

Abstract
Changes in the neuromuscular system affecting the ageing motor unit manifest structurally as a reduction in motor unit number secondary to motor neuron loss; fibre type grouping due to repeating cycles of denervation‐reinnervation; and instability of the neuromuscular junction that may be due to either or both of a gradual perturbation in postsynaptic signalling mechanisms necessary for maintenance of the endplate acetylcholine receptor clusters or a sudden process involving motor neuron death or traumatic injury to the muscle fibre. Functionally, these changes manifest as a reduction in strength and coordination that precedes a loss in muscle mass and contributes to impairments in fatigue. Regular muscle activation in postural muscles or through habitual physical activity can attenuate some of these structural and functional changes up to a point along the ageing continuum. On the other hand, regular muscle activation in advanced age (>75 years) loses its efficacy, and at least in rodents may exacerbate age‐related motor neuron death. Transgenic mouse studies aimed at identifying potential mechanisms of motor unit disruptions in ageing muscle are not conclusive due to many different mechanisms converging on similar motor unit alterations, many of which phenocopy ageing muscle. Longitudinal studies of ageing models and humans will help clarify the cause and effect relationships and thus, identify relevant therapeutic targets to better preserve muscle function across the lifespan.
Abbreviations
- AChR
acetylcholine receptor
- MHC
myosin heavy chain
- MU
motor unit
Introduction
The motor unit, consisting of a motor neuron and the myofibres it innervates, undergoes profound changes with ageing. Indeed, deterioration of neuromuscular junction morphology was reported in aged rodents at least as far back as 1966 (Gutmann & Hanzlikova, 1966) and this was confirmed in elderly humans nearly 20 years later (Oda, 1984). There is also strong support for repeating cycles of denervation–reinnervation causing remodelling of the motor unit and fibre type grouping (Kanda & Hashizume, 1989; Lexell & Downham, 1991), motor neuron death causing motor unit loss (Tomlinson & Irving, 1977; McNeil et al. 2005), sporadic myofibre atrophy and angular fibre shape (Lexell & Taylor, 1991; Rowan et al. 2011), and expression of multiple myosin heavy chains (MHCs) within a given myofibre (MHC co‐expression) (Andersen et al. 1999) that has been linked to denervation in advanced age (Rowan et al. 2012). Since these motor unit changes contribute to many of the most functionally relevant changes in ageing muscle, including muscle atrophy and increased susceptibility to falls, in recent years there has been a significant push to understand the mechanisms causing neuromuscular junction instability and the persistence of denervated myofibres in ageing muscle (Rudolf et al. 2014). On this basis, this review will focus upon the structural and functional manifestations of neuromuscular alterations in ageing muscle and current thinking about their mechanistic basis.
Neurophysiological manifestations in ageing muscle morphology
Ageing causes progressive skeletal muscle atrophy and weakness, and other functional impairments, where neurophysiological alterations play a key role in driving these changes. The evolving nature of alterations in neurophysiological processes affecting the motor unit with advancing age and how these alterations manifest at the muscle morphological level are depicted in Fig. 1 and summarized below.
Figure 1. Morphological impact of motor unit alterations in aging muscle .
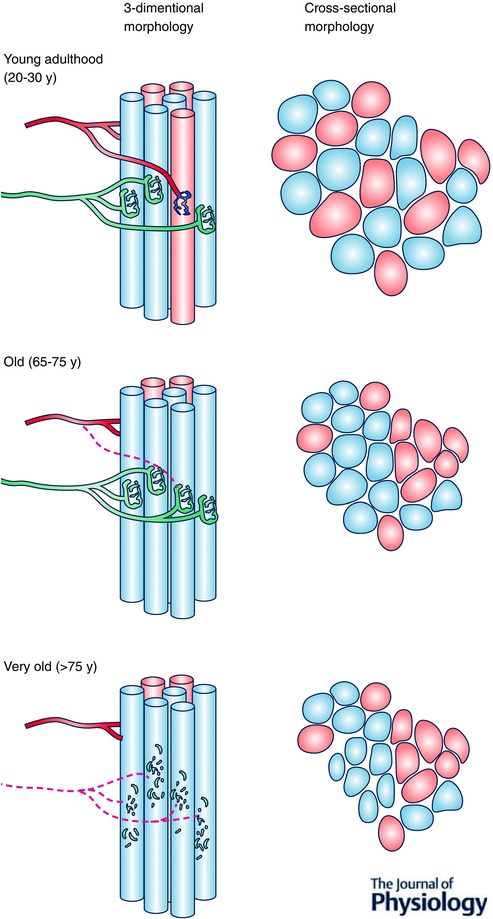
Young adulthood is characterized by an intermingling of fibres belonging to different motor units. This yields a mosaic distribution of fibre types when muscle fibres are viewed in cross‐section. Adulthood to old age is characterized by repeating cycles of denervation–‐reinnervation that result in fibres of the same type being beside one another (fibre type grouping) when viewed in cross‐section. Very old age is characterized by increasing frequency of axonal degeneration and/or motor neuron death leading to grouped fibre atrophy when viewed in cross‐section.
For much of adult life muscle undergoes repeating cycles of denervation and reinnervation. These cycles of denervation–reinnervation involve a transient disconnect of an individual muscle fibre from its motor neuron, followed by reinnervation by the original motor axon (if still intact) or through collateral sprouting of an adjacent motor neuron axon. This process of denervation–reinnervation in ageing skeletal muscle presents at the single myocyte level as significant disruption of the neuromuscular junction components, including decreased overlap of the pre‐ and postsynaptic structures; narrowing of the terminal axons; altered distribution of laminin, acetylcholine receptors and other postsynaptic membrane proteins; and extensive sprouting of terminal axons (Balice‐Gordon, 1997; Jang & Van Remmen, 2010; Chai et al. 2011; Samuel et al. 2012). Importantly, these changes in neuromuscular junction structure have been shown in rat to occur at an earlier age than myofibre atrophy (Deschenes et al. 2010).
Current hypotheses proposed to account for the transient denervation events in ageing muscle include (i) a gradual process in which the signalling network necessary for maintaining the neuromuscular junction becomes perturbed (Balice‐Gordon, 1997; Samuel et al. 2012; Personius & Parker, 2013), and (ii) a sudden change related to motor neuron death and/or axon degeneration (Faulkner et al. 2007), or traumatic injury causing myofibre necrosis and subsequent deterioration of endplate morphology (Li et al. 2011). Interestingly, one recent study showed in ageing female mice that marked disruption of the endplate morphology occurred in the absence of a loss of spinal cord motor neurons. This suggests that at least in female mice, denervation events in ageing must primarily occur as a function of changes within the muscle (e.g. impaired neuromuscular junction signalling, axon degeneration, and/or traumatic myofibre injury). Whether this also applies to male rodents has not been determined and there are no parallel studies in humans. What has been established from human cadaveric samples is that there is a progressive loss of spinal cord motor neuron cell bodies that becomes significant in the seventh decade of life and exhibits considerable individual‐to‐individual variation (Tomlinson & Irving, 1977), suggesting some individuals may be more vulnerable to motor neuron loss with ageing. Regardless of this latter possibility, current data indicate that a loss of motor neurons contributes significantly to denervation events in human muscle, especially from the seventh decade onwards.
The result of denervation–reinnervation cycles in muscle is a progressive collapse of the motor unit spatial domain, causing clustering of fibres within a given motor unit (Larsson, 1995) and the characteristic fibre type grouping seen in ageing muscle (Kanda & Hashizume, 1989; Lexell & Downham, 1991). Because the phenomenon of fibre type grouping also occurs in models in which only the stability of the neuromuscular junction is perturbed (axon and motor neuron remain intact; see below, Mechanisms of neuromuscular junction instability and link to ageing muscle atrophy) (Butikofer et al. 2011; Kulakowski et al. 2011), further studies are required to determine the relative contributions of traumatic injury, motor neuron death, axon degeneration and neuromuscular junction instability to denervation–reinnervation and resulting fibre type grouping with ageing, and whether the relative contributions change along the continuum of the ageing process. Of interest too is whether habitual activity patterns (exercise) can modify this essentially degenerative process.
It is important to note that a fraction of denervated fibres are not successfully reinnervated (for reasons that are unclear) and such fibres will progressively atrophy, becoming more angular in the process (Baloh et al. 2007). Most of these long‐term denervated fibres eventually will be lost entirely. Notably, a loss of muscle fibres is also seen in some models of neuromuscular junction instability (Butikofer et al. 2011), suggesting that motor neuron loss is not essential for fibre loss to occur. Although the relative contribution of neuromuscular junction instability to fibre loss with ageing remains unclear, the marked accumulation of muscle fibres with small size and angular shape in advanced age in both human muscle (Scelsi et al. 1980; Lexell & Taylor, 1991) and rodent muscle (Rowan et al. 2011; Purves‐Smith et al. 2012; Rowan et al. 2012) is strongly suggestive of failed reinnervation at these more advanced ages.
Another striking feature of ageing muscle is grouped fibre atrophy, particularly in advanced age (Lindboe & Torvik, 1982) when muscle atrophy accelerates (Rowan et al. 2011) and becomes more clinically relevant in terms of causing mobility impairment, falls and physical frailty. Grouped fibre atrophy is common in neuromuscular diseases involving axonal degeneration (e.g. spinal muscular atrophy diseases; Ruidnik‐Schoneborn et al. 2004) and occurs, firstly, because repeating cycles of denervation and reinnervation have caused fibres belonging to the same motor unit to cluster together, and secondly, either because an axon plugging into multiple muscle fibres within the motor unit degenerates or because an entire motor neuron dies, causing atrophy of all of the fibres previously linked to this axon or motor neuron. Thus, when whole muscle atrophy accelerates in very advanced age this is characterized morphologically by significant grouped fibre atrophy and accelerated muscle fibre loss (Lexell et al. 1988) and is coincident with a significant loss of motor neuron cell bodies in the spinal cord (Tomlinson & Irving, 1977; Faulkner et al. 2007; Rowan et al. 2012). On this basis, although early changes in the neuromuscular junction that cause fibre type grouping can occur prior to significant loss of motor neurons (Chai et al. 2011), it seems likely that motor neuron death becomes a much more significant driver of muscle atrophy at advanced age. Furthermore, the grouped atrophy may contribute to failure of reinnervation of denervated fibres that are surrounded by other denervated fibres by increasing the distance to the nearest intact axon available for collateral sprouting.
Changes in motor unit numbers and properties in ageing human muscle
There is some limited and indirect support for declining neurological function and aspects of the denervation–reinnervation process in human cross‐sectional studies. The majority of those studies related to the neuromuscular system have compared younger adults (∼20–35 years) with usually one group of older adults, whose ages range from 60–65 years to over 90 years depending on the study. When compared collectively, the gradual decline in function (strength) that begins around 30 years of age is accelerated after ∼60 years of age (Rice & Cunningham, 2001), consistent with the model depicted in Fig. 1 and described above. It is also interesting that some reports find the reduction in strength with ageing occurs prior to significant muscle loss, and even after muscle loss occurs the reduction in strength is greater than can be accounted for by muscle atrophy alone (e.g. Goodpaster et al. (2006). In addition, declining motor nerve function in humans is associated with strength declines with ageing, and importantly, those who have greater declines in motor nerve function also have greater declines in strength (Ward et al. 2015). Collectively, therefore, it appears that neurological impairments contribute to strength declines with ageing prior to changes in muscle mass.
Estimates of motor unit (MU) numbers using electrophysiological techniques from various upper and lower limb muscles reveal that, generally, MUs are lost with age and the loss may be as much as 70% when comparing those in their third decade with those in their ninth decade (Campbell et al. 1973). In one study, McNeil et al. (2005) compared numbers of MUs in the tibilais anterior among three groups: young adults (∼25 years), older adults (∼65 years) and very old adults (∼80 years). Estimated MU numbers were ∼40% lower in the older group compared with the young who differed in age by about 40 years, but the very old group representing only a further 15 years age difference had ∼33% fewer MUs compared with the older group. Thus, MU estimates in ageing humans support the notion that there is a more precipitous decline in muscle mass and function in advanced age (beyond the seventh decade) likely to be secondary to motor neuron loss.
Notwithstanding the morphological evidence of endplate disruption in aged human cadaver muscles (Oda, 1984), evidence of altered neuromuscular transmission function that may involve the neuromuscular junction in humans is limited and is necessarily indirect. Electrophysiology measures obtained from quantitative needle electomyographic techniques that reflect properties of the MU potentials have provided indices of transmission or junctional stability (‘jiggle’, where less stability is reflected in more jiggle) and collateral re‐innervation (‘jitter’, where higher fibre number per MU produces less jitter) (Stalberg & Sonoo, 1994). These techniques have been applied in various neuromuscular diseases and have indicated less junctional stability, for example in patients with amyotrophic lateral sclerosis (Stalberg & Sonoo, 1994; Campos et al. 2000) and diabetic neuropathy (Allen et al. 2015). In relation to ageing, one recent study reported that jiggle values were significantly greater in older adults when compared with younger adults in two lower limb muscles (tibialis anterior and vastus medialis), an observation suggesting neuromuscular junction instability in ageing human muscle (Hourigan et al. 2015). Collectively, therefore, these human studies support the idea that MU loss occurs with ageing, that it accelerates beyond the seventh decade of life, and that these changes are accompanied by neuromuscular junction instability. As noted above, however, these studies are cross‐sectional in design and thus additional studies involving repeated measures on the same subjects across a relevant period of the lifespan (e.g. between the ages of 65 and 75 years and between 75 and 85 years), perhaps combined with biopsy measures of muscle morphology, would provide important insights into the individual evolution of neurological deficits and their impact on ageing human muscle.
Functional consequences of age‐related motor unit remodelling
The MU is the elemental unit of contractile function and through the processes of recruitment and derecruitment of MUs, and modulation of their firing rate patterns, muscle force is controlled and modified for appropriate movements. Through age‐related remodelling (described above) the neuromuscular system of older adults performs with fewer, but larger, motor units and the high‐end range of average MU firing rates recorded during isometric voluntary contractions is diminished to variable relative degrees compared with younger adults, effects that vary depending on the muscle (Connelly et al. 1999; Dalton et al. 2008; Power et al. 2013). In addition to loss of muscle mass, force per unit of muscle is diminished and, generally speaking, velocity of contractions is reduced. Thus, absolute force is lower, and contractile speeds and rates of force development and relaxation are slowed in aged compared with young adults (Power et al. 2013). It is likely that these impairments in contractile response with ageing are due to intrinsic changes in the myocyte contractile machinery (e.g. oxidative damage to proteins involved in cross‐bridge cycling; Li et al. 2015) and a shift to greater relative numbers of slow‐type MUs (Lexell & Downham, 1991), and to extrinsic factors namely blunted or altered activation of the fibres by the motor neuron and/or impaired junctional transmission.
It has been speculated that due to the strong matching of motor neuron properties and constitutive muscle fibre properties (Gardiner & Kernell, 1990), MU firing rates in aged humans are lower to maintain the speed‐match with the slower contractile properties (Connelly et al. 1999). This speculation may be overly simplistic because subsequently it has been shown that although firing rates from a variety of limb muscles are generally lower with adult ageing, the degree is variable among muscles and does not always match contractile slowing, or there is little contractile slowing in muscles that have lower firing rates (Connelly et al. 1999; Roos et al. 1999; Dalton et al. 2009; Dalton et al. 2010 b). Thus, interpretations about the functional significance from these relatively few cross‐sectional studies in humans, in which other factors such as life‐long environmental or physical activity patterns (see below) are influential, are currently limited.
These findings noted in the preceding paragraph underscore the challenge in the field of how to determine whether age‐related changes in contractile quality and motor neuron output are coincidental, in that intrinsic changes in the muscle and motor neuron may occur independently, or that the processes related to MU remodelling may have dictated compensatory alterations in contractile function. Although recruitment order does not seem altered, recruitment thresholds may be slightly lower in older compared with younger adults, reflecting a greater percentage of slower MU types (Erim et al. 1999). There is some indirect evidence of small changes in aspects of intrinsic motor neuron (Pascoe et al. 2011) and axonal excitability properties (Jankelowitz et al. 2007), which may affect thresholds and integration of synaptic drive and indeed may contribute to the well‐established reduction in average firing rates reaching the muscle for the generation of high force (see above). The activation pattern of often recorded doublets (i.e. brief pairs or triads of action potentials at the start of a train of potentials) to enhance rates of force development when a unit is recruited also may be blunted in an aged system (Christie & Kamen, 2006). Despite lower maximal firing rates of MUs with ageing, the exact mechanism(s) contributing to this feature are not known, but it does not seem to be due to impaired voluntary activation from spinal (Jakobi & Rice, 2002) or supraspinal levels (Molenaar et al. 2013).
It is important to appreciate that intramuscular electomyographic signals from which MU firing rates have been recorded in studies cited above do not necessarily directly reflect axonal efferent traffic. Thus, microneurographic recordings of motor axon rates of action potentials during voluntary contractions (or stimulation of MUs at varying rates), in conjunction with intramuscular electomyographic recordings and force recordings, would be useful to assess the capacity and fidelity of the neuromuscular junction in ageing humans. Regardless of this current gap in knowledge, on the basis of available evidence it seems likely that there is progressive disruption at multiple levels within the ageing neuromuscular system and that this results in a mismatch at multiple levels with advancing age.
The functional consequences of an altered system of MUs with ageing have been most frequently considered in the context of causing reduced force output (strength), and slower contractile capacity. From the perspective of functional relevance, particularly to activities of daily living, at least in the early stages of ageing the loss of absolute strength per se is not as detrimental as loss of muscle control and contractile quality. Importantly, when a loss of strength and contractile speed are combined, work and power capacity become critically affected in the aged adult (Power et al. 2013). These features are manifest from minimally complicated tasks such as force steadiness, to more complex integrative whole‐body tasks such as balance and gait, which when affected contribute to falls and loss of autonomy.
When a person is asked to target a force level or to follow a simple muscle activation pattern, it has been proposed that an increased force variability (loss of steadiness) is related to MU remodelling in that smooth graded control is affected negatively by having fewer larger units, a greater concentration of one type of unit, and increased MU train variability in firing rates (Christou, 2011). These same features may then translate into problems with balance, consistent with evidence that postural sway is greater in older adults and that their recovery from postural perturbations is blunted (Enoka et al. 2003; Dalton et al. 2014; Kanekar & Aruin, 2014). An impaired ability to rapidly initiate and generate muscle force, whether isometrically or dynamically, will result in a neuromuscular response that is insufficient to withstand a perturbation or to adequately compensate before task failure. A common example in aged adults is attempting, but failing, to recover from a trip that then leads to a fall (Takacs et al. 2013). Thus, as the neuromuscular system is affected by ageing, including marked changes in the MU noted above, critical functional thresholds are reached whereby insufficient power is available to recover from an accidental trip (and prevent falls) or to undertake common daily activities such as rising from a chair, walking or stair‐climbing (Rice & Cunningham, 2001; Kato et al. 2015).
When muscles are repeatedly activated, parts of the neuromuscular system will eventually be compromised or fail (neuromuscular fatigue), leading to performance cessation. Because of substantial structural and functional alterations to the system with ageing, neuromuscular fatigue may under some circumstances be exacerbated with ageing. For isometric tasks on a relative basis, healthy older adults may be more resistant to fatigue or similar to younger adults (Kent‐Braun, 2009; Christie et al. 2011), although very old adults (> 75 years) are more fatigable than younger adults (Justice et al. 2014). The greater failure of the system at advanced ages compared with younger adults may indeed relate to the increased cumulative effects of MU remodelling. In contrast, dynamic contractions more consistently reveal greater fatigability in older compared with younger adults (Petrella et al. 2005; McNeil & Rice, 2007; Dalton et al. 2010 c, 2015), due to the combined impact of a reduction in velocity and contractile strength. Whereas exacerbated neuromuscular fatigue with ageing is often suggested to be secondary to down‐regulation of muscle energetics (see Christie et al. 2014; Layec et al. 2014), the role for the motor output side of the system is not clear, partly because studies are few in number at the MU level on this topic. For example, although it is generally well‐accepted that in younger individuals MU firing rates decline transiently after moderate to high intensity fatiguing tasks (Bigland‐Ritchie et al. 1995), in older subjects it seems that rates are also depressed (Rubinstein & Kamen, 2005; Dalton et al. 2010 a); however, it is unclear whether the degree is similar for old and young, or under what tasks. On the other hand, in only one study available, recovery of firing rates was delayed in the older group after fatigue compared with the young adults (Dalton et al. 2010 a). Notwithstanding the limited data available, in view of the features of age‐related remodelling described above, it seems likely that fatigue‐related feedback for control of motor neuron output, coupled with reduced reserve for action potential transmission along the axon and across the neuromuscular junction, would contribute to exacerbate neuromuscular fatigue with ageing. Cleary, further exploration is required on these aspects under different paradigms to stress various sites in the system.
Mechanisms of neuromuscular junction instability and link to ageing muscle atrophy
The elemental components of the neuromuscular junction comprise the presynaptic terminal axons of the motor neuron, the perisynaptic Schwann cell which envelopes the axon terminals at the endplate, and the acetylcholine receptor cluster on the postsynaptic membrane of the muscle fibre. The stability of the adult neuromuscular junction is maintained in part by an elegant signalling cascade involving communication from the terminal motor neuron to the myofibre to maintain the accurate targeting of the motor neuron to the endplate. Amongst these signalling molecules is neural agrin (Connor & Smith, 1994; Bowen et al. 1998; Mars et al. 2003), a protein released from the terminal axon, which in turn activates a muscle‐specific tyrosine kinase in the postsynaptic membrane called MuSK, via its interactions with low‐density lipoprotein receptor‐related protein 4 (LRP4; acting as a co‐receptor for neural agrin) (Wu et al. 2010). In the absence of neural agrin (e.g. using antibodies against agrin or over‐expressing its endogenous protease, neurotrypsin) neuromuscular junction disassembly occurs within days (Mars et al. 2003; Bolliger et al. 2010). The basis for this is that agrin‐activated (i.e. phosphorylated) MuSK signals the acetylcholine receptors (AChRs) to cluster in a configuration often likened to that of a pretzel, a process that is also dependent upon the docking protein downstream‐of‐tyrosine‐kinase 7 (Dok7), the membrane‐associated rapsyn protein (Wu et al. 2010), protein kinase A and myosin Va (Roder et al. 2010) (Fig. 2). This clustered formation of the AChRs permits the terminal axon to correctly target the postsynaptic endplate to maintain a functional neuromuscular junction. Additional evidence of the importance of MuSK signalling to neuromuscular junction maintenance is shown by the form of congenital myasthenia gravis in which auto‐antibodies are produced against MuSK (Selcen et al. 2004; Punga et al. 2011). Interestingly, this MuSK‐dependent form of myasthenia gravis has been likened to premature ageing in reference to its impact on the neuromuscular junction (Shigemoto et al. 2010), but experimental evidence examining the agrin–MuSK pathway in ageing muscle is presently sparse. Furthermore, although the perisynaptic Schwann cells are also essential to the integrity of the neuromuscular junction, and are particularly important in coordinating axonal sprouting during reinnervation (Auld & Robitaille, 2003; Sugiura & Lin, 2011), how their function may become perturbed with ageing is unknown.
Figure 2. The maintenance of the adult neuromuscular junction depends upon several proteins .
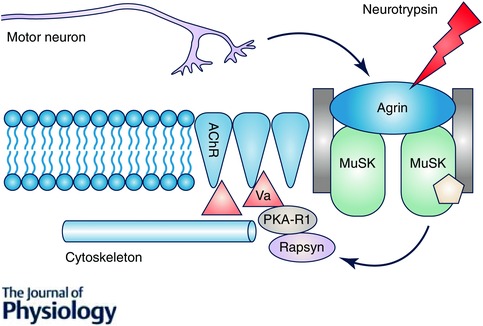
Neural agrin released from the terminal motor neurons activates muscle‐specific kinase (MuSK) which in turn recruits rapsyn to the subsynaptic domain where it acts in conjunction with protein kinase A R1 (PKAR1) and myosin Va (Va) to anchor the acetylcholine receptors (AChR) to the cytoskeleton. This signalling network thus ensures that the terminal axons of the motor neuron directly oppose the postsynaptic membrane acetylcholine receptors to maintain a functional synapse. Note that at the neuromuscular junction agrin is inactivated by the endogenous protease, neurotryspin.
In regard to the impact of ageing on the agrin–MuSK signalling axis, Hettwer et al. (2012) have previously reported that a subclass of elderly with severe muscle atrophy exhibit high circulating levels of cleaved agrin fragment, which they interpreted as evidence for hyperactivity of neurotrypsin that would in turn reduce MuSK activation with ageing. In addition, we have previously reported a marked reduction in MuSK protein at the neuromuscular junction in very old rat muscle (Gouspillou et al. 2013). Interestingly, a mouse engineered with a defect in autophagy, the ATG7−/− mouse (described in the next section) exhibits perturbed endplate morphology that is also associated with a reduction in MuSK. However, an important caveat is that it is unclear whether reduced MuSK causes (precedes) endplate disruption or whether it occurs as a secondary consequence of denervation due to some other cause in ageing muscle (e.g. traumatic myofibre damage leading to denervation; Li et al. 2011). Underscoring the challenges currently facing the field in trying to understand the mechanisms causing neuromuscular junction instability in ageing muscle, generally speaking many mouse models that have been developed to test the involvement of specific mechanisms in deterioration of the motor unit with ageing show a frustrating similarity in muscle phenotypes, as will be discussed in the next section (see below, Murine models recapitulating neurological features of ageing muscle).
Regardless of the lack of clarity about the causes of neuromuscular junction instability with ageing, when this instability yields a denervation event it can be an important cause of muscle atrophy with ageing. Specifically, experimental studies indicate that surgical denervation causes a marked upregulation of several so‐called atrogenes, including atrogin‐1/muscle atrophy F‐box (MAFbx), muscle ring finger protein 1 (MuRF1) (Bodine et al. 2001), and the recently discovered muscle ubiquitin ligase of the SCF complex in atrophy‐1 (MuSA1) (Sartori et al. 2013) and specific of muscle atrophy and regulated by transcription (SMART) (Milan et al. 2015). In all of these cases, the atrogenes target specific proteins and function as ubiquitin ligases to identify the proteins for degradation by the proteasome. It is also important to note that there are likely to be many other ubiquitin ligases involved in denervation that have yet to be identified since each ubiquitin ligase is believed to have a limited and very specific number of protein targets. In the context of ageing, we have previously shown that muscle fibres positive for a marker of denervation, the sodium channel isoform Nav1.5, exhibit an up‐regulation of MAFbx and MuRF1 immunostaining and are markedly smaller than Nav1.5‐negative (innervated) fibres (Rowan et al. 2012), providing strong evidence of the importance of this pathway in atrophy of denervated muscle fibres with ageing. In addition to the proteasomal degradation pathway, denervation also stimulates the autophagy‐lysosome pathway, to collectively provide a means of degrading proteins, organelles and cytoplasm in response to denervation (Schiaffino et al. 2013). Although many details remain to be understood, denervation causes mitochondrial fission and an increase in mitochondrial reactive oxygen species production (Muller et al. 2007; Romanello et al. 2010), which then activates both the proteosomal and autophagy‐lysosome pathways through nuclear translocation of forkhead box (Fox) O transcription factors (Bhattacharya et al. 2009; Romanello et al. 2010; Milan et al. 2015).
Murine models recapitulating neurological features of ageing muscle
Recent years have seen a marked increase in the number of mouse models that reproduce key features of neurological impairment in ageing muscle, including gross motor changes causing weakness and gait impairment, and cellular changes such as neuromuscular junction instability, fibre type grouping, fibre loss, MHC co‐expression and sporadic fibre atrophy (Table 1). However, despite producing strikingly similar outcomes for muscle, these models produce these changes by very different mechanisms. Specifically, the SOD1−/− mouse exhibits marked oxidative stress and mitochondrial impairment that causes neuromuscular junction instability and motor neuron dysfunction (Jang et al. 2010; Sakellariou et al. 2014; Shi et al. 2014); the ATG7−/− mouse has a defect in autophagy that results in impaired AChR recycling (Carnio et al. 2014); the TrkB+/− mouse has impaired neurotrophin signalling due to reduced muscle receptor density for brain‐derived neurotrophic factor and neurotrophin 4/5 (Kulakowski et al. 2011); the neurotrypsin over‐expressing mouse has impaired agrin–MuSK signalling resulting in unstable neuromuscular junctions (Butikofer et al. 2011); and the laminin A‐null mouse (LMNA−/−) exhibits disruption of the nuclear lamina leading to dramatically altered gene expression that in turn causes myelin depletion and other motor axonopathic features (De Sandre‐Giovannoli et al. 2002). Notably, there are also many neuromuscular disease models that phenocopy aspects of neurally driven changes in muscle ageing, including models of spinal muscular atrophy characterized by motor neuron dysfunction and dropout (Thomas et al. 2006; Ramzan et al. 2015), and models of amyotrophic lateral sclerosis such as the SOD1G93A mouse associated with the accumulation of denervated myofibres and unstable neuromuscular junctions (Gordon et al. 2009; Perez‐Garcia & Burden, 2012).
Table 1.
Mouse models phenocopying neurological features of ageing muscle
| Genetic alteration | Muscle atrophy | Altered NMJ | MN loss | Fibre type grouping | MHC co‐expression | Denervation |
|---|---|---|---|---|---|---|
| SOD1−/− | Y | Y | ? | Y | Y | Y |
| ATG7−/− | Y | Y | ? | Y | ||
| TrkB+/− | Y | Y | Y | Y | ||
| Neurotrypsin Tg | Y | Y | N | Y | Y | Y |
| SOD1G93A | Y | Y | Y | Y | Y | Y |
| LMNA−/− | Y | Y | Y | Y |
NMJ, neuromuscular junction. Neurotrypsin Tg: neurotrypsin over‐expressing mouse.
Although these different models produce striking recapitulation of many key features of neurological impairment in ageing muscle and are also associated with contractile dysfunction and muscle atrophy, the lack of specificity of the phenotypic and functional outcomes currently precludes discerning which mechanisms account for the defects in ageing motor units. Indeed, the reduced MuSK level at the neuromuscular junction seen in ageing muscle and its association with fragmentation of the aged neuromuscular junction (Gouspillou et al. 2013) is also seen with autophagy impairment (Carnio et al. 2014). It is also striking that the downstream effector of activated MuSK, rapsyn, is reduced in muscle of mice with exacerbated oxidative stress secondary to SOD1 knockout (Jang et al. 2010). Therefore, based upon the fact that a loss of MuSK and downstream AChR clustering signals can occur as a consequence of denervation, it remains unclear whether the reduction in MuSK is a cause or effect of age‐related denervation.
Attenuation of neurological changes in ageing muscle by physical activity
Physical activity (muscle activation) is amongst the most effective interventions known to slow the progression of ageing muscle affects, including the neurological aspects. Indeed, for a portion of the lifespan exercise training may also be able to reverse some of these changes. For example, Valdez and colleagues have previously shown that one month of voluntary wheel running in 22‐month‐old inbred mice attenuated endplate fragmentation; however, benefits for the presynaptic structures were of lesser magnitude (Valdez et al. 2010). Similarly, Deschenes and colleagues showed that 10 weeks of treadmill exercise training in 20‐month‐old inbred rats partially reversed the age‐related dispersion of postsynaptic endplate structures in the soleus muscle (Deschenes et al. 2011). On the other hand, there is also evidence that the plasticity of the neuromuscular junction to physical activity is attenuated with ageing, and denervation may become exacerbated by exercise training in very old age. For example, treadmill exercise training in young adult (8 months old) inbred rats reduces the size of the postsynaptic endplate area in plantaris muscle, but this was not seen in exercise‐trained 24‐month‐old inbred rats (Deschenes et al. 2011). Furthermore, long term exercise training (7 months) initiated in old (29 months old) Fischer 344 x Brown Norway F1‐hybrid rats (a model that lives considerably longer with less pathology than inbred strains of rats; Lipman et al. 1999) was associated with greater muscle atrophy (Betik et al. 2009), and more severe grouped fibre atrophy and accumulation of severely atrophied angular fibres in very old rats (36 months old) (Thomas et al. 2010). These latter findings suggest that initiating exercise at a point in the lifespan where significant MU remodelling has already occurred (e.g. individual MUs are already expanded due to motor neuron loss and collateral reinnervation by surviving MUs) may overload the surviving MUs and cause a post‐polio‐like dropout of remaining motor neurons in advanced age. Whether this negative effect of physical activity in very advanced age can be prevented through other adjuncts to the training (e.g. nutritional interventions that reduce oxidative stress) has not been determined.
Studies in humans, although less direct by necessity, generally support an attenuation of neurological alterations in ageing muscle by physical activity. For example, the soleus muscle, which is chronically active with any upright posture or activity in humans (Dalton et al. 2009) and which is composed largely of slow twitch fibres (> 80%) (Johnson et al. 1973), exhibits only a marginal reduction in MU numbers in men aged ∼75 years compared with those aged ∼27 years (Dalton et al. 2008), which contrasts with findings from other limb muscles summarized above (see above, Changes in motor unit numbers and properties in ageing human muscle). However, even the postural slow twitch muscle exhibits a marked loss of MUs in men in their 10th decade compared to younger adult men (Vandervoort & McComas, 1986), showing that muscle activation can only delay, not prevent, MU loss with ageing. This leads to the speculation that because of the unique properties of the soleus (> 80% slow twitch fibres), in combination with life‐long activation through postural maintenance, age‐related MU loss is delayed until reaching a very old age.
Further support for the idea that regular physical activity may delay or modify alterations in MU properties in humans with ageing was demonstrated in the tibialis anterior muscles of chronic endurance runners aged ∼65 years (Power et al. 2010). The older runners had similar numbers of MUs to young adults (∼25 years) and both groups were about 35% greater than the numbers found in age‐matched healthy, but non‐running older adults. In a follow‐up study in these runners, their biceps brachii muscles (which presumably did not have the same degree of chronic activation) were not ‘protected’ from losses of MUs (Power et al. 2012). Thus, although regular physical activity is proposed to have a whole‐body neuroprotective effect with ageing (Valdez et al. 2010), there is likely to be some degree of a muscle‐specific (motor neuron pool specific) neuroprotective effect due to habitual activation (i.e. lesser benefit to motor neuron pools beyond the principle ones activated).
In addition to the protection of MU numbers, seniors who maintain a high level of physical activity have been shown to exhibit greater type I fibre grouping, which, in combination with their greater muscle mass, has been interpreted as evidence for greater reinnervation capacity in physically active seniors (Mosole et al. 2014). Thus, collectively the human data support age‐related MU protection seen in rodent models in showing that physical activity is beneficial in promoting survival of motor neurons, and in facilitating reinnervation of muscle fibres that become denervated secondary to impaired neuromuscular junction stability. The potential for physical activity to have adverse impacts when initiated in old age has not been addressed in humans, but should be considered based upon the aforementioned data in very old rats (Betik et al. 2009; Thomas et al. 2010).
Conclusions
Ageing is associated with marked alterations in both quantity and quality of MUs. These changes include motor neuron loss, neuromuscular junction instability and repeating cycles of denervation and reinnervation leading to fibre type grouping and expansion of surviving MUs. Functionally, remodelling of MUs with ageing not only reduces strength and power but likely also causes impaired coordination that collectively contributes to some of the most severe outcomes of ageing for the neuromuscular system, including falls and physical frailty. The mechanisms contributing to these changes in the ageing MU, including neuromuscular junction instability, axonal degeneration and motor neuron death, remain unclear, in part owing to the fact that multiple mechanisms can produce very similar consequences in the MU. Whilst physical activity (muscle activation) is beneficial in promoting MU survival, reinnervation and neuromuscular junction morphology, the ageing MU has diminished plasticity in response to physical activity, particularly in advanced age where exercise training might over‐burden the remodelled surviving MUs and exacerbate their demise. Future studies involving longitudinal measures in both animal models and humans will clarify individual variation in susceptibility to age‐related changes in the MU. Furthermore, study of animal models with superior MU ageing properties or unique human populations with superior neuromuscular function with ageing (e.g. competitive masters athletes at ages associated with marked MU loss and functional impairment in non‐athletes; > 75 years of age) could yield novel insights into mechanisms for promoting healthy ageing of the MU and, thus, attenuate the burden of these changes for the individual, their families and the healthcare system.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
Funding support from the Canadian Institutes of Health Research (MOP119583 and MOP125986 to R.T.H.) and the Natural Sciences and Engineering Research Council (grant no. 180970 to C.L.R.) is gratefully acknowledged.
Biography
Russell T. Hepple is a Professor in the Department of Kinesiology & Physical Education and at the Research Institute of the McGill University Health Centre at McGill University, Montreal, Canada. He is also Director for the McGill Research Centre on Physical Activity and Health. His research interests have focused on the mechanisms of atrophy and contractile dysfunction in ageing muscle using both animal models and clinical populations. Charles L. Rice, PhD is a professor in the School of Kinesiology and the Department of Anatomy & Cell Biology at The University of Western Ontario, London, Canada. He is also Research Director for the Canadian Centre for Activity and Aging, London, Canada. Research interests are focused at the motor unit level in humans under conditions of health, disease and fatigue. Many studies during his career have centred on adult ageing and the relationship between sarcopenia and motor unit structure and function.

References
- Allen MD, Stashuk DW, Kimpinski K, Doherty TJ, Hourigan ML & Rice CL (2015). Increased neuromuscular transmission instability and motor unit remodelling with diabetic neuropathy as assessed using novel near fibre motor unit potential parameters. Clin Neurophysiol 126, 794–802. [DOI] [PubMed] [Google Scholar]
- Andersen JL, Terzis G & Kryger A (1999). Increase in the degree of coexpression of myosin heavy chain isoforms in skeletal muscle fibers of the very old. Muscle Nerve 22, 449–454. [DOI] [PubMed] [Google Scholar]
- Auld DS & Robitaille R (2003). Perisynaptic Schwann cells at the neuromuscular junction: nerve‐ and activity‐dependent contributions to synaptic efficacy, plasticity, and reinnervation. Neuroscientist 9, 144–157. [DOI] [PubMed] [Google Scholar]
- Balice‐Gordon RJ (1997). Age‐related changes in neuromuscular innervation. Muscle Nerve Suppl 5, S83–87. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Rakowicz W, Gardner R & Pestronk A (2007). Frequent atrophic groups with mixed‐type myofibers is distinctive to motor neuron syndromes. Muscle Nerve 36, 107–110. [DOI] [PubMed] [Google Scholar]
- Betik AC, Thomas MM, Wright KJ, Riel CD & Hepple RT (2009). Exercise training from late middle age until senescence does not attenuate the declines in skeletal muscle aerobic function. Am J Physiol Regul Integr Comp Physiol 297, R744–755. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, Jang YC, Ran Q & Van Remmen H (2009). Denervation induces cytosolic phospholipase A2‐mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem 284, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigland‐Ritchie B, Rice CL, Garland SJ & Walsh ML (1995). Task‐dependent factors in fatigue of human voluntary contractions. Adv Exp Med Biol 384, 361–380. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VKM, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD & Glass DJ (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. [DOI] [PubMed] [Google Scholar]
- Bolliger MF, Zurlinden A, Luscher D, Butikofer L, Shakhova O, Francolini M, Kozlov SV, Cinelli P, Stephan A, Kistler AD, Rulicke T, Pelczar P, Ledermann B, Fumagalli G, Gloor SM, Kunz B & Sonderegger P (2010). Specific proteolytic cleavage of agrin regulates maturation of the neuromuscular junction. J Cell Sci 123, 3944–3955. [DOI] [PubMed] [Google Scholar]
- Bowen DC, Park JS, Bodine S, Stark JL, Valenzuela DM, Stitt TN, Yancopoulos GD, Lindsay RM, Glass DJ & DiStefano PS (1998). Localization and regulation of MuSK at the neuromuscular junction. Dev Biol 199, 309–319. [DOI] [PubMed] [Google Scholar]
- Butikofer L, Zurlinden A, Bolliger MF, Kunz B & Sonderegger P (2011). Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J 25, 4378–4393. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, McComas AJ & Petito F (1973). Physiological changes in ageing muscles. J Neurol Neurosurg Psychiatry 36, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos C, Malanda A, Gila L, Segura V, Lasanta I & Artieda J (2000). Quantification of jiggle in real electromyographic signals. Muscle Nerve 23, 1022–1034. [DOI] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R & Sandri M (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai RJ, Vukovic J, Dunlop S, Grounds MD & Shavlakadze T (2011). Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie A & Kamen G (2006). Doublet discharges in motoneurons of young and older adults. J Neurophysiol 95, 2787–2795. [DOI] [PubMed] [Google Scholar]
- Christie A, Snook EM & Kent‐Braun JA (2011). Systematic review and meta‐analysis of skeletal muscle fatigue in old age. Med Sci Sports Exerc 43, 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie AD, Tonson A, Larsen RG, DeBlois JP & Kent JA (2014). Human skeletal muscle metabolic economy in vivo: effects of contraction intensity, age, and mobility impairment. Am J Physiol Regul Integr Comp Physiol 307, R1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou EA (2011). Aging and variability of voluntary contractions. Exerc Sport Sci Rev 39, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly DM, Rice CL, Roos MR & Vandervoort AA (1999). Motor unit firing rates and contractile properties in tibialis anterior of young and old men. J Appl Physiol (1985) 87, 843–852. [DOI] [PubMed] [Google Scholar]
- Connor EA & Smith MA (1994). Retrograde signaling in the formation and maintenance of the neuromuscular junction. J Neurobiol 25, 722–739. [DOI] [PubMed] [Google Scholar]
- Dalton BH, Blouin JS, Allen MD, Rice CL & Inglis JT (2014). The altered vestibular‐evoked myogenic and whole‐body postural responses in old men during standing. Exp Gerontol 60, 120–128. [DOI] [PubMed] [Google Scholar]
- Dalton BH, Harwood B, Davidson AW & Rice CL (2009). Triceps surae contractile properties and firing rates in the soleus of young and old men. J Appl Physiol (1985) 107, 1781–1788. [DOI] [PubMed] [Google Scholar]
- Dalton BH, Harwood B, Davidson AW & Rice CL (2010. a). Recovery of motoneuron output is delayed in old men following high‐intensity fatigue. J Neurophysiol 103, 977–985. [DOI] [PubMed] [Google Scholar]
- Dalton BH, Jakobi JM, Allman BL & Rice CL (2010. b). Differential age‐related changes in motor unit properties between elbow flexors and extensors. Acta Physiol (Oxf) 200, 45–55. [DOI] [PubMed] [Google Scholar]
- Dalton BH, McNeil CJ, Doherty TJ & Rice CL (2008). Age‐related reductions in the estimated numbers of motor units are minimal in the human soleus. Muscle Nerve 38, 1108–1115. [DOI] [PubMed] [Google Scholar]
- Dalton BH, Power GA, Paturel JR & Rice CL (2015). Older men are more fatigable than young when matched for maximal power and knee extension angular velocity is unconstrained. Age (Dordr) 37, 9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton BH, Power GA, Vandervoort AA & Rice CL (2010. c). Power loss is greater in old men than young men during fast plantar flexion contractions. J Appl Physiol (1985) 109, 1441–1447. [DOI] [PubMed] [Google Scholar]
- De Sandre‐Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D & Levy N (2002). Homozygous defects in LMNA, encoding lamin A/C nuclear‐envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot‐Marie‐Tooth disorder type 2) and mouse. Am J Hum Genet 70, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes MR, Roby MA, Eason MK & Harris MB (2010). Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp Gerontol 45, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes MR, Roby MA & Glass EK (2011). Aging influences adaptations of the neuromuscular junction to endurance training. Neuroscience 190, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoka RM, Christou EA, Hunter SK, Kornatz KW, Semmler JG, Taylor AM & Tracy BL (2003). Mechanisms that contribute to differences in motor performance between young and old adults. J Electromyogr Kinesiol 13, 1–12. [DOI] [PubMed] [Google Scholar]
- Erim Z, Beg MF, Burke DT & de Luca CJ (1999). Effects of aging on motor‐unit control properties. J Neurophysiol 82, 2081–2091. [DOI] [PubMed] [Google Scholar]
- Faulkner JA, Larkin LM, Claflin DR & Brooks SV (2007). Age‐related changes in the structure and function of skeletal muscles. Clin Exp Pharmacol Physiol 34, 1091–1096. [DOI] [PubMed] [Google Scholar]
- Gardiner PF & Kernell D (1990). The “fastness” of rat motoneurones: time‐course of afterhyperpolarization in relation to axonal conduction velocity and muscle unit contractile speed. Pflugers Arch 415, 762–766. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M & Newman AB (2006). The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Gordon T, Ly V, Hegedus J & Tyreman N (2009). Early detection of denervated muscle fibers in hindlimb muscles after sciatic nerve transection in wild type mice and in the G93A mouse model of amyotrophic lateral sclerosis. Neurol Res 31, 28–42. [DOI] [PubMed] [Google Scholar]
- Gouspillou G, Picard M, Godin R, Burelle Y & Hepple RT (2013). Role of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) in denervation‐induced atrophy in aged muscle: facts and hypotheses. Longev Healthspan 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann E & Hanzlikova V (1966). Motor unit in old age. Nature 209, 921–922. [DOI] [PubMed] [Google Scholar]
- Hettwer S, Dahinden P, Kucsera S, Farina C, Ahmed S, Fariello R, Drey M, Sieber CC & Vrijbloed JW (2012). Elevated levels of a C‐terminal agrin fragment identifies a new subset of sarcopenia patients. Exp Gerontol 48, 69–75. [DOI] [PubMed] [Google Scholar]
- Hourigan ML, McKinnon NB, Johnson M, Rice CL, Stashuk DW & Doherty TJ (2015). Increased motor unit potential shape variability across consecutive motor unit discharges in the tibialis anterior and vastus medialis muscles of healthy older subjects. Clin Neurophysiol 126, 2381–2389. [DOI] [PubMed] [Google Scholar]
- Jakobi JM & Rice CL (2002). Voluntary muscle activation varies with age and muscle group. J Appl Physiol 93, 457–462. [DOI] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC & Van Remmen H (2010). Age‐associated alterations of the neuromuscular junction. Exp Gerontol 46, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankelowitz SK, McNulty PA & Burke D (2007). Changes in measures of motor axon excitability with age. Clin Neurophysiol 118, 1397–1404. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Polgar J, Weightman D & Appleton D (1973). Data on the distribution of fibre types in thirty‐six human muscles. An autopsy study. J Neurol Sci 18, 111–129. [DOI] [PubMed] [Google Scholar]
- Justice JN, Mani D, Pierpoint LA & Enoka RM (2014). Fatigability of the dorsiflexors and associations among multiple domains of motor function in young and old adults. Exp Gerontol 55, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda K & Hashizume K (1989). Changes in properties of the medial gastrocnemius motor units in aging rats. J Neurophysiol 61, 737–746. [DOI] [PubMed] [Google Scholar]
- Kanekar N & Aruin AS (2014). The effect of aging on anticipatory postural control. Exp Brain Res 232, 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Islam MM, Young KC, Rogers ME & Takeshima N (2015). Threshold of chair stand power necessary to perform activities of daily living independently in community‐dwelling older women. J Geriatr Phys Ther 38, 122–126. [DOI] [PubMed] [Google Scholar]
- Kent‐Braun JA (2009). Skeletal muscle fatigue in old age: whose advantage? Exerc Sport Sci Rev 37, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakowski SA, Parker SD & Personius KE (2011). Reduced TrkB expression results in precocious age‐like changes in neuromuscular structure, neurotransmission, and muscle function. J Appl Physiol 111, 844–852. [DOI] [PubMed] [Google Scholar]
- Larsson L (1995). Motor units: remodeling in aged animals. J Gerontol A Biol Sci Med Sci 50 Spec No, 91–95. [DOI] [PubMed] [Google Scholar]
- Layec G, Trinity JD, Hart CR, Kim SE, Groot HJ, Le Fur Y, Sorensen JR, Jeong EK & Richardson RS (2014). In vivo evidence of an age‐related increase in ATP cost of contraction in the plantar flexor muscles. Clin Sci (Lond) 126, 581–592. [DOI] [PubMed] [Google Scholar]
- Lexell J & Downham DY (1991). The occurrence of fibre‐type grouping in healthy human muscle: a quantitative study of cross‐sections of whole vastus lateralis from men between 15 and 83 years. Acta Neuropathol (Berl) 81, 377–381. [DOI] [PubMed] [Google Scholar]
- Lexell J & Taylor CC (1991). Variability in muscle fibre areas in whole human quadriceps muscle: effects of increasing age. J Anat 174, 239–249. [PMC free article] [PubMed] [Google Scholar]
- Lexell J, Taylor CC & Sjostrom M (1988). What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15‐ to 83‐year‐old men. J Neurol Sci 84, 275–294. [DOI] [PubMed] [Google Scholar]
- Li M, Ogilvie H, Ochala J, Artemenko K, Iwamoto H, Yagi N, Bergquist J & Larsson L (2015). Aberrant post‐translational modifications compromise human myosin motor function in old age. Aging Cell 14, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lee Y & Thompson WJ (2011). Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J Neurosci 31, 14910–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindboe CF & Torvik A (1982). The effects of ageing, cachexia and neoplasms on striated muscle. Quantitative histological and histochemical observations on an autopsy material. Acta Neuropathol 57, 85–92. [DOI] [PubMed] [Google Scholar]
- Lipman RD, Dallal GE & Bronson RT (1999). Effects of genotype and diet on age‐related lesions in ad libitum fed and calorie‐restricted F344, BN, and BNF3F1 rats. J Gerontol A Biol Sci Med Sci 54, B478–B491. [DOI] [PubMed] [Google Scholar]
- Mars T, King MP, Miranda AF, Walker WF, Mis K & Grubic Z (2003). Functional innervation of cultured human skeletal muscle proceeds by two modes with regard to agrin effects. Neuroscience 118, 87–97. [DOI] [PubMed] [Google Scholar]
- McNeil CJ, Doherty TJ, Stashuk DW & Rice CL (2005). Motor unit number estimates in the tibialis anterior muscle of young, old, and very old men. Muscle Nerve 31, 461–467. [DOI] [PubMed] [Google Scholar]
- McNeil CJ & Rice CL (2007). Fatigability is increased with age during velocity‐dependent contractions of the dorsiflexors. J Gerontol A Biol Sci Med Sci 62, 624–629. [DOI] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA & Sandri M (2015). Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 6, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JP, McNeil CJ, Bredius MS & Gandevia SC (2013). Effects of aging and sex on voluntary activation and peak relaxation rate of human elbow flexors studied with motor cortical stimulation. Age (Dordr) 35, 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosole S, Carraro U, Kern H, Loefler S, Fruhmann H, Vogelauer M, Burggraf S, Mayr W, Krenn M, Paternostro‐Sluga T, Hamar D, Cvecka J, Sedliak M, Tirpakova V, Sarabon N, Musaro A, Sandri M, Protasi F, Nori A, Pond A & Zampieri S (2014). Long‐term high‐level exercise promotes muscle reinnervation with age. J Neuropathol Exp Neurol 73, 284–294. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A & Van Remmen H (2007). Denervation‐induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293, R1159–1168. [DOI] [PubMed] [Google Scholar]
- Oda K (1984). Age changes of motor innervation and acetylcholine receptor distribution on human skeletal muscle fibres. J Neurol Sci 66, 327–338. [DOI] [PubMed] [Google Scholar]
- Pascoe MA, Holmes MR & Enoka RM (2011). Discharge characteristics of biceps brachii motor units at recruitment when older adults sustained an isometric contraction. J Neurophysiol 105, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Garcia MJ & Burden SJ (2012). Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep 2, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personius KE & Parker SD (2013). TrkB expression at the neuromuscular junction is reduced during aging. Muscle Nerve 47, 532–538. [DOI] [PubMed] [Google Scholar]
- Petrella JK, Kim JS, Tuggle SC, Hall SR & Bamman MM (2005). Age differences in knee extension power, contractile velocity, and fatigability. J Appl Physiol 98, 211–220. [DOI] [PubMed] [Google Scholar]
- Power GA, Dalton BH, Behm DG, Doherty TJ, Vandervoort AA & Rice CL (2012). Motor unit survival in lifelong runners is muscle dependent. Med Sci Sports Exerc 44, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Power GA, Dalton BH, Behm DG, Vandervoort AA, Doherty TJ & Rice CL (2010). Motor unit number estimates in masters runners: use it or lose it? Med Sci Sports Exerc 42, 1644–1650. [DOI] [PubMed] [Google Scholar]
- Power GA, Dalton BH & Rice CL (2013). Human neuromuscular structure and function in old age: A brief review. J Sport Health 2, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punga AR, Lin S, Oliveri F, Meinen S & Ruegg MA (2011). Muscle‐selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol 230, 207–217. [DOI] [PubMed] [Google Scholar]
- Purves‐Smith FM, Solbak NM, Rowan SL & Hepple RT (2012). Severe atrophy of slow myofibers in aging muscle is concealed by myosin heavy chain co‐expression. Exp Gerontol 47, 913–918. [DOI] [PubMed] [Google Scholar]
- Ramzan F, McPhail M, Rao P, Mo K, Halievski K, Swift‐Gallant A, Mendoza‐Viveros L, Cheng HY & Monks DA (2015). Distinct etiological roles for myocytes and motor neurons in a mouse model of Kennedy's disease/spinobulbar muscular atrophy. J Neurosci 35, 6444–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CL & Cunningham DA (2001). Aging of the neuromuscular system: Influences of gender and physical activity In Gender, Physical Activity and Aging, ed. Shephard RJ, pp. 121–150. CRC Press, Boca Raton, FL, USA. [Google Scholar]
- Roder IV, Choi KR, Reischl M, Petersen Y, Diefenbacher ME, Zaccolo M, Pozzan T & Rudolf R (2010). Myosin Va cooperates with PKA RIα to mediate maintenance of the endplate in vivo. Proc Natl Acad Sci USA 107, 2031–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R & Sandri M (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 29, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos MR, Rice CL, Connelly DM & Vandervoort AA (1999). Quadriceps muscle strength, contractile properties, and motor unit firing rates in young and old men. Muscle Nerve 22, 1094–1103. [DOI] [PubMed] [Google Scholar]
- Rowan SL, Purves‐Smith FM, Solbak NM & Hepple RT (2011). Accumulation of severely atrophic myofibers marks the acceleration of sarcopenia in slow and fast twitch muscles. Exp Gerontol 46, 660–669. [DOI] [PubMed] [Google Scholar]
- Rowan SL, Rygiel K, Purves‐Smith FM, Solbak NM, Turnbull DM & Hepple RT (2012). Denervation causes fiber atrophy and myosin heavy chain co‐expression in senescent skeletal muscle. PLoS One 7, e29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein S & Kamen G (2005). Decreases in motor unit firing rate during sustained maximal‐effort contractions in young and older adults. J Electromyogr Kinesiol 15, 536–543. [DOI] [PubMed] [Google Scholar]
- Rudolf R, Khan MM, Labeit S & Deschenes MR (2014). Degeneration of neuromuscular junction in age and dystrophy. Front Aging Neurosci 6, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruidnik‐Schoneborn S, De Visser M & Zerres K (2004). Spinal muscle atrophies In Myology, 3rd edn, ed. Engel AG. & Franzini‐Armstrong C, pp. 1845–1864. McGraw‐Hill, New York. [Google Scholar]
- Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A & Brooks SV (2014). Neuron‐specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD‐knockout mice. FASEB J 28, 1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Valdez G, Tapia JC, Lichtman JW & Sanes JR (2012). Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One 7, e46663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H & Sandri M (2013). BMP signaling controls muscle mass. Nat Genet 45, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Scelsi R, Marchetti C & Poggi P (1980). Histochemical and ultrastructural aspects of m. vastus lateralis in sedentary old people (aged 65–89 years). Acta Neuropathol 51, 99–105. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Dyar KA, Ciciliot S, Blaauw B & Sandri M (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280, 4294–4314. [DOI] [PubMed] [Google Scholar]
- Selcen D, Fukuda T, Shen XM & Engel AG (2004). Are MuSK antibodies the primary cause of myasthenic symptoms? Neurology 62, 1945–1950. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ivannikov MV, Walsh ME, Liu Y, Zhang Y, Jaramillo CA, Macleod GT & Van Remmen H (2014). The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS One 9, e100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto K, Kubo S, Mori S, Yamada S, Akiyoshi T & Miyazaki T (2010). Muscle weakness and neuromuscular junctions in aging and disease. Geriatr Gerontol Int 10 Suppl 1, S137–147. [DOI] [PubMed] [Google Scholar]
- Stalberg EV & Sonoo M (1994). Assessment of variability in the shape of the motor unit action potential, the “jiggle,” at consecutive discharges. Muscle Nerve 17, 1135–1144. [DOI] [PubMed] [Google Scholar]
- Sugiura Y & Lin W (2011). Neuron‐glia interactions: the roles of Schwann cells in neuromuscular synapse formation and function. Biosci Rep 31, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs J, Carpenter MG, Garland SJ & Hunt MA (2013). The role of neuromuscular changes in aging and knee osteoarthritis on dynamic postural control. Aging Dis 4, 84–99. [PMC free article] [PubMed] [Google Scholar]
- Thomas MM, Khan W, Betik AC, Wright KJ & Hepple RT (2010). Initiating exercise training in late middle age minimally protects muscle contractile function and increases myocyte oxidative damage in senescent rats. Exp Gerontol 45, 856–867. [DOI] [PubMed] [Google Scholar]
- Thomas PS Jr, Fraley GS, Damian V, Woodke LB, Zapata F, Sopher BL, Plymate SR & La Spada AR (2006). Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X‐linked spinal and bulbar muscular atrophy. Hum Mol Genet 15, 2225–2238. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE & Irving D (1977). The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci 34, 213–219. [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Kang H, Clemenson GD Jr, Gage FH, Lichtman JW & Sanes JR (2010). Attenuation of age‐related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci U S A 107, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandervoort AA & McComas AJ (1986). Contractile changes in opposing muscles of the human ankle joint with aging. J Appl Physiol 61, 361–367. [DOI] [PubMed] [Google Scholar]
- Ward RE, Boudreau RM, Caserotti P, Harris TB, Zivkovic S, Goodpaster BH, Satterfield S, Kritchevsky S, Schwartz AV, Vinik AI, Cauley JA, Newman AB & Strotmeyer ES; Health ABC Study (2015). Sensory and motor peripheral nerve function and longitudinal changes in quadriceps strength. J Gerontol A Biol Sci Med Sci 70, 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Xiong WC & Mei L (2010). To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137, 1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]