

Abstract
Phosphatidyl-myo-inositol mannosyltransferase A (PimA) is an essential glycosyltransferase that initiates the biosynthetic pathway of phosphatidyl-myo-inositol mannoside, lipomannan, and lipoarabinomannan, which are key glycolipids/lipoglycans of the mycobacterial cell envelope. PimA belongs to a large family of membrane-associated glycosyltransferases for which the understanding of the molecular mechanism and conformational changes that govern substrate/membrane recognition and catalysis remains a major challenge. Here, we determined that PimA preferentially binds to negatively charged phosphatidyl-myo-inositol substrate and non-substrate membrane model systems (small unilamellar vesicle) through its N-terminal domain, inducing an important structural reorganization of anionic phospholipids. By using a combination of single-point mutagenesis, circular dichroism, and a variety of fluorescence spectroscopy techniques, we determined that this interaction is mainly mediated by an amphipathic α-helix (α2), which undergoes a substantial conformational change and localizes in the vicinity of the negatively charged lipid headgroups and the very first carbon atoms of the acyl chains, at the PimA-phospholipid interface. Interestingly, a flexible region within the N-terminal domain, which undergoes β-strand-to-α-helix and α-helix-to-β-strand transitions during catalysis, interacts with anionic phospholipids; however, the effect is markedly less pronounced to that observed for the amphipathic α2, likely reflecting structural plasticity/variability. Altogether, we propose a model in which conformational transitions observed in PimA might reflect a molten globule state that confers to PimA, a higher affinity toward the dynamic and highly fluctuating lipid bilayer.
Keywords: enzyme, glycolipid, glycosyltransferase, membrane enzyme, structure-function
Introduction
Phosphatidyl-myo-inositol mannosyltransferase A (PimA), is an essential GT-B enzyme involved in the biosynthesis of phosphatidyl-myo-inositol mannosides (PIMs),3 lipomannan, and lipoarabinomannan, key structural elements and virulence factors of Mycobacterium tuberculosis (1–6). PimA is an amphitropic/peripheral enzyme that catalyzes the transfer of a Manp residue from GDP-Man to the 2-position of phosphatidyl-myo-inositol (PI) to form phosphatidyl-myo-inositol monomannoside on the cytoplasmic side of the plasma membrane (Fig. 1A) (5, 7, 8). The term amphitropic emphasizes the affinity of certain proteins for both aqueous and hydrophobic environments, suggesting that they can reversibly and transiently interact with the lipid bilayer (9). The crystal structure of PimA was solved in different states of its catalytic cycle as follows: the apo-form, the PimA-GDP-Man and PimA-GDP complexes, and that of PimA in complex with GDP and 1,2-dioctanoyl-sn-glycero-3-phospho-inositol (diC8PI), a PI analog with shorter acyl chains (Fig. 1B) (10–13). This structural information revealed unprecedented conformational changes that appear to be critical during substrate/membrane binding and catalysis, including the reshuffling of secondary structural elements and interdomain (open-to-closed) motions associated with GDP-Man binding between the N- and C-terminal domains, which markedly stabilize the enzyme (12, 14, 15). Specifically, the nucleotide moiety of GDP-Man seems to be the main driving force to trigger the open-to-closed motion, with the β-PO4 group playing a key role in the following: (i) providing the high affinity binding of the nucleotide sugar to PimA and (ii) the generation of a competent active site. Interestingly, the apo-form of PimA crystallized with four monomers in the asymmetric unit, capturing two different conformations of the enzyme (12). In the apo “compact” conformation, a helical hairpin consisting of helices α4 (residues 134–145) and α5 (residues 149–157) folds back against the core of the N-terminal domain. In contrast, in the apo “extended” the helical hairpin protrudes away from the core, allowing the exposure of a significant hydrophobic surface to the solvent. The apo-extended conformation might account for an additional lobe observed in small angle x-ray scattering studies (Fig. 1B) (15). However, the most striking observation is the reshuffling of secondary structure elements on the N-terminal domain of the PimA-GDP-Man complex (11, 12). This conformational switch involves both β-strand-to-α-helix and α-helix-to-β-strand transitions. The largest differences are localized in the flexible region corresponding to residues 118–163, which represent a substantial portion of the N-terminal “Rossmann-fold” domain. The extended and compact apo-forms of PimA display a conserved anti-parallel β-strand (β6) and the two flexible α-helices (α4 and α5), whereas the PimA-GDP-Man conformation exhibits the equivalent β-strand in a parallel orientation and three α-helices, α41 (residues 122–132), α42 (residues 134–140), and α5 (residues 148–160), with a novel structural arrangement (Fig. 1, B and C). Importantly, a detailed inspection of the crystal structures of the apo-forms of PimA suggested that they correspond to an inactive conformation. This interpretation was supported by mutagenesis studies in which the two different conformations, apo-form and GDP-Man-bound forms, have been locked with disulfide bridges and then used for isothermal titration calorimetry and enzyme activity assays in the presence and absence of reducing agents (12).
FIGURE 1.

Biosynthesis of PIMs in mycobacteria. A, PimA transfers a Manp residue from GDP-Man to the 2-position of the myo-inositol ring of PI to form phosphatidyl-myo-inositol monomannoside (PIM1). The reaction occurs with retention of the anomeric configuration of the sugar donor. B, schematic representation showing the crystal structures of PimA, including its apo compact, apo extended, PimA-GDP-Man, and PimA-GDP-diC8PI complexes (11, 12). The dynamic N-terminal region is shown in rainbow-colored schematic representation. C, secondary structure elements of a selected region corresponding to residues 110–170, as observed in the apo compact, apo extended, PimA-GDP-Man, and PimA-GDP-diC8PI complexes. D, location of Trp-82, Trp-154, and Trp-349 in the N-terminal Rossmann domain of the GT-B mannosyltransferase PimA. E, activity measurements showing that PimA-Trp-82, PimA-Trp-154, and PimA-Trp-349 mutant variants are functional proteins. The enzymatic activities of the three double mutants are shown as a percentage of the wild type activity. Data represent mean ± S.D. from three independent experiments.
Another key aspect of the mode of action of PimA is its interaction with the lipid acceptor PI and the mycobacterial membrane. A close association of PimA with the lipid bilayer is likely to be a strict requirement for PI modification (5). Despite the fact that sugar transfer is catalyzed between the mannosyl group of the GDP-Man donor and the myo-inositol ring of PI (11), the enzyme strictly requires the two fatty acid chains of the acceptor substrate for the transfer reaction to take place. Interestingly, PimA was able to interact with a mono-disperse PI analog with shorter acyl chains (1,2-dioctanoyl-sn-glycero-3-phosphoinositol), for which the critical micellar concentration (CMC) has been experimentally reported as 60 μm (16). However, its transferase activity was stimulated by diC8PI at a concentration of 300 μm (i.e. five times the CMC) or high concentrations of non-substrate anionic surfactants, such as cardiolipin or 1,2-dipalmitoyl-sn-glycero-3-phosphate, but not with the zwitterion phosphatidylethanolamine or 1,2-dipalmitoyl-sn-glycerol, indicating that the reaction requires a lipid-water interface (11, 14). Inspection of the crystal structure of PimA suggested that the presence of an amphipathic α-helix (α2-helix; residues 72–87) and surface-exposed hydrophobic residues in the N-terminal domain of the protein could mediate the association of PimA with anionic phospholipids via electrostatic interactions (11).
The crystal structure of PimA in the presence of GDP and diC8PI at 500 μm (∼10 times its CMC) revealed remarkable structural differences located in the dynamic region of the N-terminal domain, suggesting that diC8PI micelles could act as an inducer of the observed conformational changes. The near-UV CD spectrum of PimA in solution exhibited a broad positive signal characteristic of constrained aromatic side chains in the protein. The addition of diC8PI at sub-CMC concentrations did not change the spectrum. In contrast, the addition of diC8PI micelles induced a loss of the tertiary structure constraints on aromatic side chains, as indicated by the absence of dichroic signal. This result clearly indicates that PimA aromatic side chains gain freedom in the presence of diC8PI micelles. This lipid-induced destabilization was also observed in thermal unfolding experiments followed by CD in the far-UV region (12). Such a loss of cooperativity upon diC8PI binding is characteristic of membrane-bound proteins lacking tertiary structure interactions (17, 18). In line with these results, limited proteolysis experiments in the presence of diC8PI micelles or anionic SUVs increased the susceptibility of PimA to proteolysis (12). To further advance the molecular mechanism of PimA association to the lipid acceptor PI and to the membrane, we have used a combination of single-point mutagenesis, circular dichroism, and several spectroscopy techniques, including microscale technology (MST), intrinsic tryptophan fluorescence, tryptophan fluorescence anisotropy, and fluorescence resonance energy transfer (FRET). We propose a plausible model for PI and membrane interaction, which might prove useful for the comprehension of the peripheral membrane-associated GT-Bs at the molecular level.
Experimental Procedures
Materials and Methods
Recombinant PimA from Mycobacterium smegmatis was produced in Escherichia coli and purified to apparent homogeneity as described previously (10, 11, 14). The double mutant variants of PimA, W82F/W154F, W82F/W349F, and W154F/W349F, were obtained from GenScript by using pET29a-pimA (14) as the DNA template and expressed and purified as described previously (14). The purified W82F/W154F, W82F/W349F, and W154F/W349F PimA variants displayed enzymatic activity, demonstrating that they are functional (Fig. 1, D and E). Enzymatic activity was measured as described previously (5, 14). Lipids were purchased from Avanti Polar Lipids (Alabaster, AL).
Preparation of Small Unilamellar Vesicles (SUVs)
The lipid vesicles were prepared in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) for neutral vesicles, and DOPC and bovine liver phosphatidylinositol (PI) or phosphatidylglycerol (DOPG) at a 50:50 molar ratio. SUVs were prepared by reverse phase evaporation,filtration, and sonication (19). Hexanoyl-1-hexadecanoyl-sn-glycero-3-phosphocholine (HPC) labeled with 2-(6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino) (NBD) at C6 (NBD-C6-HPC) or C12 (NBD-C12-HPC) positions along the acyl chain were used for fluorescence anisotropy experiments. Stock solutions at 12.5 mm of each NBD-HPC were prepared in 100% ethanol. NBD-C6-HPC or NBD-C12-HPC was incorporated at 0.5% into 2 mm DOPC, DOPC/DOPG, or DOPC/PI preformed SUVs, vortexing vigorously, and incubating for 30 min at room temperature. To perform FRET experiments, N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (dansyl-DHPE; Invitrogen), was mixedwith methanol and dissolved by sonication to give a 12.5 mm stock solution. SUVs containing dansyl were prepared from dansyl-DHPE/DOPC at 9.5:0.5 and DOPC/DOPG and DOPC/PI at a 4.25:4.25:0.5 molar ratio.
MST Technology
MST analyses were carried out with a Monolith NT LabelFree instrument (Nano Temper, Munich, Germany). A titration series was prepared, in which the concentration of PimA WT was kept constant at 1 μm. The concentrations of the titrants (DOPC, DOPC/DOPG (50:50), and DOPC/PI (50:50) SUVs) were varied (60 nm to 2 mm) in 50 mm Tris, pH 8.0, 33 mm NaCl. MST measurements were performed at room temperature after 5 min of incubation. Standard treated capillaries (K002 Monolith NT.115) were loaded with the samples, and all measurements were performed three times. For each measurement, the laser was switched on for 30 s and off for 5 s. Binding curves were obtained from the thermophoresis T-jump phase, with IR laser power at 10%. The average Kd values were calculated after analysis of the data with KaleidaGraph software.
Intrinsic Tryptophan Fluorescence
Measurements were performed with an FP-6200 spectrofluorometer (Jasco, Japan) equipped with a Peltier-thermostated cell holder, using 0.1-ml samples in 1-cm path length quartz cells (111-QS from Hellma). A bandwidth of 5 nm was used for the excitation and emission beams. The excitation wavelength was fixed at 290 nm, and the emission spectra were recorded at 25 °C from 300 to 400 nm at a scan rate of 125 nm/min. PimA concentration was 1 μm in 50 mm Tris, pH 8.0, 150 mm NaCl, and it was added to 1 mm SUVs prepared in the same buffer from DOPC for neutral vesicles, and DOPC/PI or DOPC/DOPG at a 50:50 molar ratio. For the three double mutants a 3 μm concentration was used to have similar intensity signals compared with those from the PimA wild type. The ratio of fluorescence intensities emitted at 320 and 360 nm (FIR320/360) represents the average of three replicates obtained from emission spectra that were previously corrected for buffer effects.
Tryptophan Fluorescence Anisotropy
Measurements were performed in the same equipment and in the same experimental conditions described under “Intrinsic Tryptophan Fluorescence.” A bandwidth of 5 nm was used for the excitation and emission beams. For the Trp excitation the wavelength was fixed at 290 nm, and the emission spectra were recorded at 25 °C with a scan rate of 500 nm/min. Emission spectra were recorded from 300 to 400 nm. PimA and three double mutant concentration was 5 μm in 50 mm Tris, pH 8.0, 150 mm NaCl, and it was added to 1 mm SUVs.
Steady-state fluorescence anisotropy experiments were performed with vertically (V) and horizontally (H) polarized beam light using FDP-223 polarizers at both excitation (x) and emission (m) apertures. Anisotropy (r) was calculated as shown in Equation 1,
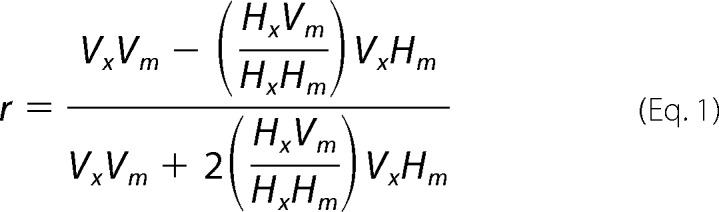 |
The r values represent the average of three replicates obtained from emission spectra that were previously corrected for buffer effects. An unpaired t test (p < 0.0001) was performed between data sets. The intensity values were recorded from 300 to 400 nm with one value per wavelength and in triplicate, hence n = 303.
Fluorescence Resonance Energy Transfer (FRET)
The kinetic mode was used to follow the intensity of maximum emission for the dansyl group at 515 nm during 1,000 s. A bandwidth of 5 nm was used for the excitation and emission beams. The excitation wavelength was fixed at 280 nm to excite the Trp, and the emission was recorded at 25 °C. The intensity coming from dansylated SUVs at 1 mm lipid concentration was recorded during 200 s. The intensity coming from 1 mm dansylated SUVs was recorded during 200 s. After signal stabilization, 4 μm protein was added in buffer 50 mm Tris, pH 8.0, 30 mm NaCl. After 500 s, the NaCl concentration was increased ≈×10 as shown in Equations 2 and 3,
where It 190 is the dansyl intensity at 190 s after stabilization, and It 220 is the dansyl intensity after buffer or protein injection at 220 s. Likewise, Δdansyl intensity for NaCl addition values are obtained for both the blank and protein from Equation 4,
where It 690 is the dansyl intensity at 690 s before NaCl addition, and It 720 is the dansyl intensity after NaCl injection at 720 s. Finally, The ΔFRET values represent the average of three replicates obtained from emission spectra, previously corrected for buffer effects and for both dilution of protein and NaCl addition. The values were multiplied by 100 for an easier reading as shown in Equation 5,
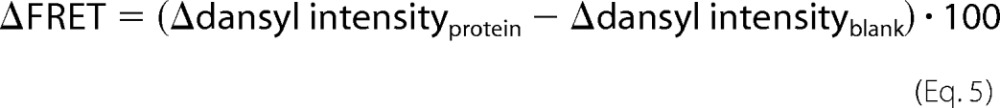 |
NBD C6-HPC and NBD C12-HPC SUVs Anisotropy
5 μm PimA in 25 mm Tris-HCl, pH 8.0, 150 mm NaCl was added to labeled SUVs at a lipid concentration of 1 mm. The excitation wavelength was 465 nm, and the emission spectra were recorded from 500 to 550 nm. Steady-state fluorescence anisotropy experiments were performed with vertically (V) and horizontally (H) polarized beam light using FDP-223 polarizers at both excitation (x) and emission (m) apertures. Anisotropy (r) was calculated as described under “Tryptophan Fluorescence Anisotropy.” The Δr values represent the average of three replicates obtained from emission spectra that were previously corrected for buffer effects. To confirm whether the difference between means was statistically significant, a t test unpaired (p < 0.0001) was performed between data sets. The n values depend on the range of wavelengths chosen for each anisotropy experiment, one value per wavelength, and NBD-HPC lipid anisotropy values were recorded from 500 to 550 nm in triplicate, n = 103. The Δr values for the NBD-HPC lipids come from Equation 6,
Values are multiplied by 1000 for easier reading.
Far-UV Circular Dichroism Analysis
Spectra were acquired in a J-810 CD spectropolarimeter (Jasco Corp., Tokio, Japan) by using Hellma 110-QS quartz cuvettes with a 1-mm optical path. Spectra were recorded in continuous mode with 1-nm bandwidth, 1-s response, and a scan speed of 100 nm/min−1. Samples were 4.61 μm PimA in 10 mm Tris-HCl, pH 7.5. Substrates were added in a 1:10 ratio, and 30 scans were accumulated to obtain the final spectra, which were further corrected for the baseline signal. Spectra were recorded in the 195–250-nm range at 20 °C.
Temperature Scans
The J-810 CD spectropolarimeter (Jasco Corp., Tokio, Japan) has a Peltier thermal device, allowing the temperature control during the experiments. Thermal dependences of the ellipticity were monitored in a range from 10 to 90 °C at 222 nm. Temperature was increased stepwise by 1°/min. Protein concentration, buffer composition, and cuvette were as described under “Far-UV Circular Dichroism Analysis.”
Near-UV Circular Dichroism Analysis
Spectra were acquired in a J-810 CD spectropolarimeter (Jasco Corp., Tokio, Japan) by using Hellma 105.200-QS quartz cuvettes with a 1-cm optical path. Spectra were recorded in a continuous mode with a 1-nm bandwidth, 1-s response, and a scan speed of 100 nm/min−1. Samples were 10 μm PimA in 10 mm Tris-HCl, pH 7.5. Substrates were added in a 1:1.4 ratio, and 25 scans were accumulated to obtain the final spectra, which were further corrected for the baseline signal. Spectra were recorded in the 250–300-nm range at 20 °C.
Results and Discussion
Anionic Phospholipids Selectively Trigger a Conformational Change in PimA
To study the binding affinity of PimA to different SUVs, we successfully applied the MST technology, providing the apparent dissociation constants for DOPC, DOPG, and PI SUVs (20). SUVs containing the acceptor substrate PI and the non-substrate anionic phospholipid DOPG were seen to bind PimA with dissociation constants Kd of 80 and 100 μm, respectively. In contrast, much lower binding of DOPC could be detected to PimA, with a Kd >1.5 mm. To investigate the impact of phospholipids binding into the conformation of PimA, we monitored the tertiary structure changes of PimA induced by the addition of SUVs made of DOPC, DOPG, and PI, by near-UV CD (Fig. 2A). The near-UV CD spectrum of PimA in solution exhibited a broad positive signal characteristic of aromatic side chains, namely tyrosines and tryptophans, constrained in the protein. The addition of DOPC liposomes did not change the near-UV spectrum. In contrast, the addition of PI or DOPG SUVs induced a loss of the constraints on aromatic side chains, as indicated by the absence of a dichroic signal. This result clearly indicates that PimA aromatic side chains gain freedom in the presence of anionic phospholipids, thus, at least partially, losing the tertiary structure. As shown in Fig. 2B, this lipid-induced destabilization was also observed during the thermal unfolding experiments followed by CD in the far-UV region. Such a loss of cooperativity upon binding of PI or DOPG liposomes is characteristic of membrane-bound proteins in molten globule-like (MG-like) states, i.e. displaying native-like secondary structures but lacking a stable tertiary structure (17, 18, 21).
FIGURE 2.

Circular dichroism spectra for PimA in solution and in the presence of SUVs. A, near-UV CD spectra reveal a loss of dichroic signal only in the presence of anionic SUVs. B, thermally induced unfolding process followed by CD signal at 222 nm shows the loss of cooperativity in the unfolding process when anionic SUVs interact with PimA.
PimA Induces an Important Structural Reorganization of Anionic SUVs
The effect of PimA interaction to different SUVs can be followed by the anisotropy of labeled lipids. Depending on the protein binding mechanism, it is expected that SUV properties will change, having direct impact on the rotational diffusion of an embedded probe and therefore on its anisotropy. To this end, HPC labeled with NBD at C6 or C12 positions along the acyl chain were incorporated into the DOPC, DOPG, or PI SUVs (see “Experimental Procedures” for details). This fluorophore has the maximum of excitation at 463 nm and a maximum of emission at 536 nm. As depicted in Table 1, the anisotropy values for DOPC and DOPG SUVs revealed that PimA induced slight constraints on the lipid acyl chains. In contrast, the PI SUVs containing the NBD at C6 and C12 positions displayed a significant increase of anisotropy as a consequence of the introduction of structural constraints in the vicinity of the probe due to the presence of PimA. The effect was particularly apparent for PI liposomes containing the lipid NBD probe located at the C6 position. Overall, the experimental data indicate the following: (i) PimA induces an important structural reorganization of the SUVs containing the acceptor substrate PI; (ii) the binding of PimA to PI SUVs markedly affects the vesicle fluidity in the acyl region close to the lipid headgroup; and (iii) the effect on the deeper hydrophobic core is less pronounced.
TABLE 1.
NBD-C6-HPC and NBDC12-HPC anisotropy changes upon PimA interaction
The Δr value represents the difference between the mean and the standard deviation (n = 153; for an easier interpretation, the values are multiply by 1000). A t test was performed for anisotropy values in the absence versus presence of PimA WT. All are statistically significant (n = 153; p < 0.0001) except DOPC/NBD-HPC C12 versus DOPC/NBD-HPC C12 in the presence of PimA WT (n = 153; p = 0.6557).
| Labeled SUV type | Δr C6 · ± S.D. | Δr C12 · ± S.D. |
|---|---|---|
| DOPC | −5.5 ± 2.5 | −0.5 ± 1.1 |
| DOPC/DOPG | −1.6 ± 1.1 | −8.0 ± 1.3 |
| DOPC/PI | 31.1 ± 1.9 | 5.8 ± 1.2 |
N-terminal Domain Mediates the Interaction of PimA with Anionic SUVs
PimA contains three tryptophan residues located in different regions within the N-terminal domain (Fig. 3). Trp-82 is located in the amphipathic α-helix 2 (residues 68–79), facing the hydrophobic core of the enzyme (11, 14). Trp-154 is located in the mobile helical hairpin (residues 134–157) of the N terminus that undergoes the secondary structural reshuffling (12), whereas Trp-349 is placed in the hinge between both N- and C-terminal Rossmann-fold domains (residues 343–352). This structural configuration of the tryptophan residues in PimA placed us in a favorable position to study its molecular mechanism of association to the membrane by using a combination of fluorescence spectroscopy techniques. To investigate the interaction of the N-terminal domain of PimA to phospholipids, FRET studies were performed with SUVs labeled with dansyl-DHPE. It is worth noting that the dansyl group, which acts as acceptor of the fluorescence energy transfer, is attached to the polar head of DHPE (see “Experimental Procedures” for details). As depicted in Table 2, both PI and DOPG anionic SUVs showed a significant increase of FRET from wild type PimA compared with that observed with neutral DOPC liposomes, indicating that the N-terminal domain of PimA is clearly involved in the association. Therefore, the interaction between the positively charged residues mainly located in the amphipathic α2-helix of the N-terminal domain and the negatively charged residues located at the surface of the lipids seems to be the determinant for the interaction to take place (11, 14). To test this hypothesis, upon protein addition, the ionic strength was increased 10 times to generate an electrostatic competition with the SUVs. As shown in Table 2, no significant changes were observed when PimA was incubated with neutral DOPC SUVs. However, the addition of NaCl once PimA was bound to the anionic DOPG and PI SUVs resulted in a drastic decrease of FRET, confirming that the association of the N-terminal domain of PimA to the surface of anionic SUVs is mainly due to attractive electrostatic interactions.
FIGURE 3.

Model for PimA membrane association. Trp-82 in yellow is located in the amphipathic helix 2 shown in blue. Trp-154 in orange is in the mobile helical hairpin at the reshuffling region. The Trp-349 is shown in red between the N-and C-terminal domains.
TABLE 2.
ΔFRET between PimA and dansyl-DHPE at low and high NaCl concentration
The mean (n = 3) and the standard deviation are represented. For an easier interpretation, values are multiplied ×100.
| PimA WT | ΔFRET·100 ± S.D., low ionic strength | ΔFRET·100 ± S.D., high ionic strength |
|---|---|---|
| SUV type | ||
| DOPC | −1.6 ± 5.0 | 0.7 ± 1.0 |
| DOPC/DOPG | 17.2 ± 1.4 | −8.0 ± 3.0 |
| DOPC/PI | 16.5 ± 4.0 | −3.2 ± 2.5 |
| PimA Trp-82 | ||
| DOPC | 0.4 ± 1.4 | 1.4 ± 1.6 |
| DOPC/DOPG | 16.1 ± 10.3 | −3.8 ± 2.4 |
| DOPC/PI | 14.1 ± 0.7 | 0.0 ± 0.6 |
| PimA Trp-154 | ||
| DOPC | −4.5 ± 1.3 | 1.0 ± 3.0 |
| DOPC/DOPG | 6.8 ± 8.1 | −0.8 ± 2.2 |
| DOPC/PI | 3.0 ± 2.2 | 0.7 ± 0.5 |
| PimA Trp-349 | ||
| DOPC | 0.2 ± 4.6 | 1.5 ± 1.2 |
| DOPC/DOPG | 3.9 ± 5.6 | −1.8 ± 2.8 |
| DOPC/PI | 4.4 ± 1.4 | −0.1 ± 1.8 |
Dissecting the Structural Determinants of PimA Interaction with SUVs
To study in detail the regions within the N-terminal domain involved in the interaction of PimA with anionic SUVs, we designed three double mutants in which the other tryptophan residues were replaced by phenylalanine. The mutant PimA W154F/W349F, where the only tryptophan residue present is Trp-82, was named as the PimA-Trp-82 variant, because we determined the contribution of this particular residue to the SUV association. Therefore, the double mutants PimA W82F/W439F and PimA W82F/W154F were named PimA-Trp-154 and PimA-Trp-349 variants, respectively.
Amphipathic α-helices represent a common membrane-binding motif in proteins. According to the currently accepted “helical anchor” model, an amphipathic α-helix packs its aliphatic side against the hydrophobic protein core in the soluble state and rotates it to present this side to the membrane in the bound state while the charge side of the helix faces the lipid headgroup region (22–26). We have previously postulated that the amphipathic α2-helix is an important element to determine the binding of PimA to the membrane (11, 14). To further investigate the implication of this amphipathic α2-helix in the membrane interaction process, we performed fluorescence spectroscopy studies with the PimA-Trp-82 variant. The FIR 310/340 of PimA-Trp-82 strongly increased in the presence of DOPG and PI SUVs, indicating that the surrounding of Trp-82 reaches an apolar environment (Table 3). Moreover, a high FRET was established between the Trp-82 residue and anionic dansylated DOPG and PI liposomes (Table 2). Upon NaCl addition, the FRET values decreased but were still detectable. The intrinsic anisotropy data revealed that Trp-82 is particularly constrained mainly in the presence of PI SUVs (Table 4). Altogether, the experimental data indicate that Trp-82 undergoes a strong anionic SUV-dependent conformational change that localizes it in a more apolar environment, probably in the vicinity of the negatively charged lipid headgroups and the very first carbon atoms of the acyl chains. Anionic SUVs were able to induce this interaction, but only PI seems to stabilize/bury the local structure surrounding the Trp-82, including the amphipathic α2-helix.
TABLE 3.
Intrinsic fluorescence parameter FIR
The mean (n = 3) and the standard deviation are represented. The FIR parameter is calculated from the intensities at 320 and 360 nm for all the proteins.
| FIR ± S.D. | |
|---|---|
| PimA WT | |
| In solution | 1.30 ± 0.01 |
| DOPC | 1.35 ± 0.01 |
| DOPC/DOPG | 1.25 ± 0.04 |
| DOPC/PI | 1.43 ± 0.01 |
| PimA Trp-82 | |
| In solution | 1.03 ± 0.01a |
| DOPC | 1.04 ± 0.03a |
| DOPC/DOPG | 1.18 ± 0.01a |
| DOPC/PI | 1.29 ± 0.04a |
| PimA Trp-154 | |
| In solution | 1.15 ± 0.05 |
| DOPC | 1.23 ± 0.04 |
| DOPC/DOPG | 1.31 ± 0.01 |
| DOPC/PI | 1.35 ± 0.03 |
| PimA Trp-349 | |
| In solution | 1.70 ± 0.01 |
| DOPC | 1.74 ± 0.004 |
| DOPC/DOPG | 1.66 ± 0.005 |
| DOPC/PI | 1.84 ± 0.02 |
a Noteworthy, the FIR parameter for PimA Trp-82 is calculated using the intensities at 310 and 340 nm due to a large change in the shape of the spectra as a consequence of the strong shift in the maximum of emission.
TABLE 4.
Tryptophan anisotropy changes upon SUV interaction
The mean (n = 303) and standard deviations are represented. The t test showed no statistical significance between Trp-82 in solution versus the presence of DOPC SUVs (p = 0.344). Likewise, PimA Trp-154 t test showed no statistical significance between Trp-154 in solution versus the presence of DOPC SUVs (p = 0.3051). Nevertheless, PimA Trp-349 values revealed significant differences between in solution versus DOPC and/or DOPC/DOPG. However, the values obtained in the presence of PI-containing liposomes were not statistically significant (n = 303; p = 0.0589).
| r ± S.D. | |
|---|---|
| PimA-Trp-2 | |
| In solution | 0.12 ± 0.01 |
| DOPC | 0.12 ± 0.01 |
| DOPC/DOPG | 0.12 ± 0.01 |
| DOPC/PI | 0.14 ± 0.04 |
| PimA-Trp-54 | |
| In solution | 0.10 ± 0.01 |
| DOPC | 0.10 ± 0.01 |
| DOPC/DOPG | 0.12 ± 0.03 |
| DOPC/PI | 0.12 ± 0.01 |
| PimA-Trp-349 | |
| In solution | 0.13 ± 0.02 |
| DOPC | 0.12 ± 0.02 |
| DOPC/DOPG | 0.11 ± 0.02 |
| DOPC/PI | 0.13 ± 0.04 |
Trp-154 residue is located in a region of the N-terminal domain of PimA (residues 129–163), exhibiting important conformational variability, including the reshuffling of secondary structure elements in the form of α-helix-to-β-strand and β-strand-to-α-helix transitions (12, 15). The FIR 320/360 of the PimA-Trp-154 ratio of fluorescence intensities indicates that Trp-154 is exposed to a more apolar environment in the presence of DOPG or PI-containing SUVs (Table 3). However, the effect is less pronounced to that observed with Trp-82. The intrinsic anisotropy data suggest that Trp-154 is also constrained in the presence of PI SUVs (Table 4). Interestingly, the FRET values revealed a lower energy transfer between Trp-154 and anionic liposomes, compared with that measured with Trp-82. Altogether the experimental data suggest that the Trp-154 surrounding region is in contact with the anionic phospholipids; however, this behavior is less apparent than that observed for the Trp-82 surrounding region. This effect might reflect the structural plasticity of the reshuffling region, which adopts several transient conformations at the membrane surface.
Finally, the crystal structures of PimA do not predict the hinge region between the N- and C-terminal domains to be directly involved in lipid acceptor recognition or membrane association. Supporting this notion, the FIR 320/360 of PimA-Trp-349 revealed that the tryptophan residue is rather buried in the protein structure. Specifically, no significant changes were detected with DOPC or DOPG SUVs. In addition, a slight shift to more apolar localization in the presence of PI liposomes was observed (Table 3). Moreover, FRET studies revealed a low energy transfer only between Trp-349 and anionic dansylated SUVs (Table 2). The intrinsic anisotropy data revealed that Trp-349 is less constrained in the presence of liposomes, reflecting a global effect on the protein structure upon addition of liposomes (Table 4).
Model for PimA Membrane Interaction
The importance of nonspecific electrostatic and apolar interactions as driving forces for membrane association of peripheral/monotopic membrane proteins has been well established (27, 28). Our experimental data clearly demonstrate that PimA preferentially binds to non-substrate anionic phospholipids, which are able to stimulate catalysis (11). From a thermodynamic point of view, major contributions to membrane interaction free energies arise from (i) the electrostatic attraction between protein basic residues and membrane acidic phospholipids and from (ii) the desolvation of protein and membrane during the association process. Inspection of the PimA crystal structures suggests that a region located in the N-terminal domain, adjacent to the substrate binding cleft, could fulfill these criteria to mediate protein-membrane interaction. Specifically, PimA exhibits a high isoelectric point (pI = 8.1) at the N-terminal domain, reflecting the presence of positively charged residues at its surface (11, 29–33). These positively charged residues are mostly localized in the amphipathic α2-helix, including Arg-77, Lys-78, Lys-80, and Lys-81. Interestingly, the substitution of this cluster by serine residues completely inactivated PimA and drastically impaired the ability of the mutant to bind phospholipids (11). Our fluorescent spectroscopy data indicate that the α2-helix is positioned in the vicinity of the negatively charged lipid headgroups and the very first carbon atoms of the acyl chains. Moreover, theoretical calculations also suggested that the N-terminal domain of PimA displays the minimal desolvation energies of the entire protein (11, 12, 14). We propose that, due to its intrinsic amphipathicity, PimA first orients the N-terminal domain facing the membrane (Fig. 3).
To determine the conformational state of PimA upon/during membrane interaction, the “protein trinity” paradigm represents a useful tool to understand the protein-membrane association mechanism. In contrast to the traditional lock-and-key models, based on relatively rigid and well defined structures, the protein trinity paradigm propose that order-to-disorder and disorder-to-order transitions could lead a native disorder protein to adopt a wide range of conformations suitable for the interaction with different targets (34, 35). This hypothesis might explain the process by which the N-terminal domain of PimA associates with the membrane. Similarly to the well known case of cytochrome c, PimA can follow a coupled reaction mechanism where either the exothermic energy provided by (i) the electrostatic interactions between the positively charged protein residues and the negatively charged lipid headgroups, and (ii) the weakly denaturing conditions of the negatively charged membranes, non-bulk pH and dielectric constant, are responsible for the generation of an MG-like state (25, 36–38). Specifically, the formation of a PimA-MG-like state upon anionic membrane interaction is experimentally supported by our structural and biophysical approaches. First, the crystal and solution structures of PimA revealed several unprecedented conformational changes, including β-strand-to-α-helix and α-helix-to-β-strand transitions, reflecting its inherent plasticity/variability. Second, single molecule force spectroscopy indicates that PimA not only exhibits weak mechanical stability but also unfolds the following heterogeneous multiple step mechanical unfolding pathways akin to MG-like states (15). Finally, the combination of near UV CD and thermally induced unfolding followed by CD and differential scanning calorimetry revealed (i) a loss of tertiary structure and (ii) a loss of the cooperativity during the thermally induced unfolding process in the presence of non-substrate anionic vesicles (herein and see Ref. 14). These characteristics are typical of membrane-bound proteins lacking tertiary structure interactions, presumably as in MG-like states (17, 39).
How does PimA specifically recognize the lipid acceptor substrate PI? The answer to this question remains a challenge in part because no direct structural information is available for PimA in the presence of PI. Molecular docking approaches combined with site-directed mutagenesis activity assays support the location of the polar head of PI in a well defined pocket at the N-terminal domain. This cavity comprises the connecting loops β1-α1, β3-α2, and β6-α4. As a consequence, the inositol phosphate ring places with its O2 atom in close contact with the mannose ring of GDP-Man (11). It is worth noting that PimA requires the substrate acyl chains for catalysis, because glycerophosphoryl-myo-inositol (GPI, the fully deacylated form of PI) was not a substrate of the enzyme either in the absence or in the presence of anionic surfactants and (ii) no binding of GPI could be detected to PimA (Kd >100 μm). Our fluorescence spectroscopy data clearly indicate that the binding of PimA to PI introduces structural constraints in a probe located at the C6 position, strongly suggesting that PimA markedly affects the lipid bilayer fluidity in the acyl region close to the PI headgroup.
Author Contributions
P. M. A., A. C., and M. E. G. conceived the project. A. R.-U., M. A. M., and N. C. performed the biochemical and biophysical studies. A. R.-U., P. M. A., A. C., and M. E. G. wrote the manuscript. All authors reviewed the results and approved th final version of the manuscript.
Acknowledgments
We gratefully acknowledge Pedro Arrasate (Unit of Biophysics, Spain) for technical assistance. We also thank all members of the Structural Glycobiology Group for valuable scientific discussions.
This work was supported by the European Commission Contract HEALTH-F3-2011-260872, the Spanish Ministry of Economy and Competitiveness Contract BIO2013-49022-C2-2-R, the Basque Government (to M. E. G.), and Institut Pasteur (to A. C. and P. M. A.). The authors declare that they have no conflicts of interest with the contents of this article.
- PIM
- phosphatidyl-myo-inositol mannoside
- LAM
- lipoarabinomannan
- PI
- phosphatidyl-myo-inositol
- diC8PI
- 1,2-dioctanoyl-sn-glycero-3-phosphoinositol
- NBD
- 2-(6-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)
- SUV
- small unilamellar vesicle
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphoglycerol
- HPC
- hexanoyl-1-hexadecanoyl-sn-glycero-3-phosphocholine
- CMC
- critical micellar concentration
- MST
- MicroScale Thermophoresis
- MG
- molten globule
- DHPE
- dihexadecanoyl-sn-glycero-3-phosphoethanolamine
- dansyl-DHPE
- N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine.
References
- 1. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., and Jackson M. (2009) Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Angala S. K., Belardinelli J. M., Huc-Claustre E., Wheat W. H., and Jackson M. (2014) The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 49, 361–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guerin M. E., Korduláková J., Alzari P. M., Brennan P. J., and Jackson M. (2010) Molecular basis of phosphatidyl-myo-inositol mannoside biosynthesis and regulation in mycobacteria. J. Biol. Chem. 285, 33577–33583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morita Y. S., Fukuda T., Sena C. B., Yamaryo-Botte Y., McConville M. J., and Kinoshita T. (2011) Inositol lipid metabolism in mycobacteria: biosynthesis and regulatory mechanisms. Biochim. Biophys. Acta 1810, 630–641 [DOI] [PubMed] [Google Scholar]
- 5. Korduláková J., Gilleron M., Mikusova K., Puzo G., Brennan P. J., Gicquel B., and Jackson M. (2002) Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis. PimA is essential for growth of mycobacteria. J. Biol. Chem. 277, 31335–31344 [DOI] [PubMed] [Google Scholar]
- 6. Boldrin F., Ventura M., Degiacomi G., Ravishankar S., Sala C., Svetlikova Z., Ambady A., Dhar N., Kordulakova J., Zhang M., Serafini A., Vishwas K. G., Vishwas V. G., Kolly G. S., Kumar N., et al. (2014) The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J. Bacteriol. 196, 3441–3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guerin M. E., Kaur D., Somashekar B. S., Gibbs S., Gest P., Chatterjee D., Brennan P. J., and Jackson M. (2009) New insights into the early steps of phosphatidylinositol mannoside biosynthesis in mycobacteria: PimB′ is an essential enzyme of Mycobacterium smegmatis. J. Biol. Chem. 284, 25687–25696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Albesa-Jové D., Giganti D., Jackson M., Alzari P. M., and Guerin M. E. (2014) Structure-function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology 24, 108–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burn P. (1988) Amphitropic proteins: a new class of membrane proteins. Trends Biochem. Sci. 13, 79–83 [DOI] [PubMed] [Google Scholar]
- 10. Guerin M. E., Buschiazzo A., Korduláková J., Jackson M., and Alzari P. M. (2005) Crystallization and preliminary crystallographic analysis of PimA, an essential mannosyltransferase from Mycobacterium smegmatis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 518–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guerin M. E., Kordulakova J., Schaeffer F., Svetlikova Z., Buschiazzo A., Giganti D., Gicquel B., Mikusova K., Jackson M., and Alzari P. M. (2007) Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 282, 20705–20714 [DOI] [PubMed] [Google Scholar]
- 12. Giganti D., Albesa-Jové D., Urresti S., Rodrigo-Unzueta A., Martínez M. A., Comino N., Barilone N., Bellinzoni M., Chenal A., Guerin M. E., and Alzari P. M. (2015) Secondary structure reshuffling modulates glycosyltransferase function at the membrane. Nat. Chem. Biol. 11, 16–18 [DOI] [PubMed] [Google Scholar]
- 13. Brodhun F., and Tittmann K. (2015) Membrane enzymes: transformers at the interface. Nat. Chem. Biol. 11, 102–103 [DOI] [PubMed] [Google Scholar]
- 14. Guerin M. E., Schaeffer F., Chaffotte A., Gest P., Giganti D., Korduláková J., van der Woerd M., Jackson M., and Alzari P. M. (2009) Substrate-induced conformational changes in the essential peripheral membrane-associated mannosyltransferase PimA from mycobacteria: implications for catalysis. J. Biol. Chem. 284, 21613–21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giganti D., Alegre-Cebollada J., Urresti S., Albesa-Jové D., Rodrigo-Unzueta A., Comino N., Kachala M., López-Fernández S., Svergun D. I., Fernández J. M., and Guerin M. E. (2013) Conformational plasticity of the essential membrane-associated mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 288, 29797–29808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rebecchi M. J., Eberhardt R., Delaney T., Ali S., and Bittman R. (1993) Hydrolysis of short acyl chain inositol lipids by phospholipase C-δ1. J. Biol. Chem. 268, 1735–1741 [PubMed] [Google Scholar]
- 17. Lindeberg M., Zakharov S. D., and Cramer W. A. (2000) Unfolding pathway of the colicin E1 channel protein on a membrane surface. J. Mol. Biol. 295, 679–692 [DOI] [PubMed] [Google Scholar]
- 18. Mosbahi K., Walker D., Lea E., Moore G. R., James R., and Kleanthous C. (2004) Destabilization of the colicin E9 endonuclease domain by interaction with negatively charged phospholipids: implications for colicin translocation into bacteria. J. Biol. Chem. 279, 22145–22151 [DOI] [PubMed] [Google Scholar]
- 19. Karst J. C., Barker R., Devi U., Swann M. J., Davi M., Roser S. J., Ladant D., and Chenal A. (2012) Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 287, 9200–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., et al. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bychkova V. E., Pain R. H., and Ptitsyn O. B. (1988) The ‘molten globule’ state is involved in the translocation of proteins across membranes? FEBS Lett. 238, 231–234 [DOI] [PubMed] [Google Scholar]
- 22. Chenal A., Vernier G., Savarin P., Bushmarina N. A., Gèze A., Guillain F., Gillet D., and Forge V. (2005) Conformational states and thermodynamics of α-lactalbumin bound to membranes: a case study of the effects of pH, calcium, lipid membrane curvature and charge. J. Mol. Biol. 349, 890–905 [DOI] [PubMed] [Google Scholar]
- 23. Man P., Montagner C., Vernier G., Dublet B., Chenal A., Forest E., and Forge V. (2007) Defining the interacting regions between apomyoglobin and lipid membrane by hydrogen/deuterium exchange coupled to mass spectrometry. J. Mol. Biol. 368, 464–472 [DOI] [PubMed] [Google Scholar]
- 24. Montagner C., Perier A., Pichard S., Vernier G., Ménez A., Gillet D., Forge V., and Chenal A. (2007) Behavior of the N-terminal helices of the diphtheria toxin T domain during the successive steps of membrane interaction. Biochemistry 46, 1878–1887 [DOI] [PubMed] [Google Scholar]
- 25. Halskau Ø., Muga A., and Martínez A. (2009) Linking new paradigms in protein chemistry to reversible membrane-protein interactions. Curr. Protein Pept. Sci. 10, 339–359 [DOI] [PubMed] [Google Scholar]
- 26. Subrini O., Sotomayor-Pérez A. C., Hessel A., Spiaczka-Karst J., Selwa E., Sapay N., Veneziano R., Pansieri J., Chopineau J., Ladant D., and Chenal A. (2013) Characterization of a membrane-active peptide from the Bordetella pertussis CyaA toxin. J. Biol. Chem. 288, 32585–32598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. White S. H., and Wimley W. C. (1999) Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28, 319–365 [DOI] [PubMed] [Google Scholar]
- 28. Johnson J. E., and Cornell R. B. (1999) Amphitropic proteins: regulation by reversible membrane interactions (review). Mol. Membr. Biol. 16, 217–235 [DOI] [PubMed] [Google Scholar]
- 29. Schwartz R., Ting C. S., and King J. (2001) Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 11, 703–709 [DOI] [PubMed] [Google Scholar]
- 30. Edman M., Berg S., Storm P., Wikström M., Vikström S., Ohman A., and Wieslander A. (2003) Structural features of glycosyltransferases synthesizing major bilayer and nonbilayer-prone membrane lipids in Acholeplasma laidlawii and Streptococcus pneumoniae. J. Biol. Chem. 278, 8420–8428 [DOI] [PubMed] [Google Scholar]
- 31. Li L., Storm P., Karlsson O. P., Berg S., and Wieslander A. (2003) Irreversible binding and activity control of the 1,2-diacylglycerol 3-glucosyltransferase from Acholeplasma laidlawii at an anionic lipid bilayer surface. Biochemistry 42, 9677–9686 [DOI] [PubMed] [Google Scholar]
- 32. Ge C., Georgiev A., Öhman A., Wieslander Å., and Kelly A. A. (2011) Tryptophan residues promote membrane association for a plant lipid glycosyltransferase involved in phosphate stress. J. Biol. Chem. 286, 6669–6684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rocha J., Sarkis J., Thomas A., Pitou L., Radzimanowski J., Audry M., Chazalet V., de Sanctis D., Palcic M. M., Block M. A., Girard-Egrot A., Maréchal E., and Breton C. (2016) Structural insights and membrane binding properties of MGD1, the major galactolipid synthase in plants. Plant J. 85, 622–633 [DOI] [PubMed] [Google Scholar]
- 34. Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., et al. (2001) Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 35. Dyson H. J., and Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 36. Muga A., Mantsch H. H., and Surewicz W. K. (1991) Membrane binding induces destabilization of cytochrome c structure. Biochemistry 30, 7219–7224 [DOI] [PubMed] [Google Scholar]
- 37. Heimburg T., and Marsh D. (1993) Investigation of secondary and tertiary structural changes of cytochrome c in complexes with anionic lipids using amide hydrogen exchange measurements: an FTIR study. Biophys. J. 65, 2408–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chenal A., Savarin P., Nizard P., Guillain F., Gillet D., and Forge V. (2002) Membrane protein insertion regulated by bringing electrostatic and hydrophobic interactions into play. A case study with the translocation domain of diphtheria toxin. J. Biol. Chem. 277, 43425–43432 [DOI] [PubMed] [Google Scholar]
- 39. Chenal A., Prongidi-Fix L., Perier A., Aisenbrey C., Vernier G., Lambotte S., Haertlein M., Dauvergne M. T., Fragneto G., Bechinger B., Gillet D., Forge V., and Ferrand M. (2009) Deciphering membrane insertion of the diphtheria toxin T domain by specular neutron reflectometry and solid-state NMR spectroscopy. J. Mol. Biol. 391, 872–883 [DOI] [PubMed] [Google Scholar]