

Abstract
B cell lymphoma gene 2 (Bcl-2) family proteins are key regulators of programmed cell death and important targets for drug discovery. Pro-apoptotic and anti-apoptotic Bcl-2 family proteins reciprocally modulate their activities in large part through protein interactions involving a motif known as BH3 (Bcl-2 homology 3). Nur77 is an orphan member of the nuclear receptor family that lacks a BH3 domain but nevertheless binds certain anti-apoptotic Bcl-2 family proteins (Bcl-2, Bfl-1, and Bcl-B), modulating their effects on apoptosis and autophagy. We used a combination of NMR spectroscopy-based methods, mutagenesis, and functional studies to define the interaction site of a Nur77 peptide on anti-apoptotic Bcl-2 family proteins and reveal a novel interaction surface. Nur77 binds adjacent to the BH3 peptide-binding crevice, suggesting the possibility of cross-talk between these discrete binding sites. Mutagenesis of residues lining the identified interaction site on Bcl-B negated the interaction with Nur77 protein in cells and prevented Nur77-mediated modulation of apoptosis and autophagy. The findings establish a new protein interaction site with the potential to modulate the apoptosis and autophagy mechanisms governed by Bcl-2 family proteins.
Keywords: apoptosis, autophagy, B cell lymphoma 2 (Bcl-2) family, NMR, protein-protein interaction, Bcl-b, Bfl-1, NR4A1, Nur77
Introduction
In metazoans, Bcl-25 family proteins regulate several cellular processes, most prominently apoptosis (programmed cell death) and also autophagy (1, 2). Based on primary amino acid sequence similarity, members of the Bcl-2 protein family share at least one of four modular components, termed Bcl-2 homology (BH) domains. Of these, BH3 has an amphipathic α-helical structure and plays a pivotal role in both the activation of pro-apoptotic and the suppression of anti-apoptotic members of the family. For example, the BH3 domains of Bim, Bid, and Puma trigger conformational changes in the executioners Bax and Bak, leading to their oligomerization and permeation of the outer mitochondrial membrane. Downstream consequences of Bax and Bak activation include release of cytochrome c and, ultimately, the activation of caspase-9 and other cell death proteases. Conversely, the BH3 domains of these and other BH3-carrying proteins bind anti-apoptotic Bcl-2 family proteins and inhibit their ability to prevent Bax/Bak oligomerization in the outer mitochondrial membrane.
The BH3 domain of pro-apoptotic proteins operates analogous to a ligand to interact with a predominantly hydrophobic receptor-like binding crevice on the surfaces of anti-apoptotic Bcl-2 family proteins. In addition to regulating apoptosis, the BH3-binding pocket on anti-apoptotic Bcl-2 family proteins has been reported to bind a BH3-like domain in the autophagy protein Beclin-1, and this interaction has been shown to play an important role in regulating autophagy (3). Prior work has dissected the binding affinity of various BH3 peptides (ligands) to anti-apoptotic Bcl-2 proteins (receptors), and three-dimensional structural data are available for several of these pairs (reviewed in Ref. 4).
Targeting the BH3-binding cleft of anti-apoptotic Bcl-2 proteins with chemical compounds restores apoptosis sensitivity and is a promising strategy for cancer therapeutics (5). Progress has been made in recent years toward generating chemical inhibitors of Bcl-2 that mimic BH3-derived peptides found in naturally occurring apoptosis sensitizers (6). Examples are found in compounds designed to target Bcl-2, such as venetoclax (ABT199/GDC-0199), or compounds that bind both Bcl-2 and Bcl-X(L), such as ABT-737 and its orally available analog navitoclax (ABT-263) (7–9). However, only a subset of cancers is sensitive to these selective Bcl-2 inhibitors because of redundancy caused by simultaneous expression of multiple anti-apoptotic Bcl-2 family members. For example, because ABT-737 and ABT-263 selectively bind Bcl-2, Bcl-X(L), and Bcl-W, elevated cellular levels of Mcl-1 or Bfl-1 expression have been shown to cause resistance (10–14).
Another strategy for attacking anti-apoptotic Bcl-2 family proteins is to target non-canonical interaction sites apart from the BH3-binding crevice. Although structural details are lacking, several proteins have been reported to interact with anti-apoptotic Bcl-2 family members in a BH3-independent manner, including p53 (15), the nutrient deprivation autophagy factor 1 protein (NAF-1) (16), and the nuclear orphan receptors NR4A1 (Nur77) (17–18) and NR4A3 (Nor-1) (19). Binding of Nur77 to anti-apoptotic Bcl-2 proteins at the surface of mitochondria has been proposed to induce a conformational change that exposes the BH3 domain residues in Bcl-2 helix α2 (18), converting Bcl-2 from cytoprotective to pro-apoptotic. However, this evidence is indirect and relies on co-immunoprecipitation (co-IP) analysis of Bcl-2 mutants with extended deletions and by epitope-specific antibody binding.
Here we used a Nur77 peptide as a molecular probe to delineate the interaction interface between Nur77 and the anti-apoptotic proteins Bcl-2, Bfl-1, and Bcl-B using NMR-based methods. We found that the Nur77 peptide-binding interface flanks the canonical BH3 binding cleft. Using the anti-apoptotic Bcl-B protein as a model, we generated several mutants within the identified Nur77-binding site that showed either improved binding or loss of binding to Nur77 protein in cells, correlating with differences in autophagy and cell death regulation. These Nur77-binding site mutants of Bcl-B continue to bind Bax and protect against Bax-induced apoptosis, thus confirming that the BH3 and Nur77 binding sites are distinct and that their regulatory functions can be independent. Our results reveal that Nur77 binding requires a novel protein interaction site on anti-apoptotic Bcl-2 family proteins, adjacent to but distinct from the BH3 binding site. The findings may have implications for understanding mechanisms of apoptosis and autophagy regulation and also possibly for future small-molecule drug discovery strategies.
Experimental Procedures
Reagents and Antibodies
Rapamycin (R5000) was purchased from LC Laboratories, and leupeptin (1187125) was from RandD Systems. Benzyloxycarbonyl-VAD(O-methyl)-fluoromethyl ketone (Z-VAD(OMe)-fmk) (BML P416) was purchased from Enzo Life Sciences and protease inhibitor mixture from Roche Applied Science. NaF and sodium orthovanadate (Na3VO4) were purchased from Sigma-Aldrich. Hank's Balanced Salt Solution (HBSS) (SH30238.01) was from Hyclone, and PEI (number 23966) was from Polysciences. The BCA protein assay was from Pierce, BSA from Fischer Scientific, and Megnosphere MS300 protein G beads from JSR Life Sciences. ECL Western blotting detection reagents were from GE Healthcare. We used the following antibodies: rabbit antibodies to LC3 (L8981, Sigma); mouse antibodies to p62 (catalog no. 610833, BD Biosciences), c-Myc (catalog no. 11 667 203 001, Roche Applied Science), the HA epitope (catalog no. 11583816001, Roche Applied Science), and β-Actin (8H10D10, catalog no. 3700, Cell Signaling Technologies); and rat antibody to HA (catalog no. 11867431001, Roche Applied Science). The following secondary antibodies were used: HRP-conjugated anti-mouse (NA931V, GE Healthcare), HRP-conjugated anti-rabbit (NA934, GE Healthcare), and HRP-conjugated anti-rat (NA935, GE Healthcare).
Cloning, Expression, and Purification of Bcl-2 Proteins
The human Bcl-2 protein used in this work was prepared as a chimera containing a Bcl-X(L) loop substitution and lacking the last 31 residues encoding the membrane-anchoring segment (residues 212–233) (20). This strategy allowed us to obtain samples for NMR experiments that did not require a new NMR assignment. The Bcl-2/Bcl-X(L) NMR assignment was provided by Dr. Stephen Fesik. The Bcl-2/Bcl-X(L) chimera gene was inserted into a pET15b vector (Novagen) between NdeI and XhoI restriction sites, in-frame with a poly-histidine tag and a thrombin cleavage sequence. This construct was used to transform Escherichia coli BL21star (DE3) (Life Technologies). To prepare 15N-labeled samples, a starting culture in Lysogeny Broth medium was used to inoculate 3 liters of minimal isotopically labeled medium containing 1.0 g/liter [15N]ammonium chloride (Cambridge Isotope Laboratories). This culture was grown at 37 °C until it reached an A600 nm of ∼0.8. To induce the expression of Bcl-2/Bcl-X(L), 0.5 mm isopropyl β-d-thiogalactopyranoside was added for an additional 3 h. The cells were collected by centrifugation at 4000 × g for 15 min, resuspended in 50 ml of 50 mm NaH2PO4, 500 mm NaCl, and 20 mm imidazole (pH 8.0) containing a protease inhibitor mixture (Roche), and loaded to a nickel-nitrilotriacetic acid column (Life Technologies). The 15N-labeled Bcl-2/Bcl-X(L) chimera was purified in a gradient of 20–250 mm imidazole, pooled, and dialyzed in 25 mm Tris-HCl, 150 mm NaCl (pH 8.0) buffer containing 1 mm tris(2-carboxyethyl)phosphine and 1 mm sodium azide. In the removal of the His6 tag, 20 mg of protein was mixed with 20 units of human thrombin in a total of 8 ml of reaction volume. This reaction was carried out at 20 °C overnight. The following day, the 15N-labeled Bcl-2/Bcl-X(L) chimera was purified by size exclusion chromatography using a Sephacryl S-100 column (GE Healthcare) equilibrated with 20 mm Tris-HCl, 5 mm DTT (pH 7.8). Protein fractions were pooled, concentrated to 0.5 mm, and stored at −80 °C until use.
To prepare Bfl-1 samples, a cDNA corresponding to the human BCL2A1 gene (encoding Bfl-1 protein) was inserted into a pTYB-12 vector between EcoRI and XhoI restriction sites (New England Biolabs) in-frame with an amino-terminal chitin-intein tag. The amino acid sequence matched Uniprot entry Q16548; however, we deleted the last 22 amino acids encoding for the C-terminal helix that is responsible for targeting Bfl-1 to the outer mitochondrial membrane. This construct was used to transform E. coli strain BL21(DE3) cells propagated in LB medium containing 35 μg/ml carbenicillin. A starting culture grown to saturation in LB medium at 37 °C overnight was used to inoculate 3 liters of minimal isotopically labeled medium containing 1.0 g/liter 15N-ammonium chloride and 2.0 g/liter [13C]glucose (Cambridge Isotope Laboratories). Because only 1% (v/v) of cells (in LB medium) were used to start this culture, the amount of unlabeled proteins added was insignificant. When cells reached an optical cell density measured at 600 nm of ∼1.0, the culture was cooled to 18 °C, 0.5 mm isopropyl β-d-thiogalactopyranoside was added, and cells were allowed to grow for an additional 15 h at 18 °C. The cell pellet was harvested by centrifugation at 4000 × g for 15 min at 4 °C and then resuspended in 60 ml of phosphate-buffered saline (pH 7.4) containing 1 mm PMSF (buffer A) and disrupted by sonication on ice. Debris was removed by centrifugation at 15,000 × g for 30 min, followed by filtration in 0.45-μm pore filters. The cleared lysate was loaded to a pre-equilibrated chitin-Sepharose column (New England Biolabs) washed in 600 ml buffer A at a constant flow rate of 1 ml/min. To remove the chitin-intein tag, we first incubated the resin in buffer A containing 50 mm DTT for 15 h at room temperature and later eluted Bfl-1 in buffer A containing 50 mm DTT. For NMR experiments, the sample buffer was exchanged by dialysis in phosphate-buffered saline (pH 7.4) with NaCl added to 200 mm, 2 mm tris(2-carboxyethyl)phosphine, and 1 mm sodium azide (buffer B) or buffer B in 99.5% D2O (buffer C). Samples were concentrated to 0.5 mm by ultracentrifugation using an Amicon Ultra-15 device with a 10-kDa molecular weight cutoff (Millipore). To minimize protein precipitation or denaturation, we limited spin cycles to 5-min bursts, followed by gentle mixture with a pipette. 4,4-Dimethyl-4-silapentane-1-sulfonic acid was added to a final concentration of 0.1 mm as well as 7% D2O in buffer B. All of our buffers were extensively degassed and always prepared just before use.
Bcl-B protein (a product of the human BCL2L10 gene) lacking its last 20 carboxyl-terminal amino acids was subcloned into a pGEX-4T-1 vector (GE Healthcare). The resulting plasmid was used for the transformation of E. coli BL21star (DE3) and expression after cell growth at 37 °C in LB medium containing 50 μg/ml carbenicillin to a cell density of 0.8–1.0 (600 nm), followed by a 4-h induction with 0.4 mm isopropyl β-d-thiogalactopyranoside. Cells were harvested by centrifugation at 4000 × g for 20 min and stored at −20 °C until use. For protein purification, the cell pellet was resuspended in PBS buffer containing 1 mg/ml lysozyme and 1 mm PMSF. After incubation for 30 min at 4 °C, this mixture was submitted to sonication and centrifugation at 12,000 × g for 30 min to clear all debris. Soluble proteins were purified in glutathione-Sepharose 4B (GE Healthcare) using standard conditions, and the GST tag was removed with thrombin as described by the manufacturer. To prepare 15N-labeled samples, we used the same procedure as described above for the expression of 15N-labeled Bfl-1 proteins.
A cDNA comprising the open reading frame encoding Bcl-B was subcloned into the pcDNA3-Myc plasmid to produce the pcDNA3-Myc-Bcl-B expression vector. Site-directed mutagenesis of Bcl-B was performed to generate the double substitution mutations using the pcDNA3-Myc-Bcl-B plasmid as the DNA template and the mutagenic primers for R47A/E99A (5′-GCGGCCGCCAGGTTAGCGCAGATTCACCGGTCC-3′ and 5′-GGACCGGTGAATCTGCGCTAACCTGGCGGCCGC-3′; 5′-GCAGGGACGCTGCTGGCCAGAGGGCCGCTGGTG-3′ and 5′-CACCAGCGGCCCTCTGGCCAGCAGCGTCCCTGC-3′), for R47E/E99R (5′-GCGGCCGCCAGGTTAGAGCAGATTCACCGGTCC-3′ and 5′-GGACCGGTGAATCTGCTCTAACCTGGCGGCCGC-3′; 5′-GGGACGCTGCTGCGGAGAGGGCCGCTG-3′ and 5′-CAGCGGCCCTCTCCGCAGCAGCGTCCC-3′), for R51A/S55Y (5′-CGGCAGATTCACGCGTCCTTTTTCTACGCCTACCTCGGC-3′ and 5′-GCCGAGGTAGGCGTAGAAAAAGGACGCGTGAATCTGCCG-3′), for Y19F/A44L 5′-GACTACCTGGGGTTCTGCGCCCGGGAAC-3′ and 5′-GTTCCCGGGCGCAGAACCCCAGGTAGTC-3′; 5′-CGCTCCGCGGCCCTCAGGTTACGGCAG-3′ and 5′-CTGCCGTAACCTGAGGGCCGCGGAGCG-3′), and for L58D/L103S (5′-CTCCGCCTACGACGGCTACCCCGG-3′ and 5′-CCGGGGTAGCCGTCGTAGGCGGAG-3′; 5′-GAGAGAGGGCCGTCGGTGACCGCCCGG-3′ and 5′-CCGGGCGGTCACCGACGGCCCTCTCTC-3′) and verified by DNA sequencing. Bcl-B residues were numbered according to the protein sequence deposited in UNIPROT (entry Q9HD36). The previously described Nur77-ΔDBD (18) was subcloned into the pcDNA3-HA plasmid to produce the pcDNA3-HA-Nur77-ΔDBD expression vector.
Bfl-1 Main Chain Assignment
NMR data were recorded at 298 K on a Bruker DRX-800 spectrometer equipped with a cryoprobe. 1H, 13C, and 15N sequential resonance assignments were obtained using the following experiments: HNCA, CBCA(CO)NH, and 1H,15N HSQC (sample in buffer B); 1H,13C HSQC, and HCCH-TOCSY (sample in buffer C). Proton chemical shifts were referenced to the nine identical methyl protons present in 2,2-dimethyl-2-silapentanesulfonic acid, whereas 13C and 15N resonances were calibrated according to the IUPAC recommendations (21). Data were processed in TOPSPIN 2.1 (Bruker Biospin). Peak search was carried out in Auremol (22) and Sparky (23). A partial backbone assignment was generated by MARS (24). Results were manually inspected and validated with minor corrections, and additional peaks were added to the final backbone assignment.
Chemical Shifts with Nur77 Peptide Titration
To experimentally determine the region where Nur77 interacts with Bcl-2 family members, the 15N-labeled Bcl-2/Bcl-X(L) chimera (as described in Ref. 20), 15N-labeled Bfl-1 (amino acids 1–154), and 15N-labeled Bcl-B (amino acids 1–167) were used in 1H,15N HSQC experiments with a Nur77 9-mer peptide (termed Nu-BCP-9 as described in Ref. 25) as a molecular probe. We tested peptides with a poly-arginine membrane-translocating peptide (FSRSLHSLLGXR8) or without it (FSRSLHSLL). All experiments were recorded at 298 K on a 500 MHz Bruker Avance spectrometer. Data were processed in TOPSPIN 2.1 (Bruker Biospin), and peak searches and plots were carried out in Sparky. Both Nur77 peptides (with or without a poly-arginine tag) triggered the same chemical shifts in the Bcl-2/Bcl-X(L) chimera and Bfl-1 proteins (data not shown). For titration experiments, the Bcl-2/Bcl-X(L) chimera and Bfl-1 were used from 0.2 mm up to 1 mm, respectively, whereas peptides were added to 0, 0.05, 0.1, 0.2, 0.5, and 1 mm concentration. Chemical shifts were calculated as described in Ref. 26. The chemical shift threshold cutoff values were set by calculating the chemical shift change standard deviation (σ) of all data and then excluding from this dataset all values that were higher than 3σ and chemical shifts in buried residues to recalculate an adjusted standard deviation (σA). The threshold cutoff we used was 3σA to identify only true positives (less sensitive but more specific) (27). We used the structure of the Bcl-2/Bcl-X(L) chimera (PDB code 1GJH) (20) and Bfl-1, excluding the BH3 peptide from BIM (PDB code 2VM6) (28) to calculate the surface-exposed area by residue, considering buried all residues that are 5% exposed or less (29).
Immunoprecipitation and Protein Analysis
Myc-tagged Bcl-B- and HA-tagged Nur77-transfected cells in 10-cm plates were suspended in lysis buffer (50 mm Tris-Cl (pH 7.4), 150 mm NaCl, 20 mm EDTA, 50 mm NaF, 0.5% Nonidet P-40, and 0.1 mm Na4VO3) containing protease inhibitor mixture at 4 °C for 45 min on a wheel rotor. The lysates were cleared of cell debris by centrifugation at 10,000 × g for 15 min, and the total protein content was quantified by BCA assay. Protein G magnetic beads (15 μl/sample) were incubated with 100 μg/ml of anti-HA rat-specific antibody in TBS buffer with gentle agitation for 4 h at 4 °C. Following collection of the beads with a magnet, the supernatant was removed, and cell lysates were added to the beads and incubated at 4 °C overnight. Magnetic beads were washed three times in TBS containing 0.1% Tween 20 (TBST20), resuspended in Laemmli sample buffer containing β-mercaptoethanol, and boiled for 5 min to release bound proteins. Proteins were loaded onto SDS-PAGE (4–20%) and subjected to immunoblotting. For direct immunoblot analysis using cell lysates, cells were lysed with radio-immunoprecipitation assay buffer (50 mm Tris-Cl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.1% SDS, 0.5% deoxycholic acid, 1% Triton X-100, 50 mm NaF, and 0.1 mm Na3VO4) containing protease inhibitor mixture at 4 °C for 30 min on a wheel rotor. The lysates were centrifuged at 10,000 × g for 15 min, and the total protein content was quantified by BCA assay. Proteins were separated by (15% or 4–20%) SDS-PAGE gel electrophoresis and subjected to immunoblotting.
Cell Culture and Transfections
Human embryonic kidney cancer cells (293T) and brain neuroglioma cells (H4) were maintained in DMEM (Mediatech) with 10% FBS (Sigma), penicillin (100 IU), and 100 μg/ml streptomycin at 37 °C in 5% CO2, 95% air. Transient transfections were performed using PEI.
For immunoprecipitation experiments, 293T cells were transfected with various Myc-tagged Bcl-B plasmids (WT and R47A/E99A; R47E/E99R; R51A/S55Y; Y19F/A44L; and L58D/L103S double mutants) using 6 μg of DNA/10-cm dish with or without HA-Nur77-ΔDBD at 6 μg of DNA/10-cm dish for 24 h in the presence of 15 μm Z-VAD-fmk.
For immunoblot assays, H4 cells were transfected with various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y; Y19F/A44L and L58D/L103S double mutants) using 1 μg of DNA/6-well plate with or without HA-Nur77-ΔDBD at 0.33 μg of DNA/6-well plate in the presence or absence of 15 μm Z-VAD-fmk for 24 h. For LC3 flux assays, following 24-h incubation, cells were treated with 100 μm leupeptin and 20 mm NH4Cl lysosomal inhibitors for either 2 or 4 h.
For cell viability determinations in 96-well microtiter plates, Myc-Bcl-B-WT or mutant Bcl-B plasmids (R51A/S55Y; Y19F/A44L, and L58D/L103S double mutants) were co-transfected into cells using 0.075 μg of DNA/96-well plate together with HA-Nur77-ΔDBD at 0.03 μg of DNA/96-well plate. Cells were cultured for 32 h in complete medium.
Cell Viability Assays
The Cell Titer-Glo luminescent cell viability assay (Promega) was used to measure cellular ATP concentrations for cell viability estimation. Cells were seeded at a density of 2.5 × 104 cells/well in 100 μl of culture medium in 96-well flat-bottom plates. Transfections with plasmids encoding WT or mutant Bcl-B proteins were performed as described above. In some cases, cell cultures were supplemented with 100 μm leupeptin and 20 mm NH4Cl. Cell cultures were incubated at 37 °C/5% CO2 for 32 h. Then plates were set aside for 10 min to equilibrate to room temperature, and 100 μl of Cell Titer Glo solution was added to each well and placed in the dark for 15 min. Luminescence was measured with a luminometer (Luminoskan Ascent, Thermo Electron Corp.) at 1-s integration time per sample.
Cell viability was also evaluated by trypan blue dye exclusion assay upon collection of both floating and adherent cells (recovered by trypsin-mediated release) as described previously (30) or by reverse phase microscopy direct inspection of cells in culture dishes. Photographs of cells were taken using an EVOS microscope from AMS (Life Technologies).
Results
Nur77 Peptide Associates with a Novel Interaction Surface on Bcl-2 Family Proteins
The Nur77 protein has been reported to bind the anti-apoptotic proteins Bcl-2, Bcl-B, and Bfl-1 but not Bcl-X(L), Mcl-1, or Bcl-W (30–31). Previous studies of Nur77 mapped the Bcl-2-binding region to a nine-amino acid peptide derived from the ligand binding domain and showed that this peptide is sufficient to bind Bcl-2 and Bcl-B (31–32). We therefore sought to use an unlabeled Nur77 peptide as a probe to explore its interaction with the surface of uniformly 15N-labeled Bcl-2 family proteins by NMR-based chemical shift perturbation analysis.
Of the Bcl-2 family members, the 9-mer peptide reportedly binds tightest to Bcl-B (Kd of about 20 nm as measured by fluorescence polarization assay) (30–31). However, Bcl-B was not usable for these NMR experiments because it did not produce a well dispersed 1H,15N HSQC spectrum, suggesting that Bcl-B has a tendency to form larger molecular arrangements in solution under the conditions we tested (data not shown). Bcl-2 was also not an ideal choice because NMR spectra have only been obtained to date using chimeric Bcl-2/Bcl-X(L) protein because of solubility challenges (20). We therefore focused on Bfl-1.
We first assigned the amide backbone resonances of Bfl-1 by multidimensional NMR. Approximately 94% of the HN, N, Hα, Cα, Hβ, and Cβ resonances were assigned, excluding the N-terminal amino acids 1–7 that contain residues with broadened amide resonances. Almost all NMR-visible residues were identified, excluding amide side chains (Fig. 1). Unexpectedly, we also assigned a set of 15 strong peaks in addition to the Bfl-1 protein resonances. These residues matched N-extein peptide derived from the expression vector, with the exception of the first two amino acids (MKIEEGKLVIGSLEG). N-extein is a byproduct of the N-terminal chitin-intein tag used for expression and purification in bacteria. It is not covalently bound to the protein and thus should have been removed during dialysis. Additional experiments were carried out using an isotopically labeled His6-tagged Bfl-1 construct as an alternative to the chitin-intein tagged version, showing negligible differences in the 1H,15N HSQC spectrum of Bfl-1 (data not shown) and thus validating the original assignments.
FIGURE 1.
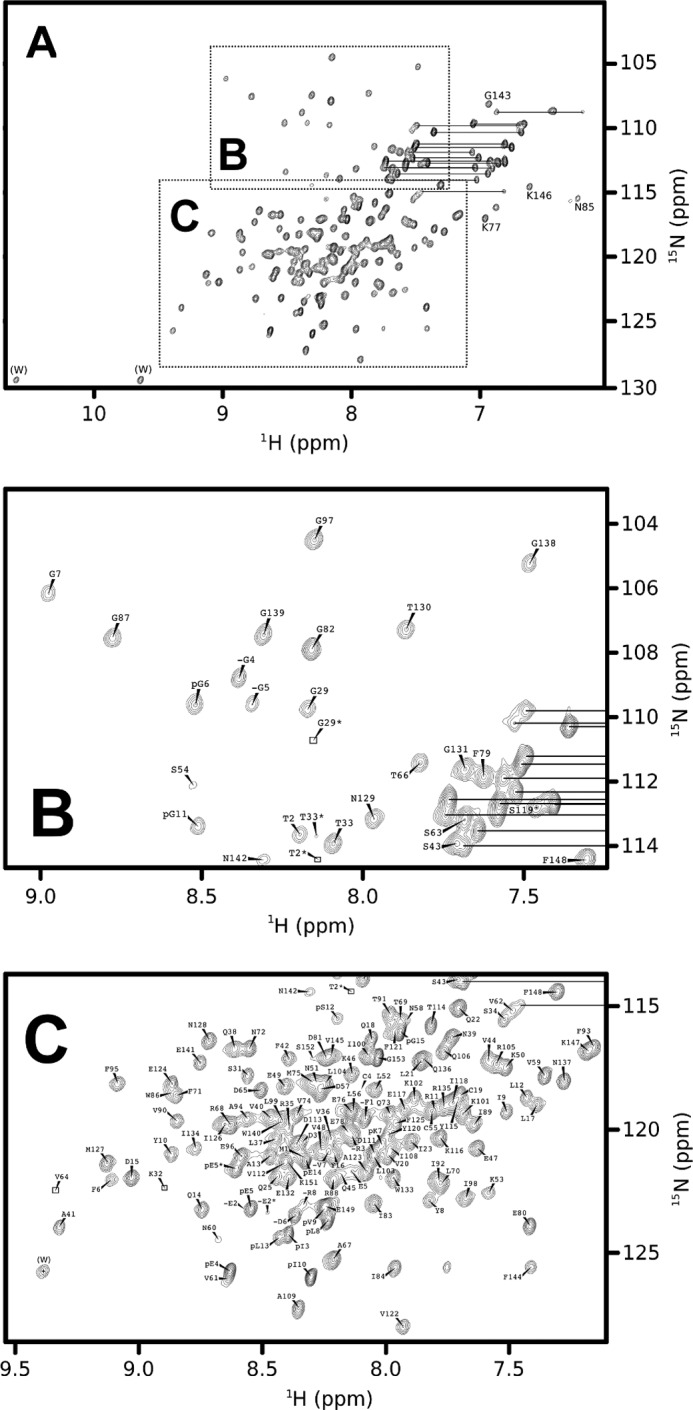
1H,15N HSQC spectra of Bfl-1. A–C, overview (A) with detailed regions as indicated (B and C). Data were acquired at 298 K at a proton frequency of 800 MHz using a protein sample concentrated to 300 μm in phosphate-buffered saline with NaCl to 200 mm and 2 mm tris(2-carboxyethyl)phosphine (pH 7.4). The reduced state of the cysteine residues was confirmed by Cβ chemical shifts at around 28 ppm compared with 41 ppm typical for cysteine residues.
Using 15N-Bfl-1 and the Nur77 9-mer peptide probe, chemical shift mapping experiments were conducted (Fig. 2). These experiments revealed that the Nur77 peptide interacts with three distinct segments of the Bfl-1 protein chain (Fig. 3). These segments comprise residues distributed along helices α2, α3, α4, and α5, corresponding to a crevice on the Bfl-1 fold that shares a wall with the BH3-binding pocket.
FIGURE 2.
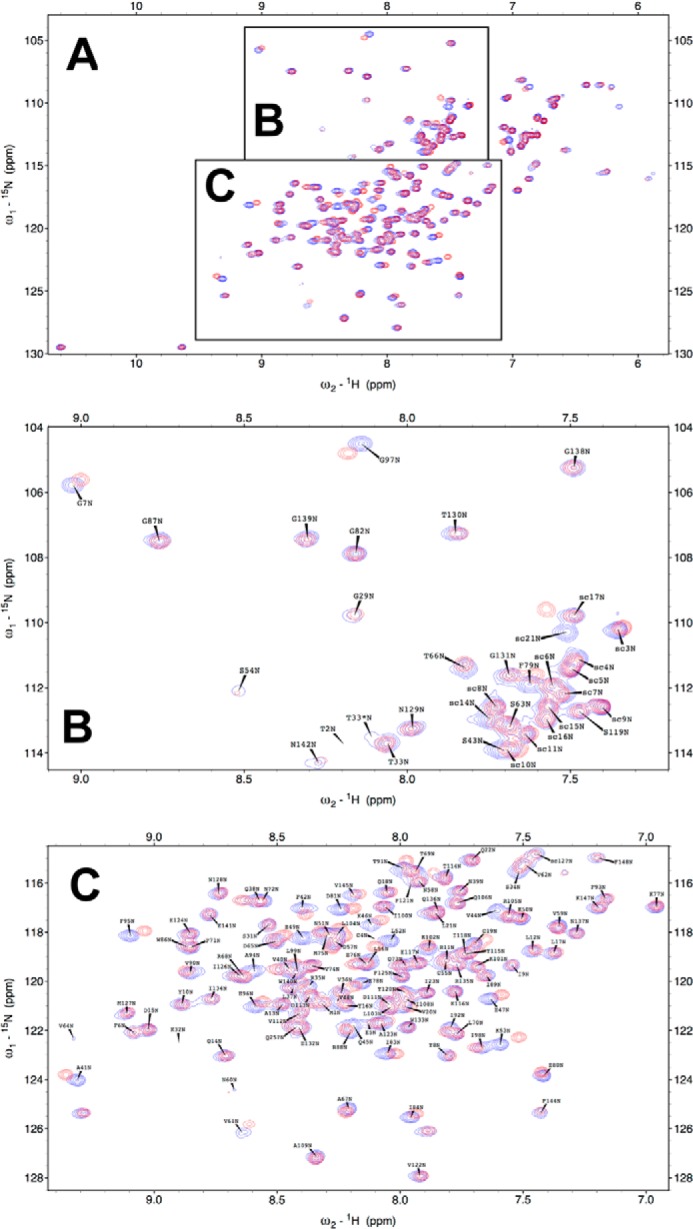
Superposition of 1H,15N HSQC spectra of Bfl-1 with versus without Nur77 peptide. A–C, superposition of 15N-labeled Bfl-1 in the absence (blue) and presence of Nur77 9-mer peptide (FSRSLHSLL) in 5× molar excess. An overview is presented (A) as well as detailed regions as indicated (B and C). Data were acquired at 298 K at a proton frequency of 500 MHz using 0.2 mm Bfl-1 (His tag construct) in phosphate-buffered saline with NaCl to 200 mm and 1 mm tris(2-carboxyethyl)phosphine (pH 7.3). Assignment labels were added for the protein-only spectrum.
FIGURE 3.

Chemical shift perturbations induced by Nur77 peptide binding. A and B, a 9-mer peptide from Nur77 was used as a molecular probe to evaluate the effect of chemical shift perturbations on the main chain NMR resonances of the Bcl-2/Bcl-X(L) chimera (A) and Bfl-1 (B). Chemical shift perturbations (Δ) were calculated as follows: Δ = √(DdH)2 + (0.2DdN)2, where Dd = (d2 − d1) corresponds to the resonance difference of 0.2 mm Bfl-1 alone and Bfl-1 in the presence of 1 mm Nur77 peptide, sampled for both backbone nitrogen (N) and its associated proton (H) chemical shifts. The dashed lines in A and B represent our threshold cutoff (3σA), whereas squares show solvent accessibility by residue, calculated as described under “Experimental Procedures.” C and D, the most perturbed residues are indicated by numbered boxes relative to the three-dimensional structure of Bcl-2/Bcl-X(L) (C) and Bfl-1 (D), leading to the proposed Nur77 binding site, which is distinct from the BH3 binding site, as indicated by dashed circles.
The chemical shift mapping was also conducted using a Bcl-2/Bcl-X(L) chimeric protein for which the residue assignments for the 1H,15N HSQC spectrum had been accomplished previously (20), again suggesting the same corresponding interaction surface. Using a Bcl-2/Bcl-X(L) chimera model (20) and the structure of Bfl-1 (28), we observed that the most perturbed amides in both the Bfl-1 and the Bcl-2/Bcl-X(L) chimeric proteins flank the BH3 binding crevice. Perturbations in α5, however, show that the Nur77 peptide interaction occurs on a distinct surface, where α2-α3 serves as a wall dividing the BH3 and Nur77 peptide sites (Fig. 4).
FIGURE 4.

Identification of the putative Nur77 binding site on the surface of the anti-apoptotic Bfl-1 protein. A–C, water-accessible surface representation of Bfl-1 colored by electrostatic potential (± 5 kT/e−) showing the Nur77 binding site within the area enclosed by a circle (A), after a 90° rotation over the x axis to show a very distinct BH3 binding site (B), and after a 90° rotation over the y axis (C). The Nur77 peptide, presumed to be partially helical (depicted as a red cylindrical object), and the BH3 peptide ligand (depicted as a blue cylindrical object) bind at distant sites separated by a common wall formed by helices α2 and α3 (dashed line).
Mutating Residues within the Predicted Binding Site of Bcl-2 Family Proteins Modulates Interactions with Nur77 Protein
We sought to use site-directed mutagenesis to confirm that the binding site for the Nur77 peptide identified in vitro is indeed relevant to the mode of binding of the Nur77 protein in cells. To this end, mutations were generated in the putative Nur77-binding crevice of Bcl-2 family proteins, and we tested their binding by co-IP assays, where proteins were expressed with epitope tags in HeLa or HEK293T cells. For these experiments, a fragment of the Nur77 protein was expressed lacking the DNA-binding domain (DBD), which is devoid of nuclear targeting sequences and thus is found constitutively in the cytoplasm. Nur77ΔDBD operates as a constitutively active variant (gain of function mutant), not requiring cellular stimulation to induce its nuclear export for interactions with Bcl-2 family members (17–18). Because Nur77 protein binds Bcl-B far better than Bfl-1 or Bcl-2 in cellular co-IP and in vitro protein interaction pulldown assays (data not shown), we conducted a mutagenesis analysis using Bcl-B, guided by the previously reported three-dimensional structure of Bcl-B (33) superimposed on the structures of the Bcl-2/Bcl-X(L) chimera and Bfl-1. Cells were cultured in the presence of the irreversible caspase inhibitor Z-VAD-fmk to preclude interference with protein expression because of cell death.
The residue changes introduced in Bcl-B included elimination of charged or polar residues (R47A, E99A, and Y19F) as well as charge reversals (R47E and E99R) and replacement of hydrophobic residues with charged or polar amino acids (L58D, L103S) (Fig. 5). None of these amino acid substitutions were predicted to be essential for maintaining the overall Bcl-B protein fold. Consistent with previous reports, WT Bcl-B protein co-immunoprecipitated with Nur77. All single point Bcl-B mutants were also able to bind Nur77 (data not shown). Therefore, double mutations in the putative Nur77-binding pocket of Bcl-B were generated, which then revealed effects on Nur77 binding. Interaction of Nur77 protein with the Bcl-B double mutants R47A/E99A (charge eliminations), Y18F/A44L (increased hydrophobicity), and L58D/L103S (increased polarity/hydrophilicity) was greatly impaired (Fig. 6A, first panel). The Bcl-B double mutant R47E/E99R (charge reversals) appeared to be unstable and did not accumulate to detectable levels in HEK293T cells (Fig. 6A, third panel). Interestingly, the Bcl-B mutant R51A/S55Y (increased hydrophobicity) showed stronger binding to Nur77 than WT Bcl-B in these co-IP assays. The Nur77 and Bcl-B protein band densities from multiple repeats of co-IP experiments were quantified, confirming that combined mutagenesis of Bcl-B at residues R51A and S55Y enhances Bcl-B/Nur77 protein interactions, whereas combined mutagenesis of Bcl-B at residues Y19F/A44L or L58D/L103S prevented the ability of Bcl-B to bind to Nur77 (Fig. 6B).
FIGURE 5.

Bcl-B residues chosen for mutagenesis. A, three-dimensional representation of Bcl-B (blue schematic) superposed with Bfl-1 (gray schematic) and a BH3 peptide from BIM (gray surface). Amino acid residues chosen for mutagenesis are shown in stick mode with indicated labels. The most perturbed Bfl-1 backbone amines in NMR experiments are shown in red. Note that Bfl-1 and Bcl-B have marked structural differences in the length of helix α2 (Ala-44 and Arg-47 mutations) and positioning of helix α3 (Ser-55 mutation) as well as the C-terminal ending of helix α5 (Glu-99 mutations). B, surface representation of Bcl-B (blue) with mutation positions (red). Images were generated in PyMOL using Bcl-B in complex with BIM peptide (PDB code 4B4S) and Bfl-1 in complex with a peptide from Bak (PDB code 3I1H).
FIGURE 6.

Identification of residues involved in the Bcl-B/Nur77 protein interaction. A, HEK293T cells were co-transfected with either the pcDNA3-Myc vector and pcDNA3-HA vector (first lane) or various Myc-tagged Bcl-B plasmids (WT and the R47A/E99A, R47E/E99R, R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) with or without HA-Nur77ΔDBD (second to seventh lanes) for 24 h in the presence of 15 μm Z-VAD-fmk. Cell lysates were immunoprecipitated using rat anti-HA antibody, and the immunoprecipitated proteins were analyzed by immunoblotting using mouse anti-HA to detect Nur77 and mouse anti-Myc to detect Bcl-B proteins. The inputs ( of lysates used for immunoprecipitation) were analyzed by immunoblotting using mouse anti-Actin as a loading control. WB, Western blotting. B, levels of co-immunoprecipitated HA-Nur77 or Myc-tagged Bcl-B proteins were quantified using scanning densitometry (mean ± S.E.; n = 3; *, p < 0.05 in comparison with Bcl-B-WT + Nur77).
Next we tested these mutants of Bcl-B for their ability to bind Bax, which binds via a BH3-dependent mode. To avoid cell death induced by overexpression of Bax in HEK293T cells, Z-VAD-fmk was added to cultures. Variations were observed among the tested Bcl-B mutants, with Y19F/A44L retaining binding similar to wild-type Bcl-B and R51A/S55Y and L58D/L103S showing reduced interaction with Bax by co-IP assay (Fig. 7A). However, all Bcl-B mutants retained their ability to protect against cell death induced by Bax overexpression (Fig. 7B). To achieve more physiologically relevant levels of expression of Bcl-B variants from transfected plasmids, the cell viability assays were conducted using HCT116 (human colon cancer) cells rather than HEK293T (human neurofetal) cells, which express SV40 large T antigen, causing replication of transfected plasmids and thus leading to supraphysiological protein expression (which facilitates protein-protein interaction assessments). Taken together, these findings suggest that the residues lining the putative Nur77-binding crevice of Bcl-B can mediate interaction with Nur77 via a BH3-independent mode of binding but also suggest the possibility of some degree of potential “cross-talk” between the BH3- and Nur77-binding sites.
FIGURE 7.

Bcl-B double mutants protect against Bax-induced cell death. A, 293T cells were co-transfected with either the pcDNA3-Myc vector and pcDNA3-HA vector (first lane) or various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) with or without HA-Bax (second to seventh lanes) for 24 h in the presence of 15 mm Z-VAD-fmk. Cell lysates were immunoprecipitated using rat anti-HA antibody, and the immunoprecipitated proteins were analyzed by immunoblotting using mouse (mo) anti-HA to detect Bax and mouse anti-Myc to detect Bcl-B proteins. The inputs ( of lysates used for immunoprecipitation) were analyzed by immunoblotting using mouse anti-α-tubulin as a loading control. WB, Western blotting. B, HCT116 cells were co-transfected with either the pcDNA3-HA vector (first lane), pcDNA3-Myc vector (second lane), or various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) with or without HA-Bax (second to sixth lanes) for 24 h. Cell viability was assessed by exclusion of trypan blue (mean ± S.D.; n = 3).
Cellular Analysis of the Nur77-binding Site Confirms a Role in Modulating Cell Survival and Autophagy
To correlate the protein interaction analysis data with cellular function, we compared the impact of WT and mutant Bcl-B proteins using cellular assays where Nur77ΔDBD shows readily measurable phenotypes. To ensure more physiologically relevant levels of protein expression, we used the H4 cell line (human glioblastoma) for these cell biology experiments rather than HEK293T cells (human neurofetal), which express SV40 large T antigen (see explanation above). Association of Bcl-B and Nur77 reportedly converts the phenotype of Bcl-B from anti-apoptotic to pro-apoptotic (30). Indeed, when transfected H4 cells were cultured in the absence of Z-VAD-fmk, a significant loss of cells was consistently observed by light microscopy following co-expression of WT Bcl-B with the cytosolic, constitutively active Nur77ΔDBD protein (Fig. 8A). Similar results were produced with the Bcl-B mutant R51A/S55Y, which retains Nur77 binding. In contrast, the numbers of viable cells were not diminished in cultures transfected with plasmids expressing the Bcl-B mutants that cannot bind to Nur77 (Y18F/A44L and L58D/L103S). These findings by microscopy were corroborated using assays measuring cellular ATP levels as a surrogate indicator of cell viability (Fig. 8B). The conversion of Bcl-B from anti-apoptotic to pro-apoptotic through interaction with Nur77 has been associated previously with increases in caspase activity and increases in the percentage of cells displaying apoptotic nuclei visualized by DAPI staining (30). Overexpression of the double mutant Bcl-B variants that do not associate with Nur77 (Y19F/A44L and L58D/L103S) had little effect on cell viability (Fig. 8B).
FIGURE 8.

The Nur77 interaction site on Bcl-B modulates the cell death phenotype. A, photographs from a representative experiment showing H4 cells co-expressing HA-Nur77ΔDBD with various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) after 32 h. CTRL, control. B, H4 cells were co-transfected with various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) with or without HA-Nur77ΔDBD and cultured in the presence (gray columns) or absence (black columns) of the lysosomal inhibitors leupeptin (100 μm) and NH4Cl (20 mm) for 32 h. Following incubation, the cells were lysed, and ATP levels were measured using Cell Titer Glo luminescence as a surrogate indicator of the relative number of viable cells (mean ± S.E., n = 3). *, p < 0.005; **, p < 0.001 in comparison with treatment with lysosomal inhibitors.
Interestingly, supplementing cell cultures with lysosome inhibitors (leupeptin and NH4Cl) partially restored cell viability in H4 cells co-expressing Nur77 with either WT or R51A/S55Y Bcl-B (Fig. 8B), suggesting a role for lysosomes in this cell death mechanism. These findings suggest that excessive autophagy may contribute to the cell death that results from Bcl-B and Nur77 protein interaction.
Bcl-B binds Beclin-1 and has recently been implicated in preventing autophagy-dependent cell death (34). Conversely, Nur77 has been reported to promote autophagy through effects on mitochondria (35, 36). To explore the role in autophagy regulation of the predicted Nur77 protein binding site on Bcl-B, we studied markers of autophagic flux in H4 cells co-expressing Nur77 with Bcl-B WT or mutants that fail to bind Nur77. Given that p62 undergoes lysosomal degradation upon induction of autophagy (37), we first compared steady-state levels of p62 protein by immunoblotting of cell lysates from H4 cells overexpressing Bcl-B and Nur77. Overexpressing WT Bcl-B or Nur77ΔDBD alone had no significant effect on p62 degradation (data not shown). When co-expressed in H4 cells, the net effect of the combination of WT Bcl-B and Nur77ΔDBD was a reduction in the level of endogenous p62 protein compared with H4 cells expressing the empty vectors (Fig. 9A, top panel), suggesting a net increase in autophagy. Co-expression of Nur77ΔDBD with the Bcl-B mutant R51A/S55Y, which retains binding to Nur77, likewise resulted in a net reduction in p62 levels. In contrast, the Bcl-B mutants Y19F/A44L and L58D/L103S, which do not associate with Nur77, demonstrated elevated levels of endogenous p62 protein even above that observed in untreated H4 cells (Fig. 9A, top panel), suggesting reduced autophagy. As a positive control, H4 cells treated with the autophagy inducer rapamycin (mTOR inhibitor) also resulted in decreased p62 protein levels (Fig. 9A, top panel).
FIGURE 9.

The Nur77 binding site on Bcl-B modulates the autophagy phenotype. A, the levels of p62 were assessed in H4 cells treated with the autophagy inducer rapamycin (25 μg/ml) for 16 h (second lane) or in H4 cells in which Myc-tagged-Bcl-B WT or mutants were co-expressed with HA-tagged Nur77ΔDBD in the presence of 15 μm Z-VAD-fmk for 24 h (third to sixth lanes). An equal amount of protein from each cell lysate was analyzed by immunoblotting using anti-p62, anti-HA, and anti-Myc antibodies. Actin served as a loading control. WB, Western blotting. B, H4 cells were co-transfected with various Myc-tagged Bcl-B plasmids (WT and the R51A/S55Y, Y19F/A44L, and L58D/L103S double mutants) with HA-Nur77ΔDBD for 24 h. Cells were then treated with the lysosomal inhibitors leupeptin (100 μm) and NH4Cl (20 mm) for 2 or 4 h. The levels of LC3-I and LC3-II were analyzed by immunoblotting of total cell lysates with anti-LC3 antibody in addition to anti-myc, anti-HA, and anti-actin to verify equal protein expression and loading. C, the LC3-II bands in B were quantified using scanning densitometry. The levels of LC3-II after 2 h of leupeptin and NH4Cl treatment were divided by the level of LC3-II without lysosomal inhibitor treatment to calculate LC3 flux (mean ± S.E., n = 5). *, p < 0.05 in comparison with Bcl-B-WT + Nur77.
Additionally, we assessed an alternative marker of autophagy, LC3 (ATG8). After proteolytic cleavage of pro-LC3, lipid conjugation converts LC3-I to LC3-II, which becomes integrated into autophagosomal membranes that eventually fuse with lysosomes (38). To examine LC3 flux, we cultured H4 cells in the presence or absence of the lysosomal inhibitors leupeptin and ammonium chloride (NH4Cl). Immunoblot analysis of cell lysates prepared at various times thereafter showed modest increases in LC3-II levels in control cells, as expected. However, in cells transfected with plasmids to co-express Nur77ΔDBD and Bcl-B WT, striking increases in LC3-II levels were observed (Fig. 9B, top panel), consistent with an increase in autophagy. The double mutant of Bcl-B (R51A/S55Y), which retains the ability to interact with Nur77, produced similar results. However, under the same culture conditions, no changes in LC3-II levels were detected in cells co-expressing Nur77ΔDBD together with the loss-of-function Bcl-B mutants Y19F/A44L or L58D/L103S, which fail to bind Nur77 (Fig. 9B). Conversely, overexpression of Bcl-B-WT alone (without Nur77) did not cause an increase in LC3-II levels in the presence of lysosomal inhibitors but, rather, reduced LC3-II accumulation (data not shown), consistent with previous reports documenting autophagy suppression by Bcl-B in the absence of Nur77 (34).
The LC3-II protein band densities from multiple experiments were quantified, and the ratio of LC3-II levels at 2 h versus 0 h of exposure to lysosomal inhibitors was calculated to determine LC3 flux. Although cells expressing Nur77ΔDBD with either WT Bcl-B or the mutant R51/S55Y Bcl-B protein demonstrated increases in LC3 flux (consistent with an increase in autophagy), co-expressing Nur77ΔDBD with non-binding mutants of Bcl-B (Y19F/A44L and L58D/L103S) did not (Fig. 9C). Taken together, these experiments provide further support for the hypothesis that the interaction surface mapped by NMR corresponds to a bona fide Nur77 binding site on Bcl-2 family proteins.
Discussion
We used a Nur77-derived peptide as a molecular probe to identify a new regulatory site in the anti-apoptotic Bcl-2, Bfl-1, and Bcl-B proteins. Our analysis of the putative Nur77 binding site of Bcl-B indicates that two pockets reside in regions close to the amino acids most perturbed in our NMR experiments. These pockets are separated by a trio of amino acids (Arg-47, Thr-95, and Glu-99) that form a network of hydrogen bonds stabilizing helices α2 and α3. Mutation of Arg-47 and Glu-99 to disrupt these interactions resulted in a Bcl-B molecule that is unstable in cells. The Y19F/A44L mutant was designed to increase hydrophobicity and hinder access to the pocket closest to the BH3 domain sequence in α2. As a consequence, we observed a loss in the ability of this mutant to bind to Nur77. The combined mutations of R51A and S55Y in the adjacent pocket removed hydrophilic amino acids to explore whether hydrogen bonding might play a role in Nur77 binding. However, we found that this change actually enhanced the Bcl-B/Nur77 protein-protein interaction, suggesting that hydrophobic interactions dominate the molecular basis for Nur77 binding to Bcl-2 family proteins. Additionally, we found that modifying residues further away from the most perturbed residues in NMR experiments, but still in the vicinity of the putative Nur77 binding site, L58D/L103S (which increases polarity/hydrophilicity), prevented Bcl-B interaction with Nur77ΔDBD. These results confirm our structural predictions and help to delimit the Nur77 binding interface in Bcl-2 family members.
Previous data suggested that a long (67-amino acid) unstructured segment between helices α1 and α2 (herein referred to as the “loop”) of Bcl-2 is responsible for binding to Nur77 (30, 39). However, we measured significant chemical shift perturbations in other regions of Bfl-1 and the Bcl-2/Bcl-X(L) chimera. Fluorescence polarization assay and CD methods were used in the previous analysis of loop binding, reporting affinities of 0.2–2 μm (21). With respect to our data, it is important to recognize that the Bcl-2/Bcl-X(L) chimera we employed has part of the loop of Bcl-X(L) instead of Bcl-2. (Note that Bcl-X(L) is incapable of binding Nur77, unlike Bcl-2) (30). Also, compared with Bcl-2, the corresponding loop segment of Bfl-1 is much shorter (only nine amino acids) and is rich in proline residues that are not resolved in 1H,15N HSQC spectra. Although we cannot disregard the involvement of the unstructured loop for the binding of Nur77, the Bcl-2/Bcl-X(L) chimera protein lacking the loop of Bcl-2 was still able to bind the Nur77 peptide. Thus, the participation of the loop segment of Bcl-2 family proteins in binding of Nur77 may assist in stabilization of the complex in the context of the full-length proteins in addition to the binding site we identified in this work. Alternatively, we cannot entirely exclude the possibility that the Nur77 peptide interacts with the loop regions of Bcl-2 family proteins and causes the loop segment to secondarily interact with or alter the confirmation of the site we identified by chemical shift mapping. In either case, however, our mutagenesis analysis shows that the site we identified is functionally important.
Mutations in Bcl-B involving the predicted Nur77 interaction site modulated Nur77 protein binding in cells, as assessed by co-IP assays. Of these Bcl-B mutants, two abolished binding to Nur77, and one enhanced association of Bcl-B to Nur77, but all retained variable ability to bind Bax in co-IP experiments, and all retained the ability to protect HCT116 cells from Bax-induced cell death. Importantly, these results demonstrate that the BH3 and Nur77 binding sites are distinct. However, the variations in the efficiency of Bax binding among the Bcl-B mutants suggests the possibility of some degree of cross-talk between the Nur77 and BH3 binding sites, which might be expected given that they are adjacent and share a bordering “wall” that separates the two crevices.
A function for the anti-apoptotic Bcl-B protein as a modulator of autophagy was described recently (27). Similar to other Bcl-2 family members, Bcl-B prevents autophagy by binding to and inhibiting Beclin-1, an essential mediator of autophagy, which is mediated by interaction of Bcl-B with the BH3 domain of Beclin. We found that, when Bcl-B was occupied by an excess of Nur77 (achieved by overexpressing the cytosolic ΔDBD construct), autophagy was induced, as assessed by reductions in p62 protein levels and by the ratio of lipid conjugated to unconjugated LC3. In contrast, in cells expressing Bcl-B mutants that fail to bind Nur77, less autophagy was observed, as demonstrated by elevated p62 levels and a reduction in LC3 lipid conjugation. These observations are consistent with the idea that these Bcl-B mutants are free to bind endogenous Beclin-1 even in the presence of high levels of Nur77, thus suppressing autophagy. However, we were unable to experimentally confirm this hypothesis for technical reasons because, when Nur77/ΔDBD was expressed in cells, Beclin protein partitioned into the insoluble fraction of cell extracts, thus precluding analysis of Bcl-B binding by co-immunoprecipitation experiments.
Interaction of Bcl-B with Nur77 has been shown to convert Bcl-B from displaying anti-apoptotic properties to a pro-apoptotic phenotype (30). Mutations in the proposed Nur77 binding site of Bcl-B that successfully disrupted protein binding were sufficient to confer an increase in cell viability in co-expression studies. We also observed that lysosomal inhibitors reduced at least partly the cell death induced by Bcl-B and Nur77/ΔDBD co-expression, suggesting the involvement of autophagic machinery in this cell death mechanism. Knockdown of Bcl-B has been shown to trigger both caspase-dependent and caspase-independent cell death mechanisms, and Beclin-1 reportedly contributes to the mediation of autophagy-dependent cell death induced by Bcl-B silencing (30). Nonetheless, we did not see complete rescue by lysosomal inhibitors. Therefore, we surmise that Bcl-B and Nur77 binding likely elicits a combination of apoptotic and autophagic cell death mechanisms.
The mechanism previously proposed for how Nur77 converts Bcl-2 into a pro-apoptotic protein envisions a conformational change that causes exposure of the BH3 peptide of Bcl-2 (18), which is normally buried in the interior of the folded protein. We did not observe overall conformational changes in Bcl-2 family proteins by NMR analysis of Nur77 peptide binding. However, it is likely that the extensive conformational changes required to expose the BH3 domain would require interactions of Bcl-2 family proteins with membranes so that exposed hydrophobic regions could be stabilized by membrane association, as proposed also for the ion channel activity that has been measured for several Bcl-2 family proteins (40). A recent NMR study (41) showed that membrane-anchored Bcl-X(L) displays a somewhat stronger affinity for a BH3 peptide, highlighting the importance of Bcl-2 protein interactions with intracellular membranes. Thus, in the cellular context, it is possible that Nur77 binding to Bcl-2 family proteins is transient, helping to promote conformational states that trigger opening of the three-dimensional protein fold to encourage association of the hydrophobic surface of the amphipathic α-helices of these proteins with intracellular membranes. Alternatively, because Nur77 typically exists as a heterodimer with RXR-α in cells, perhaps its dimerization partner is required to promote the conformational change of Bcl-2 family proteins. Additional but hypothetical possibilities for the Nur77 interaction with Bcl-2 family proteins include Nur77-induced allosteric release of proteins bound to the adjacent BH3 cleft or blocking access of other proteins to the adjacent BH3 crevice by steric interference.
In summary, we show that Nur77 binding requires a previously unrecognized regulatory site on anti-apoptotic Bcl-2 family proteins. This knowledge could foster an improved understanding of the molecular basis for apoptosis and autophagy regulation and could also suggest new strategies for generating novel classes of chemical compounds targeting Bcl-2, Bfl-1, and Bcl-B to mimic or antagonize interactions with Nur77 for modulating apoptosis and autophagy with intended therapeutic benefits.
Author Contributions
P. H. C. G. prepared the protein samples and performed the biochemical analysis. P. H. C. G. and C. E. M. contributed to the NMR analysis. R. P. W. G., A. H., R. S., and H. Y. prepared the cell-based assays and analysis. R. P. W. G., Y. M., and H. Y. prepared the DNA constructs. Y. C. prepared the peptides. Y. Y., F. M. M., and H. R. K. contributed to the design of NMR experiments, data analysis, and interpretation of results. P. H. C. G., R. P. W. G., S. M., and J. C. R. wrote the manuscript. All authors contributed to the design of experiments, data analysis, and interpretation of results.
Acknowledgments
We thank the Structural Genomics Consortium for the Bfl-1 cDNA cloned into a His-tagged vector (PDB code 2VM6). The Bcl-2/Bcl-X(L) chimera protein NMR assignment used in this work was provided by Dr. Stephen Fesik.
This work was supported by California Tobacco-related Disease Research Program Fellowship 16FT-0056 (to P. H. C. G.) and National Institutes of Health Grants AI-091967 (to S. M.), CA-179087 (to F. M. M.), CA-55164 (to J. C. R.), and CA-81543 (to J. C. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Bcl
- B cell lymphoma
- BH
- Bcl-2 homology
- IP
- immunoprecipitation
- Z-VAD-fmk
- benzyloxycarbonyl-VAD-fluoromethyl ketone
- DBD
- DNA-binding domain
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Moldoveanu T., Follis A. V., Kriwacki R. W., and Green D. R. (2014) Many players in BCL-2 family affairs. Trends Biochem. Sci. 39, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang R., Zeh H. J., Lotze M. T., and Tang D. (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., and Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]
- 4. Chipuk J. E., Moldoveanu T., Llambi F., Parsons M. J., and Green D. R. (2010) The BCL-2 family reunion. Mol. Cell 37, 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang J., Fairbrother W., and Reed J. C. (2015) Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev. Hematol. 8, 283–297 [DOI] [PubMed] [Google Scholar]
- 6. Vogler M., Dinsdale D., Dyer M. J., and Cohen G. M. (2009) Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 16, 360–367 [DOI] [PubMed] [Google Scholar]
- 7. Wendt M. D. (2008) Discovery of ABT-263, a Bcl-family protein inhibitor: observations on targeting a large protein-protein interaction. Expert Opin. Drug Discov. 3, 1123–1143 [DOI] [PubMed] [Google Scholar]
- 8. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., et al. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 [DOI] [PubMed] [Google Scholar]
- 9. Khaw S. L., Mérino D., Anderson M. A., Glaser S. P., Bouillet P., Roberts A. W., and Huang D. C. (2014) Both leukaemic and normal peripheral B lymphoid cells are highly sensitive to the selective pharmacological inhibition of prosurvival Bcl-2 with ABT-199. Leukemia 28, 1207–1215 [DOI] [PubMed] [Google Scholar]
- 10. Brien G., Trescol-Biemont M.-C., and Bonnefoy-Bérard N. (2007) Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene 26, 5828–5832 [DOI] [PubMed] [Google Scholar]
- 11. Konopleva M., Contractor R., Tsao T., Samudio I., Ruvolo P. P., Kitada S., Deng X., Zhai D., Shi Y. X., Sneed T., Verhaegen M., Soengas M., Ruvolo V. R., McQueen T., Schober W. D., et al. (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388 [DOI] [PubMed] [Google Scholar]
- 12. van Delft M. F., Wei A. H., Mason K. D., Vandenberg C. J., Chen L., Czabotar P. E., Willis S. N., Scott C. L., Day C. L., Cory S., Adams J. M., Roberts A. W., and Huang D. C. (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vogler M., Butterworth M., Majid A., Walewska R. J., Sun X. M., Dyer M. J., and Cohen G. M. (2009) Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 113, 4403–4413 [DOI] [PubMed] [Google Scholar]
- 14. Yecies D., Carlson N. E., Deng J., and Letai A. (2010) Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagn F., Klein C., Demmer O., Marchenko N., Vaseva A., Moll U. M., and Kessler H. (2010) BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J. Biol. Chem. 285, 3439–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tamir S., Rotem-Bamberger S., Katz C., Morcos F., Hailey K. L., Zuris J. A., Wang C., Conlan A. R., Lipper C. H., Paddock M. L., Mittler R., Onuchic J. N., Jennings P. A., Friedler A., and Nechushtai R. (2014) Integrated strategy reveals the protein interface between cancer targets Bcl-2 and NAF-1. Proc. Natl. Acad. Sci. U.S.A. 111, 5177–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H., Kolluri S. K., Gu J., Dawson M. I., Cao X., Hobbs P. D., Lin B., Chen G., Lu J., Lin F., Xie Z., Fontana J. A., Reed J. C., and Zhang X. (2000) Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science 289, 1159–1164 [DOI] [PubMed] [Google Scholar]
- 18. Lin B., Kolluri S. K., Lin F., Liu W., Han Y.-H., Cao X., Dawson M. I., Reed J. C., and Zhang X.-K. (2004) Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor TR3/NGFI-B/Nur77. Cell 116, 527–540 [DOI] [PubMed] [Google Scholar]
- 19. Thompson J., and Winoto A. (2008) During negative selection, Nur77 family proteins translocate to mitochondria where they associate with Bcl-2 and expose its proapoptotic BH3 domain. J. Exp. Med. 205, 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petros A. M., Medek A., Nettesheim D. G., Kim D. H., Yoon H. S., Swift K., Matayoshi E. D., Oltersdorf T., and Fesik S. W. (2001) Solution structure of the antiapoptotic protein Bcl-2. Proc. Natl. Acad. Sci. U.S.A. 98, 3012–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Markley J. L., Bax A., Arata Y., Hilbers C. W., Kaptein R., Sykes B. D., Wright P. E., and Wüthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids: IUPAC-IUBMB-IUPAB inter-union task group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. J. Biomol. NMR 12, 1–23 [DOI] [PubMed] [Google Scholar]
- 22. Gronwald W., and Kalbitzer H. (2004) Automated structure determination of proteins by NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 44, 33–96 [Google Scholar]
- 23. Goddard T. D., and Kneller D. G.. SPARKY 3, University of California, San Francisco [Google Scholar]
- 24. Jung Y.-S., and Zweckstetter M. (2004) Mars: robust automatic backbone assignment of proteins. J. Biomol. NMR 30, 11–23 [DOI] [PubMed] [Google Scholar]
- 25. Kolluri S. K., Zhu X., Zhou X., Lin B., Chen Y., Sun K., Tian X., Town J., Cao X., Lin F., Zhai D., Kitada S., Luciano F., O'Donnell E., Cao Y., et al. (2008) A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell 14, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ziarek J. J., Peterson F. C., Lytle B. L., and Volkman B. F. (2011) Binding site identification and structure determination of protein-ligand complexes by NMR. Methods Enzymol. 493, 241–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williamson M. P. (2013) Using chemical shift perturbation to characterize ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 28. Herman M. D., Nyman T., Welin M., Lehtiö L., Flodin S., Trésaugues L., Kotenyova T., Flores A., and Nordlund P. (2008) Completing the family portrait of the anti-apoptotic Bcl-2 proteins: crystal structure of human Bfl-1 in complex with Bim. FEBS Lett 582, 3590–3594 [DOI] [PubMed] [Google Scholar]
- 29. Miller S., Janin J., Lesk A. M., and Chothia C. (1987) Interior and surface of monomeric proteins. J. Mol. Biol. 5, 641–656 [DOI] [PubMed] [Google Scholar]
- 30. Luciano F., Krajewska M., Ortiz-Rubio P., Krajewski S., Zhai D., Faustin B., Bruey J. M., Bailly-Maitre B., Lichtenstein A., Kolluri S. K., Satterthwait A. C., Zhang X. K., and Reed J. C. (2007) Nur77 converts phenotype of Bcl-B, an antiapoptotic protein expressed in plasma cells and myeloma. Blood 109, 3849–3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar M., Gupta D., Singh G., Sharma S., Bhat M., Prashant C. K., Dinda A. K., Kharbanda S., Kufe D., and Singh H. (2014) Novel polymeric nanoparticles for intracellular delivery of peptide cargos: antitumor efficacy of the BCL-2 conversion peptide NuBCP-9. Cancer Res. 74, 3271–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yip K. W., Godoi P. H., Zhai D., Garcia X., Cellitti J. F., Cuddy M., Gerlic M., Chen Y., Satterthwait A., Vasile S., Sergienko E., and Reed J. C. (2008) A TR3/Nur77 peptide-based high-throughput fluorescence polarization screen for small molecule Bcl-B inhibitors. J. Biomol. Screen. 13, 665–673 [DOI] [PubMed] [Google Scholar]
- 33. Rautureau G. J., Yabal M., Yang H., Huang D. C., Kvansakul M., and Hinds M. G. (2012) The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 3, e443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robert G., Gastaldi C., Puissant A., Hamouda A., Jacquel A., Dufies M., Belhacene N., Colosetti P., Reed J. C., Auberger P., and Luciano F. (2012) The anti-apoptotic Bcl-B protein inhibits BECN1-dependent autophagic cell death. Autophagy 8, 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bouzas-Rodríguez J., Zárraga-Granados G., Sánchez-Carbente Mdel R., Rodríguez-Valentín R., Gracida X., Anell-Rendón D., Covarrubias L., and Castro-Obregón S. (2012) The nuclear receptor NR4A1 induces a form of cell death dependent on autophagy in mammalian cells. PLoS ONE 7, e46422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang W. J., Wang Y., Chen H. Z., Xing Y. Z., Li F. W., Zhang Q., Zhou B., Zhang H. K., Zhang J., Bian X. L., Li L., Liu Y., Zhao B. X., Chen Y., Wu R., et al. (2014) Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat. Chem. Biol. 10, 133–140 [DOI] [PubMed] [Google Scholar]
- 37. Ichimura Y., Kominami E., Tanaka K., and Komatsu M. (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 38. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferlini C., Cicchillitti L., Raspaglio G., Bartollino S., Cimitan S., Bertucci C., Mozzetti S., Gallo D., Persico M., Fattorusso C., Campiani G., and Scambia G. (2009) Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 69, 6906–6914 [DOI] [PubMed] [Google Scholar]
- 40. Volkmann N., Marassi F. M., Newmeyer D. D., and Hanein D. (2013) The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 21, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yao Y., Fujimoto L. M., Hirshman N., Bobkov A. A., Antignani A., Youle R. J., and Marassi F. M. (2015) Conformation of BCL-XL upon membrane integration. J. Mol. Biol. 427, 2262–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]