

Abstract
Age is one of the major risk factors associated with cardiovascular disease (CVD). About one‐fifth of the world population will be aged 65 or older by 2030, with an exponential increase in CVD prevalence. It is well established that environmental factors (overnutrition, smoking, pollution, sedentary lifestyles) may lead to premature defects in mitochondrial functionality, insulin signalling, endothelial homeostasis and redox balance, fostering early senescent features. Over the last few years, molecular investigations have unveiled common signalling networks which may link the ageing process with deterioration of cardiovascular homeostasis and metabolic disturbances, namely insulin resistance. These different processes seem to be highly interconnected and their interplay may favour adverse vascular and cardiac phenotypes responsible for myocardial infarction, stroke and heart failure. In the present review, we carefully describe novel molecular cues underpinning ageing, metabolism and CVD. In particular, we describe a dynamic interplay between emerging pathways such as FOXOs, AMPK, SIRT1, p66Shc, JunD and NF‐kB. This overview will provide the background for attractive molecular targets to prevent age‐driven pathology in the vasculature and the heart.
Abbreviations
- ADL
activity of daily living
- AGE
advanced glycation end products
- AICAR
aminoimidazole carboxamide ribonucleotide
- ALDH‐2
aldehyde dehydrogenase‐2
- AMPK
AMP‐activated protein kinase
- AP‐1
activator protein‐1
- CVD
cardiovascular disease
- ECM
extracellular matrix
- eNOS
endothelial nitric oxide synthase
- Erk
extracellular signal‐regulated kinase
- FFA
free fatty acids
- FOXO
Forkhead transcription factor
- HUVECs
human‐umbilical‐vein‐endothelial‐cells
- ILVM
inappropriate left ventricular mass
- LV
left ventricular
- MnSOD
manganese superoxide dismutase
- NAD
nicotinamide adenine dinucleotide
- Nampt
nicotinamide phosphoribosyltransferase
- NF‐kB
nuclear factor‐kappa‐B
- PGI2
prostaglandin I2
- PKCβII
protein kinase C βII
- ROS
reactive oxygen species
- SA‐β‐gal
senescence‐associated β‐galactosidase
- TOR
target of rapamycin
- VEGF‐A
vascular endothelial growth factor‐A
- VSMCs
vascular smooth muscle cells
- miR
microRNA
- WT
wild type
Ageing and cardiovascular disease
Ageing is perhaps the most important risk factor affecting cardiovascular homeostasis (Kovacic et al. 2011). Advances in the treatment of cardiovascular disease (CVD) and particularly acute myocardial infarction have prolonged median life‐expectancy, with an estimated fourfold increase of people aged more than 75 years (Heidenreich et al. 2011; Nichols et al. 2014). Accordingly, about one‐fifth of the world population will be aged 65 or older by 2030, with an exponential increase in the prevalence of CVD due to the fact that additional 27 million people will have hypertension, 8 million coronary heart disease, 4 million stroke and 3 million heart failure (Heidenreich et al. 2011). Moreover, the prevalence of metabolic disturbances, namely metabolic syndrome and diabetes, is significantly increased in the elderly population and further contributes to CV morbidity and mortality (Fadini et al. 2011). These numbers may explain why approximately 40% of the deaths in people aged >65 are caused by atherosclerotic disease and its complications (Heidenreich et al. 2011). Furthermore, the cost to treat CVD will triple over the next few years. By 2018, CVD costs among those aged 65–79 years are expected to exceed CVD costs among those aged 45–64 years (Heidenreich et al. 2011; Tarride et al. 2009; Fig. 1). This scenario strengthens the importance of understanding the molecular cues underlying the aetiology of the ageing process and its link with cardiovascular disease phenotypes. So far, the fields of cardiovascular disease and ageing have remained largely separated. More recently, it has been postulated that ageing and CVD are highly interconnected and may share common pathways (Fadini et al. 2011). Consistently, researchers have found that many of the factors underlying age‐related changes in the arteries are also implicated in the development of CVD (Kovacic et al. 2011). In the present article, we provide an overview of emerging molecular pathways implicated in the interplay between senescence, atherosclerotic disease and metabolic disturbances. These novel targets might be considered for the design and development of mechanism‐based therapeutic strategies to combat CVD burden over the next decades.
Figure 1. Projections of crude cardiovascular disease prevalence (%), 2010 –2030 in the United States .
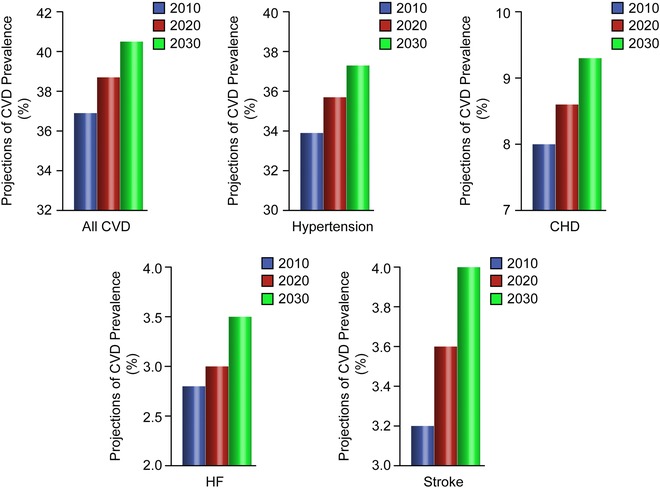
Modified.
Pathological features of cardiovascular ageing
Ageing is accompanied by changes in vascular structure and function, especially in the large arteries (Safar et al. 2010; Kovacic et al. 2011). Age‐related impairment of vascular function is the result of phenotypic alterations of different cell types, such as endothelial cells, smooth muscle cells and pericytes (Sawabe et al. 2010). Morphological changes are in most cases organ‐specific and include vascular wall thickening, collagen deposition, perivascular fibrosis and vessel dilatation (Fig. 2). Progressive myointimal thickening is generally due to enhanced elastin degradation and collagen deposition in the vascular media as well as intimal hyperplasia (Scioli et al. 2014). Autopsy studies of perfusion‐fixed human arteries have shown that thickening is mostly confined to the non‐load‐bearing intima, rather than the load‐bearing‐media. Other studies show that both intima and media are involved (O'Rourke et al. 2007). Thickening is a key hallmark of the aged vasculature promoting arterial stiffness (Lee et al. 2010). Changes in arterial stiffness are also due to an altered vascular tone, which results from the imbalance between vasoconstriction and vasorelaxation (Zieman et al. 2005). Arterial stiffness is a major determinant of vascular impedance, which affects the pulsatile ejection of blood from the heart. The decrease in aortic distensibility that is associated with ageing creates a mismatch between ventricular ejection and aortic flow energies, which results in increased aortic systolic pressure, changes in aortic pressure contour, pulse wave reflection, and characteristic aortic impedance. Experimental work examining arterial mechanical changes during ageing showed that both incremental passive and active circumferential stiffness are significantly enhanced in 23‐month‐old compared with 6‐month‐old rats (Gaballa et al. 1998). Moreover, ageing was found to increase media thickness, collagen content and the collagen/elastin ratio by 12%, 21% and 38%, respectively (Gaballa et al. 1998). By contrast, elastin density and the number of smooth muscle cell nuclei were decreased by 20% and 31%, respectively, with ageing (Gaballa et al. 1998). Another established notion is that proximal arteries such as the central aorta and carotid artery dilate with age, resulting in increased lumen diameter. Importantly, adverse vascular remodelling is accompanied by key alterations of endothelial homeostasis. Occurrence of endothelial dysfunction is a major cause of morbidity and mortality (Flammer et al. 2012). Endothelium‐dependent vasorelaxation is impaired in aged vessels and this phenomenon is associated with increased vascular permeability and inflammation as well as impaired angiogenesis (Kung et al. 1995; Kovacic et al. 2011). Moreover, endothelial cells become more heterogeneous in size, shape and axial orientation, so that intraluminal blood flow may be less laminar and the number of sites for lipid deposition may increase, thus fostering the formation of atherosclerotic plaques (Chiu et al. 2011).
Figure 2. Structural changes of the aged vasculature .
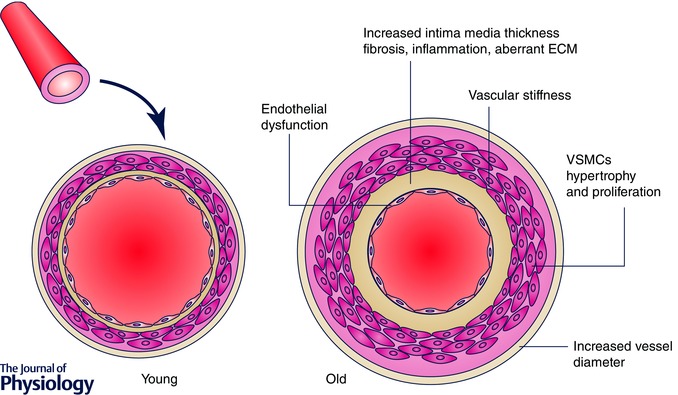
ECM, extracellular matrix; VSMCs, vascular smooth muscle cells.
Besides its effect on the vascular system, from Heidenreich et al. 2011 ageing has a remarkable effect on the heart (Stern et al. 2003). Heart weight increases with age whereas the number of cardiac myocytes is progressively reduced. This continued loss of functional cardiac cells is paralleled by a decline in regenerative activity from 1% per year at age 20 to 0.4% at 75 years (Dai et al. 2012). Myocardial fibrosis in the aged myocardium is an important determinant of impaired diastolic and systolic function (Biernacka et al. 2011). In this setting, a reduction in cardiac output due to decline in function stimulates the myocardium to compensate by increasing muscle mass (Cheitlin et al. 2003). Increased myocardial hypertrophy is an efficient short‐term mechanism, but as a compensatory mechanism it is detrimental over the long term since it directly impairs cardiac performance (Isoyama et al. 1987). In elderly subjects, the increase in left ventricular (LV) hypertrophy significantly exceeds the predicted values which are based on cardiac workload. In other words, a large part of left ventricular mass is not generated to offset an increase in afterload but rather reflects pathological alterations of cardiac structure likely to be dictated by myocardial fibrosis and inflammation (de Simone et al. 2002). This has led to the definition of inappropriate left ventricular mass (ILVM), to indicate an adverse phenotype of LV remodelling. Interestingly, ILVM strongly correlates with systo‐diastolic dysfunction as well as cardiovascular mortality (de Simone et al. 2002). In the aged heart, fibrosis serves as a pathological substrate for bradi‐ and tachyarrhythmias (de Jong et al. 2011). For example, heart rate is influenced not only by the loss of cells in the sinoatrial node (responsible for controlling heart rate) but also by structural changes in the heart, including fibrosis and hypertrophy, which slow propagation of electric impulse (Csepe et al. 2015). A recent work has clearly demonstrated that age‐dependent mitochondrial DNA damage is an important substrate underpinning the pathophysiology of cardiac arrhythmias (Baris et al. 2015). In this study, genetically modified mice with accelerated accumulation of mtDNA deletions in the myocardium accumulated few randomly distributed cardiomyocytes with compromised mitochondrial function, which led to spontaneous ventricular premature contractions and atrio‐ventricular blocks at the age of 18 months. These symptoms were not caused by a general mitochondrial dysfunction in the entire myocardium, and were not observed in mice at 12 months with significantly lower numbers of dysfunctional cells. These results suggest that the disposition to arrhythmia typically found in the aged human heart might be due to the random accumulation of mtDNA deletions and the subsequent mosaic respiratory chain deficiency (Baris et al. 2015). Another important pathological feature associated with ageing is the calcification of aortic and mitral valves which triggers stenosis/insufficiency resulting in cardiac pressure/volume overload (Freeman et al. 2005).
Molecular pathways linking senescence, metabolic alterations and cardiovascular phenotypes
Growing evidence is supporting the concept that ageing is able to derail pathways leading to adverse metabolic profile, high blood pressure and altered lipid metabolism. On the other hand, metabolic conditions, namely obesity, diabetes and insulin resistance, are associated with premature features of vascular and cardiac senescence. These aspects highlight a dynamic interplay between ageing, metabolism and cardiovascular disease (Fig. 3).
Figure 3. Schematic representation of the molecular pathways linking ageing, metabolism and cardiovascular disease .
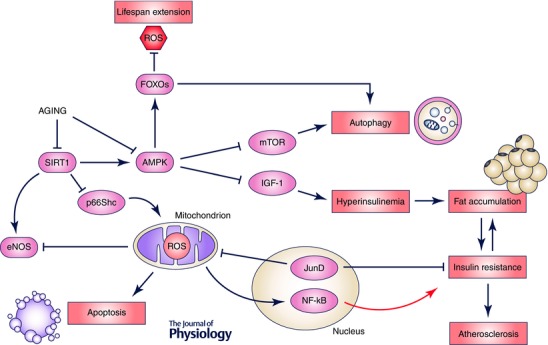
ROS, reactive oxygen species; eNOS, endothelial nitric oxide synthase; NF‐kB, nuclear factor‐kappa B; AMPK, AMP‐activated protein kinase; FOXOs, Forkhead box O; IGF‐1, insulin growth factor‐1; mTOR, mammalian target of rapamycin.
Mitochondrial adaptor p66Shc
Mechanistic studies over the last decade have demonstrated that the mitochondrial adaptor p66Shc is an important molecular effector which may explain how ageing is connected with metabolic and cardiovascular disease. This enzyme plays a major role in the generation of reactive oxygen species (ROS) (Paneni et al. 2012). Several stimuli activate protein kinase C βII (PKCβII) isoform to induce Ser‐36 phosphorylation of p66Shc, allowing transfer of the protein from the cytosol to the mitochondrion where it fosters ROS accumulation by oxidizing cytochrome c (Giorgio et al. 2005). This latter event leads to mitochondrial disruption, release of solutes and water, and subsequent apoptotic programmes. Intracellular free radicals are reduced in cells lacking the p66Shc gene (p66Shc −/− cells), and both systemic and intracellular free radicals are diminished in p66Shcc −/− mouse models exposed to high oxidative stress (Giorgio et al. 2005; Camici et al. 2008). An interesting observation from our lab was that endothelium‐dependent relaxation in response to acetylcholine is age‐dependently impaired in wild‐type but not in p66Shcc −/− mice (Francia et al. 2004). This phenomenon was largely explained by preservation of nitric oxide availability in mice lacking the p66Shc gene (Francia et al. 2004). Further work has demonstrated that p66Shc activation is critically involved in different processes including adipogenesis, insulin resistance and diabetes‐related cardiovascular complications (Camici et al. 2007; Berniakovich et al. 2008; Paneni et al. 2014). More recently, we found that vascular p66Shc levels are increased in genetically obese mice and participate in endothelial insulin resistance (Paneni et al. 2014). In this study, endothelium‐dependent relaxations to insulin were blunted in obese as compared to WT mice. Interestingly, in vivo gene silencing of p66Shc restored insulin response via the IRS‐1–Akt–eNOS pathway (Paneni et al. 2014). Furthermore, p66Shc knockdown in endothelial cells isolated from obese mice attenuated ROS production, free fatty acids (FFA) oxidation and prevented dysregulation of redox‐sensitive pathways such as nuclear factor‐kappa‐B (NF‐kB), advanced glycation end products (AGE) precursor methylglyoxal and prostaglandin I2 (PGI2) synthase (Paneni et al. 2014). Activation of p66Shc also interferes with the acquisition of the heart senescent phenotype and the development of heart failure in diabetic mice. Indeed, diabetic p66Shcc −/− mice were protected against myocardial oxidative stress, apoptosis and telomere shortening. Moreover, ablation of the p66Shc gene in cardiac stem cells preserved the growth reserve of the heart (Rota et al. 2006). The clinical relevance of p66Shc is supported by the notion that p66Shc gene expression is increased in mononuclear cells obtained from patients with type 2 diabetes and coronary artery disease (Pagnin et al. 2005; Franzeck et al. 2012). Moreover, a recent study showed that p66Shc expression is higher in fibroblasts isolated from centenarians (Pandolfi et al. 2005). This finding probably indicates that p66Shc expression may increase in a time‐dependent manner. Taken together, these findings show that increased p66Shc expression during the life‐course may foster ROS accumulation with subsequent deregulation of pathways implicated in mitochondrial dysfunction, fat accumulation, insulin resistance and diabetes.
AMP‐activated protein kinase
The functional AMP‐activated protein kinase (AMPK) is a heterotrimer consisting of a catalytic alpha (α), a regulatory gamma (γ) and a scaffolding beta (β) subunit and is activated by low cellular energy status (Salminen et al. 2012). AMPK activation orchestrates many biochemical events including glucose uptake, glycolysis, oxidation of free fatty acids (FFAs) and mitochondrial biogenesis (Towler et al. 2007). These processes significantly contribute to raise ATP levels and restore myocardial contractile efficiency and vascular responses. AMPK also activates endothelial nitric oxide synthase (eNOS), and promotes autophagy and mitophagy, thus preventing mitochondrial insufficiency, inflammation and cellular death (Alers et al. 2012). Autophagy is a major intracellular degradation process recognized to play a central role in cell survival and longevity (He et al. 2009). In recent years, this phenomenon has emerged as a unifying downstream feature of several evolutionarily conserved anti‐ageing interventions including both dietary restrictions and reduced target of rapamycin (TOR) signalling (Jung et al. 2010). A recent study found that upregulation of AMPK in the adult Drosophila nervous system induces autophagy both in the brain and in the intestinal epithelium. Interestingly, induction of autophagy was linked to improved intestinal homeostasis during ageing and increased lifespan by 30% (Ulgherait et al. 2014). AMPK is also a master regulator of key molecular effectors involved in metabolic processes, longevity and cardiovascular homeostasis. It modulates mTOR signalling by directly phosphorylating the TSC1/2 complex, regulates the IGF‐1 pathway through the extracellular signal‐regulated kinase (Erk) cascade, and controls sirtuin activity by regulating the abundance of nicotinamide adenine dinucleotide (NAD) and nicotinamide phosphoribosyltransferase (Nampt) (Salminen et al. 2012). Old mice (28–30 months) display reduced arterial AMPK expression compared to 3‐ to 6‐month‐old animals. Pharmacological activation of AMPK by aminoimidazole carboxamide ribonucleotide (AICAR) for 2 weeks increased arterial AMPK and reversed ROS‐driven endothelial dysfunction (Lesniewski et al. 2012). Moreover, AMPK activation has shown to diminish senescence‐associated β‐galactosidase (SA‐β‐gal) staining while restoring proliferation of vascular smooth muscle cells (Sung et al. 2011). Interestingly, recent evidence indicates that AMPK is amenable to pharmacological intervention and, hence, represents a potentially ‘druggable’ target to prevent ageing‐related features. Experimental work has indeed shown that dietary curcumin administration for 1 month remarkably restored cerebrovascular endothelium‐dependent vasorelaxation in aged rats. In cerebral arteries from old animals and cultured endothelial cells, curcumin promoted AMPK phosphorylation and reduced ROS production. Interestingly, the beneficial effects of curcumin were no longer seen following AMPK inhibition (Pu et al. 2013). Meformin, a biguanide often used in the treatment of diabetes, is capable of inducing AMPK activation by phosphorylating a key regulatory site (Thr‐172) on the catalytic (α) subunit. Administration of metformin before, during or after myocardial ischaemia has been shown to prevent ischaemia–reperfusion injury and adverse remodelling of the left ventricle (El Messaoudi et al. 2011). Consistently, metformin has been shown to preserve insulin secretion by promoting the AMPK‐dependent autophagic response in pancreatic beta cells (Jiang et al. 2014). However, in a recent randomized trial chronic treatment with metformin did not affect the p‐AMPK/total AMPK ratio in peripheral blood mononuclear cells isolated from prediabetic subjects (Vigili de Kreutzenberg et al. 2015). Moreover, high dose resveratrol stimulated differentiation of vascular smooth muscle cells (VSMCs) through AMPK‐mediated inhibition of the mTORC1 pathway, allowing activation of the Akt kinase. These latter findings hint that pharmacological activation of AMPK may play a role in VSMC phenotypic plasticity, thus promoting vessel maintenance, repair and adaptation to vascular changes associated with ageing (Thompson et al. 2014).
SIRT1
The SIRT1 gene belongs to the family of nicotinamide adenine dinucleotide (NAD)‐dependent proteins and is considered a major gatekeeper against oxidative stress, inflammation and cardiovascular ageing (Pillarisetti et al. 2008). SIRT1 protects the heart from senescent features, ischaemia/reperfusion injury, hypertrophy and cardiomyocyte apoptosis (Alcendor et al. 2007). SIRT1 overexpression leads to reduced myocardial hypertrophy, interstitial fibrosis, oxidative stress and senescent markers such as p15INK4b and p19ARF (Sundaresan et al. 2011). Moreover, pharmacological activation of SIRT1 by resveratrol attenuates ageing‐induced elevations of fibrotic collagen deposition and markers of oxidative damage including 4HNE and nitrotyrosine (Sin et al. 2014). These changes were associated with a significant improvement in cardiac function, as assessed by ejection fraction and fractional shortening. Moreover, SIRT1 ameliorates endothelial function, prevents macrophage foam cell formation and calcification of vascular smooth muscle cells (Stein et al. 2011). Age‐dependent downregulation of SIRT1 favours acetylation of nuclear factor (NF)‐kB p65, leading to its nuclear translocation and transcription of inflammatory genes (Yeung et al. 2004). Recent work has shown that sirtuin expression can be modulated by epigenetic changes, namely increased DNA methylation and non‐coding RNAs such as miR‐200a and miR‐34a (Eades et al. 2011; Sahin et al. 2014; Yu et al. 2015). The maintenance of SIRT1 homeostasis is fundamental to repression of detrimental pathways of arterial ageing such as the Forkhead transcription factor (FOXO) pathway (Brunet et al. 2004). Specifically, SIRT1 has the ability to deacetylate FOXO 1, 3 and 4 in the nucleus thus preventing DNA damage, cell cycle arrest and oxidative stress (Brunet et al. 2004; van der Horst et al. 2004; Kobayashi et al. 2005). Moreover, SIRT1 deacetylates LKB1 leading to activation of the final effector enzyme AMPK, a central energy regulator involved in glucose homeostasis, maintenance of cellular ATP levels and endothelial integrity via regulation of eNOS activity and autophagy (Mattagajasingh et al. 2007). Perturbation of the SIRT1–LKB1–AMPK pathway leads to energy imbalance, cellular stress and activation of the apoptotic machinery, thus contributing to vascular ageing (Pillarisetti et al. 2008). A randomized, placebo‐controlled trial including 38 prediabetic subjects, showed that treatment with metformin (1500 mg day–1) significantly restored the SIRT1–mTOR–AMPK axis while suppressing senescent markers such as p53 and p21 in peripheral blood mononuclear cells (Vigili de Kreutzenberg et al. 2015). SIRT1 inhibition also affected telomere length in these patients (Vigili de Kreutzenberg et al. 2015). Another important mechanism whereby SIRT1 prevents cardiovascular ageing is the activation of endothelial nitric oxide synthase (eNOS) (Stein et al. 2011). In human umbilical vein endothelial cells (HUVECs), SIRT1 activation protects against ROS‐induced premature senescence by increasing eNOS protein expression and activity (Ota et al. 2007). These data were further strengthened by the observation that microRNA (miR)‐217, an endogenous inhibitor of SIRT1, triggers endothelial senescence by suppressing SIRT1‐dependent eNOS functionality (Menghini et al. 2009). Taken together, these results indicate that the SIRT1–eNOS axis could be a potential target against vascular ageing and age‐related vascular diseases. Pharmacological approaches to inhibition of SIRT1 have been shown to prevent maladaptive pathways promoting ageing, and metabolic and cardiovascular disease (Winnik et al. 2015). Among different compounds, resveratrol is of particular importance. This is a polyphenol naturally present in several plants that is identified as a small‐molecule activator of SIRT1. Modulation of SIRT1 activity by resveratrol suppresses oxidant and inflammatory genes by altering the epigenetic status of their promoter (Fernandez et al. 2011). Indeed, SIRT1‐induced deacetylation of histone tails reduce chromatin accessibility to transcription factors, thereby reducing the expression of detrimental genes such as the adaptor p66Shc (Paneni et al. 2013 b; Costantino et al. 2015). Beside epigenetic regulation, resveratrol also has effects on mitochondrial metabolism and insulin sensitivity by activating the AMPK–SIRT1–PGC‐1α axis (Lagouge et al. 2006). Chronic treatment with resveratrol improves endothelial function and prevents metabolic disturbances in obese individuals (Beaudeux et al. 2010). Furthermore, resveratrol improved diastolic function in patients with coronary artery disease (Magyar et al. 2012). Many experimental studies have shown that resveratrol prolongs lifespan in different animal models. In mammals, controversial results have also been found and they seem to depend on the composition of the diet supplied with the resveratrol supplementation (Fernandez et al. 2011). However, it has been claimed that resveratrol may have other targets than sirtuins and the direct activation of sirtuins by resveratrol is currently a matter of debate (Costantino et al. 2015). Further research is needed to fully understand the potential and clinical application of resveratrol as a SIRT1‐activating approach.
Forkhead transcription factors (FOXOs)
Forkhead transcription factors (FOXOs) regulate the expression of genes involved in cell growth, proliferation, differentiation and longevity (Ronnebaum et al. 2010). FOXOs are also modulated by SIRT1‐mediated deacetylation and IGF‐1 signalling (Giannakou et al. 2004; Gan et al. 2005). Inactivation of sirtuins during ageing favours FOXO acetylation and subsequent transcription of FOXO‐dependent genes favouring cellular apoptosis, cell cycle arrest, accumulation of ROS and metabolic derangements. Moreover, FOXOs control the expression of many autophagy‐related genes and are important for lifespan extension (van der Horst et al. 2007; Webb et al. 2014). A role for the FOXO family in autophagy was first described in murine models of muscle atrophy, an age‐related condition to which several degradative pathways contribute, especially autophagy (Lapierre et al. 2015). In the muscle, FOXO1 and FOXO3 elevate the autophagic flux by increasing the expression of autophagy genes mainly working as part of the core machinery and additionally increase protein degradation via the proteasomal pathway (Sanchez et al. 2014). In particular, FOXO3 increases the capacity of the lysosome to degrade incoming cargo, indicating a role for lysosomal function in muscle atrophy (Zhao et al. 2007). Other FOXOs (FOXO1, FOXO4, and FOXO6) also play roles in proteostasis and autophagy (Lapierre et al. 2015).
FOXOs are key downstream effectors of the PI3K–Akt pathway (Ronnebaum et al. 2010). Following stimulation of PI3K–Akt signalling by growth factors, Akt phosphorylates FOXOs on three conserved residues, which leads to their cytoplasmic sequestration and inactivation (Huang et al. 2007). By contrast, Akt is inactive in the aged vasculature, thus promoting FOXOs‐dependent transcriptional programmes culminating with endothelial senescent phenotypes (Ronnebaum et al. 2010). Expression of FOXO3a in mouse hearts resulted in reduction of cardiomyocyte size, suggesting that this FOXO factor functions to reduce hypertrophy (Ronnebaum et al. 2010). Indeed, FOXO1 or FOXO3 in cardiomyocytes attenuate calcineurin phosphatase activity and inhibit agonist‐induced hypertrophic growth (Ni et al. 2006). FOXO3a deficiency has been shown to increase eNOS expression and enhances postnatal vessel formation and maturation (Salih et al. 2008). Loss of insulin signalling is emerging as an important factor underpinning impaired lifespan (Paneni et al. 2015). A very recent study found that insulin signalling via phosphorylation of FOXO‐1 and consequent eNOS stimulation was selectively impaired in visceral adipose tissue and endothelial cells of obese subjects (Karki et al. 2015). Interestingly, pharmacological antagonism of FOXO1 by AS184256 and gene silencing reversed insulin resistance and restored endothelial eNOS functionality in endothelial cells from obese subjects (Karki et al. 2015). Further work has demonstrated that FOXO3 is required for lifespan extension in mice undergoing dietary restriction. Indeed, low caloric intake was associated with increased survival in wild‐type (WT) mice but not in Foxo3‐knockout heterozygous (+/−) and homozygous (−/−) animals (Shimokawa et al. 2015). In line with these data, FOXO3A single nucleotide polymorphisms (SNPs) were significantly associated with cognitive function, handgrip strength, activity of daily living (ADL), and self‐rated health in a cohort of 1088 oldest‐old Danes (age 92–93 years) (Soerensen et al. 2015). Understanding the biology of FOXOs may have important implications for the prevention of ageing phenotypes.
Nuclear factor kappa‐B
Nuclear factor kappa‐B (NF‐kB) is an important transcription factor expressed in all mammalian cell types (Brasier et al. 2006). It is responsible for regulating gene expression of factors that control cell adhesion, proliferation, inflammation, redox state, and tissue‐specific enzymes (Baker et al. 2011). Activation of NF‐kB mediates vascular and myocardial inflammation in metabolic and age‐related diseases. A recent study clearly demonstrated that endothelial suppression of NF‐kB prolongs lifespan in mice and ameliorates obesity‐induced endothelial insulin resistance. Hasegawa and co‐workers have shown that transgenic mice with endothelium‐specific overexpression of the inhibitory NF‐kB subunit IkBα (E‐DNIkB) were protected against insulin resistance in adipose tissue and skeletal muscle (Hasegawa et al. 2012). In these mice, obesity‐induced macrophage infiltration of adipose tissue and plasma oxidative stress markers were reduced whereas blood flow and muscle mitochondrial content were increased. Of note, capillary recruitment and subsequent insulin delivery were explained by restoration of NO levels in E‐DNIkB animals (Hasegawa et al. 2012). Impaired insulin signalling is indeed an important hallmark linking metabolic disease with premature ageing of the CV system (Rask‐Madsen et al. 2011). The importance of inflammation in the pathogenesis of metabolic disturbances is strengthened by the observation that proinflammatory endothelial activation detected by molecular imageing in obese non‐human primates coincides with onset of insulin resistance and progressively increases with duration of altered glucose homeostasis (Chadderdon et al. 2014). The relevance of these findings is supported by the notion that NF‐κB protein is elevated in vascular endothelial cells isolated from obese and aged adults compared with normal‐weight and young controls (Seals et al. 2011). Moreover, age‐dependent NF‐κB activation was associated with systemic inflammation and impaired endothelial dependent dilatation (Tabit et al. 2013). NF‐kB is also a potent mediator of age‐induced myocardial inflammation and fibrosis. Accordingly, NF‐kB suppression, using direct gene delivery of short hairpin p65 RNA, attenuates remodelling and cardiac hypertrophy (Tas et al. 2009). These data validate NF‐kB as a therapeutic target to prevent cardiac disease in the elderly.
Transcription factor JunD
Growing evidence is supporting the notion that the activator protein‐1 (AP‐1) transcription factor JunD is a key molecule implicated in age‐related diseases, mostly by modulating oxidative stress levels. The transcription factor AP‐1 is a heterodimeric complex which is composed of several proteins belonging to the c‐Fos, c‐Jun, ATF and JDP families (Hirai et al. 1989). AP‐1 modulates gene expression in response to a variety of stimuli, including bacterial and viral infections, stress, cytokines and growth factors. Moreover, this molecular complex regulates transcriptional programmes implicated in cellular differentiation, proliferation, and apoptosis (Hirai et al. 1989). Over the last few years, several investigations have unveiled a critical role for the AP‐1 component JunD in cell growth and survival as well as in the regulation of redox signalling by modulating genes involved in antioxidant defence and ROS production (Gerald et al. 2004). In this regard, we have recently reported that JunD is highly relevant for cardiovascular integrity during the life course (Paneni et al. 2013 a). An interesting experimental observation from this study is that vascular JunD expression progressively declines with ageing, thus altering the balance between pro‐oxidant (NADPH oxidase) and antioxidant enzymes (manganese superoxide dismutase (MnSOD) and aldehyde dehydrogenase‐2 (ALDH‐2)), with subsequent accumulation of free radicals. Indeed, genetic deletion of JunD in young mice (6 months old) was associated with premature disturbances of redox signalling, mitochondrial disruption and endothelial dysfunction. Moreover, young JunD−/− mice displayed premature features of vascular senescence which were comparable with those observed in aged animals (22 months old) (Paneni et al. 2013 a). We found that age‐dependent downregulation of JunD was the result of epigenetic changes occurring on its promoter, namely increased CpG methylation (Paneni et al. 2013 a). This latter finding is in line with the notion that postgenomic mechanisms (i.e. DNA methylation, histone modifications and non‐coding RNAs) may significantly alter the expression of genes relevant to senescence, metabolic disorders and cardiovascular damage (Lopez‐Otin et al. 2013). The relevance of JunD in the context of vascular ageing is supported by the observation that in vivo overexpression of the transcription factor was able to rescue endothelial dysfunction in aged mice (Paneni et al. 2013 a). Furthermore, we have translated these findings to human subjects by showing that JunD expression is reduced in peripheral blood monocytes isolated from aged compared to young healthy volunteers. Taken together, the results of our recent work imply that JunD can be regarded as an attractive molecular target to prevent or delay age‐driven cardiovascular diseases. In agreement with our observations, genetic disruption of JunD promotes pressure overload‐induced apoptosis, hypertrophic growth, and angiogenesis in the heart (Ricci et al. 2005). By contrast, adenoviral over‐expression of wild‐type JunD blunted phenylephrine‐mediated cardiomyocyte hypertrophy by negatively regulating AP‐1 transcriptional activity (Hilfiker‐Kleiner et al. 2006). Notably, JunD protein levels are decreased in patients with end‐stage heart failure, suggesting that the transcription factor may protect against age‐related cardiac dysfunction (Hilfiker‐Kleiner et al. 2005). Furthermore, reduced JunD levels may affect longevity by controlling pathways relevant to angiogenesis and insulin signalling. Insulin–IGF‐1 signalling is constitutively stimulated in mice lacking JunD, leading to inactivation of FOXO1, a positive regulator of longevity (Laurent et al. 2008). The importance of insulin homeostasis is outlined by experimental observations in C. elegans showing that suppression of insulin–IGF‐1 signalling increases lifespan. Likewise, substantial increases in lifespan are associated with mutations that reduce insulin–IGF‐1 signalling in the fruit fly Drosophila melanogaster (van Heemst et al. 2010). JunD−/− mice display hyperinsulinaemia, most probably resulting from enhanced pancreatic islet vascularization due to chronic oxidative stress. Indeed, accumulation of free radicals in beta‐cells enhances vascular endothelial growth factor‐A (VEGF‐A) transcription, which in turn increases pancreatic angiogenesis and insulin secretion (Laurent et al. 2008). Interestingly enough, long‐term treatment with an antioxidant rescued the metabolic disturbances observed in JunD−/− mice (Laurent et al. 2008). Indeed, dietary antioxidant supplementation was protective against pancreatic angiogenesis, hyperinsulinaemia, and subsequent activation of insulin signalling cascades in peripheral tissues. Collectively, these data establish a pivotal role for oxidative stress in systemic regulation of insulin and define a key role for the JunD protein in longevity.
Conclusions
In the present review, we have reported novel molecular mechanisms implicated in the interplay between ageing, metabolism and cardiovascular disease. The importance of such emerging signalling pathways is that they may represent common molecular targets to prevent a spectrum of comorbidities driven by senescence, metabolic defects and impaired cardiovascular homeostasis. Premature deregulation of genes involved in oxidative stress, insulin signalling, autophagy and inflammation may anticipate pathological features of the heart and vessels. Of note, most of these pathways are dysregulated in human ageing and a deep understanding of these networks may foster the development of mechanism‐based therapeutic approaches to slow down cardiovascular senescence.
Additional information
Competing interests
None declared.
Funding
Research discussed in this manuscript was supported by the Italian Ministry of Education, University and Research, PRIN 2010–2011 (F.C.); European Foundation for the Study of Diabetes (EFSD) (F.C.); Hjärt‐Lungfonden (Swedish Heart‐Lung Foundation) (F.C.) ; Research Grant 20140360 from Center for Gender Medicine. (S.C.); Kvinnor & Halsas Forskning Stipendier 2015 (F.P.).
Biographies
Sarah Costantino obtained her PhD in Pharmacology and Experimental Medicine at the Second University of Naples, Italy. Then, she worked as postdoctoral research fellow in the Cardiovascular Research team of Professors Cosentino and Luscher at the University of Zurich, Switzerland. Since 2014, she has been a Research Associate in the Molecular Cardiology of Karolinska Institute, Stockholm, under the supervision of Professor Francesco Cosentino. Her main research topics are: chromatin modifications, microRNAs, vascular disease, diabetic cardiomyopathy, redox signalling, diabetes, and obesity.
Francesco Paneni obtained his MD degree in 2006 and then started his clinical training in Cardiology at the University of Rome ‘Sapienza’. In 2014, he obtained a PhD in Experimental Medicine at the University of Rome ‘Sapienza’. From 2011 to 2013 he worked as Research Associate in the Cardiovascular Research team of Professors Cosentino and Luscher at the University of Zurich, Switzerland. He is currently Research Associate at the Unit of Cardiology and Centre for Molecular Medicine, Karolinska Institute, Stockholm. His main research topics are cardiometabolic diseases, endothelial dysfunction, ageing, oxidative stress, epigenetics, inflammation, hypertension, coronary artery disease and heart failure.
Professor Francesco Cosentino obtained his MD degree in 1987 and completed clinical training in Internal Medicine and Cardiovascular Disease at the University of Rome. In 1991 he obtained a PhD in Biomedical sciences – cardiovascular pharmacology at the Mayo Clinic & Foundation. From 1995 to 2014, he held research, teaching and professorial positions at the Cardiovascular Division at the University Hospital of Bern, the Division of Cardiology of Zurich University Hospital and at the University of Rome ‘Sapienza’. Currently, he is professor of Cardiovascular Medicine at the Unit of Cardiology of Karolinska University Hospital, Stockholm, Sweden. Professor Cosentino has investigated the role of endothelium‐derived nitric oxide (NO) as a regulator of vascular homeostasis and signal transduction pathways involved in diabetes‐induced vascular dysfunction and oxidative stress. A novel research line of his group focuses on epigenetic mechanisms underpinning oxidative stress and inflammation in the context of cardiometabolic disease and ageing.

References
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF & Sadoshima J (2007). Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100, 1512–1521. [DOI] [PubMed] [Google Scholar]
- Alers S, Loffler AS, Wesselborg S & Stork B (2012). Role of AMPK‐mTOR‐Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RG, Hayden MS & Ghosh S (2011). NF‐kappaB, inflammation, and metabolic disease. Cell Metab 13, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baris OR, Ederer S, Neuhaus JF, von Kleist‐Retzow JC, Wunderlich CM, Pal M, Wunderlich FT, Peeva V, Zsurka G, Kunz WS et al (2015). Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab 21, 667–677. [DOI] [PubMed] [Google Scholar]
- Beaudeux JL, Nivet‐Antoine V & Giral P (2010). Resveratrol: a relevant pharmacological approach for the treatment of metabolic syndrome? Curr Opin Clin Nutr Metab Care 13, 729–736. [DOI] [PubMed] [Google Scholar]
- Berniakovich I, Trinei M, Stendardo M, Migliaccio E, Minucci S, Bernardi P, Pelicci PG & Giorgio M (2008). p66Shc‐generated oxidative signal promotes fat accumulation. J Biol Chem 283, 34283–34293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A & Frangogiannis NG (2011). Aging and cardiac fibrosis. Aging Dis 2, 158–173. [PMC free article] [PubMed] [Google Scholar]
- Brasier AR (2006). The NF‐kappaB regulatory network. Cardiovasc Toxicol 6, 111–130. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al (2004). Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Camici GG, Cosentino F, Tanner FC & Luscher TF (2008). The role of p66Shc deletion in age‐associated arterial dysfunction and disease states. J Appl Physiol (1985) 105, 1628–1631. [DOI] [PubMed] [Google Scholar]
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin‐Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P et al (2007). Genetic deletion of p66Shc adaptor protein prevents hyperglycemia‐induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci USA 104, 5217–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderdon SM, Belcik JT, Bader L, Kirigiti MA, Peters DM, Kievit P, Grove KL & Lindner JR (2014). Proinflammatory endothelial activation detected by molecular imaging in obese nonhuman primates coincides with onset of insulin resistance and progressively increases with duration of insulin resistance. Circulation 129, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheitlin MD (2003). Cardiovascular physiology – changes with aging. Am J Geriatr Cardiol 12, 9–13. [DOI] [PubMed] [Google Scholar]
- Chiu JJ & Chien S (2011). Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91, 327–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino S, Paneni F & Cosentino F (2015). Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr Pharm Biotechnol 16, 531–543. [DOI] [PubMed] [Google Scholar]
- Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J & Fedorov VV (2015). Fibrosis: a structural modulator of sinoatrial node physiology and dysfunction. Front Physiol 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Chen T, Johnson SC, Szeto H & Rabinovitch PS (2012). Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 16, 1492–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong S, van Veen TA, van Rijen HV & de Bakker JM (2011). Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol 57, 630–638. [DOI] [PubMed] [Google Scholar]
- de Simone G, Verdecchia P, Pede S, Gorini M & Maggioni AP (2002). Prognosis of inappropriate left ventricular mass in hypertension: the MAVI Study. Hypertension 40, 470–476. [DOI] [PubMed] [Google Scholar]
- Eades G, Yao Y, Yang M, Zhang Y, Chumsri S & Zhou Q (2011). miR‐200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)‐like transformation in mammary epithelial cells. J Biol Chem 286, 25992–26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messaoudi S, Rongen GA, de Boer RA & Riksen NP (2011). The cardioprotective effects of metformin. Curr Opin Lipidol 22, 445–453. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Ceolotto G, Pagnin E, de Kreutzenberg S & Avogaro A (2011). At the crossroads of longevity and metabolism: the metabolic syndrome and lifespan determinant pathways. Aging Cell 10, 10–17. [DOI] [PubMed] [Google Scholar]
- Fernandez AF & Fraga MF (2011). The effects of the dietary polyphenol resveratrol on human healthy aging and lifespan. Epigenetics 6, 870–874. [DOI] [PubMed] [Google Scholar]
- Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Luscher TF, Shechter M, Taddei S et al (2012). The assessment of endothelial function: from research into clinical practice. Circulation 126, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia P, delli Gatti C, Bachschmid M, Martin‐Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M & Cosentino F (2004). Deletion of p66shc gene protects against age‐related endothelial dysfunction. Circulation 110, 2889–2895. [DOI] [PubMed] [Google Scholar]
- Franzeck FC, Hof D, Spescha RD, Hasun M, Akhmedov A, Steffel J, Shi Y, Cosentino F, Tanner FC, von Eckardstein A et al (2012). Expression of the aging gene p66Shc is increased in peripheral blood monocytes of patients with acute coronary syndrome but not with stable coronary artery disease. Atherosclerosis 220, 282–286. [DOI] [PubMed] [Google Scholar]
- Freeman RV & Otto CM (2005). Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation 111, 3316–3326. [DOI] [PubMed] [Google Scholar]
- Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B & Goldman S (1998). Large artery remodeling during aging: biaxial passive and active stiffness. Hypertension 32, 437–443. [DOI] [PubMed] [Google Scholar]
- Gan L, Han Y, Bastianetto S, Dumont Y, Unterman TG & Quirion R (2005). FoxO‐dependent and ‐independent mechanisms mediate SirT1 effects on IGFBP‐1 gene expression. Biochem Biophys Res Commun 337, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M & Mechta‐Grigoriou F (2004). JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 118, 781–794. [DOI] [PubMed] [Google Scholar]
- Giannakou ME & Partridge L (2004). The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 14, 408–412. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M et al (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T et al (2012). Blockade of the nuclear factor‐kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125, 1122–1133. [DOI] [PubMed] [Google Scholar]
- He C & Klionsky DJ (2009). Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich PA, Trogdon JG, Khavjou OA et al on behalf of American Heart Association Advocacy Coordinating Committee , Stroke Council , Council on Cardiovascular Radiology and Intervention , Council on Clinical Cardiology , Council on Epidemiology and Prevention , Council on Arteriosclerosis, Thrombosis and Vascular Biology , Council on Cardiopulmonary, Critical Care Perioperative, Resuscitation , Council on Cardiovascular Nursing , Council on the Kidney in Cardiovascular Disease , Council on Cardiovascular Surgery and Anesthesia , and Interdisciplinary Council on Quality of C & Outcomes R (2011). Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 123, 933–944. [DOI] [PubMed] [Google Scholar]
- Hilfiker‐Kleiner D, Hilfiker A, Castellazzi M, Wollert KC, Trautwein C, Schunkert H & Drexler H (2006). JunD attenuates phenylephrine‐mediated cardiomyocyte hypertrophy by negatively regulating AP‐1 transcriptional activity. Cardiovasc Res 71, 108–117. [DOI] [PubMed] [Google Scholar]
- Hilfiker‐Kleiner D, Hilfiker A, Kaminski K, Schaefer A, Park JK, Michel K, Quint A, Yaniv M, Weitzman JB & Drexler H (2005). Lack of JunD promotes pressure overload‐induced apoptosis, hypertrophic growth, and angiogenesis in the heart. Circulation 112, 1470–1477. [DOI] [PubMed] [Google Scholar]
- Hirai SI, Ryseck RP, Mechta F, Bravo R & Yaniv M (1989). Characterization of junD: a new member of the jun proto‐oncogene family. EMBO J 8, 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H & Tindall DJ (2007). Dynamic FoxO transcription factors. J Cell Sci 120, 2479–2487. [DOI] [PubMed] [Google Scholar]
- Isoyama S, Wei JY, Izumo S, Fort P, Schoen FJ & Grossman W (1987). Effect of age on the development of cardiac hypertrophy produced by aortic constriction in the rat. Circ Res 61, 337–345. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X, Zhou Z & Zhang J (2014). Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK‐dependent autophagy. Int J Biol Sci 10, 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM & Kim DH (2010). mTOR regulation of autophagy. FEBS Lett 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, Farb MG, Ngo DT, Myers S, Puri V, Hamburg NM, Carmine B, Hess DT & Gokce N (2015). Forkhead Box O‐1 modulation improves endothelial insulin resistance in human obesity. Arterioscler Thromb Vasc Biol 35, 1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Furukawa‐Hibi Y, Chen C, Horio Y, Isobe K, Ikeda K & Motoyama N (2005). SIRT1 is critical regulator of FOXO‐mediated transcription in response to oxidative stress. Int J Mol Med 16, 237–243. [PubMed] [Google Scholar]
- Kovacic JC, Moreno P, Hachinski V, Nabel EG & Fuster V (2011). Cellular senescence, vascular disease, and aging: part 1 of a 2‐part review. Circulation 123, 1650–1660. [DOI] [PubMed] [Google Scholar]
- Kung CF & Luscher TF (1995). Different mechanisms of endothelial dysfunction with aging and hypertension in rat aorta. Hypertension 25, 194–200. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart‐Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P et al (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC‐1alpha. Cell 127, 1109–1122. [DOI] [PubMed] [Google Scholar]
- Lapierre LR, Kumsta C, Sandri M, Ballabio A & Hansen M (2015). Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11, 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, Solari F, Mateescu B, Karaca M, Castel J, Bourachot B, Magnan C, Billaud M & Mechta‐Grigoriou F (2008). Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab 7, 113–124. [DOI] [PubMed] [Google Scholar]
- Lee HY & Oh BH (2010). Aging and arterial stiffness. Circ J 74, 2257–2262. [DOI] [PubMed] [Google Scholar]
- Lesniewski LA, Zigler MC, Durrant JR, Donato AJ & Seals DR (2012). Sustained activation of AMPK ameliorates age‐associated vascular endothelial dysfunction via a nitric oxide‐independent mechanism. Mech Ageing Dev 133, 368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magyar K, Halmosi R, Palfi A, Feher G, Czopf L, Fulop A, Battyany I, Sumegi B, Toth K & Szabados E (2012). Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin Hemorheol Microcirc 50, 179–187. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K & Irani K (2007). SIRT1 promotes endothelium‐dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA 104, 14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa‐Nicotera M, Ippoliti A, Novelli G, Melino G et al (2009). MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 120, 1524–1532. [DOI] [PubMed] [Google Scholar]
- Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, Cheng J, Lu G, Morris DJ, Castrillon DH et al (2006). Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols M, Townsend N, Scarborough P & Rayner M (2014). Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J 35, 2929. [DOI] [PubMed] [Google Scholar]
- O'Rourke MF & Hashimoto J (2007). Mechanical factors in arterial aging: a clinical perspective. J Am Coll Cardiol 50, 1–13. [DOI] [PubMed] [Google Scholar]
- Ota H, Akishita M, Eto M, Iijima K, Kaneki M & Ouchi Y (2007). Sirt1 modulates premature senescence‐like phenotype in human endothelial cells. J Mol Cell Cardiol 43, 571–579. [DOI] [PubMed] [Google Scholar]
- Pagnin E, Fadini G, de Toni R, Tiengo A, Calo L & Avogaro A (2005). Diabetes induces p66shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab 90, 1130–1136. [DOI] [PubMed] [Google Scholar]
- Pandolfi S, Bonafe M, Di Tella L, Tiberi L, Salvioli S, Monti D, Sorbi S & Franceschi C (2005). p66shc is highly expressed in fibroblasts from centenarians. Mech Ageing Dev 126, 839–844. [DOI] [PubMed] [Google Scholar]
- Paneni F & Cosentino F (2012). p66Shc as the engine of vascular aging. Curr Vasc Pharmacol 10, 697–699. [DOI] [PubMed] [Google Scholar]
- Paneni F, Costantino S & Cosentino F (2014). p66Shc‐induced redox changes drive endothelial insulin resistance. Atherosclerosis 236, 426–429. [DOI] [PubMed] [Google Scholar]
- Paneni F, Costantino S & Cosentino F (2015). Role of oxidative stress in endothelial insulin resistance. World J Diabetes 6, 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Osto E, Costantino S, Mateescu B, Briand S, Coppolino G, Perna E, Mocharla P, Akhmedov A, Kubant R et al (2013. a). Deletion of the activated protein‐1 transcription factor JunD induces oxidative stress and accelerates age‐related endothelial dysfunction. Circulation 127, 1229–1240. [DOI] [PubMed] [Google Scholar]
- Paneni F, Volpe M, Luscher TF & Cosentino F (2013. b). SIRT1, p66Shc, and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes 62, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillarisetti S (2008). A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat Cardiovasc Drug Discov 3, 156–164. [DOI] [PubMed] [Google Scholar]
- Pu Y, Zhang H, Wang P, Zhao Y, Li Q, Wei X, Cui Y, Sun J, Shang Q, Liu D & Zhu Z (2013). Dietary curcumin ameliorates aging‐related cerebrovascular dysfunction through the AMPK/uncoupling protein 2 pathway. Cell Physiol Biochem 32, 1167–1177. [DOI] [PubMed] [Google Scholar]
- Rask‐Madsen C & King GL (2011). Endothelium‐dependent delivery of insulin to muscle interstitium. Cell Metab 13, 236–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci R, Eriksson U, Oudit GY, Eferl R, Akhmedov A, Sumara I, Sumara G, Kassiri Z, David JP, Bakiri L et al (2005). Distinct functions of junD in cardiac hypertrophy and heart failure. Genes Dev 19, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnebaum SM & Patterson C (2010). The FoxO family in cardiac function and dysfunction. Annu Rev Physiol 72, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota M, LeCapitaine N, Hosoda T, Boni A, De Angelis A, Padin‐Iruegas ME, Esposito G, Vitale S, Urbanek K, Casarsa C et al (2006). Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res 99, 42–52. [DOI] [PubMed] [Google Scholar]
- Safar ME (2010). Arterial aging – hemodynamic changes and therapeutic options. Nat Rev Cardiol 7, 442–449. [DOI] [PubMed] [Google Scholar]
- Sahin K, Yilmaz S & Gozukirmizi N (2014). Changes in human sirtuin 6 gene promoter methylation during aging. Biomed Rep 2, 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA & Brunet A (2008). FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 20, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A & Kaarniranta K (2012). AMP‐activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11, 230–241. [DOI] [PubMed] [Google Scholar]
- Sanchez AM, Candau RB & Bernardi H (2014). FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci 71, 1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawabe M (2010). Vascular aging: from molecular mechanism to clinical significance. Geriatr Gerontol Int 10 (Suppl. 1), S213–S220. [DOI] [PubMed] [Google Scholar]
- Scioli MG, Bielli A, Arcuri G, Ferlosio A & Orlandi A (2014). Ageing and microvasculature. Vasc Cell 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL & Donato AJ (2011). Aging and vascular endothelial function in humans. Clin Sci (Lond) 120, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa I, Komatsu T, Hayashi N, Kim SE, Kawata T, Park S, Hayashi H, Yamaza H, Chiba T & Mori R (2015). The life‐extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 14, 707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Rudd JA & Siu PM (2014). Modulating effect of SIRT1 activation induced by resveratrol on Foxo1‐associated apoptotic signalling in senescent heart. J Physiol 592, 2535–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soerensen M, Nygaard M, Dato S, Stevnsner T, Bohr VA, Christensen K & Christiansen L (2015). Association study of FOXO3A SNPs and aging phenotypes in Danish oldest‐old individuals. Aging Cell 14, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S & Matter CM (2011). Protective roles of SIRT1 in atherosclerosis. Cell Cycle 10, 640–647. [DOI] [PubMed] [Google Scholar]
- Stern S, Behar S & Gottlieb S (2003). Cardiology patient pages. Aging and diseases of the heart. Circulation 108, e99–e101. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Pillai VB & Gupta MP (2011). Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J Mol Cell Cardiol 51, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JY, Woo CH, Kang YJ, Lee KY & Choi HC (2011). AMPK induces vascular smooth muscle cell senescence via LKB1 dependent pathway. Biochem Biophys Res Commun 413, 143–148. [DOI] [PubMed] [Google Scholar]
- Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N et al (2013). Protein kinase C‐beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 127, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarride JE, Lim M, DesMeules M, Luo W, Burke N, O'Reilly D, Bowen J & Goeree R (2009). A review of the cost of cardiovascular disease. Can J Cardiol 25, e195–e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas SW, Vervoordeldonk MJ & Tak PP (2009). Gene therapy targeting nuclear factor‐kappaB: towards clinical application in inflammatory diseases and cancer. Curr Gene Ther 9, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AM, Martin KA & Rzucidlo EM (2014). Resveratrol induces vascular smooth muscle cell differentiation through stimulation of SirT1 and AMPK. PLoS One 9, e85495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler MC & Hardie DG (2007). AMP‐activated protein kinase in metabolic control and insulin signaling. Circ Res 100, 328–341. [DOI] [PubMed] [Google Scholar]
- Ulgherait M, Rana A, Rera M, Graniel J & Walker DW (2014). AMPK modulates tissue and organismal aging in a non‐cell‐autonomous manner. Cell Rep 8, 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst A & Burgering BM (2007). Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 8, 440–450. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries‐Smits LM, Frye RA, Medema RH & Burgering BM (2004). FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1 . J Biol Chem 279, 28873–28879. [DOI] [PubMed] [Google Scholar]
- van Heemst D (2010). Insulin, IGF‐1 and longevity. Aging Dis 1, 147–157. [PMC free article] [PubMed] [Google Scholar]
- Vigili de Kreutzenberg S, Ceolotto G, Cattelan A, Pagnin E, Mazzucato M, Garagnani P, Borelli V, Bacalini MG, Franceschi C, Fadini GP & Avogaro A (2015). Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr Metab Cardiovasc Dis 25, 686–693. [DOI] [PubMed] [Google Scholar]
- Webb AE & Brunet A (2014). FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 39, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnik S, Auwerx J, Sinclair DA & Matter CM (2015). Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 36, 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA & Mayo MW (2004). Modulation of NF‐kappaB‐dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23, 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zhang L, Wen G, Zhao H, Luong LA, Chen Q, Huang Y, Zhu J, Ye S, Xu Q, Wang W & Xiao Q (2015). Upregulated sirtuin 1 by miRNA‐34a is required for smooth muscle cell differentiation from pluripotent stem cells. Cell Death Differ 22, 1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH & Goldberg AL (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6, 472–483. [DOI] [PubMed] [Google Scholar]
- Zieman SJ, Melenovsky V & Kass DA (2005). Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25, 932–943. [DOI] [PubMed] [Google Scholar]