

Abstract
Cryptochromes constitute a group of flavin-binding blue light receptors in bacteria, fungi, plants, and insects. Recently, the response of cryptochromes to light was extended to nearly the entire visible spectral region on the basis of the activity of the animal-like cryptochrome aCRY in the green alga Chlamydomonas reinhardtii. This finding was explained by the absorption of red light by the flavin neutral radical as the dark state of the receptor, which then forms the anionic fully reduced state. In this study, time-resolved UV-visible spectroscopy on the full-length aCRY revealed an unusually long-lived tyrosyl radical with a lifetime of 2.6 s, which is present already 1 μs after red light illumination of the flavin radical. Mutational studies disclosed the tyrosine 373 close to the surface to form the long-lived radical and to be essential for photoreduction. This residue is conserved exclusively in the sequences of other putative aCRY proteins distinguishing them from conventional (6–4) photolyases. Size exclusion chromatography showed the full-length aCRY to be a dimer in the dark at 0.5 mm injected concentration with the C-terminal extension as the dimerization site. Upon illumination, partial oligomerization was observed via disulfide bridge formation at cysteine 482 in close proximity to tyrosine 373. The lack of any light response in the C-terminal extension as evidenced by FTIR spectroscopy differentiates aCRY from plant and Drosophila cryptochromes. These findings imply that aCRY might have evolved a different signaling mechanism via a light-triggered redox cascade culminating in photooxidation of a yet unknown substrate or binding partner.
Keywords: algae, biophysics, cryptochrome, flavoprotein, infrared spectroscopy (IR spectroscopy), photoreceptor, ultraviolet-visible spectroscopy (UV-Vis spectroscopy)
Introduction
Cryptochromes represent a group of diverse sensory photoreceptors present in all kingdoms of life (1, 2). Together with the UV-light-dependent DNA repair enzymes, the photolyases (3), they constitute the cryptochrome/photolyase family. Members of this family share a highly conserved photolyase homology region (PHR)2, which comprises ∼500 amino acids and carries a non-covalently bound flavin adenine dinucleotide (FAD) as a chromophore. The C-terminal extension (CCT) present in many cryptochromes and photolyases is strongly variable in amino acid composition and length and has been shown to be crucial for signal transduction in the Arabidopsis cryptochrome AtCRY1 (4). The diverse subfamilies of cryptochromes comprise proteins acting as central blue light sensors in bacteria, fungi, plants, and insects (animal type I CRY) (1). Moreover, CRYs are also found as the light-independent, central part of the oscillator of the biological clock in mammals (animal type II CRY) (5) and as a mediator for light-dependent magnetosensitivity in flies (6). Opposed to these findings, DASH cryptochromes have been found to repair lesions in single-stranded DNA and double-stranded loop-structured DNA in vitro (7, 8). Therefore, DASH cryptochromes are more similar in terms of functionality to photolyases than cryptochromes. Among the photolyases, two different types are separated depending on their ability to either repair cyclobutane pyrimidine dimers (CPD) (9) or (6–4) photoproducts (10). Animal type II CRY are closely related to eukaryotic (6–4) photolyases whereas plant cryptochromes are homologous to CPD photolyases. In contrast, prokaryotic (6–4) photolyases (11) and bacterial cryptochromes (12) form a distant subfamily.
The paradigm of cryptochromes and other flavoproteins as classical blue light receptors has been challenged recently by the finding that the animal-like cryptochrome (aCRY) from the green alga Chlamydomonas reinhardtii strongly influences gene expression not only in response to blue but also to yellow and red light in vivo (13). These genes code for proteins involved in chlorophyll and carotenoid biosynthesis, light-harvesting complexes, nitrogen metabolism, cell cycle control, and the circadian clock. The extended spectral sensitivity was explained by the presence of the flavin neutral radical (FADH•) in the dark form of the receptor (Fig. 1A) (13) as opposed to the oxidized flavin (FADox) found in plant cryptochromes (14, 15). The absorption spectrum of FADH• covers almost the full visible spectrum of light extending up to 680 nm in agreement with the action spectrum of aCRY (13). In vitro, the predominant part of aCRY was found in the oxidized state after purification. Accordingly, formation of FADH• requires a pre-illumination with blue light which was not applied in the in vivo experiments (13). However, FTIR spectroscopy revealed structural changes in the protein moiety only in the transition from FADH• to the anionic fully reduced state (FADH−) of aCRY providing further support for FADH• as the dark state of the chromophore (16). Strikingly, these changes in turn structures were not detected in the closely related Xenopus laevis (6–4) photolyase (17). The lifetime of FADH• state in vitro was strongly sensitive to alterations of the pH but not of the oxygen level in contrast to other cryptochromes (18–20).
FIGURE 1.

Photoreaction sequence and position of selected residues of aCRY and its response to pre-illumination with blue light. A, FAD neutral radical (FADH•, top left) represents the dark state in vivo and is photoreduced by light of λ < 680 nm into the anionic fully reduced state of FAD (FADH−, top right). In vitro, FADH• slowly decays into the oxidized state (bottom right). It is regenerated by pre-illumination with light of λ < 510 nm, possibly via the flavin anion radical (bottom left). B, position of residues tyrosine 373, tyrosine 393, and cysteine 482 investigated in this study relative to the FAD chromophore in aCRY. Tryptophan residues form the classical tryptophan triad. The structural model was derived from the crystal structure of Arabidopsis (6–4) photolyase (51). C, pre-illumination of aCRY in vitro with blue light (450 nm) for 10 s generates FADH• from FADox. An additional band is present at 416 nm directly after illumination which is lost within 30 s in the dark. The corresponding absorption difference spectrum is shown in the inset.
The molecular mechanisms underlying these extraordinary characteristics of aCRY have remained undisclosed up to now because of missing time-resolved information on the early events after red light illumination. Previously, time-resolved UV-visible (UV-vis) spectroscopy on cryptochromes and photolyases has revealed the involvement of transient radical species formed from FAD, tryptophan, and tyrosine as part of the electron transfer cascade (21–27). Tryptophan neutral radicals (Trp•) exhibit a broad band centered at around 510 nm (28), whereas tyrosyl radicals (TyrO•) are characterized by two sharp, adjacent bands at 388 and 408 nm (29, 30). Moreover, the formation of these amino acid radicals in the electron transfer cascade has been disclosed by time-resolved EPR studies (31, 32). However, to date the photoreduction process of the FADH• has not been studied time-resolved in the cryptochrome as well as (6–4) photolyase families.
In this study, we focused on resolving the events in aCRY starting from the FADH• state by its selective induction using illumination with red light, because the blue light-induced conversion of FADox in aCRY is not considered to be physiologically relevant (13). To elucidate the red light-induced processes, we employed transient UV-vis spectroscopy, size exclusion chromatography (SEC), and FTIR difference spectroscopy on the full-length aCRY as well as on point mutants Y393A, Y373F, C482A (Fig. 1B), and aCRYΔCCT lacking the 99 residues of the CCT. We identified the contribution from a remarkably long-lived TyrO• formed by Tyr-373 close to the surface (Fig. 1B), which is conserved in aCRY homologues but not in any other subfamily of cryptochromes and photolyases. Furthermore, we investigated the role of the CCT in the light-induced structural response and the oligomerization of aCRY.
Experimental Procedures
Generation of aCRY Mutants
aCRYΔCCT coding for amino acids 1 to 496 of the aCRY gene, lacking the 99 amino acids of its CCT, was codon-adapted for E. coli (synthesized by Geneart) and cloned into pET28a(+) (Novagen), providing a 6x His-Tag at the C terminus by using the restriction enzyme sites NcoI and HindIII. Mutation Y393A was inserted into the full-length codon-adapted sequence of aCRY (synthesized by Geneart (13)) by the replacement of a 739 bp BstEII and HindIII fragment with a fragment containing the codon-adapted sequence for the substituted amino acid Y393A. Full-length aCRY-Y393A was ligated via NcoI and HindIII restriction sites into vector pET28a(+). The Y373F and C482A mutations were inserted into the full-length aCRY gene in the pET28a(+) vector using phosphorylated back-to-back primers, of which one primer contained the mutation. To amplify the whole plasmid, Phusion DNA polymerase (New England Biolabs) was used in the polymerase chain reaction. The reaction products were ligated afterward. The amino acid exchanges in all resulting plasmids were verified by dideoxy sequencing.
Expression and Purification
Expression and purification of aCRY and its variants were conducted following published procedures (13). Finally, the proteins were obtained in a 50 mm sodium phosphate buffer, pH 7.0, 100 mm NaCl, 20% (v/v) glycerol.
Millisecond Time-resolved UV-Vis Spectroscopy
The concentration of the sample was adjusted to A447 = 0.3. An HR2000+ spectrometer with DH-2000-BAL light source (Ocean Optics) was used for experiments in the millisecond to second time regime modified with a mesh filter with 35% transmission to avoid sample conversion by the probe light. For illumination, a 451 nm LED (Luxeon Star, Lumileds) with an intensity of 67 milliwatt/cm2 (full width at half maximum (FWHM) of 20 nm) and a 632 nm LED (Luxeon Star, Lumileds) with an intensity of 95 milliwatt/cm2 (FWHM of 15 nm) at the sample were attached to the sample holder perpendicular to the measuring beam. For the generation of FADH•, aCRY was illuminated for 10 s at 451 nm in a 2 × 10 mm fluorescence cuvette (Helma). FADH− was produced by illuminating the sample for 2 s or 10 s at 632 nm. A continuous series of spectra were recorded before, during and after illumination with an integration time of 2 ms and a time resolution of ∼40 ms. Difference spectra were calculated and summarized on a logarithmic time scale to increase the signal-to-noise ratio using MATLAB (The Mathworks).
Nanosecond Time-resolved UV-Vis Spectroscopy
The concentration of the sample was adjusted to A447 = 0.5. The experimental setup for time-resolved UV-vis spectroscopy on slowly recovering systems has been described previously (29). aCRY was pre-illuminated for 15 s under stirring to generate FADH• using a 455 nm LED (Luxeon Star, Lumileds) with an intensity of 10 milliwatt/cm2 at the sample (FWHM of 20 nm). For excitation of FADH•, a 630 nm pulse with 10 ns duration and 2 mJ/cm2 energy density was generated by a tunable optical parametric oscillator (Opta), which was pumped by the 355-nm third harmonic of a Nd:YAG laser (Quanta-Ray GCR-12, Spectra Physics). Laser pulses with a repetition rate of (1.6 s)−1 were selected by a shutter. Multiple excitations were minimized by rotating a magnetic stirring bar inside the cuvette for 450 ms after each detection. A fast sample exchange within a total volume of 2.5 ml was ensured by a horizontal geometry of the excitation beam. Moreover, contributions from previous excitations were avoided by alternating the recording of reference and signal spectra. All experiments were conducted at 20 °C. Spectra were recorded at 1 μs, 10 μs, 100 μs, and 20 ms after excitation with an integration time of 1 μs, 5 μs, 100 μs, and 100 μs, respectively. Each spectrum was obtained by averaging 10–61 separate experiments, in which each sample was excited 15 times.
For the comparison of the wild type and the Y373F mutant of aCRY, ascorbic acid was added as a reducing agent to a final concentration of 3 mm. The samples were pre-illuminated with blue light for 1 s and 45 s for wild type and Y373F mutant, respectively. For excitation, the 532 nm second harmonic of a Nd:YAG laser (Ultra 100, Quantel) was used at a repetition rate of (1.6 s)−1 with a pulse duration of 10 ns and an energy density of 15 mJ/cm2 and 30 mJ/cm2 for wild type and mutant, respectively. The integration time was set to 500 ns for the difference spectra recorded at 500 ns. Each spectrum of aCRY was obtained by averaging 8–20 separate experiments, in which each sample was excited 15 times.
FTIR Experiments
The samples were concentrated to an A447 ∼27 by ultrafiltration using Vivaspin 500 filter devices (Sartorius, 50 kDa cutoff). During centrifugation at 15,000 × g, the protein was washed three times with 20 mm sodium phosphate buffer, pH 7.8, 100 mm NaCl, 1% (v/v) glycerol. A 1.8-μl droplet of the sample solution was applied to a BaF2 window (20 mm diameter) and kept at 20 °C and atmospheric pressure for up to 30 s to gently reduce the water content. The samples were sealed with a second BaF2 window. Thus, a well-hydrated film with an absorbance ratio of amide I/water (1650 cm−1) to amide II (1550 cm−1) of 2.3–2.5 was obtained. An appropriate hydration of the sample is essential to ensure that the full extent of changes in secondary structure of the protein is detected.
IR experiments were performed on an IFS 66v spectrometer (Bruker) equipped with a photoconductive mercury cadmium telluride (MCT) detector at a spectral resolution of 2 cm−1. The difference spectra were obtained with a long wave pass filter (OCLI) cutting off infrared light above 2256 cm−1. The experiments were performed at 20 °C. The blue light response of aCRY was induced by illumination for 1 s with a 451-nm LED equipped with a diffusion disc and yielding an intensity of 32 milliwatt/cm2 at the sample. Red light illumination was conducted for 10 s with a 632-nm LED with an intensity of 40 milliwatt/cm2 at the sample. To obtain a representative difference spectrum, 512 scans were averaged.
Size Exclusion Chromatography
SEC was performed using an Äkta purifier (GE Healthcare) with a Superdex200 10/300 GL column (GE Healthcare) at 4 °C. For equilibration and elution, 50 mm phosphate buffer at pH 7.0 and 150 mm NaCl were used. Aliquots of 100 μl with a protein concentration of 0.5 mm were centrifuged at 21,400 × g for 10 min at 4 °C. For the investigation of aCRY carrying FADox and FADH•, loading of the samples and the SEC were performed in the dark. FADH• was obtained by illuminating the sample for 10 s with two 451 nm LEDs (Luxeon Star, Lumileds) with an intensity of 67 milliwatt/cm2 each at the sample. The elution profiles were recorded at 447 and 630 nm to ensure that only protein with the flavin bound contributes. For the generation of FADH−, samples were illuminated for 45 s with the two 451 nm LEDs followed by 30 s in darkness and illumination for 20 s with a 451 nm LED and a 632 nm LED with an intensity of 64 milliwatt/cm2. Alternatively, the latter two LEDs were used to illuminate the sample for 45 s. This variation in illumination did not have any detectable effect on the elution profiles of the proteins recorded at 447 and 370 nm. Standard marker proteins (GE Healthcare) were used to determine the apparent molecular mass of the sample by calibration. Representative traces are shown from the experiments, which were repeated at least three times for each of 2–3 independent preparations.
Phylogenetic Analysis
Phylogenetic analyses were conducted using the MEGA software package version 6 (33). The protein sequences used for the alignment were selected according to the results of the NCBI BLASTP 2.3.0 (34) using the sequences with the highest identity to aCRY. Additionally, sequences of closely related and well characterized cryptochromes and photolyases were included (supplemental Table S1). The protein sequences were aligned with the ClustalW algorithm.
Results
The UV-vis spectrum of aCRY recorded after purification revealed FADox to be present in aCRY in vitro (Fig. 1C). Blue light illumination then generated FADH• with characteristic bands at 585 and 633 nm. We noticed an additional small band at 416 nm with a shoulder at 410 nm in the absorption spectrum after illumination, which was only present for a few seconds (Fig. 1C). Such contribution is indicative for transient accumulation of an amino acid radical, but usually detected on a much shorter time scale. Therefore, this finding was scrutinized in a time-resolved manner in the context of the physiologically more relevant reduction of FADH• to FADH−.
Microsecond Time-resolved Response to Red Light of aCRY
Time-resolved UV-vis experiments have revealed the involvement of tryptophan and tyrosyl radicals in the photoreaction of cryptochromes in vitro starting from FADox. Here, flash photolysis was applied to detect possible amino acid radicals involved in the red light-induced photoreduction of FADH• in aCRY. Previous steady-state experiments have shown only the formation of FADH− (16).
Samples containing FADox were pre-illuminated with blue light for 15 s to generate a sufficient amount of FADH•, which was stabilized by adjusting the pH to 7.0 (16). To selectively convert FADH• to FADH−, nanosecond laser pulses at 630 nm were used. Time-resolved UV-vis difference spectra were recorded at time points from 1 μs to 2 ms (Fig. 2A). All difference spectra show the characteristic negative band pattern of the bleaching of FADH• with two broad maxima at 590 nm and 630 nm. Additionally, a small positive contribution from the formation of FADH− is detected at 400 nm. For comparison, spectra after illumination for 2 s with an LED were obtained in the absence and in the presence of 0.1 mm ascorbic acid. This strong reducing agent quenches all contributions by amino acid radicals within a few microseconds. The time-resolved spectra show a pronounced positive band at 416 nm, which is still present 2 s after illumination but is missing in the presence of ascorbic acid. This additional contribution is present to an equal extent relative to flavin at all time points.
FIGURE 2.
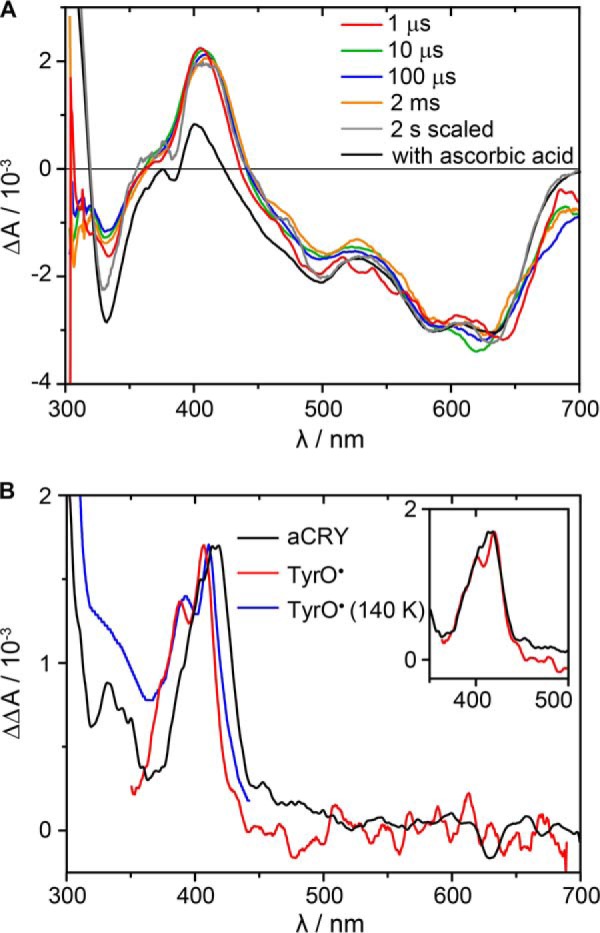
Microsecond time-resolved UV-vis spectra of the red light response in aCRY. A, pre-illuminated samples were excited with 630 nm laser pulses to selectively convert FADH• to FADH−. Spectra were recorded at indicated time points. For comparison, a steady state spectrum of the FADH• to FADH− conversion in the presence of 0.1 mm ascorbic acid is shown. B, double difference spectrum of the 1 μs spectrum minus the steady state spectrum was calculated. The comparison to reference spectra of TyrO• in water (red) (29) and at 140 K (blue) (30) reveals a red shift. A complete match is obtained by shifting the reference spectrum by 9 nm (inset).
To reveal the identity of the species contributing at 416 nm, the double difference spectrum of the 1 μs spectrum minus the steady-state spectrum was calculated. The resulting spectrum shows a well-defined peak with a maximum at 416 nm and a shoulder at ∼400 nm (Fig. 2B). The comparison to reference spectra (29, 30) revealed that such a band is characteristic for TyrO•. In contrast to the reference spectra obtained in water, TyrO• in aCRY is red-shifted by the protein environment by 9 nm (inset, Fig. 2B). Any hint for the presence of a tryptophan radical at 1 μs or later was not found.
Kinetic Analysis of the Decay of TyrO• in aCRY after Red Light Illumination
Tyrosyl radicals in proteins usually exhibit maximal lifetimes in the range of a few milliseconds (21, 23, 29, 35). Here, the time-resolved studies on aCRY indicated a much higher lifetime. Therefore, the decay of TyrO• and FADH− after red light illumination was analyzed on a time scale of seconds at 20 °C. The sample was pre-illuminated with blue light and then exposed to 632 nm for 2 s. A continuous series of difference spectra with a time resolution of ∼40 ms was recorded for 15 s (Fig. 3). The decay of FADH− was analyzed indirectly using the negative marker bands of the recovery of FADH•. The absorbance at 615–643 nm was averaged and subsequently analyzed with a monoexponential fit yielding a time constant of 2.6 s. For the decay of TyrO•, changes in absorbance at 395–420 nm were averaged. The monoexponential fit yielded a time constant of 2.6 s for TyrO•, implying a mutual decay with FADH−.
FIGURE 3.
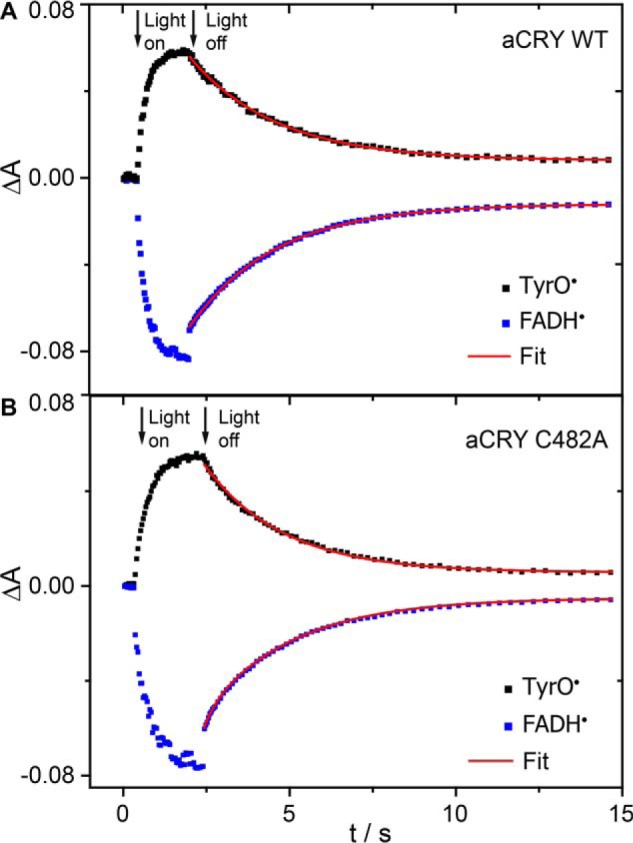
Kinetic analysis of the red light response in aCRY. A, pre-illuminated sample of wild-type aCRY was illuminated for 2 s with 632 nm to selectively convert FADH• to FADH−. The recovery of FADH• and the decay of TyrO• were analyzed with a monoexponential fit using the marker bands at 615–643 nm and 395–420 nm, respectively. Both processes show the same time constant of 2.6 s. B, these time constants change only slightly to 2.3 s in the C482A mutant of aCRY speaking against an efficient pathway of thiol oxidation by TyrO•.
Candidates for the Tyrosine Residue Involved in the Red Light Response of aCRY
To reveal the identity of the tyrosine residue contributing to the difference spectra, two different mutants of aCRY, Y373F and Y393A were produced. The residues Tyr-373 and Tyr-393 were selected because they are not conserved in other cryptochrome or photolyase families and might therefore provide a basis for the unique photoreceptor properties of aCRY (Fig. 1B). Both mutants were expressed and purified as soluble yellow proteins. Samples were pre-illuminated for 10 s with 451 nm to generate FADH•, but in Y373F, FADH• was not formed. Difference spectra were then recorded after illumination of the samples for 10 s with 632 nm (Fig. 4A). For comparison, the same procedure was repeated with wild-type aCRY. Both the wild type and the Y393A mutant showed the typical pattern of the conversion of FADH• to FADH− accompanied by the formation of TyrO•. In contrast, the Y373F mutant did not show any light-induced difference in absorption. These findings lead to the conclusion that in aCRY-Y373F the light sensitivity is strongly reduced by the exchange of the tyrosine residue, whereas Tyr-393 seems not to have any influence on the photoreaction. However, it remained unclear whether Tyr-373 is the residue forming the radical or whether the mutation negatively affected the structural integrity of aCRY.
FIGURE 4.
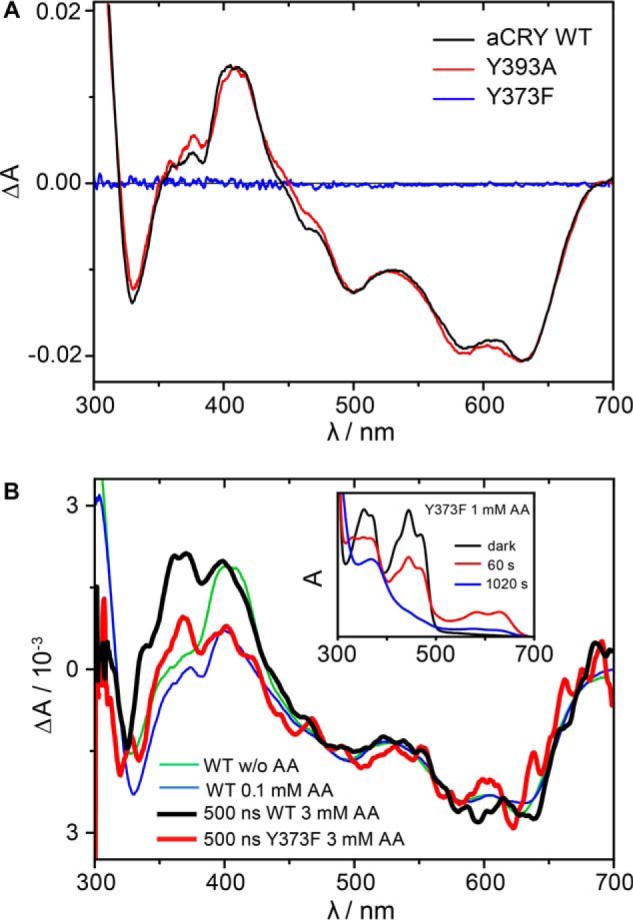
Effects of mutations in tyrosine residues on the red-light response of aCRY. A, red light-induced UV-vis difference spectra of the wild type, the Y393A and Y373F mutants of aCRY. Pre-illuminated samples were illuminated for 10 s with 632 nm. In both the wild type and the Y393A mutant, FADH• is converted to FADH− accompanied by the formation of TyrO•. The Y373F mutant does not show any light response. B, time-resolved absorption spectra at 500 ns of the Y373F mutant and wild type aCRY in the presence of 3 mm ascorbic acid. Both the Y373F mutant and the wild type show the conversion of FADH• to FADH−. In contrast to the wild type, the Y373F mutant does not show an additional contribution of TyrO• at 416 nm identifying Tyr-373 as the origin of this absorption. The conversion of FADox to FADH• and FADH− is observed in aCRY-Y373F only in the presence of ascorbic acid (inset).
Identification of Tyr-373 Forming the Long-lived Radical
We have investigated whether there are conditions under which a photoreduction of the Y373F mutant was observable. The protein could be photoreduced from FADox to FADH• and further to FADH− by the addition of ascorbic acid, albeit after a much longer light exposure (Fig. 4B, inset). However, under these harsh conditions TyrO• in the wild type was quenched preventing a direct comparison with the mutant (Fig. 2). Therefore, time-resolved experiments were conducted to capture TyrO• in the wild type before its reaction with ascorbic acid. Conditions had to be established under which the Y373F mutant was sufficiently photoactivated while the wild type showed the signal of TyrO•, resulting in time-resolved spectra at 500 ns in the presence of 3 mm ascorbic acid. After pre-illumination with blue light, both samples were excited with laser pulses at 532 nm. The intensity was adjusted so that the difference spectra of the wild type and the Y373F mutant showed both the formation of the FADH• to a similar extent (Fig. 4B). The spectrum of the wild type showed the same band pattern as a sample in the absence of ascorbic acid besides an additional band at around 370 nm originating from a reaction intermediate in the oxidation of ascorbic acid. The Y373F mutant, however, does not produce the band at 416 nm of TyrO•. In conclusion, Tyr-373 was identified to form the long-lived radical in aCRY.
Size Exclusion Chromatography on aCRY and Its Variants
The oligomerization state of aCRY and its dependence on the redox state of the flavin was studied by SEC at injected concentrations of 0.5 mm. aCRY containing FADH• was analyzed using blue light-illuminated samples. Absorbance was recorded at 630 nm to selectively detect protein with bound FADH•. The SEC showed a single peak with a maximum at an elution volume of 13.2 ml (Fig. 5A), which corresponds to an apparent molecular mass of 130 kDa. Considering a theoretical molecular mass of 66.4 kDa, the peak can be assigned to a dimer of aCRY. To resolve the functional role of the CCT, aCRY was truncated after amino acid 496. In contrast to the full-length receptor, aCRYΔCCT showed a peak at an elution volume of 14.8 ml, which corresponds to an apparent molecular mass of 60 kDa (Fig. 5A). With a theoretical mass of 57.7 kDa of aCRYΔCCT, the peak can be assigned to a monomer. This difference points to the fact that the CCT is necessary for the dimerization of aCRY carrying FADH•.
FIGURE 5.
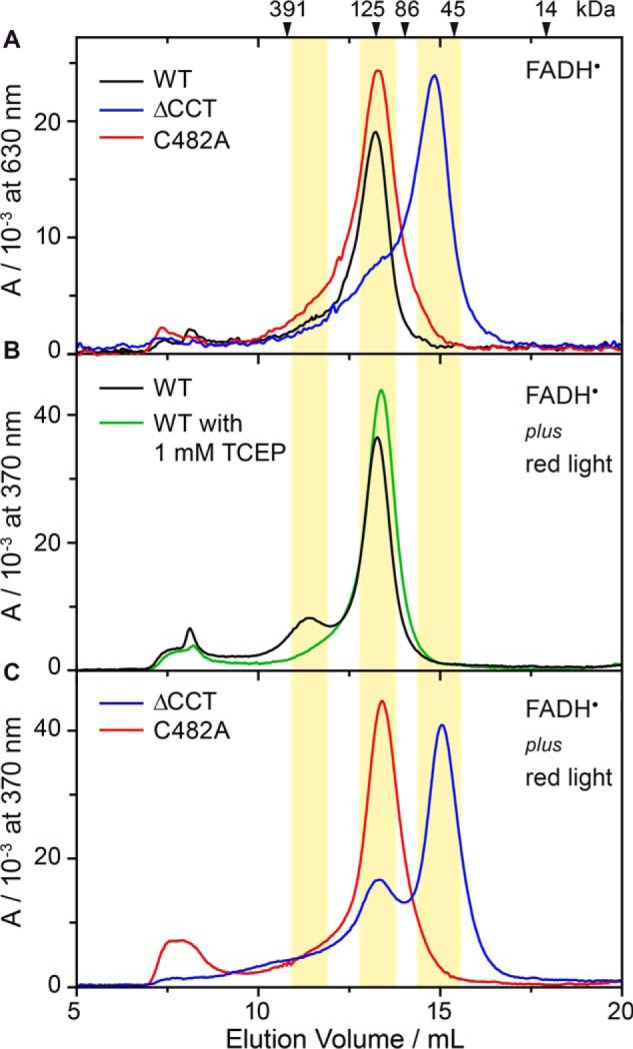
Size exclusion chromatography of wild-type aCRY, aCRYΔCCT and the C482A mutant at injected concentrations of 0.5 mm. A, samples were illuminated for 10 s with 451 nm to convert FADox to FADH•, which was selectively detected at 630 nm. Wild-type aCRY and the C482A mutant eluted at 13.2 ml, which corresponds to an apparent molecular mass of 130 kDa. Considering a theoretical molecular mass of 66.4 kDa of aCRY, the bands can be assigned to dimers. aCRYΔCCT predominantly eluted at 14.8 ml, which corresponds to an apparent molecular mass of 60 kDa and can accordingly be assigned to a monomer. B, samples were illuminated for 45 s with 451 nm and 630 nm to convert FADox to FADH• and FADH−, which were detected at 370 nm. Wild-type aCRY shows an additional, small peak at 11.4 ml (314 kDa), which indicates a tetrameric species. This light-induced oligomerization can be abolished by the addition of the reducing agent TCEP. C, samples were treated as in B. In the C482A mutant, light-induced oligomerization is not observed. In contrast, aCRYΔCCT shows an additional peak at 13.3 ml, which can be assigned to a dimeric species. Therefore, some light-induced oligomerization was found for both wild-type aCRY and aCRYΔCCT.
The effects of the formation of FADH− in aCRY were investigated using blue and red light-illuminated samples. Due to the decay of FADH− back to FADH• within a few seconds during the SEC, the detected absorbance at 370 nm mainly represents FADH• in aCRY, but FADH− and FADox might absorb as well. Interestingly, aCRY showed the same profile as before, but with an additional peak at 11.4 ml (314 kDa) possibly from a tetrameric species (Fig. 5B). Similarly, aCRYΔCCT showed an additional peak at 13.3 ml (122 kDa) after the conversion, which is assignable to the formation of a dimer (Fig. 5C). Therefore, it can be concluded that the red light-induced changes in aCRY are persistent even after the fast recovery of FADH• from FADH−. The formation of FADH− is accompanied by light-induced oligomerization of aCRY to some extent. This process is abolished by the presence of 1 mm of the strong reducing agent TCEP (Fig. 5B). As TCEP is known to reduce disulfide bonds, a potential candidate for such an intermolecular bond was identified as Cys-482 on the basis of its close proximity to the reactive tyrosine residue Tyr-373 (Fig. 1B) as well as its position close to the protein surface. The replacement of Cys-482 with alanine led to a disappearance of the additional peak in the SEC profile of aCRY after red-light illumination (Fig. 5C), whereas the profile of aCRY without red light treatment was not altered in the C482A mutant (Fig. 5A). Therefore, light-induced oligomerization in aCRY most likely proceeds via disulfide bridge formation at Cys-482.
Red Light-induced FTIR Difference Spectroscopy on aCRYΔCCT
The previously recorded FTIR difference spectrum of the wild type (16) showed changes in turn structures in the light-induced conversion from FADH• to FADH− that are not present in (6–4) photolyase (17). To further investigate a possible involvement of the CCT in the red light response of aCRY, difference spectra of aCRYΔCCT were taken after red light exposure. The reference was recorded after the sample was pre-illuminated for 1 s with blue light. Immediately afterward the sample was illuminated for 10 s at 632 nm to induce the formation of FADH− (Fig. 6A). In the resulting difference spectrum, only bands of vibrational modes changing upon red light illumination are detectable. The negative bands represent the dark state with bound FADH• in good agreement with the spectrum of the wild type (Fig. 6B), showing marker bands of FADH• at 1663, 1651, and 1534 cm−1 (16). The positive bands in the difference spectrum of aCRYΔCCT are also in good agreement with those of the wild type, most of which can be assigned to FADH−. Marker bands for FADH− (16) are present in the spectrum of aCRYΔCCT at 1626, 1620, 1515, and 1395 cm−1. The positive band assigned to a change in turn elements is also present in aCRYΔCCT at 1676 cm−1. Only the band at 1564 cm−1 originating from both flavin and the amide II band shows an upshift of 5 cm−1 as compared with the wild type spectrum. In conclusion, the difference spectrum of aCRYΔCCT is almost identical to that of the wild type taking into account a slight shift of several bands by ∼1 cm−1. It can be concluded that the CCT in aCRY does not respond to light by a change in secondary structure, in contrast to the CCT in plant cryptochromes (36, 37). So far, structural changes detected in aCRY are therefore limited to the PHR domain.
FIGURE 6.

Red light-induced FTIR difference spectra of aCRYΔCCT as compared with wild-type aCRY. Samples were pre-illuminated with blue light and then exposed to 632 nm for 10 s to selectively convert FADH• to FADH−. Contributions identified in the analysis to originate from changes in secondary structure are highlighted in gray. Both samples show very similar responses excluding the CCT as the origin of these structural changes.
Discussion
Unusual Stability of TyrO• in aCRY
Tyrosyl radicals have been observed as part of the electron transfer cascade in other cryptochromes and photolyases than aCRY (21, 23, 29, 32). Lifetimes of these tyrosyl radicals have been determined to range from 1 ms up to >140 ms (21, 23, 29). In contrast, an unusually long-lived TyrO• with a lifetime of 2.6 s is formed in aCRY, implying a high stability of the radical.
A very similar lifetime of TyrO• has been achieved in α3Y, a de novo model protein specifically designed for investigating the stabilizing effect of the protein matrix on the lifetime of tyrosyl radicals (38). α3Y does not contain any cofactors and places the reactive tyrosine in a three helix bundle scaffold. Similar to aCRY, αY3 does not carry a covalent modification of tyrosine or a metal center for stabilization of TyrO• (39). However, the residue is buried deeply in a hydrophobic environment in α3Y without any hydrogen bonding partner or access to the solvent (38). Therefore, the surrounding protein matrix is rather different compared with the one present in aCRY, considering the relatively exposed position of Tyr-373 in a cavity on the surface of the protein, as derived from the modeled structure (16).
Interestingly, a system more similar to aCRY was found in the D2 subunit of photosystem II (PSII). Here, TyrD forms a radical that is stable for hours under physiological conditions without a metal cluster in close proximity (40). TyrD was reported to be deprotonated upon oxidation, because phenolic compounds become strong acids upon oxidation. Such a proton-coupled electron transfer has also been found for other tyrosyl radicals (39). For TyrD, it is believed that the kinetics of reprotonation is of considerable importance for the stability of TyrO• (41). The radical is found in a very well-ordered hydrogen bonding network with rather hydrophobic residues, which was postulated to prevent any uncontrolled protonation (41). Additional stability might be provided by the movement of either the proton acceptor (41) or the tyrosine residue away from the proton-donating network upon oxidation, as found for the stable TyrO• coupled to a diiron oxo site in subunit R2 of class I ribonucleotide reductases (RNR) (42). Moreover, it might be important to consider that after oxidation and deprotonation, the phenolic oxygen of the radical might be hydrogen-bonded to an adjacent residue or a coordinated water molecule (43, 44). This hydrogen bond may provide an additional stabilization by delocalizing the spin density of the oxygen, as it has been suggested for the rather hydrophilic environment in mouse RNR (44).
Transferring these findings to aCRY, it seems plausible that TyrO• is also coordinated in a very stable, non-flexible hydrogen bonding network comparable to the one found for TyrD in PSII, even if there are not that many adjacent hydrophobic residues in aCRY. In PSII, arginine and aspartate close to TyrD form a salt bridge, facilitating the proton transfer during oxidation (41). In the modeled structure of aCRY (16), arginine (Arg-485) and aspartate (Asp-321) also form a salt bridge close to Tyr-373, possibly creating a similar environment as in PSII. Still, it is unclear whether an amino acid residue or a coordinated water molecule is the proton acceptor in aCRY. Furthermore, it needs to be resolved if the phenolic oxygen of TyrO• is also hydrogen bonded as in mouse RNR and PSII-D2. In summary, the lifetime of TyrO• in aCRY can be considered to be unusually long because the protein does not provide the classical environment for stabilization such as a metal center or hydrophobic residues. Furthermore, TyrO• in aCRY does not benefit from possibly stabilizing effects of a membrane-bound complex such as PSII.
Red Shift of TyrO• Absorbance
Another striking feature of TyrO• found in aCRY is its red-shifted absorption pattern. Typically the radical shows absorption maxima at ∼390 and ∼410 nm (29, 30). In contrast, the maxima in aCRY are found at ∼400 and 416 nm, similar to what has been reported previously in mouse RNR (45). An explanation for the shift in both aCRY and the mammalian RNR might be the existence of the hydrogen bond between the phenolic oxygen and a hydrogen bond donor. Interestingly, TyrO• in the Escherichia coli RNR is not hydrogen-bonded and shows a non-shifted absorption spectrum in the direct comparison (45). A covalent crosslink of the cysteine to the tyrosine as observed in galactose oxidase (46) can be ruled out as the absorption difference spectrum of the aCRY-C482A mutant is identical to that of the wild type (data not shown).
Formation of TyrO• from Trp•
Another surprising aspect of the time-resolved UV-vis spectra is the absence of any bands from tryptophan radicals after 1 μs. In other UV-vis-spectroscopic studies on cryptochromes, significant Trp• contributions have been identified in this time region up to milliseconds (23, 24). To our knowledge, the only similar finding has been reported for Xenopus laevis (6–4) photolyase, in which TyrO• is already formed within 100 ns after illumination (32). For aCRY, the question arises whether the Trp• decays ultrafast into TyrO• or if the Trp• is not formed in the first place. The latter suggestion is unlikely because a tryptophan triad (23) (Trp-322, Trp-376, Trp-399) is also conserved in aCRY. As such a triad is considered to represent an optimal electron transfer pathway, the electron is most probably guided via this “electron wire” over the distance of ∼16 Å, as derived from the model, from the Tyr-373 on the surface to the chromophore. Therefore, the most likely reason for the Trp• band not to be present in the spectra after 1 μs is its ultrafast decay in a time range of pico- to nanoseconds. This short lifetime might be the consequence of a high reactivity of Tyr-373.
Oligomerization State of aCRY
In the dark, SEC at 0.5 mm injected concentration revealed aCRY to form a homodimer whereas the truncated ΔCCT mutant remains predominantly as a monomer. These findings indicate that the CCT may represent the oligomerization site of aCRY in the dark. In the light, SEC revealed some oligomerization for both proteins upon FADH− formation at a concentration of 0.5 mm. Therefore, the CCT does not play a significant role during red light-induced oligomerization. This conclusion is underlined by the FTIR experiments which do not show any significant differences between red light-induced difference spectra of the wild type and the ΔCCT mutant. These findings are in contrast to events observed in other cryptochromes. In the Arabidopsis cryptochrome AtCRY1, large conformational changes upon photoexcitation were noted by a rearrangement of the CCT (36, 37). In the Drosophila cryptochrome dCRY, an increase of proteolytic susceptibility of the CCT upon illumination indicates the involvement of this domain in the light response (47).
Further SEC experiments imply that the light-induced oligomerization originates from the formation of disulfide bridges most likely by Cys-482. From the structural model, Cys-482 was derived to be in close proximity with a 4 Å distance to the reactive Tyr-373. It can be concluded that red light illumination of aCRY triggers a redox cascade with a partial electron transfer from a cysteine to TyrO•. Such transfer has already been observed in class I RNR, in which the cascade does not result in disulfide bridge formation but in the reduction of ribonucleotides to deoxyribonucleotides (48). In aCRY this reaction is inefficient, because the lifetime of TyrO• is not increased in the C482A mutant but even slightly shortened (Fig. 3B). Therefore, the formation of disulfide bridges might be regarded as a non-physiological reaction or as a consequence of a missing in vivo redox substrate/partner, which might explain the low extent of oligomerization. Alternatively, we may speculate that the covalent linkage of the aCRY dimers constitutes a pathway present under oxidative stress and high light conditions only, which might lead to an inactivation of aCRY signaling by inhibiting any binding to its signaling partner (49).
Tyr-373 in the Context of Other Cryptochromes and Photolyases
The postulate of a central role of Tyr-373 in the sensory mechanism of aCRY would imply that this residue is conserved in other potential red light receptors. A BLAST search (Fig. 7, supplemental Table S1) revealed that indeed the two closest relatives of aCRY, the putative photolyases from the algae Volvox carteri and Dunaliella salina share this residue as well as a sequence from Coccomixa subellipsoidea but not from Chlorella variabilis. These sequences form a subgroup that is well separated in the phylogenetic analysis from other candidates for conventional (6–4) photolyases. Even more convincingly, the next 40 candidates in the alignment of the aCRY homologues and all well characterized members of the cryptochrome/photolyase family to our knowledge do not possess a tyrosine at this position. We consider this finding to be significant as a further support for the crucial role of Tyr-373 in the function of aCRY as light receptor with an extended sensitivity range.
FIGURE 7.
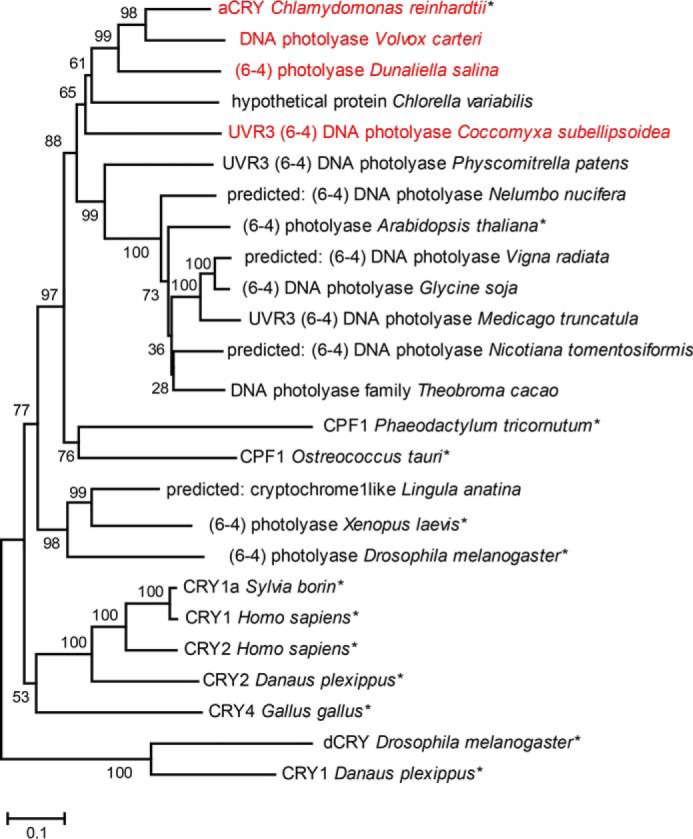
Phylogenetic classification of aCRY and conservation of the tyrosine residue 373. Three uncharacterized proteins homologous to aCRY were found by BLASTP that all carry the tyrosine residue 373 of aCRY (red). These three sequences are among the four closest to aCRY and share more than 63% sequence identity, hinting toward the presence of aCRY-like receptors in these organisms. All other presented sequences lack this tyrosine. Characterized members of other cryptochrome and photolyase subfamilies have been marked with an asterisk. Bootstrap values are given after 1000 replications. The scale bar indicates amino acid substitutions per site.
Conclusions
To date, only small conformational changes have been identified for aCRY in the red-light induced photoreduction. In contrast to other full-length cryptochromes, these changes resulting from illumination do not involve the CCT. This finding raises the question of how the signal proceeds from the flavin to the protein surface in aCRY if not by such conformational changes. An alternative model would involve a redox cascade starting with the reduction of flavin and proceeding to the surface, where a signal partner or a substrate may then be converted (50).
This study provides insights into the early events of the red light response of aCRY (Fig. 8). We revealed the rapid formation within 1 μs of a tyrosine radical with an unusually long lifetime of 2.6 s, which decays concomitant with FADH−. Tyrosine 373 was disclosed to be responsible for the radical formation. This residue is only conserved within a small group of proteins that might be regarded as a new subfamily of the cryptochrome/photolyase superfamily. Partial light-induced oligomerization of the protein was observed and attributed to disulfide bridge formation via cysteine 482 in close proximity to tyrosine 373. These unexpected findings point to an important role of redox reactions in the signaling process of aCRY and imply that aCRY might have evolved a signaling mechanism via a light-triggered redox cascade, which culminates in a photooxidation of a yet unknown substrate or binding partner.
FIGURE 8.
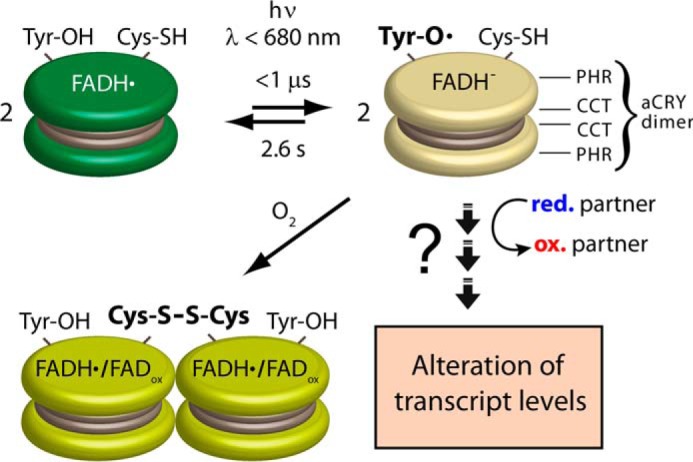
Model of the response to red light of aCRY in vitro. aCRY forms a homodimer (represented by two disks) via its CCT. aCRY carrying FADH• is reduced to FADH− by light with λ < 680 nm. As a result, TyrO• is formed by Tyr-373 within 1 μs with a red-shifted absorption in aCRY compared with water. The lifetime of 2.6 s of TyrO• is unusually high and might allow for an efficient oxidation of a signaling partner in vivo finally leading to the changes in the levels of multiple transcripts. The decay of TyrO• proceeds concomitant with that of FADH−. Some of the light-activated aCRY oligomerizes via disulfide bridge formation at Cys-482 in the absence of external reductant.
Author Contributions
T. K., M. M., and L. O. E. designed the study. S. O. performed and analyzed all the experiments with advice by T. K. S. F. and S. W. generated plasmids of aCRY variants. S. O. and T. K. wrote the manuscript with advice by S. F., S. W., L. O. E., and M. M. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Benedikt Beel for help in constructing aCRYΔCCT and Mona Lisa Remmers for preliminary experiments with SEC. T. K. thanks Thomas Hellweg for generous support.
This work was supported by the Deutsche Forschungsgemeinschaft within the framework of Research Group 1261 (Grants KO3580/1-2, MI373/12-1, MI373/12-2, and ES152/12) and by a Heisenberg Fellowship (KO3580/4-1). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
- PHR
- photolyase homology region
- FADox
- oxidized flavin adenine dinucleotide
- CCT
- C-terminal extension
- AtCRY1
- Arabidopsis thaliana cryptochrome 1
- CPD
- cyclobutane pyrimidine dimers
- aCRY
- animal-like cryptochrome
- FADH•
- FAD neutral radical
- FADH−
- anionic fully reduced state of FAD
- UV-vis
- UV-visible
- Trp•
- tryptophan neutral radical
- TyrO•
- tyrosyl radical
- SEC
- size exclusion chromatography
- RNR
- ribonucleotide reductase
- PSII
- photosystem II.
References
- 1. Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L. O., van der Horst G. T., Batschauer A., and Ahmad M. (2011) The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 [DOI] [PubMed] [Google Scholar]
- 2. Losi A., and Gärtner W. (2012) The evolution of flavin-binding photoreceptors: an ancient chromophore serving trendy blue-light sensors. Annu. Rev. Plant Biol. 63, 49–72 [DOI] [PubMed] [Google Scholar]
- 3. Sancar A. (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2237 [DOI] [PubMed] [Google Scholar]
- 4. Yang H. Q., Wu Y. J., Tang R. H., Liu D., Liu Y., and Cashmore A. R. (2000) The C termini of Arabidopsis cryptochromes mediate a constitutive light response. Cell 103, 815–827 [DOI] [PubMed] [Google Scholar]
- 5. Etchegaray J. P., Lee C., Wade P. A., and Reppert S. M. (2003) Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421, 177–182 [DOI] [PubMed] [Google Scholar]
- 6. Gegear R. J., Casselman A., Waddell S., and Reppert S. M. (2008) Cryptochrome mediates light-dependent magnetosensitivity in Drosophila. Nature 454, 1014–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pokorny R., Klar T., Hennecke U., Carell T., Batschauer A., and Essen L. O. (2008) Recognition and repair of UV lesions in loop structures of duplex DNA by DASH-type cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 105, 21023–21027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selby C. P., and Sancar A. (2006) A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc. Natl. Acad. Sci. U.S.A. 103, 17696–17700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rupert C. S. (1960) Photoreactivation of transforming DNA by an enzyme from bakers' yeast. J. Gen. Physiol. 43, 573–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Todo T., Takemori H., Ryo H., Ihara M., Matsunaga T., Nikaido O., Sato K., and Nomura T. (1993) A new photoreactivating enzyme that specifically repairs ultraviolet light-induced (6–4) photoproducts. Nature 361, 371–374 [DOI] [PubMed] [Google Scholar]
- 11. Zhang F., Scheerer P., Oberpichler I., Lamparter T., and Krauss N. (2013) Crystal structure of a prokaryotic (6–4) photolyase with an Fe-S cluster and a 6,7-dimethyl-8-ribityllumazine antenna chromophore. Proc. Natl. Acad. Sci. U.S.A. 110, 7217–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geisselbrecht Y., Frühwirth S., Schroeder C., Pierik A. J., Klug G., and Essen L. O. (2012) CryB from Rhodobacter sphaeroides: a unique class of cryptochromes with new cofactors. EMBO Rep. 13, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beel B., Prager K., Spexard M., Sasso S., Weiss D., Müller N., Heinnickel M., Dewez D., Ikoma D., Grossman A. R., Kottke T., and Mittag M. (2012) A flavin binding cryptochrome photoreceptor responds to both blue and red light in Chlamydomonas reinhardtii. Plant Cell 24, 2992–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Banerjee R., Schleicher E., Meier S., Viana R. M., Pokorny R., Ahmad M., Bittl R., and Batschauer A. (2007) The signaling state of Arabidopsis cryptochrome 2 contains flavin semiquinone. J. Biol. Chem. 282, 14916–14922 [DOI] [PubMed] [Google Scholar]
- 15. Bouly J. P., Schleicher E., Dionisio-Sese M., Vandenbussche F., Van Der Straeten D., Bakrim N., Meier S., Batschauer A., Galland P., Bittl R., and Ahmad M. (2007) Cryptochrome blue light photoreceptors are activated through interconversion of flavin redox states. J. Biol. Chem. 282, 9383–9391 [DOI] [PubMed] [Google Scholar]
- 16. Spexard M., Thöing C., Beel B., Mittag M., and Kottke T. (2014) Response of the sensory animal-like cryptochrome aCRY to blue and red light as revealed by infrared difference spectroscopy. Biochemistry 53, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 17. Yamada D., Zhang Y., Iwata T., Hitomi K., Getzoff E. D., and Kandori H. (2012) Fourier-transform infrared study of the photoactivation process of Xenopus (6–4) photolyase. Biochemistry 51, 5774–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berndt A., Kottke T., Breitkreuz H., Dvorsky R., Hennig S., Alexander M., and Wolf E. (2007) A novel photoreaction mechanism for the circadian blue light photoreceptor Drosophila cryptochrome. J. Biol. Chem. 282, 13011–13021 [DOI] [PubMed] [Google Scholar]
- 19. Immeln D., Schlesinger R., Heberle J., and Kottke T. (2007) Blue light induces radical formation and autophosphorylation in the light-sensitive domain of Chlamydomonas cryptochrome. J. Biol. Chem. 282, 21720–21728 [DOI] [PubMed] [Google Scholar]
- 20. Müller P., and Ahmad M. (2011) Light-activated cryptochrome reacts with molecular oxygen to form a flavin-superoxide radical pair consistent with magnetoreception. J. Biol. Chem. 286, 21033–21040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aubert C., Mathis P., Eker A. P., and Brettel K. (1999) Intraprotein electron transfer between tyrosine and tryptophan in DNA photolyase from Anacystis nidulans. Proc. Natl. Acad. Sci. U.S.A. 96, 5423–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aubert C., Vos M. H., Mathis P., Eker A. P., and Brettel K. (2000) Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 586–590 [DOI] [PubMed] [Google Scholar]
- 23. Giovani B., Byrdin M., Ahmad M., and Brettel K. (2003) Light-induced electron transfer in a cryptochrome blue-light photoreceptor. Nat. Struct. Biol. 10, 489–490 [DOI] [PubMed] [Google Scholar]
- 24. Langenbacher T., Immeln D., Dick B., and Kottke T. (2009) Microsecond light-induced proton transfer to flavin in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 131, 14274–14280 [DOI] [PubMed] [Google Scholar]
- 25. Brazard J., Usman A., Lacombat F., Ley C., Martin M. M., Plaza P., Mony L., Heijde M., Zabulon G., and Bowler C. (2010) Spectro-temporal characterization of the photoactivation mechanism of two new oxidized cryptochrome/photolyase photoreceptors. J. Am. Chem. Soc. 132, 4935–4945 [DOI] [PubMed] [Google Scholar]
- 26. Müller P., Yamamoto J., Martin R., Iwai S., and Brettel K. (2015) Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6–4) photolyases. Chem. Commun. 51, 15502–15505 [DOI] [PubMed] [Google Scholar]
- 27. Paulus B., Bajzath C., Melin F., Heidinger L., Kromm V., Herkersdorf C., Benz U., Mann L., Stehle P., Hellwig P., Weber S., and Schleicher E. (2015) Spectroscopic characterization of radicals and radical pairs in fruit fly cryptochrome - protonated and nonprotonated flavin radical-states. FEBS J. 282, 3175–3189 [DOI] [PubMed] [Google Scholar]
- 28. Solar S., Getoff N., Surdhar P. S., Armstrong D. A., and Singh A. (1991) Oxidation of tryptophan and N-methylindole by N3., Br2-, and (SCN)2- radicals in light-water and heavy-water solutions - A pulse-radiolysis study. J. Phys. Chem. 95, 3639–3643 [Google Scholar]
- 29. Thöing C., Oldemeyer S., and Kottke T. (2015) Microsecond deprotonation of aspartic acid and response of the α/β subdomain precede C-terminal signaling in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 137, 5990–5999 [DOI] [PubMed] [Google Scholar]
- 30. Proshlyakov D. A. (2004) UV optical absorption by protein radicals in cytochrome c oxidase. Biochim. Biophys. Acta 1655, 282–289 [DOI] [PubMed] [Google Scholar]
- 31. Biskup T., Schleicher E., Okafuji A., Link G., Hitomi K., Getzoff E. D., and Weber S. (2009) Direct observation of a photoinduced radical pair in a cryptochrome blue-light photoreceptor. Angew. Chem. Int. Ed. 48, 404–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weber S., Kay C. W., Mögling H., Möbius K., Hitomi K., and Todo T. (2002) Photoactivation of the flavin cofactor in Xenopus laevis (6–4) photolyase: observation of a transient tyrosyl radical by time-resolved electron paramagnetic resonance. Proc. Natl. Acad. Sci. U.S.A. 99, 1319–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tamura K., Dudley J., Nei M., and Kumar S. (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 24, 1596–1599 [DOI] [PubMed] [Google Scholar]
- 34. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gauden M., van Stokkum I. H. M., Key J. M., Lührs D. Ch., van Grondelle R., Hegemann P., and Kennis J. T. (2006) Hydrogen-bond switching through a radical pair mechanism in a flavin-binding photoreceptor. Proc. Natl. Acad. Sci. U.S.A. 103, 10895–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kondoh M., Shiraishi C., Müller P., Ahmad M., Hitomi K., Getzoff E. D., and Terazima M. (2011) Light-induced conformational changes in full-length Arabidopsis thaliana cryptochrome. J. Mol. Biol. 413, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Partch C. L., Clarkson M. W., Ozgür S., Lee A. L., and Sancar A. (2005) Role of structural plasticity in signal transduction by the cryptochrome blue-light photoreceptor. Biochemistry 44, 3795–3805 [DOI] [PubMed] [Google Scholar]
- 38. Glover S. D., Jorge C., Liang L., Valentine K. G., Hammarström L., and Tommos C. (2014) Photochemical tyrosine oxidation in the structurally well-defined α3Y protein: proton-coupled electron transfer and a long-lived tyrosine radical. J. Am. Chem. Soc. 136, 14039–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoganson C. W., and Tommos C. (2004) The function and characteristics of tyrosyl radical cofactors. Biochim. Biophys. Acta 1655, 116–122 [DOI] [PubMed] [Google Scholar]
- 40. Pujols-Ayala I., and Barry B. A. (2004) Tyrosyl radicals in photosystem II. Biochim. Biophys. Acta 1655, 205–216 [DOI] [PubMed] [Google Scholar]
- 41. Saito K., Rutherford A. W., and Ishikita H. (2013) Mechanism of tyrosine D oxidation in Photosystem II. Proc. Natl. Acad. Sci. U.S.A. 110, 7690–7695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lendzian F. (2005) Structure and interactions of amino acid radicals in class I ribonucleotide reductase studied by ENDOR and high-field EPR spectroscopy. Biochim. Biophys. Acta 1707, 67–90 [DOI] [PubMed] [Google Scholar]
- 43. Campbell K. A., Peloquin J. M., Diner B. A., Tang X. S., Chisholm D. A., and Britt R. D. (1997) The τ-nitrogen of D2 histidine 189 is the hydrogen bond donor to the tyrosine radical YD• of photosystem II. J. Am. Chem. Soc. 119, 4787–4788 [Google Scholar]
- 44. van Dam P. J., Willems J. P., Schmidt P. P., Potsch S., Barra A. L., Hagen W. R., Hoffman B. M., Andersson K. K., and Graslund A. (1998) High-frequency EPR and pulsed Q-Band ENDOR studies on the origin of the hydrogen bond in tyrosyl radicals of ribonucleotide reductase R2 proteins from mouse and herpes simplex virus type 1. J. Am. Chem. Soc. 120, 5080–5085 [Google Scholar]
- 45. Thelander M., Gräslund A., and Thelander L. (1985) Subunit M2 of mammalian ribonucleotide reductase. Characterization of a homogeneous protein isolated from M2-overproducing mouse cells. J. Biol. Chem. 260, 2737–2741 [PubMed] [Google Scholar]
- 46. Ito N., Phillips S. E., Stevens C., Ogel Z. B., McPherson M. J., Keen J. N., Yadav K. D., and Knowles P. F. (1991) Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 350, 87–90 [DOI] [PubMed] [Google Scholar]
- 47. Vaidya A. T., Top D., Manahan C. C., Tokuda J. M., Zhang S., Pollack L., Young M. W., and Crane B. R. (2013) Flavin reduction activates Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 20455–20460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minnihan E. C., Nocera D. G., and Stubbe J. (2013) Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 46, 2524–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lokhandwala J., Hopkins H. C., Rodriguez-Iglesias A., Dattenböck C., Schmoll M., and Zoltowski B. D. (2015) Structural biochemistry of a fungal LOV domain photoreceptor reveals an evolutionarily conserved pathway integrating light and oxidative stress. Structure 23, 116–125 [DOI] [PubMed] [Google Scholar]
- 50. Oztürk N., Song S. H., Selby C. P., and Sancar A. (2008) Animal type 1 cryptochromes. Analysis of the redox state of the flavin cofactor by site-directed mutagenesis. J. Biol. Chem. 283, 3256–3263 [DOI] [PubMed] [Google Scholar]
- 51. Hitomi K., DiTacchio L., Arvai A. S., Yamamoto J., Kim S. T., Todo T., Tainer J. A., Iwai S., Panda S., and Getzoff E. D. (2009) Functional motifs in the (6–4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc. Natl. Acad. Sci. U.S.A. 106, 6962–6967 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.