 kg and 40 kg, and has a higher capillary density than other tissues (with exception of the heart), estimates are that at least 400 m2 of the ECL is present in the skeletal muscle microvasculature, again with the largest surface area in the dense network of capillaries (Wolinsky, 1980). The metabolic rationale behind this distribution seems to be that the ECL in muscle capillaries contains all the enzymes that facilitate the transendothelial transport of glucose, amino acids, fatty acids and insulin into the interstitial fluid that surrounds the muscle fibres and that transport capacity, therefore, can be increased by recruiting a larger endothelial surface area at times of increased metabolic demand (e.g. following meal ingestion and during exercise).
Transendothelial transport of oxygen, nutrients and insulin
ECs in the skeletal muscle microvasculature form a continuous monolayer and as such present the first potential barrier for the transport of oxygen, nutrients and insulin from the vascular compartment to the interstitial fluid surrounding the skeletal muscle fibres. The number of studies that have evaluated the impact of this endothelial barrier on the supply of oxygen and nutrients to the interstitium of skeletal muscle is surprisingly small. As far as the authors are aware it is not known today what percentage of the oxygen released from haemoglobin and the red blood cells is diffusing into the interstitium of skeletal muscle via the paracellular route (through the tight junctions between neighbouring ECs) and via the transcellular route (transendothelial transport (TET) from the luminal to the abluminal membrane through the cytosol of ECs). Nevertheless, to answer the question whether O2 diffusion from capillary red bloods cells into the muscle interstitium limits the maximum rate of O2 uptake () in healthy humans and in sedentary obese and elderly individuals with and without type 2 diabetes and/or cardiovascular disease and ischaemia, this is important information (Roca et al. 1989; Spires et al. 2012).
The review of Mann et al. (2003) comes to the conclu‐sion that glucose transport across the luminal and abluminal membrane of the endothelium of the blood–brain barrier occurs via a facilitative, energy‐independent and saturable transport process by the GLUT1 transporter. GLUT1 has also been detected in the endothelium of the blood–retinal barrier and the cornea, and in freshly isolated and cultured bovine aorta ECs (bAECs). Total glucose transport rates across the retinal endothelium exceeded the metabolic rates of the retinal endothelium by far suggesting that GLUT1 in ECs facilitates TET of glucose into the underlying tissues. The driving force for glucose transport by GLUT1 is the concentration gradient between the capillary lumen and the interstitium. Davey et al. (2007), using immunogold electron microscopy methods, have shown that in rat heart GLUT1 is the only glucose transporter present in the capillary ECs, while GLUT4 was the dominant glucose transporter in cardiomyocytes. H. Bradley and A. J. M. Wagenmakers (unpublished observations) using immunostaining microscopy observed an intense GLUT1 signal in ECs of capillaries in human vastus lateralis muscle and no GLUT4, while the reverse was seen in the muscle fibres (Bradley et al. 2014), implying that the same GLUT distribution is present in human skeletal muscle as in rat heart. These data suggest that the main transport route of glucose is through the transcellular route with glucose transport facilitated by GLUT1.
In addition Mann et al. (2003) in their review come to the conclusion that ECs lining the blood–brain barrier and blood–retinal barrier, and cultured bAECs, all express a large number of specific amino acid transport systems for practically all amino acids. There is no information on the presence of these amino acid transporters in the capillary endothelium of skeletal muscle, but the assumption is made that amino acids again are primarily transported into the skeletal muscle interstitium via the transcellular route.
Fatty acids (FAs), chylomicron triglycerides (CM‐TGs) and very low density lipoprotein triglycerides (VLDL‐TGs) are important fuels for human skeletal muscle both at rest (Bickerton et al. 2007) and during exercise, with peak oxidation rates occurring at intensities of 55–65% (Van Loon et al. 2001). Plasma FAs and TGs are again delivered to muscle fibres via the capillaries. The enzyme lipoprotein lipase (LPL) is attached to the luminal membrane of capillary ECs and is responsible for the breakdown of CM‐TGs and VLDL‐TGs to FAs. Until recently it was an open question how these FAs are subsequently transported into the skeletal muscle interstitium for subsequent uptake in skeletal muscle, although the first study that observed a high concentration of fatty acid translocase/cluster of differentiation 36 (FAT/CD36) in the endothelium of skeletal muscle, heart and adipose tissue of mice dates back to 1995 (Greenwalt et al. 1995). This observation was confirmed in 2004 with high quality confocal immunofluorescence microscopy images clearly showing that the FAT/CD36 content of the capillary endothelium is much higher than in the skeletal muscle fibres (Vistisen et al. 2004). FAT/CD36 today is regarded as the most important FA transporter responsible for FA entry into skeletal muscle (Glatz et al. 2010; McFarlan et al. 2012). This conclusion is based on experiments with giant plasma membrane vesicles suggesting that postprandial increases in insulin and exercise/contraction lead to translocation of FAT from hypothetical microsomal storage depots (similar to GLUT4 storage and translocation) to the plasma membrane of skeletal muscle fibres (reviewed by Glatz et al. 2010). However, Vistisen et al. (2004), using confocal immunofluorescence microscopy, found the amount of FAT/CD36 in the plasma membrane of skeletal muscle fibres in resting biopsies obtained in the fasted state to be only a fraction of the stain in the capillary ECs. Hagberg et al. (2010), also using immunostaining methods, observed that FA transporter proteins (FATP3 and FATP4) were expressed abundantly in the ECs of capillaries of mouse skeletal muscle, heart and adipose tissue. These proteins also facilitate the transport of FAs across the luminal and abluminal membrane. Iso et al. (2013) observed that FA binding proteins (FABP4 and FABP5) are abundantly expressed in the ECL of mouse skeletal muscle capillaries and venules (but not in arterioles, arteries and aorta). These proteins function as the transport vehicle for FAs diffusing through the cytosol from the luminal to the abluminal membrane of ECs. In skeletal muscle of FAT/CD36 knockout mice (McFarlan et al. 2012), with a low expression of FATP3 and FATP4 (Hagberg et al. 2010), and with a double knockout for FABP4 and FABP5 (Iso et al. 2013) skeletal muscle uptake of FA was substantially reduced, while the uptake of glucose and GLUT4 expression in skeletal muscle was remarkably increased. Collectively these data provide convincing evidence that the main route for transport of FAs across the ECL is via the transcellular route, that a low protein expression of FAT/CD36, FATPs and FABPs limits FA uptake in mouse skeletal muscle and that capillary ECs act as the gate‐keepers for uptake of FAs into skeletal muscle and as such may play a very important role in the distribution of lipids between skeletal muscle, subcutaneous adipose tissue and ectopic fat stores and therefore the mechanisms leading to insulin resistance of the skeletal muscle fibres (Fig. 3).
Figure 3. Activation of the insulin‐signalling cascade in skeletal muscle .
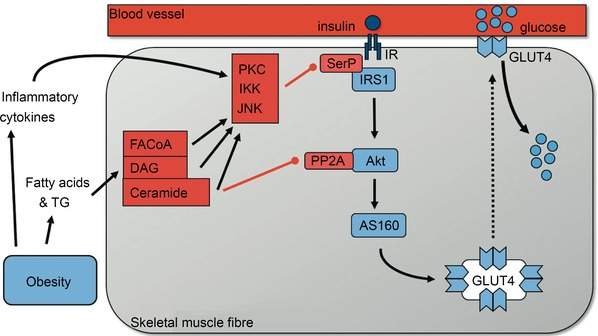
In healthy trained individuals increases in the insulin concentration in the interstitial fluid surrounding muscle fibres leads to increased activation of the insulin signalling cascade and translocation of GLUT4 (main glucose transporter in muscle) to the plasma membrane. As muscle is the main tissue responsible for glucose uptake in the period after meal ingestion this will keep the rise in blood glucose concentration following ingestion of a meal relatively modest. As such healthy trained individuals have firm control on their blood glucose levels, both in the fasted period and after food intake. In sedentary, obese or elderly individuals, and those with metabolic syndrome and type 2 diabetes, high blood levels of fatty acids and triglycerides contribute to an excessive accumulation of lipid metabolites such as long chain fatty acyl‐CoA (FACoA), diacylglycerol (DAG) and ceramide in skeletal muscle. This together with increased plasma levels of inflammatory cytokines in obese individuals (Berg & Scherer, 2005) and local inflammation of the microvasculature in skeletal muscle activates the serine kinases protein kinase C (PKC), IκB kinase (IKK) and Jun amino‐terminal kinase (JNK) (Wagenmakers et al. 2006). These phosphorylate insulin receptor substrate 1 (IRS1) on serine residues SerP leading to inactivation of IRS1 and downstream inactivation of the insulin signalling cascade. Ceramide accumulation also activates the phosphatase PP2A which dephosphorylates and inactivates Akt. These mechanisms collectively prevent activation of the insulin signalling cascade in skeletal muscle and therefore reduce insulin‐mediated activation of Akt substrate of 160 kDa (AS160) and GLUT4 translocation and skeletal muscle glucose uptake. Reproduced with permission from Shaw & Wagenmakers (2012), The Biochemist, © the Biochemical Society.
Transport(Wagenmakers et al. 2006 of insulin from the capillary lumen into the skeletal muscle interstitium has been investigated in great detail in a series of elegant studies by the research group of Professor Eugene Barrett, primarily in cultured bAECs (Wang et al. 2008, 2009, 2011, 2012, 2013) and in rat skeletal muscle in vivo (Wang et al. 2006). These studies are also placed in the context of the related human literature in reviews of Barrett et al. (2009, 2011). Both in the in vivo study investigating the rectus muscle of the rat and in in vitro studies with bAECs, ECs rapidly took up fluorescein isothiocyanate (FITC)‐labelled insulin. The fluorescent label allowed quantification of insulin uptake with confocal fluorescence microscopy. In the in vivo study in rats (Wang et al. 2006) the insulin concentration in ECs by far exceeded the concentration in plasma and muscle interstitium. Both the insulin receptor and the insulin‐like growth factor 1 (IGF‐1) receptor mediated insulin transit through monolayers of bAECs in a process involving the caveolae, which are present in the lipid rafts in the plasma membrane (PM) (Wang et al. 2006). Adding insulin provoked the prompt translocation of both caveolin‐1 and eNOS to the PM and led to co‐immunoprecipitation of these proteins, suggesting that insulin signalling brings these proteins together in lipid rafts in the PM (Wang et al. 2009). Insulin uptake required intact insulin signalling via both the phosphatidylinositol 3‐kinase (PI3K) and mitogen‐activated protein kinase (MAPK) signalling cascades (Wang et al. 2008). Pre‐incubation of bAECs with the pro‐inflammatory cytokines tumour necrosis factor α (TNF‐α) and interleukin 6 significantly diminished insulin uptake, at least in part by inhibiting caveolin‐1 expression (Wang et al. 2011). Wang et al. (2012) found that exposure of bAECs to insulin within 5 min induced substantial cortical actin filament remodelling. The remodelling was inhibited by inhibition of PI3K or MAPK signalling and disruption of actin microfilaments and lipid rafts with specific inhibitors. Knockdown of either caveolin‐1 or protein kinase B (Akt) led to complete elimination of the insulin‐induced cortical actin filament remodelling. The conclusion of Wang et al. (2012) was that insulin‐induced cortical actin filament remodelling is required for TET of insulin and depends both on intact PI3K/Akt signalling and the presence of caveolin‐1 in intact lipid rafts. Wang et al. (2013) observed that l‐NAME (NOS inhibitor) pretreatment of bAECs blocked FITC‐labelled insulin uptake, whereas pretreatment with l‐arginine (converted into NO by eNOS) and the NO‐donor sodium nitroprusside (SNP) enhanced insulin uptake. SNP also reversed the inhibition of insulin uptake in bAECs by l‐NAME, wortmannin (PI3K inhibitor) and TNFα. SNP increased TET of 125I‐labelled insulin with 40% in bAEC monolayers. SNP finally increased S‐nitrosylation of the protein tyrosine phosphatase PTP1B which dephosphorylates tyrosine residues on the insulin receptor and insulin receptor substrates 1 and 2 (IRS1 and IRS2). S‐nitrosylation led to inactivation of PTP1B activity and an increase in insulin signalling leading to Ser473 phosphorylation of Akt. The conclusion of Wang et al. (2013) was that a high endothelial NO concentration directly promotes insulin transport into ECs and TET of insulin via a mechanism that involves the enhancement of S‐nitrosylation, inhibition of the activity of PTP1B and enhanced insulin signalling. As enhanced insulin signalling activates eNOS (Fig. 2) and increases endothelial NO production, this implies that insulin stimulates its own uptake in ECs lining skeletal muscle capillaries (Barrett et al. 2011).
Figure 2. Insulin‐induced vasodilatation of TAs leads to increased perfusion of muscle capillaries .
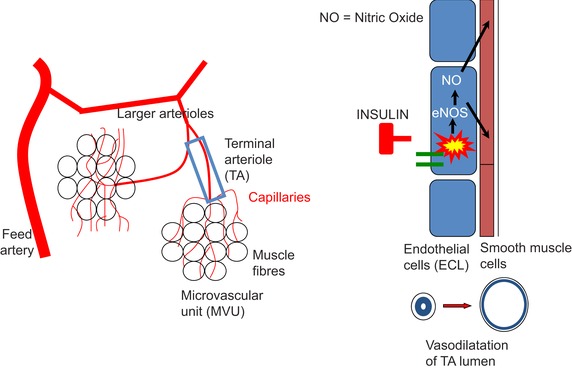
Meal‐induced increases in the plasma insulin concentration activate an insulin signalling cascade (insulin receptor/IRS1/PI3K/PDK1/Akt/eNOS) that is present in the ECL of the TAs. The end result is an increase in nitric oxide (NO) production. NO is a potent vasodilator acting upon the smooth muscle cell layer in TAs. The simple version of the underlying mechanism is that insulin‐induced increases in vasodilatation of TAs lead to recruitment of additional MVUs and capillaries that were not perfused before ingestion of the meal and therefore explain the observed increase in microvascular perfusion of skeletal muscle. The real mechanism probably is more complex as several studies have shown that the microvascular blood flow in skeletal muscle undergoes rhythmic oscillations attributable to spontaneous changes in the diameter of the TA lumen (Lund et al. 1987; Newman et al. 2009). This phenomenon is called ‘arteriolar vasomotion’. Newman et al. (2009) using laser Doppler flowmetry recently observed that a hyperinsulinaemic euglycaemic clamp in rats increased the arteriolar oscillations. The interpretation of this observation by the authors is that the insulin‐induced increase in microvascular perfusion of skeletal muscle is at least in part due to an increase in the intensity of the vasomotion that occurs in TAs of skeletal muscle. This Figure has been drawn by the authors using data from more than 20 cited references in the paragraphs explaining the anatomy of MVU's and mechanisms in control of blood flow in MVUs to include an earlier anatomical interpretation in Cohen et al. (2000).
Permeability surface area product as a measure of transendothelial glucose transport
Gudsbjörnsdóttir et al. (2003) have combined forearm arteriovenous cannulations in healthy lean men with microdialysis of the brachioradialis muscle to measure arterial, venous and interstitial glucose concentrations. Using this approach the authors were able to make a direct calculation of the permeability–surface area product (PS) of the ECL for glucose in this muscle. The PS depends both on the surface area of the ECL that is available for glucose uptake (and therefore on the number of simultaneously perfused MVUs) and the permeability of the ECL for glucose. As GLUT1 is the only transporter detected in the capillary endothelium of human skeletal muscle (H. Bradley & A. J. M. Wagenmakers, unpublished observations), it is the GLUT1 protein content that determines permeability. A traditional oral glucose tolerance test increased the PS for glucose and the glucose uptake by the muscle about 2‐fold in the 90–150 min period after glucose ingestion. During a steady state euglycaemic hyperinsulinaemic clamp PS for glucose increased 10‐fold and glucose uptake by the muscle increased 8‐ to 9‐fold. These data provide convincing evidence that oral ingestion of glucose and insulin infusion lead to substantial increases in the recruitment of endothelial surface area in the muscle microvasculature and that these lead to proportional increases in muscle glucose uptake. As glucose transport across the ECL is mediated by the insulin‐insensitive GLUT1 transporter it is unlikely that increases in endothelial permeability make a major contribution to the observed increases in PS, although increases by other currently unknown activation mechanisms cannot be excluded.
Gudsbjörnsdóttir et al. (2005) measured the PS for glucose and insulin during a steady‐state euglycaemic hyperinsulinaemic clamp in male individuals with type 2 diabetes (T2D) and age‐ and weight‐matched obese controls with normal fasting glucose. The PS for glucose was 2.2‐fold lower in the patients with T2D than the obese controls and muscle glucose uptake was 1.7‐fold lower in the patients. The absolute value for the PS for glucose in the lean men during the steady‐state phase of the euglycaemic hyperinsulinaemic clamp was 2.3 ± 0.9 ml min−1 (100 g) in the study of Gudsbjörnsdóttir et al. (2003) and the equivalent values in the obese controls and type 2 diabetes patients in Gudsbjörnsdóttir et al. (2005) were 1.1 ± 0.2 and 0.5 ± 0.1 ml min−1 (100 g). The corresponding muscle glucose uptake rates were 4.4 ± 1.2, 3.0 ± 0.4 and 1.8 ± 0.3 μmol min−1 (100 g) in the lean healthy men, obese controls and type 2 diabetes patients, respectively. These data confirm that the patients with type 2 diabetes have by far the lowest insulin‐stimulated capacity for transcapillary passage of glucose into the muscle interstitium and that obese men have a lower capacity than lean men. The differences are likely, at least in part, to be the result of the reduction in the surface area resulting from reduced insulin‐stimulated increases in microvascular dilatation and blood volume (Keske et al. 2009), but differences in endothelial GLUT1 content between these groups can also make a contribution.
Spires et al. (2012) have used experimental measure‐ments in combination with a computational model to calculate the endothelial PS during incremental moderate intensity exercise and observed a gradual increase in PS, which showed a linear relationship with the increase in blood flow through the skeletal muscle capillary bed. How can we translate this into recruitment of MVUs and capillaries in skeletal muscle? For reasons explained in the previous sections it is not known today what percentage of the MVUs in human (or animal) skeletal muscle is perfused in the resting state at a given time. The occurrence of vasomotion with large differences in phase and frequency between different MVUs also implies that the MVUs are constantly switching between the closed and open position in resting muscles. However, given the fact that oxygen consumption by the combined skeletal muscles and blood flow through the muscle capillary bed increases 80‐ to 100‐fold going from rest to maximal exercise (Andersen & Saltin, 1985) it seems reasonable to assume that the percentage of MVUs in the open position is close to 100 during maximal exercise and much lower (maybe as low as 5–10%) in the resting state. However, over the course of minutes it is likely that all MVUs have been in the open position part of the time. This then implies that only a fraction of the ECL surface area will be available for transport of oxygen, glucose and FAs in the resting state, while most of this surface area will be recruited during maximal exercise. A gradual increase in the number of MVUs that are constantly open would provide a powerful mechanism to ensure that the supply of blood‐borne fuels and oxygen meets the growing energy demand of the contracting muscle fibres. The authors of this review want to make the reader aware here that Poole et al. (2013) have proposed that also at rest all capillaries in skeletal muscle are simultaneously perfused, but only over a fraction of their length with longitudinal recruitment occurring during exercise. It is not clear to the authors of this review, however, how such a recruitment pattern can be made compatible with the blood flow patterns, including the vasomotion that has been observed in MVUs by, among others, Lund et al. 1987 and Delashaw & Duling (1988).
Shear stress signal function of the endothelial glycocalyx
The luminal surface of the endothelium is covered with a brush‐like glycocalyx layer (glycoproteins and proteoglycans), which is approximately 0.5 μm thick in capillaries and 4.5 μm in arteries (Reitsma et al. 2007; Weinbaum et al. 2007). An important function of the glycocalyx is the translation of haemodynamic forces (shear forces exerted by flowing blood and individual blood cells passing through the narrow lumen of a TA) into vasodilatory responses during exercise. As such, mechanical forces exerted on the glycocalyx are likely to be early signalling events leading to eNOS activation and exercise training‐induced increases in skeletal muscle microvascular density.
The molecular mechanisms by which insulin increases muscle capillary blood flow
There is today compelling evidence both in rat and humans that an insulin‐mediated increase in microvascular perfusion of skeletal muscle is a conditional early event leading to increases of the transendothelial transport (TET) of both glucose and insulin into the interstitial fluid that surrounds the muscle fibres (reviewed by Barrett et al. 2009, 2011; Keske et al. 2015. Once the insulin concentration in the interstitium starts to increase (15–20 min after ingestion of a glucose load or start of a euglycaemic hyperinsulinaemic clamp, this leads to subsequent activation of the insulin signalling cascade, GLUT4 translocation to the plasma membrane and increased glucose uptake in skeletal muscle fibres (Vincent et al. 2004).
Important early information regarding the molecular mechanisms by which insulin is able to recruit additional blood flow in muscle capillaries has come from studies in cultured ECs (for references see Vincent et al. 2003; Wagenmakers et al. 2006; Muniyappa et al. 2007; Keske et al. 2015). These studies identified an insulin‐signalling cascade (insulin receptor/IRS1/PI3K/PDK1/Akt/eNOS; Fig. 2), activation of which leads to increased production of nitric oxide (NO). NO produced in vivo by the ECL is a potent vasodilator acting upon the smooth muscle cell layer in skeletal muscle arterioles. Insulin was found to activate the enzyme endothelial nitric oxide synthase (eNOS) among others by means of Ser1176 phosphorylation in cultured rat ECs and Ser1177 phosphorylation in cultured human ECs. Other signals found to activate eNOS in cultured ECs via Ser1176/1177 phosphorylation are fluid shear forces exerted on a cultured EC monolayer and exposure of cultured ECs to vascular endothelial growth factor‐A (VEGF‐A).
With the current understanding of the role played by TAs in the blood supply to MVUs, the likely eNOS activation site by which physiological increases in insulin following meal ingestion lead to the simultaneous recruitment of a larger number of MVUs are the TAs. This has been confirmed by the observation of Cocks et al. (2013 b) that 80 min after the start of a hyperinsulinaemic euglycaemic clamp in lean Zucker rats eNOS Ser1176 phosphorylation was increased by 14% (P < 0.05) in the ECL of TAs, while no increase was seen in the capillaries.
Newman et al. (2009), using laser Doppler flowmetry of skeletal muscle, observed that a hyperinsulinaemic euglycaemic clamp in rats induced an increase in the relative amplitude of the myogenic component of vasomotion which coincided with an increase in hindlimb glucose uptake. This observation suggested that the ability of insulin to recruit muscle microvascular blood flow is, at least in part, the result of increased arteriolar vasomotion in skeletal muscle. Vasomotion of TAs is also the likely mechanism to expose the ECL of a previously closed TA to the higher insulin concentration of the incoming blood. It is also possible that TET of insulin in open MVUs leads to increases in the interstitial insulin concentration and thus to the activation of the insulin receptor or IGF‐1 receptor in the abluminal membrane of closed MVUs. There is currently no information on the density of these receptors on the luminal and abluminal endothelial membrane, implying that this back‐door mechanism cannot be excluded. It also cannot be excluded that adenosine or ATP or other vasodilators play a role in giving luminal plasma insulin access to the luminal membrane of the ECL of closed TAs in the period that the plasma and interstitial insulin concentrations are rising. The adenosine may be produced by ECs, vascular smooth muscle cells and also skeletal muscle fibres in which the metabolic rate is increased by rises in interstitial insulin that are not matched by the oxygen supply as most of the MVUs serving that muscle fibre are in the closed position. Collectively these mechanisms will lead to an increase in the available capillary surface area and therefore result in increases in transport rates of insulin and glucose without the necessity of a measurable increase in total muscle blood. The latter has been observed in several studies (e.g. Raitakari et al. 1996). Insulin as mentioned before also increases its own TET via a mechanism that involves eNOS activation, enhancement of S‐nitrosylation of PTP1B inhibiting its phosphatase activity and therefore enhancing endothelial insulin signalling (Fig. 8 in Wang et al. 2013).
There is also evidence that there are impairments in the insulin‐induced activation of eNOS in animal models of insulin resistance and patients with type 2 diabetes. Cocks et al. (2013 b) did not find an increase in eNOS Ser1176 phosphorylation during a euglycaemic clamp in the ECL of TAs in obese Zucker rats, while a significant 14% increase was seen in lean Zucker rats. Tabit et al. (2013) reported an insulin‐induced increase in eNOS Ser1177 in freshly isolated venous ECs from non‐diabetic controls, but not in diabetic patients (P = 0.003), consistent with endothelial insulin resistance in type 2 diabetes. This study also provided convincing evidence that oxidative stress (nitrotyrosine levels) and inflammation (nuclear factor κB activation) was higher in ECs from the patients and that protein kinase C‐β activity was implicated in the endothelial insulin resistance. These data suggest that the mechanisms leading to insulin resistance in the ECL and in muscle fibres are quite similar (for comparison see Fig. 3; for further evidence and references see Wagenmakers et al. 2006 and Muniyappa et al. 2007).
The molecular mechanisms by which an exercise‐induced increase in blood shear stress increases muscle capillary blood flow
Endurance exercise at moderate intensities always leads to parallel increases in capillary perfusion and total muscle perfusion. Endurance exercise for 1 h at 65% led to eNOS activation by means of increased eNOS Ser1177 phosphorylation in a mixture of muscle capillaries and TAs (Cocks et al. 2012; Cocks et al. 2013 a). The increase in shear stress sensed by the endothelial glycocalyx is likely to play a role in the activation mechanism. This again will allow recruitment of more MVUs and a higher blood flow rate in the recruited MVUs by maintaining a high blood flow rate for a longer fraction of the time via increased vasomotion activity. In addition, despite the lack of published data to confirm this, such increases in eNOS phosphorylation are also likely to occur in lower order arterioles and the muscular feed arteries. This assumption is based on the observations of van Teeffelen & Segal (2000) that as the metabolic demand of the contracting muscle fibres increases, vasodilatation ascends progressively from the TAs into larger arterioles and their feed arteries. The induction of this mechanism is NO independent (Budel et al. 2003). The research of Segal & Jacobs (2001) provided evidence that electrical signals generated by ECs can travel rapidly along the ECL from TAs into their parent blood vessels, thereby leading to vasodilatation of the larger arterioles, resistance and feed arteries. This mechanism is called conductive vasodilatation (Segal & Jacobs, 2001) and contributes to the increases in skeletal muscle perfusion during exercise. The resultant increase in shear stress in these vessels is likely to lead to eNOS activation in lower order arterioles and muscular feed arteries. Acute endurance exercise has also been shown to activate eNOS via Ser1177 phosphorylation in mouse aorta (Zhang et al. 2009; Cacicedo et al. 2011).
The molecular mechanisms by which an exercise‐induced increase in the interstitial VEGF‐A concentration increases muscle capillary blood flow
Vascular endothelial growth factor‐A (VEGF‐A) is regarded as the central angiogenic factor in skeletal muscle capillary growth in response to exercise training interventions (Hoier & Hellsten, 2014). Skeletal muscle fibres are an important source of VEGF‐A in humans. Muscle fibres contain substantial VEGF‐A stores in small vesicles present between the myofibrils, with smaller amounts of VEGF‐A being present in cells located in the interstitial fluid that fills the space between capillaries and muscle fibres, including ECs and pericytes. Hoier & Hellsten (2014) in their recent review propose that VEGF‐A‐containing vesicles translocate to the plasma membrane and secrete their content into the interstitial fluid during exercise. As VEGF‐A mRNA expression is primarily increased after exercise, it is also proposed that the VEGF‐A stores lost through secretion during exercise are rapidly replenished in the 12–24 h period after exercise. As VEGF‐A is secreted during exercise it is impossible to separate the contribution made by increased blood shear stress and by VEGF‐A to the increase in eNOS Ser1177 phosphorylation seen by Cocks et al. (2012 and 2013 a) in the ECL of a mixed population of TAs and capillaries after 1 h of cycling exercise at 65% . As VEGF‐A is approaching the ECL via the skeletal muscle interstitium it may also help to activate eNOS via receptors on the abluminal membrane of TAs to thus recruit previously closed MVUs.
Activation of eNOS by increases in blood shear stress and interstitial VEGF during aerobic endurance exercise are therefore both likely to contribute to the mechanisms that lead to the recruitment of additional MVUs and maintenance of a higher mean blood flow in the recruited MVUs, but it is clear that there is an interaction between several locally produced vasodilators (NO, prostaglandins, ATP, adenosine) (for recent review see Mortensen & Saltin, 2014). There is evidence that NO and prostaglandins are the main vasodilators with a compensatory formation of the other vasodilator occurring when one is inhibited (Mortensen & Saltin, 2014).
VEGF‐B controls the expression of endothelial FA transporter proteins
Four closely related vascular endothelium growth factor (VEGF) isoforms are expressed in human skeletal muscle (VEGF A–D). VEGF‐A is best characterised because of its role in the angiogenic effect of exercise training. Until recently the role of VEGF‐B in regulating blood vessel function was not known. Hagberg et al. (2010) made the observation that VEGF‐B in mouse skeletal muscle was co‐expressed with a large cluster of nuclear genes encoding for mitochondrial proteins. This co‐expression was unique for VEGF‐B. Using a broad spectrum of molecular research methods, freshly isolated and cultured ECs and transgenic animal models, this study generated convincing evidence for the existence of a novel mechanism, which couples uptake and transendothelial transport of FAs to the capacity of mitochondrial β‐oxidation (Fig. 4). In brief VEGF‐B produced by skeletal muscle fibres is released to the interstitium and diffuses to the abluminal membrane of ECs in the capillary ECL. Here, it binds to VEGF receptor 1 and neuropilin 1 which are both expressed by the ECL. Signalling of VEGF‐B via a PI3K‐dependent mechanism then leads to the abundant expression of FATP3 and FATP4 in the ECL and incorporation into the luminal and abluminal membrane and therefore leads to increased transendothelial transport of FAs into the muscle interstitium where they diffuse to the muscle plasma membrane. Hagberg et al. (2010) also performed a study in which 14C‐oleate (a long‐chain FA) was given to wild‐type and VEGF‐B (–/–) mice by oral gavage and the incorporation of the radioactive long‐chain FA was measured in tissues. The incorporation into muscle, heart and brown adipose tissue of VEGF‐B (–/–) mice was much lower than in tissues of wild‐type mice. In the VEGF‐B (–/–) mice most of the radioactive FA accumulated in white adipose tissue pads. VEGF‐B (–/–) mice also gained more weight with age and developed obesity as evidenced by 60–90% larger fat pads and a higher body fat mass.
Figure 4. Schematic illustration of the role of VEGF‐B in matching transendothelial FA transport to skeletal muscle FA oxidation capacity .
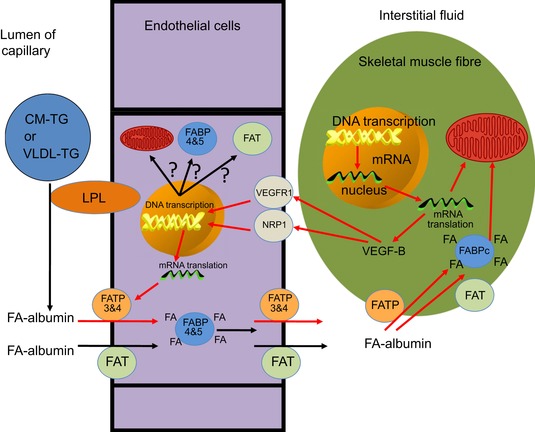
Hagberg et al. (2010) made the observation that VEGF‐B in mouse skeletal muscle was co‐expressed in a large number of different physiological conditions with a large cluster of nuclear genes encoding for mitochondrial proteins. They subsequently generated strong supportive evidence for the existence of a novel mechanism that couples uptake and transendothelial transport of FA to the capacity of mitochondrial β‐oxidation in skeletal muscle. VEGF‐B produced by skeletal muscle fibres was assumed to be released to the interstitium and then diffuses to the abluminal membrane of ECs in skeletal muscle capillaries, similar to the VEGF‐A release by skeletal muscle during exercise (Hoier & Hellsten, 2014). Here, VEGF‐B binds to its receptors (VEGF receptor 1 (VGRF1) and neuropilin 1 (NRP1)) which are both expressed by the ECs. Signalling of VEGF‐B then leads to the abundant expression of FATP3 and FATP4 in the ECs and incorporation into the luminal and abluminal membrane and is therefore assumed to lead to increased TET of FAs into the muscle interstitium where FAs are bound again to albumin to diffuse to the muscle plasma membrane and to be taken up, transported by the cytosolic fatty acid binding protein (FABPc) and oxidised in the skeletal muscle mitochondria as described by Glatz et al. (2010). The red arrows in this figure indicate the sequential steps in this novel mechanism as proposed by Hagberg et al. (2010). Other studies have shown that FAT/CD36 (Vistisen et al. 2004) and FABP4 and FABP5 (Iso et al. 2013) are also expressed abundantly in ECs and contribute to the high capacity for TET of FAs, but Hagberg et al. 2010 failed to generate evidence that the expression of their genes was under the control of VEGF‐B. It is also tempting to speculate that VEGF‐B controls the expression of the 1500 nuclear‐encoded mitochondrial genes in the ECs as the metabolic rate and protein turnover rate of ECs is potentially high because of repeated exposure to reactive oxygen species (ROS). It also cannot be excluded that some of the TET systems that help to increase endothelial permeability in the postprandial state and during exercise are energy (ATP) dependent (e.g. the insulin transport system which concentrates insulin in the ECs against a concentration gradient). This implies that ECs in trained inviduals should also have a high mitochondrial density to optimally support a high metabolic rate and the various transport functions. LPL, lipoprotein lipase.
Conclusions
The most important conclusion of this review is that the monolayer of ECs lining the lumen of skeletal muscle capillaries contains high concentrations of binding and transporter proteins, which help to facilitate the transport of glucose (GLUT1), FAs (FAT/CD36, FATP4, FATP5), insulin and amino acids across the luminal and abluminal plasma membrane and diffusion of FA (FABP3 and FABP4) through the cytosol of ECs. This implies that studies that aim to determine transport rates from the lumen of the capillary into the skeletal muscle fibre should consider the role of the ECL as a potential barrier (limiting skeletal muscle uptake) or as a facilitator (exceeding or driving the muscle uptake, e.g. the ability of the endothelial insulin transport system to create a higher insulin concentration in the cytosol of ECs than in the lumen of capillaries and in muscle interstitium). This finding also implies that studies using extracts of muscle homogenates and Western blots to measure the content of these transporters cannot assume that the measured concentration reflects the protein content of the muscle fibres if the transporter is expressed in a much higher concentration in the capillary endothelium than in the plasma membrane or cytosol of skeletal muscle. Examples of the latter are FAT/CD36 content (Vistisen et al. 2004) and FATP4 (Iso et al. 2013). Use of Western blot data in that case may confound important isomer differences between the capillary endothelium and the skeletal muscle fibres.
Another important message is that a careful review of earlier publications (Lund et al. 1987; Delashaw & Duling, 1988) using intravital microscopy of red blood cells moving in the microvasculature of the anterior tibialis muscle of hamsters confirms Microfil cast images of rat skeletal muscle (Fig. 1 C of Segal, 2005) suggesting that MVUs are the smallest functional elements that can be used to adjust capillary blood flow in response to physiological stimuli (insulin, exercise, adrenaline) and metabolic demand of muscle fibres. The blood flow patterns observed in these studies suggest that some MVUs have no flow, some undergo cyclic vasomotion and some are continuously perfused with a coordinated response occurring in each of these cases in all capillaries sharing a common TA. This pattern is not in line with recent suggestions that all capillaries in skeletal muscle are also simultaneously perfused at rest, but only over a fraction of their length with longitudinal recruitment occurring during exercise (Poole et al. 2013).
The recent observation (Hagberg et al. 2010) that the VEGF‐B gene is co‐expressed in mouse skeletal muscle and brown adipose tissue with nuclear genes encoding for mitochondrial proteins and simultaneously has a unique role in the expression of FATP3 and FATP4 in the capillary endothelium of these tissues is important for the following reasons. (1) The observation seems to provide a mechanism for the fact that meal‐derived lipids in lean, healthy and physically active humans are preferentially channelled for storage and subsequent oxidation into skeletal muscle and subcutaneous adipose tissue (Bickerton et al. 2007). (2) As VEGF‐B production, similar to VEGF‐A, is likely to be lower (and less frequent) in sedentary individuals, the latter, similar to VEGF (–/–) mice, are likely to deposit meal‐derived lipids in ectopic fat stores (visceral adipose tissue, around blood vessels and in man probably also in the liver) and develop obesity. As sedentary and obese individuals have a low capacity for fat oxidation in skeletal muscle this may also explain the accumulation of FA metabolites (diacylglycerols and ceramides) in their muscle, which is known to lead to insulin resistance (Fig. 3) and eventually to chronic disease (type 2 diabetes and cardiovascular disease). Future research should investigate these hypotheses by making comparisons between trained and sedentary humans and studying the impact of obesity, ageing and type 2 diabetes on the protein content of endothelial nutrient and insulin transporters. As VEGF‐B controls the expression of FATP3 and FATP4 via VEGF receptor 1 and neuropilin 1 this research may also create opportunities for the development of novel pharmacological interventions to reverse the pathological metabolic consequences of obesity, ageing, type 2 diabetes and cardiovascular disease.
Additional information
Competing interests
There are no competing interests.
Funding
Part of the data presented in this review were generated with support from the Insulin Dependent Diabetes Trust and Diabetes UK.
Acknowledgements
The authors thank Professor Steven S. Segal for a most useful discussion about the likely dimensions of microvascular units (MVUs) in a variety of mammalian skeletal muscles and for sharing relevant references on dimensions of MVUs and on the length of human skeletal muscle fibres.
Biography
Anton Wagenmakers is Professor of Exercise Metabolism and research group lead at the Research Institute for Sport and Exercise Sciences of Liverpool John Moores University. The focus of his current research is on (i) identification of the mechanisms by which a sedentary lifestyle leads to insulin resistance, sarcopenia, obesity, metabolic syndrome and type 2 diabetes, looking at both skeletal muscle fibres and their microvasculature, and (ii) the design of optimal exercise training interventions to counteract the underlying metabolic impairments.

This review is based on talks presented at the symposium Impact of Physical Activity, Ageing, Obesity and Metabolic Syndrome on Muscle Microvascular Perfusion and Endothelial Metabolism, which took place at Physiology 2014, the annual meeting of The Physiological Society, London, UK on 1 July 2014.
References
- Abdul‐Ghani MA & DeFronzo RA (2009). Plasma glucose concentration and prediction of future of type 2 diabetes. Diabetes Care 32, S194–S196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P & Saltin B (1985). Maximal perfusion of skeletal muscle in man. J Physiol 366, 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal S, Buring JE, Rifai N, Mora S, Sacks FM & Ridker PM (2007). Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA 298, 309–316. [DOI] [PubMed] [Google Scholar]
- Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W & Liu Z. (2009). The vascular actions of insulin control its delivery to muscle and regulate the rate‐limiting step in skeletal muscle insulin action. Diabetologia 52, 752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EJ, Wang H, Upchurch CT & Liu Z (2011). Insulin regulates its own delivery to skeletal muscle by feed‐forward actions on the vasculature. Am J Physiol Endocrinol Metab 301, E252–E263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AH & Scherer PE (2005). Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96, 939–949. [DOI] [PubMed] [Google Scholar]
- Bickerton AST, Roberts R, Fielding BA, Hodson L, Blaak EE, Wagenmakers AJM, Gilbert M, Karpe F & Frayn KN (2007). Preferential uptake of dietary fatty acids in adipose tissue and muscle in the postprandial period. Diabetes 56, 168–176. [DOI] [PubMed] [Google Scholar]
- Bradley H, Shaw CS, Worthington PL, Shepherd SO, Cocks M & Wagenmakers AJM (2014). Quantitative immunofluorescence microscopy of subcellular GLUT4 distribution in human skeletal muscle: effects of endurance and sprint interval training. Physiol Rep 2, e12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budel S, Bartlett IS & Segal SS (2003). Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch. Unmasking an NO wave. Circ Res 93, 61–68. [DOI] [PubMed] [Google Scholar]
- Cacicedo JM, Gauthier M‐S, Lebrasseur NK, Jasuja R, Ruderman NB & Ido Y (2011). Acute exercise activates AMPK and eNOS in the mouse aorta. Am J Physiol Heart Circ Physiol 301, H1255–H1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriello A (2005). Postprandial hyperglycemia and diabetes complications: is it time to treat? Diabetes 54, 1–7. [DOI] [PubMed] [Google Scholar]
- Clerk LH, Vincent MA, Jahn LA, Liu Z, Lindner JR & Barrett EJ (2006). Obesity blunts insulin‐mediated microvascular recruitment in human forearm muscle. Diabetes 55, 1436–1442. [DOI] [PubMed] [Google Scholar]
- Cocks M, Shaw CS, Shepherd SO, Fisher JP, Ranasinghe AM, Barker TA, Tipton KD & Wagenmakers AJM (2013. a). Sprint interval and endurance training are equally effective in increasing muscle microvascular density and eNOS content in sedentary males. J Physiol 591, 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks M, Shepherd SO, Shaw CS, Achten J, Costa ML & Wagenmakers AJM (2012). Immunofluorescence microscopy to assess enzymes controlling nitric oxide availability and microvascular blood flow in muscle. Microcirculation 19, 642–651. [DOI] [PubMed] [Google Scholar]
- Cocks M, Stride A, Macdonald R, Shaw CS, Marshall JM, Poucher SM & Wagenmakers AJM (2013. b). Impaired insulin‐mediated ser1176 phosphorylation of eNOS in skeletal muscle arterioles of obese Zucker rats. Obes Facts 6 (Suppl. 1), 92. [Google Scholar]
- Cohen KD, Berg BR & Sarelius IH (2000). Remote arteriolar dilations in response to muscle contraction under capillaries. Am J Physiol Heart Circ Physiol 278, H1916–H1923. [DOI] [PubMed] [Google Scholar]
- Davey KAB, Garlick PB, Warley A & Southworth R (2007). Immunogold labeling study of GLUT‐1 and GLUT‐4 in cardiac tissue following stimulation by insulin or ischemia. Am J Physiol Heart Circ Physiol 292, H2009–H2019. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Gunnarsson R, Björkman O, Olsson M & Wahren J (1985). Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin‐dependent (type II) diabetes mellitus. J Clin Invest 76, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delashaw JB & Duling BR (1988). A study of the functional elements regulating capillary perfusion in striated muscle. Microvasc Res 36, 162–171. [DOI] [PubMed] [Google Scholar]
- Glatz JFC, Luiken JJFP & Bonen A (2010). Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol Rev 90, 367–417. [DOI] [PubMed] [Google Scholar]
- Greenwalt DE, Scheck SH & Rhinehart‐Jones T (1995). Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest 96, 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald W & Sowa W (1977). Capillary structures and O2 supply to tissue. An analysis with a digital diffusion model as applied to skeletal muscle. Rev Physiol Biochem Pharmacol 77, 149–209. [PubMed] [Google Scholar]
- Gudsbjörnsdóttir S, Sjöstrand M, Strindberg L & Lönnroth P (2005). Decreased muscle capillary permeability surface area in type 2 diabetic subjects. J Clin Endocrinol Metab 90, 1078–1082. [DOI] [PubMed] [Google Scholar]
- Gudsbjörnsdóttir S, Sjöstrand M, Strindberg L, Wahren J & Lönnroth P (2003). Direct measurements of the permeability surface area for insulin and glucose in human skeletal muscle. J Clin Endocrinol Metab 88, 4559–4564. [DOI] [PubMed] [Google Scholar]
- Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, Van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone‐Elander S, Claesson‐Welsh L, Ylä‐Herttula S, Lindahl P & Eriksson U (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. [DOI] [PubMed] [Google Scholar]
- Hoier B & Hellsten Y (2014). Exercise‐induced capillary growth in human skeletal muscle and the dynamics of VEGF. Microcirculation 21, 301–314. [DOI] [PubMed] [Google Scholar]
- Iso T, Maeda K, Hanaoka H, Suga T, Goto K, Rizky M, Syamsunarno AA, Hishiki T, Nagahata Y, Matsui H, Arai M, Yamaguchi A, Abumrad NA, Sano M, Suematsu M, Endo K, Hotamisligil GS & Kurabayashi M (2013). Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arterioscler Thromb Vasc Biol 33, 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LD, Glickman MG, Rapoport S, Ferrannini E & DeFronzo RA (1983). Splanchnic and peripheral disposal of oral glucose in man. Diabetes 32, 675–679. [DOI] [PubMed] [Google Scholar]
- Keske MA, Clerk LH, Price WJ, Jahn LA & Barrett EJ (2009). Obesity blunts microvascular recruitment in human forearm muscle after a mixed meal. Diabetes Care 32, 1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keske MA, Premilovac D, Bradley EA, Dwyer RM, Richards SM & Rattigan S (2015). Muscle microvascular blood flow responses in insulin resistance and ageing. J Physiol 594, 2223–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A (1918). The number and distribution of capillaries in muscle with calculations of the oxygen pressure head necessary for supplying the tissue. J Physiol 52, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindbom L (1983). Microvascular blood flow distribution in skeletal muscle: An intravital microscopic study in the rabbit. Acta Physiol Scand Suppl 525, 1–40. [PubMed] [Google Scholar]
- Lund N, Damon DH, Damon DN & Duling BR (1987). Capillary grouping in hamster tibialis anterior muscles: flow patterns, and physiological significance. Int J Microcirc Clin Exp 5, 359–372. [PubMed] [Google Scholar]
- McFarlan JT, Yoshida Y, Jain SS, Han X‐X, Snook LA, Lally J, Smith BK, Glatz JFC, Luiken JJFP, Sayer RA, Tupling AR, Chabowski A, Holloway GP & Bonen A (2012). In vivo, fatty acid translocase (CD36) critically regulates skeletal muscle fuel selection, exercise performance, and training‐induced adaptation of fatty acid oxidation. J Biol Chem 287, 23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann GE, Yudilevich DL & Sobrevia L (2003). Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol Rev 83, 183–252. [DOI] [PubMed] [Google Scholar]
- Mortensen SP & Saltin B (2014). Regulation of the skeletal muscle blood flow in humans. Exp Physiol 99, 1552–1558. [DOI] [PubMed] [Google Scholar]
- Muniyappa R, Montagnani M, Koh KK & Quon MJ (2007). Cardiovascular actions of insulin. Endocr Rev 28, 463–491. [DOI] [PubMed] [Google Scholar]
- Newman JMB, Dwyer RM, St‐Pierre P, Richards SM, Clark MG & Rattigan S (2009). Decreased microvascular vasomotion and myogenic response in rat skeletal muscle in association with acute insulin resistance. J Physiol 587, 2579–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman RN (2013). Oxygen transport in the microcirculation and its regulation. Microcirculation 20, 117–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC, Copp SW, Ferguson SK & Musch TI (2013). Skeletal muscle capillary function: contemporary observations and novel hypotheses. Exp Physiol 98, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitakari M, Nuutila P, Ruotsalainen U, Laine H, Teräs M, Iida H, Mäkimattila S, Utriainen T, Oikonen V, Sipilä H, Haaparanta M, Solin O, Wegelius U, Knuuti J & Yki‐Järvinen H (1996). Evidence for dissociation of insulin stimulation of blood flow and glucose uptake in human skeletal muscle: studies using [15O]H2O, [18F]fluoro‐2‐deoxy‐d‐glucose, and positron emission tomography. Diabetes 45, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Rattigan S, Clark MG & Barrett EJ (1997). Hemodynamic actions of insulin in rat skeletal muscle: evidence for capillary recruitment. Diabetes 46, 1381–1388. [DOI] [PubMed] [Google Scholar]
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MA & Oude Egbrink MG (2007). The endothelial glycocalyx: composition, functions and visualisation. Pflugers Arch 454, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RS, Poole DC, Knight DR, Kurdak SS, Hogan MC, Grassi B, Johnson EC, Kendrick KF, Erickson BK & Wagner PD (1993). High muscle blood flow in man: is maximal O2 extraction compromised? J Appl Physiol 75, 1911–1916. [DOI] [PubMed] [Google Scholar]
- Roca J, Hogan MC, Story D, Bebout DE, Haab P, Gonzalez R, Ueno O & Wagner PD (1989). Evidence for tissue diffusion limitation of V O2max in normal humans. J Appl Physiol 67, 291–299. [DOI] [PubMed] [Google Scholar]
- Saltin B (1988). Capacity of blood flow delivery to exercising skeletal muscle in humans. Am J Cardiol 62, 30E–35E. [DOI] [PubMed] [Google Scholar]
- Saltin B & Gollnick PD (1983). Skeletal muscle adaptability: significance for metabolism and performance. In Handbook of Physiology, section 10, Skeletal Muscle, pp. 555–631. American Physiological Society, Bethesda, MD, USA. [Google Scholar]
- Saltin B, Henriksson J, Nygaard E, Andersen P & Jansson E (1977). Fibre types and metabolic potentials of skeletal muscle in sedentary man and endurance runners. Ann N Y Acad Sci 301, 3–29. [DOI] [PubMed] [Google Scholar]
- Segal SS (2005). Regulation of blood flow in the microcirculation. Microcirculation 12, 33–45. [DOI] [PubMed] [Google Scholar]
- Segal SS & Bearden SE (2012). Organisation and control of circulation to skeletal muscle In ACSM's Advanced Exercise Physiology, 2nd edn, ed. Farrell PA, Joyner MJ. & Caiozzo VJ, pp. 332–347. Wolters Kluwer/Lippincott Williams & Wilkins, London, Philadelphia. [Google Scholar]
- Segal SS & Jacobs TL (2001). Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CS & Wagenmakers AJM (2012). The endurance athlete: high aerobic capacity and improved longevity. Health consequences of exercise and inactivity. Biochem (Lond) 34, 20–23. [Google Scholar]
- Spires J, Gladden LB, Grassi B, Saidel GM & Lai N (2012). Model analysis of the relationship between intracellular and energy demand in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 303, R1110–R1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Mustali M, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA & Hamburg NM (2013). Protein kinase C‐β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 127, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loon LJC, Greenhaff PL, Constantin‐Teodosiu D, Saris WHM & Wagenmakers AJM (2001). The effect of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 536, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Teeffelen JW & Segal SS (2000). Effect of motor unit recruitment on functional vasodilatation in hamster retractor muscle. J Physiol 524, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S & Barrett EJ (2004). Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53, 1418–1423. [DOI] [PubMed] [Google Scholar]
- Vincent MA, Clerk LH, Lindner JR, Price WJ, Jahn LA, Leong‐Poi H & Barrett EJ (2006). Mixed meal and light exercise each recruit muscle capillaries in healthy humans. Am J Physiol Endocrinol Metab 290, E1191–E1197. [DOI] [PubMed] [Google Scholar]
- Vincent MA, Montagnani M & Quon MJ (2003). Molecular and physiological actions of insulin related to production of nitric oxide in vascular endothelium. Curr Diab Rep 3, 279–288. [DOI] [PubMed] [Google Scholar]
- Vistisen B, Roepstorf K, Roepstorf C, Bonen A, van Deurs B & Kiens B (2004). Sarcolemmal FAT/CD36 in human skeletal muscle colocalizes with caveolin‐3 and is more abundant in type 1 than in type 2 fibers. J Lipid Res 45, 603–609. [DOI] [PubMed] [Google Scholar]
- Wagenmakers AJM, Van Riel NAW, Frenneaux MP & Stewart PM (2006). Integration of the metabolic and cardiovascular effects of exercise. Essays Biochem 42, 193–210. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu Z, Li G & Barrett EJ (2006). The vascular endothelial cell mediates insulin transport into skeletal muscle. Am J Physiol Endocrinol Metab 291, E323–E332. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang AX, Aylor K & Barrett (2013). Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes 62, 4030–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang AX & Barrett EJ (2011). Caveolin‐1 is required for vascular endothelial insulin uptake. Am J Physiol Endocrinol Metab 200, E134–E144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang AX & Barrett EJ (2012). Insulin‐induced endothelial cortical actin filament remodeling: a requirement for trans‐endothelial insulin transport. Mol Endocrinol 26, 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang AX, Liu Z & Barrett EJ (2008). Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes 57, 540–547. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang AX, Liu Z, Chai W & Barrett EJ (2009). The trafficking/interaction of eNOS and caveolin‐1 induced by insulin modulates endothelial nitric oxide production. Mol Endocrinol 23, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SR, Eng CM, Smallwood LH & Lieber RL (2009). Are current measurements of lower extremity muscle architecture accurate? Clin Orthop Relat Res 467, 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER (1984). The Pathway for Oxygen: Structure and Function in the Mammalian Respiratory System. Fig. 7.3, p. 179. Harvard University Press, Cambridge, MA, USA. [Google Scholar]
- Weinbaum S, Tarbell JM & Damiano ER (2007). The structure and function of the endothelial glycocalyx layer. Ann Rev Biomed Eng 9, 121–167. [DOI] [PubMed] [Google Scholar]
- Wickiewicz TL, Roy RR, Powel PL & Edgerton VR (1983). Muscle architecture of the human lower limb. Clin Orthop Relat Res 179, 275–283. [PubMed] [Google Scholar]
- Wolinsky H (1980). A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ Res 47, 301–311. [DOI] [PubMed] [Google Scholar]
- Zhang Q‐J, McMillin SL, Tanner JM, Palionyte M, Abel ED & Symons JD (2009). Endothelial nitric oxide synthase phosphorylation in treadmill‐running mice: role of vascular signalling kinases. J Physiol 587, 3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]