

Abstract
Our previous work has demonstrated an intrinsic mRNA-specific protein synthesis salvage pathway operative in glioblastoma (GBM) tumor cells that is resistant to mechanistic target of rapamycin (mTOR) inhibitors. The activation of this internal ribosome entry site (IRES)-dependent mRNA translation initiation pathway results in continued translation of critical transcripts involved in cell cycle progression in the face of global eIF-4E-mediated translation inhibition. Recently we identified compound 11 (C11), a small molecule capable of inhibiting c-MYC IRES translation as a consequence of blocking the interaction of a requisite c-MYC IRES trans-acting factor, heterogeneous nuclear ribonucleoprotein A1, with its IRES. Here we demonstrate that C11 also blocks cyclin D1 IRES-dependent initiation and demonstrates synergistic anti-GBM properties when combined with the mechanistic target of rapamycin kinase inhibitor PP242. The structure-activity relationship of C11 was investigated and resulted in the identification of IRES-J007, which displayed improved IRES-dependent initiation blockade and synergistic anti-GBM effects with PP242. Mechanistic studies with C11 and IRES-J007 revealed binding of the inhibitors within the UP1 fragment of heterogeneous nuclear ribonucleoprotein A1, and docking analysis suggested a small pocket within close proximity to RRM2 as the potential binding site. We further demonstrate that co-therapy with IRES-J007 and PP242 significantly reduces tumor growth of GBM xenografts in mice and that combined inhibitor treatments markedly reduce the mRNA translational state of cyclin D1 and c-MYC transcripts in these tumors. These data support the combined use of IRES-J007 and PP242 to achieve synergistic antitumor responses in GBM.
Keywords: cyclin D1, glioblastoma, heterogeneous nuclear ribonucleoprotein (hnRNP), mechanistic target of rapamycin (mTOR), Myc (c-Myc), translation, IRES, ITAF, mRNA translation
Introduction
Glioblastoma (GBM)3 is one of the most common primary malignant brain tumors, and median survival is only approximately 12 months (1). The lethality of this tumor is, in part, due to the difficulties associated with complete surgical resections and the development of drug resistance (2). As a consequence of EGFR amplification or activating mutation and phosphatase and tensin homolog (PTEN) loss (3, 4), hyperactivation of the PI3K pathway is seen in nearly 90% of all GBMs (5, 6). As a result, the mechanistic target of rapamycin (mTOR) kinases, a downstream effector, are often constantly hyperactivated (7). mTOR is a central regulator of mRNA translation, metabolism, and autophagy in the cell and, consequently, controls tumor cell growth, survival, and drug resistance (8, 9).
First-generation allosteric mTOR inhibitors, such as rapamycin and CCI-779, have been unsuccessful as monotherapies in the clinic for GBM because of loss of feedback regulation leading to AKT activation (10, 11). Additionally, mTORC2 has been shown to play a crucial role in GBM growth, invasion, and rapamycin resistance (12, 13). These studies have underscored the potential role of mTOR kinase inhibitors as a promising therapeutic option in the treatment of GBM.
The interdependence between mTOR signaling complexes suggests the possibility that multiple mechanisms of mTOR inhibitor resistance exist (14–16). Our previous studies have demonstrated that both allosteric and direct kinase inhibitors of mTOR can activate a transcript-specific protein synthesis salvage pathway, maintaining the translation of crucial mRNAs involved in cell cycle progression, resulting in resistance to mTOR therapies (17–19). The activation of this intrinsic pathway is dependent on SAPK2/p38-mediated activation of IRES-dependent initiation of cyclin D1 and c-MYC mRNAs in GBM (20). Recently, we have described the therapeutic potential of targeting IRES-dependent c-MYC translation via the identification of a small molecule inhibitor that blocked c-MYC IRES-mediated translation initiation (21).
In this study, we expand on this work, demonstrating that the previously identified IRES inhibitor additionally blocks cyclin D1 IRES activity. We show that the IRES inhibitor displays strong synergistic anti-GBM activities when combined with mTOR kinase inhibitors. Structure-activity relationship studies led to the identification of an improved IRES inhibitor (IRES-J007), and further mechanistic investigation characterized the target of the inhibitors as the ITAF (IRES-trans-acting factor), hnRNP A1. In silico docking analysis predicted that IRES-J007 binds to a small pocket structure within the RRM-containing fragment of hnRNP A1, UP1. The pocket is within close proximity to RRM2, and inhibitor binding to hnRNP A1 blocked the ability of the ITAFs to associate with either the cyclin D1 or c-MYC IRESs. Furthermore, we observed a synergistic antiproliferative effect of these compounds when used in combination with PP242 in vitro and in xenografted GBM cells in mice. Finally, we show that the mRNA translational state of the cyclin D1 and c-MYC mRNAs is markedly reduced in vivo following co-therapy with PP242.
Experimental Procedures
Cell Lines, Constructs, and Transfections
The glioblastoma lines LN229 and LN18 were obtained from the ATCC (Manassas, VA). Paul Mischel (Ludwig Institute, University of California, San Diego) kindly provided the LN428 line, and the SF763 line was from the UCSF Neurosurgery Tissue Bank (University of California, San Francisco). 293T cells were kindly provided by Norimoto Yanagawa (UCLA). Normal mature human neurons were obtained from ScienCell (Carlsbad, CA). The dicistronic constructs pRF, pRCD1F, pRmycF, and pRp27F have been described previously (19). The pREMCVF construct was provided by Eric Jan (Department of Biochemistry, University of British Columbia). The pGEX-2T/hnRNP A1 (full-length hnRNP A1) and pGEX-2T/UP1 GST fusions were provided by Ronald Hay (Centre for Gene Regulation and Expression, University of Dundee) and used to generate additional deletion mutants. To generate the hnRNP A1 alanine substitution mutants, the full-length hnRNP A1-containing plasmid was mutagenized using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) using appropriate mutagenic primers according to the manufacturer. All plasmids were sequenced to verify the constructs. DNA transfections were performed using Effectene transfection reagent according to the manufacturer (Qiagen).
Recombinant Proteins, Antibodies, Reagents, and C11 Structure-Activity Relationship (SAR) Analog Preparation
Recombinant native and mutant hnRNP A1 was expressed and purified from HEK293 cells using anti-glutathione-Sepharose column chromatography as described previously (19). Antibodies were as follows: mouse IgG (Santa Cruz Biotechnology, catalog no. sc-2025, lot no. I2806), hnRNP A1 (Abcam, catalog no. ab5832, lot no. GR2405064-1, mouse origin), cyclin D1 (Cell Signaling Technology, catalog no. 2922, lot no. 3, rabbit origin), c-MYC (Cell Signaling Technology, catalog no. 9402, lot no. 11, rabbit origin), actin (Abcam, catalog no. ab3280, lot no. GR235922-1, mouse origin), eIF-4E (BD Transduction Laboratories, catalog no. 610269, lot no. 87), and anti-GST (Cell Signaling Technology, catalog no. 2622, lot no. 5, rabbit origin). PP242 and rapamycin were obtained from LC Laboratories (Woburn, MA). C11 (NSC-603707) was obtained from the Developmental Therapeutics Program repository at the NCI, National Institutes of Health. The synthetic procedures for the SAR analogs are described in detail in the supplemental Experimental Procedures.
Protein and RNA Analyses
Western blotting and quantitative RT-PCR analyses were performed as described previously (20). Briefly, for Western blotting, cells or tissues were lysed in radioimmune precipitation assay (lysis) buffer containing protease inhibitor mixture and phosSTOP phosphatase inhibitor mixture (Roche), and extracts were resolved by SDS-PAGE. Proteins were transferred to PVDF membranes and incubated with the indicated antibodies. Antigen-antibody complexes were detected using appropriate horseradish peroxidase-conjugated secondary antibodies (anti-mouse IgG-HRP, GE Healthcare, catalog no. NA931V, lot no. 9640720, sheep origin; anti-rabbit IgG-HRP, GE Healthcare, catalog no. NA934V, lot no. 9670531, donkey origin) and enhanced chemiluminescence (Amersham Biosciences, ECL Prime). Primary antibody concentrations were used as follows: hnRNP A1, 1:1000; cyclin D1, 1:500; c-MYC, 1:500; actin, 1:5000; and anti-GST, 1:1000. Secondary antibody concentrations were adjusted for each primary antibody used for signal intensity within the linear range. Band intensities were quantified by densitometry analyses via ImageJ software. Drug treatments did not affect actin protein levels, which was used as gel loading controls. For IRES reporter assays, the indicated mRNA reporters were co-transfected into cells with pSVβ-galactosidase to normalize for transfection efficiency as described previously (17). Cells were harvested 18 h following transfection, and Renilla, firefly, and β-galactosidase activities were determined using the Dual-Glo luciferase and β-galactosidase assay systems (Promega). For immunopurification of eIF4E and bound RNA (22), cells were lysed in buffer containing 50 mm Tris-HCl (pH 7.4), 300 mm NaCl, 0.5% Nonidet P-40, 10 mm NaF, 1 mm sodium orthovanadate, 1 mm PMSF, 1.5% aprotinin, and SUPERase-IN (Thermo Fisher Scientific, 0.025 units/ml) using a Dounce homogenizer at 4 °C. Extracts were cleared by centrifugation and immunoprecipitated with eIF4E or control IgG. Beads were washed, and obtained RNA was subjected to quantitative RT-PCR. For quantitative RT-PCR, extraction of RNA was performed using TRIzol (Thermo Fisher). Total RNA was then quantified, and integrity was assessed using an Agilent Technology 2100 Bioanalyzer. Total RNA was reverse-transcribed with random primers using the RETROscript kit from Thermo Fisher. SYBR Green quantitative PCR was performed in triplicate in 96-well optical plates on an ABI Prism 7000 sequence detection system (Life Technologies) according to the instructions of the manufacturer. The primer sequences for CCND1, c-MYC, and actin are available upon request. For RNA pulldown assays (19), cytoplasmic extracts were prepared by hypotonic lysis in buffer containing 10 mm HEPES (pH 7.5), 10 mm potassium acetate, 1.5 mm magnesium acetate, 2.5 mm DTT, 0.05% Nonidet P-40, 10 mm NaF, 1 mm sodium orthovanadate, 1 mm PMSF, and 1.5% aprotinin using a Dounce homogenizer. Extracts were precleared by centrifugation, and SUPERase-IN (Thermo Fisher, 0.025 units/ml) and yeast tRNA (15 μg/ml) were added and applied to an equilibrated heparin-agarose column (Bio-Rad). Eluates were further cleared with 100 μl of streptavidin-Sepharose (Sigma) for 1 h at 4 °C. Following centrifugation, 10 μg of in vitro transcribed biotinylated IRES RNA (mMESSAGE Machine T7 transcription kit, Thermo Fisher) was added to the supernatant and incubated for 1 h at 4 °C. The protein and biotinylated RNA complexes were recovered by adding 30 μl of streptavidin-Sepharose, which was incubated for 2 h at 4 °C. The complexes were washed five times in binding buffer (10 mm HEPES (pH 7.5), 90 mm potassium phosphate, 1.5 mm magnesium acetate, 2.5 mm DTT, 0.05% Nonidet P-40, 10 mm NaF, 1 mm sodium orthovanadate, 1 mm PMSF, and 1.5% aprotinin) and then boiled in SDS and resolved by gel electrophoresis. Max exon 5 alternative splicing analysis was performed as described previously (23). Briefly, alternative splicing was assayed using quantitative RT-PCR using primers designed to constitutive exons flanking alternative exons. The primers were as follows: Max (85F), 5′-tcagtcccatcactccaagg-3′; MAX(85R), 5′-gcacttgacctcgccttct-3′; hnRNPA1, 5′-acaacttcggtcgtggaggaaact-3′ (forward) and 5′-ccaaaattgcttccatcattaccaa-3′ (reverse); Actin, 5′-catgtacgttgctatccaggc-3′ (forward) and 5-ctccttaatgtcacgcacgat-3′ (reverse). Reverse primers were 32P end-labeled, and PCR reactions were amplified for 22 cycles and subsequently resolved by denaturing PAGE and imaged. RNA interference targeting hnRNP A1 was performed as described previously (18, 23) using siRNAs with the target sequence 5′-gguucuauuuggaauuuau-3′ obtained from Ambion (Applied Biosystems). Polysome analyses were performed as before (20). Briefly, cells were lysed in buffer containing 100 μg/ml cycloheximide at 4 °C. Following removal of mitochondria and nuclei, supernatants were layered onto 15–50% sucrose gradients and centrifuged at 38,000 rpm for 2 h at 4 °C in an SW40 rotor (Beckman Instruments). Gradients were fractionated into 11 1-ml fractions using a density gradient fractionator (Brandel Instruments). The profiles of the gradients were monitored at 260 nm, and RNAs from individual fractions were pooled into a nonribosomal/monosomal pool and a polysomal pool. These RNAs (100 ng) were used in real-time quantitative RT-PCR analysis for the indicated transcripts. Filter binding assays were performed as described previously (19, 24). GST-tagged hnRNP A1 or hnRNP A1 deletion mutants were added to in vitro transcribed 32P-labeled RNAs corresponding to either the cyclin D1 or c-MYC IRESs in separate reactions in a volume of 10 μl in buffer containing 5 mm HEPES (pH 7.6), 30 mm KCl, 2 mm MgCl2, 200 mm DTT, 4% glycerol, and 10 ng of yeast tRNA for 10 min at room temperature. 8 μl of each binding reaction was applied to nitrocellulose membranes on a slot blot apparatus (Minifold II, Schleicher & Schüll). Membranes were washed and dried, and signals were quantified via phosphorimaging.
Docking Analysis of IRES Inhibitors and UP1
The in silico analysis of C11, IRES-J007, and UP1 was performed using AutoDock Vina (25). The steric structure of monomeric UP1 was derived from the crystal structure deposited in the PDB (1HA1). The UP1 structure was preprocessed, and hydrogen atoms were added prior to the docking simulation. Models were visualized using PyMOL v1.5.6 (Schrödinger, LLC).
Photo-cross-linking Assays
Photo-cross-linked C11 and IRES-J007 beads were prepared as described previously (26). Briefly, activated Sepharose beads were washed three times with 1 mm aqueous HCl followed by coupling solution (100 mm NaHCO3 and 50% dioxane mixture). A solution of photoaffinity linker in coupling solution was subsequently added to the beads and incubated at 37 °C for 2 h. After washing five times with coupling solution, the beads were blocked, placed in a spin column, and washed three times with water and methanol. The beads were subsequently irradiated in a UV cross-linker at 365 nm (4 J/cm2) and washed with methanol. Purified GST-tagged native or mutant hnRNP A1 proteins were added to 20 μl of C11 or J007 cross-linked or control non-cross-linked beads. After incubating at 4 °C for 24 h, the beads were washed three times, and bound proteins were eluted in 10% SDS-PAGE sample buffer at 100 °C for 5 min. The eluted proteins were resolved by SDS-PAGE and immunoblotted using GST antibodies.
Cell Proliferation, Cell Cycle Distribution, and TUNEL Assays
Cells were plated into 96-well plates, and after culturing for various time points, cell numbers were measured by 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt (XTT) assay (Roche) as described by the manufacturer. The viability of human neurons was assessed by trypan blue exclusion. Cell cycle analysis was done by propidium iodide staining of cells and flow cytometry as described previously (20). Cells were stained using a FITC-conjugated annexin V (annexin V-FITC early apoptosis detection kit, Cell Signaling Technology) to monitor apoptosis. TUNEL staining of tumor sections was performed using the TACSXL DAB in situ apoptosis detection kit (Trevigen) according to the instructions of the manufacturer (20). The combination index (CI) values were determined by using CalcuSyn v2.0 software (Biosoft) (20).
Xenograft Studies
All animal experiments were performed under an approved institutional animal care and use committee protocol and conformed to the guidelines established by the Association for the Assessment and Accreditation of Laboratory Animal Care. Xenografts of LN229 cells were performed in female C.B.-17-scid (Taconic) mice as described previously (15). Tumors were harvested at autopsy for Western blotting analysis. Sections of paraffin-embedded tumors on slides were processed for immunohistochemistry as described previously (15, 20).
Statistical Analysis
Statistical analyses were performed with Student's t test and analysis of variance models using Systat 13 (Systat Software, Chicago, IL). p < 0.05 was considered significant.
Results
C11 Inhibits Cyclin D1 and c-Myc IRES Activity in GBM via Blockade of hnRNPA1-IRES Interactions
We previously identified C11 (Fig. 1A), which specifically blocked c-Myc IRES-mediated translation initiation by screening ∼145,000 compounds in a yeast-based, three-hybrid assay targeting disruption of the ITAF hnRNP A1-IRES interaction (21). Because the cyclin D1 IRES utilizes hnRNP A1 as an ITAF, and its ability to initiate IRES-mediated translation is regulated in a similar manner as the c-MYC IRES, we determined whether C11 would also inhibit its IRES activity. We utilized several dicistronic IRES mRNA reporter constructs (Fig. 1B) in which the indicated IRES sequences were inserted within the intercistronic region. LN229 cells transiently transfected with these constructs were assayed for Renilla and firefly luciferase activities, which are readouts of cap-dependent and IRES-mediated translation initiation, respectively (17). As shown in Fig. 1C, C11 significantly inhibited both cyclin D1 and c-MYC IRES activity, consistent with the requirement of these IRESs for hnRNP A1 function (19). C11, however, did not affect IRES-mediated initiation from either the p27Kip1 or encephalomyocarditis virus IRESs that do not utilize hnRNP A1 as an ITAF. To examine whether C11 would affect hnRNP A1 binding to the cyclin D1 or c-MYC IRESs, RNA pulldown assays were performed, utilizing cell extracts from cells treated with C11. As shown in Fig. 1D, hnRNP A1 was preferentially precipitated by either of the IRES RNAs; however, C11 treatment markedly reduced hnRNP A1 binding. We additionally examined the effects of C11 on the translational state of the cyclin D1 and c-MYC mRNAs. Polysome analysis was performed, and, as shown in Fig. 1E, C11 treatment induced a significant shift in both cyclin D1 and c-MYC mRNA to monosomal/nonribosomal fractions, whereas actin mRNA distribution was unaffected. This is consistent with previous observations that actin mRNA is translated via cap-dependent initiation (27). We also noted that C11 did not appear to alter cyclin D1 or c-MYC steady-state mRNA levels because total monosomal/nonribosomal plus polysomal mRNA content was unchanged compared with controls, suggesting that the inhibitor does not affect transcription or mRNA stability. As hnRNP A1 is also a splicing factor, we determined whether C11 affected Max splicing in GBM. EGFRvIII signaling promotes Max splicing via hnRNP A1 to generate Delta Max, a truncated version of the MAX protein, which includes exon 5 in GBM (23), and, as shown in Fig. 1F, top panel, C11 treatment did not appear to alter Max splicing in U87 cells stably expressing EGFRvIII. As a control, knockdown of hnRNP A1 expression via RNA interference in these cells markedly reduced exon 5-containing Delta Max transcript levels. To further confirm that C11 does not affect eIF-4E-mediated initiation, we immunoprecipitated eIF-4E from cells treated with C11 and assessed the relative amounts of both cyclin D1 and c-MYC mRNAs within these complexes by quantitative RT-PCR. As shown in Fig. 1F, center panel, C11 exposure did not affect either cyclin D1 or c-MYC association with eIF-4E. Finally, cyclin D1 and c-MYC protein levels following C11 exposure in LN229 and SF763 cells were markedly reduced (Fig. 1F, bottom panel). These data demonstrate that C11 inhibits both cyclin D1 and c-MYC IRES-mediated mRNA translation, leading to reductions in protein levels.
FIGURE 1.

C11 inhibits both cyclin D1 and c-MYC IRES activity in glioblastoma cells. A, chemical structure of C11. B, schematics of the dicistronic constructs used in this study. The constructs used were pRF; pRCD1F, which contains the human cyclin D1 IRES; pRmycF, containing the human c-myc IRES; pRp27F, containing the human p27Kip1 IRES; and pREMCVF, containing the IRES from the encephalomyocarditis virus. Luc, luciferase. C, relative Renilla and firefly luciferase activities obtained from LN229 GBM cells transfected with the indicated constructs in the absence or presence of the inhibitor C11. The mean ± S.D. are shown for three independent experiments. p < 0.01, significantly different from firefly and firefly + C11. D, RNA pulldown assays utilizing biotinylated cyclin D1 or c-MYC IRES RNAs. Cytoplasmic extracts of LN229 cells treated with C11 (50 nm) as indicated were incubated with biotinylated cyclin D1 or c-MYC IRES RNAs and precipitated with streptavidin-Sepharose beads. Input and bound fractions were analyzed by immunoblotting using hnRNP A1 antibodies. Data shown are representative of experiments repeated twice. con, control. E, polysome distributions of cyclin D1, c-MYC, and actin mRNAs in LN229 cells in the absence or presence of C11 (50 nm). Extracts were subjected to sucrose density centrifugation and then divided into 11 1-ml fractions that were pooled into a nonribosomal, monosomal (N, white columns), and polysomal fraction (P, black columns). Purified RNAs were subsequently used in real-time quantitative RT-PCR analysis to determine the distributions of cyclin D1, c-MYC, and actin mRNAs across the gradients. Polysome tracings are shown above values obtained from the RT-PCR analyses, which are displayed graphically below. RT-PCR measurements were performed in quadruplicate, and the mean ± S.D. are shown. *, p < 0.05, significantly different from CCND1, c-MYC and CCND1 + C11, or c-MYC + C11. F, top panel, U87EGFRvIII cells or U87EGFRvIII cells in which hnRNP A1 expression was knocked down (KD) via siRNA were treated with C11 as indicated, and RT-PCR splicing analysis for Max exon 5 was performed as described under “Experimental Procedures.” Data shown are representative of experiments repeated twice. Center panel, LN229 cells treated with C11 (50 nm) as indicated were lysed and immunoprecipitated (IP) using either eIF-4E or control IgG antibodies. Bound CCND1 or c-MYC RNAs were detected via RT-PCR. The mean ± S.D. are shown for three independent experiments. Bottom panel, cyclin D1 and c-MYC protein levels from the indicated GBM cell lines in the absence or presence of C11 at 24 h following treatment. Data shown are representative of experiments repeated twice.
C11 Inhibits mTOR Inhibitor-induced IRES Activity and Potentiates PP242 anti-GBM Responses
Because we and others (20, 28) have demonstrated that mTOR inhibitors induce the up-regulation of IRES activity as an intrinsic mechanism of resistance to this class of inhibitors, we were interested whether C11 would enhance PP242 cytotoxicity. As shown in Fig. 2A, treatment with rapamycin (left panel) or PP242 (right panel) led to dramatic induction of cyclin D1 and c-MYC IRES activity, which was significantly inhibited upon cotreatment with C11. No significant inhibition of cell proliferation was observed from C11 treatment at any of the concentrations tested, up to 10 μm in several GBM lines (Fig. 2B). This was similar to our previous findings with multiple myeloma cell lines (21). However, as shown in Fig. 2C, in the LN229, LN18, LN428, and SF763 GBM lines, treatment with C11 at 10 and 100 nm concentrations resulted in synergistic inhibition of cell proliferation over a wide range of PP242 concentrations tested (CI = 0.5 at an ED50 ratio of 1:100 (29, 30)). We also determined whether the combination of C11 with PP242 induced G1 arrest and apoptosis in the four GBM cells lines. As shown in Fig. 2D, PP242 treatment increased G1 arrest, and co-treatment with C11 markedly stimulated G1 arrest. Similarly, PP242 alone induced the percentage of apoptotic cells, and, when combined with C11, further potentiated the percentage of cells undergoing apoptosis. Cyclin D1 and c-MYC protein levels were also markedly reduced in GBM lines co-treated with PP242 and C11 (Fig. 2E). These results demonstrate that C11 significantly enhances PP242-induced G1 arrest and apoptosis in GBM lines.
FIGURE 2.

Synergistic anti-GBM effects of C11 in combination with mTOR inhibitors. A, inhibition of mTOR inhibitor-induced IRES activity in LN229 cells. Cells transiently transfected with the indicated IRES mRNA reporter constructs were treated with rapamycin (Rapa) or rapamycin + C11 (left panel) and PP242 or PP242 + C11 (right panel), and luciferase activities were determined. The results are expressed as relative -fold change in firefly (FF) luciferase activity, and the mean ± S.D. is shown for three independent experiments. B, inhibition of GBM cell line proliferation following 48 h culture in C11. Data represent mean ± S.D. of three independent experiments. C, combination analysis of PP242 and C11 inhibitors in GBM cell lines treated with the indicated doses of PP242 alone or in combination for 48 h. Cell proliferation relative to control cultures was assessed via XTT assays. Control cells were treated with DMSO vehicle. Data are mean ± S.D. (n = 3). D, cell cycle phase distributions were determined on the indicated GBM cell lines in the absence or presence of PP242 or C11 as shown. Percent apoptotic cells as determined via annexin V-FITC staining are also shown below each graph. One of three experiments with similar results is shown. E, protein levels of cyclin D1, c-Myc, and actin in GBM lines following the indicated treatments with PP242, C11, or both compounds at 24 h. Data shown are representative of experiments repeated twice.
Structure-Activity Relationship Studies Derive Active Analogs of C11
We next explored the SAR of C11 by synthesizing and testing analogs. Modifications were made to the left and right sides of the C11 molecule, and each analog was tested for its ability to decrease PP242-induced cyclin D1 and c-MYC IRES activity. Additionally, we determined whether the analogs demonstrated synergistic cytotoxic responses when combined with PP242 in LN229 cells. The degree of apoptotic cell death following co-treatment with analog and PP242 was also monitored. These results are summarized in Fig. 3. In Fig. 3A, showing analogs with modifications to the left of C11, IRES-J007, with a phthalimido group in place of the dichloromaleimide unit, demonstrated the greatest degree of IRES inhibition relative to the parent. The inhibition of IRES activity correlated with an increase in synergistic antitumor response in combination with PP242 (reduction in combination index; CI value) and a marked induction of apoptosis. Within the SAR analogs containing modifications to the right side (Fig. 3B) and those containing a six-membered ring moiety on the left side (Fig. 3C) of C11, IRES-J008, with a 4-methoxyphenyl substituent in place of the 2,4-dimethoxybenzyl unit and, to a lesser degree, IRES-J009, with an N1-methyluracil unit in place of the dichloromaleimide unit, inhibited cyclin D1 and c-MYC PP242-induced IRES activity. Both of these analogs also demonstrated significant synergistic cytotoxic effects in combination with PP242, with coordinate induction of apoptosis. Additional modifications of the IRES-J007 analog were synthesized (Fig. 3D); however, none of these compounds exhibited significantly improved properties compared with C11. We subsequently determined the in vitro cytotoxicities of analogs IRES-J007, IRES-J008, and IRES-J009 relative to C11 in human neurons (data not shown). IRES-J007 displayed the least toxicity to normal neurons, with no significant cytotoxic effects for concentrations up to 10 mm, and was therefore chosen for further study. The reduced toxicity of IRES-J007 versus C11 might possibly be due to the lack of the quite reactive dichloromaleimide unit present in C11, which is absent in IRES-J007.
FIGURE 3.

Structures and synergistic anti-GBM activities of C11 analogs in combination with PP242. A, structures of C11 analogs modified on the left side of the molecule and their PP242 synergistic anti-GBM activities. B, SAR of C11 with modifications on the right side of molecule. C, SAR of C11 with the six-membered ring moiety on the left side of the molecule. D, SAR of IRES-J007 with additional modifications to the right side of IRES-J007. The synthesis of the parent compound and its analogs is described under “Experimental Procedures.” §, -fold decrease in either PP242-induced cyclin D1 or c-MYC IRES activity in LN229 cells relative to values obtained with the parent compound C11 are shown for each analog. Data represent mean ± S.D. of three independent experiments. For each analog, the CI was calculated from combination analyses performed with PP242 and analog, as in Fig. 2C and as described in Ref. 29. CI = 1.0 (dose-additive), CI < 0.5 (synergy), CI < 0.3 (strong synergy). Percent apoptosis was determined for LN229 cells co-treated with PP242 (50 nm) and analog (100 nm) at 24 h via annexin V-FITC staining. One of three experiments with similar results is shown.
C11 or Its Analog IRES-J007 Blocks Association of UP1 to Cyclin D1 or c-MYC IRESs
To begin to investigate the mechanism of action of C11 and its analog IRES-J007, we initially determined whether the UP1 fragment of hnRNP A1 was sufficient to recapitulate C11- or IRES-J007-mediated inhibition of IRES binding to this ITAF. Several GST-tagged deletion mutants of hnRNP A1 were generated and purified as shown in Fig. 4A. The relative association between the mutant proteins and either the cyclin D1 or c-MYC IRESs was determined by filter binding assays in the absence or presence of the inhibitors (Fig. 4B). C-terminal deletion of the glycine-rich region, encompassing the RGG box and M9 domain did not affect either cyclin D1 or c-MYC IRES binding, and binding was inhibitable by C11. The UP1 fragment (amino acids 1–196) containing RRM1 and RRM2 and immediately adjacent sequences efficiently bound the IRESs, and either C11 or IRES-J007 blocked association. We also noted that the analog IRES-J007 demonstrated a modest but significant increase in its ability to block UP1/IRES RNA interactions compared with C11. These results were consistent with the relative improvement of the analog to inhibit PP242-induced IRES activity (Fig. 3). A mutant encompassing the first 102 amino acids and containing only RRM1 of hnRNP A1 did not demonstrate IRES binding. However, mutant 102–196, containing RRM2, did bind both the cyclin D and c-MYC IRESs, and both interactions were sensitive to C11 or IRES-J007. Additionally, a mutant encompassing residues 103–372, containing RRM2, bound both IRES sequences, and binding was reduced in the presence of either of the inhibitors. We then removed only RRM2 (amino acids 130–158) from hnRNP A1, and this mutant did not bind either of the IRES RNAs. These data suggest that much of the C-terminal half of hnRNP A1 is dispensable and that RRM1 alone is insufficient to mediate efficient IRES binding and inhibition by C11 or IRES-J007. The presence of RRM2 is necessary and appears to cooperate with RRM1 to mediate IRES binding that is sensitive to C11 or IRES-J007. Finally, UP1 is capable of IRES binding that is blocked by either C11 or the analog IRES-J007.
FIGURE 4.
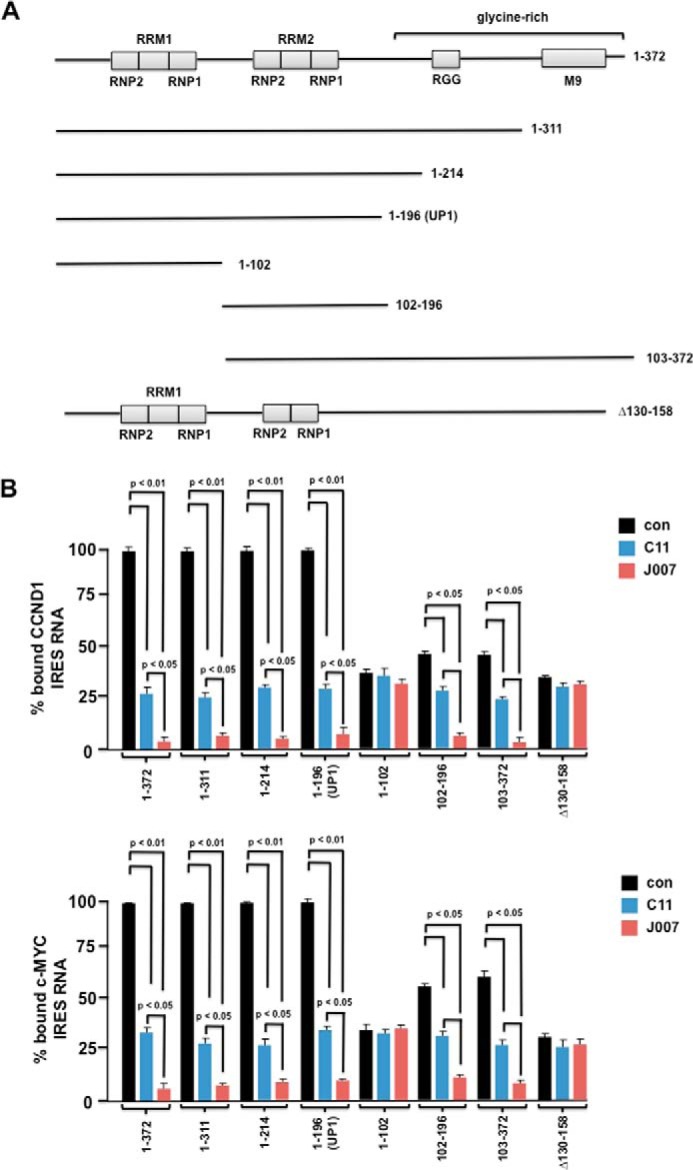
C11 and SAR-optimized IRES-J007 compounds block IRES-hnRNP A1 binding, and the UP1 fragment is sufficient to mediate inhibitor effects on IRES RNA binding in vitro. A, schematic of the various hnRNP A1 deletion mutations. Mutant 1–196 constitutes the Up1 fragment of full-length human hnRNP A1. In the Δ130–158 mutant, the sequences encompassing RRM2 have been removed. B, binding of either cyclin D1 (top panel) or c-MYC (bottom panel) IRES RNAs to GST-tagged hnRNP A1 mutants in the absence or presence of C11 or IRES-J007 as assayed by filter binding. The mean ± S.D. is shown for three independent experiments. con, control.
C11 or the Analog IRES-J007 Binds to a Small Pocket Structure within UP1
To further examine how C11 and IRES-J007 inhibit hnRNP A1 function, we used an in silico docking analysis to create a potential model for the binding of these inhibitors to UP1. To generate unbiased predictive virtual docking models, we obtained the crystal structure of monomeric UP1 from the PDB and performed docking studies using AutoDock Vina molecular modeling simulation software (25). Two potential binding sites were identified; however, the binding models that predicted the highest binding free energy (ΔG) occupied the site shown in Fig. 5A. This binding site was in close proximity to RRM2 and predicted binding to both C11 and IRES-J007 with similar binding free energies. The interaction maps for this binding site revealed that four residues (His-120, Asp-123, Tyr-124, and Asn-171) were predominantly involved in the interaction with C11 or IRES-J007. C11 and IRES-J007 occupied the same pocket with the configurations shown in Fig. 5A, inset, displaying the best binding scores, ΔG of −8.09 and −9.26 kcal/mol, respectively. To confirm the accuracy of this binding model, we initially generated C11 and IRES-J007 cross-linked affinity beads using a photo-cross-linking procedure (26). We subsequently tested whether the inhibitor-coupled beads (C11 beads or J007 beads, Fig. 5B) bound to native or mutant hnRNP A1 proteins harboring alanine substitutions at all four potential residues predicted to participate in the interactions. Recombinant GST-tagged native hnRNP A1 (A1) and mutant A1 proteins (4ΔA1) were purified by glutathione affinity methods. The purity was confirmed by SDS-PAGE followed by silver staining (Fig. 5B, top panels). The purified proteins were then incubated with control, C11, or J007 beads, and binding was analyzed by immunoblotting using anti-GST antibodies. Native GST-tagged hnRNP A1 bound to either C11 or J007 beads but not to control beads; however, the amount of mutant hnRNP A1 (4ΔA1) that bound either C11 or J007 beads was markedly reduced relative to native hnRNP A1 (Fig. 5B, bottom panel). These results suggest that C11 and IRES-J007 bind hnRNP A1 through the residues predicted by the interaction model. To further test this notion, we co-transfected either native or the mutant (4ΔA1) hnRNP A1 with the IRES mRNA reporters into cells in which endogenous hnRNP A1 had been silenced by siRNA treatment. The cells were subsequently treated with IRES-J007, and IRES activity was assessed. As shown in Fig. 5C, expression of the mutant (4ΔA1) hnRNP A1 rendered cells insensitive to IRES-J007-mediated inhibition of cyclin D1 or c-MYC IRES activity compared with cells expressing native hnRNP A1. We were also interested in determining whether C11 or the analog IRES-J007 would inhibit basal cyclin D1 or c-MYC IRES activity in 293T cells that express high endogenous levels of hnRNP A1 and show elevated IRES activity (31).4 As shown in figure 5D, 293T cells transiently transfected with the cyclin D1 and c-MYC IRES mRNA reporters and subsequently treated with either the C11 or IRES-J007 analog demonstrated reduced IRES activity. IRES-J007 inhibited cyclin D1 and c-MYC IRES activity to a greater extent compared with the parent compound C11. RNA pulldown assays in 293T cells also demonstrated an improved ability of the analog IRES-J007 to block cyclin D1 or c-MYC IRES-hnRNP A1 interactions relative to C11 (Fig. 5E).
FIGURE 5.

Model for potential binding of IRES inhibitors to UP1. A, in silico docking analysis was utilized to predict potential binding sites for C11 and IRES-J007 on UP1. The configurations with the most favorable binding energies were visualized using PyMOL v1.5.6. The electrostatic surface representation of the crystal structure of UP1 is shown (green), with RNP residues of RRM1 and RRM2 labeled in blue. In the 90°-rotated model, the inhibitor interaction pocket is shown in yellow. The inset is a close-up of C11 (magenta) and IRES-J007 (cyan) binding to the potential binding site on UP1. Residues predicted to interact with the inhibitors are labeled. B, purified GST-tagged wild-type hnRNP A1 (A1, asterisk) and mutant A1 (4ΔA1, double asterisk) proteins harboring alanine substitutions at all four potential binding sites (120, 123,124, and 171) were added to non-cross-linked and C11- and J007-cross-linked beads. Isolated wild-type (A1) and mutant (4ΔA1) proteins were resolved by SDS-PAGE and silver-stained to monitor purity (top panels). The binding of A1 to control, C11, and J007 beads was detected by immunoblotting with GST antibodies (bottom panel). Data shown are representative of experiments repeated twice. C, mutant (4ΔA1) A1 overexpression renders cells insensitive to J007-mediated inhibition of IRES activity. Wild-type hnRNP A1 or mutant A1 expression plasmids were cotransfected with cyclin D1 and c-MYC IRES reporters in LN229 cells in which endogenous hnRNP A1 had been silenced via siRNA. Cells were subsequently cultured in the absence or presence of J007, and IRES reporter activity was determined. The mean ± S.D. is shown for three independent experiments. Inset, immunoblot of endogenous hnRNP A1 levels following siRNA treatment in LN229 cells. D, inhibition of basal IRES activity in 293T cells upon treatment with C11 or IRES-J007. IRES reporter activity was assessed following 24-h treatment with 50 nm concentrations of each inhibitor as indicated. The mean ± S.D. is shown for three independent experiments. E, RNA pulldown assays utilizing biotinylated cyclin D1 or c-MYC IRES RNAs of 293T cell extracts treated with inhibitors as in Fig. 1D. Data shown are representative of experiments repeated twice.
In Vivo Effects of IRES-J007 and PP242 Combination Therapy in Xenografts
To determine whether the combination of IRES and mTOR inhibitor co-therapy would be efficacious in vivo, we conducted xenograft studies utilizing LN229 cells in mice. Mice were subcutaneously implanted with tumor cells, and when tumors were palpable and reached ∼200 mm3 in size, mice were randomized into treatment groups receiving double vehicle, PP242 (50 mg/kg/day), IRES-J007 (20 mg/kg/day), and PP242 (50 mg/kg/day) + IRES-J007 (20 mg/kg/day). As shown in Fig. 6A, xenografts receiving monotherapy with PP242 resulted in significant inhibition of tumor growth rate (36% inhibition at the end of the dosing period; tumor growth delay, 6.0 days). Tumor growth following monotherapy with IRES-J007 did not differ significantly and exhibited similar growth rates as double vehicle controls, consistent with the lack of effects of this inhibitor alone in vitro. However, the combination of PP242 and IRES-J007 was significantly more efficacious than either monotherapies alone (93% inhibition at the end of the dosing period; tumor growth delay, 20.5 days). Consistent with the effects on xenograft growth, overall survival of mice receiving combination IRES and mTOR therapy was significantly extended compared with either of the monotherapies (Fig. 6B). Notably, mice tolerated this dosing regimen without obvious short- or long-term toxicity or weight loss. The induction of apoptosis was also monitored via TUNEL staining of tumor sections from harvested tumors upon autopsy. As can be seen in Fig. 6C, left panel, significant staining was observed in tumors that received combination therapy, corroborating the increases in apoptotic cell death observed in vitro (Fig. 3). Marked reductions in cyclin D1 and c-MYC protein levels were also displayed in tumors receiving combination therapy (Fig. 6C, center and right panels).
FIGURE 6.

Combination effects of PP242 and IRES-J007 on GBM tumor growth in mice. A, tumor burden of SCID mice implanted with LN229 cells and treated with the indicated schedules of double vehicle, PP242 (blue line), J007 (red line), or a combination for 10 consecutive days. Tumor growth was assessed every 2 days following initiation of treatment (start, day 0). *, p < 0.05, significantly different from double vehicle, PP242 (50 mg/kg/day), and J007 (20 mg/kg/day). B, overall survival of subcutaneous LN229 tumors receiving the indicated treatment schedules. C, left panel, apoptotic cells were identified by TUNEL assays of sections prepared from harvested tumors at day 12 following initiation of treatment regimens. Data are expressed as the number of positive apoptotic bodies divided by high-power field (hpf, 10–12 high-power fields/tumor). Values are mean ± S.D. *, p < 0.05. Center panel, cyclin D1 protein levels in tumors. Values are mean ± S.D., *, p < 0.05, significantly different from vehicle, PP242, and J007. Right panel, c-MYC protein levels in tumors. Values are mean ± S.D. *, p < 0.05, significantly different from vehicle, PP242, and J007. Protein levels were quantified by Western blotting analyses of harvested tumors from mice with the corresponding treatments as indicated and as described under “Experimental Procedures.”
The Cyclin D1 and c-MYC mRNA Translational State in Response to IRES and mTOR Inhibitors
We have demonstrated previously that cyclin D1 and c-MYC IRES activity nearly exclusively directs mRNA translation of these determinants following mTOR inhibitor exposure (19). To discern whether alterations in cyclin D1 and c-MYC expression mediated by the inhibitor therapies in xenografted tumors were the result of actual changes in mRNA translational efficiency of these transcripts, we conducted polysome analysis of freshly harvested LN229 tumors following the last day of inhibitor dosing. Polysomes were separated via sucrose density gradient sedimentation and fractionated into heavy polysomal and nonribosomal/monosomal fractions. Spectrophoretic monitoring of fractions at 260 nm was used to identify polysome- and non-ribosome-containing fractions and monitor polysome integrity as before (Fig. 1E). As shown in Fig. 7, tumors from mice that received double vehicle treatments, cyclin D1 and c-MYC, were present in polysomal fractions at ∼45% and 50% of total cyclin D1 and c-MYC mRNA, respectively. Mice that received PP242 monotherapy exhibited significantly different cyclin D1 and c-MYC mRNA translational states, reduced to 38 and 35%, respectively. Actin mRNA polysomal distribution was also monitored and, as shown, this mRNA, whose synthesis is mediated via eIF-4E dependent initiation, was markedly redistributed to non-ribosomal/polysomal fractions, demonstrating effective inhibition of cap-dependent initiation. Mice that received IRES-J007 monotherapy displayed a significant reduction in cyclin D1 and c-MYC mRNA translational efficiency, consistent with the results of inhibiting IRES-mediated translation via C11 treatment in vitro (Fig. 1E). Actin mRNA translational efficiency was unaffected in tumors treated with IRES-J007 monotherapy. However, tumors that received PP242 and IRES-J007 co-therapy displayed a larger reduction in both cyclin D1 and c-MYC translational efficiency compared with IRES-J007 or PP242 monotherapy, with most of these transcripts being redistributed to nonribosomal/monosomal fractions (CCND1, 5% polysomal; c-MYC, 3% polysomal; *, p < 0.05). These data, taken together, suggest that the IRES-J007 inhibitor effectively inhibits cyclin D1 and c-MYC IRES-mediated protein synthesis in these tumors. Additionally, these data also suggest that the cyclin D1 and c-MYC transcripts are capable of being translated by cap-dependent (PP242-sensitive) and IRES-dependent (IRES-J007-sensitive) mechanisms.
FIGURE 7.

Cyclin D1 and c-MYC mRNA translational state in subcutaneous LN229 GBM tumors in response to combination IRES and mTOR inhibitor therapy. Polysome distributions of cyclin D1, c-MYC, and actin mRNAs from xenografted tumors harvested from mice receiving the indicated treatment schedules. Tumor extracts were subjected to sucrose density gradient centrifugation, fractionated, and pooled into a nonribosomal monosomal fraction (N, white columns) and a polysomal fraction (P, black columns). Purified RNAs were used in real-time quantitative RT-PCR analysis to determine the distributions of cyclin D1, c-MYC, and actin mRNAs across the gradients. Polysome gradient tracings are shown above each graph. Mean ± S.D. are shown for quadruplicate RT-PCR measurements. *, p < 0.05.
Discussion
Drug target identification and mechanism of action studies have critical roles in drug discovery efforts. In our previous studies, we identified a small-molecule inhibitor of c-MYC IRES-dependent protein synthesis and demonstrated its efficacy in multiple myeloma when combined with inducers of the endoplasmic reticulum stress response (21). In this report, we have evaluated the action of this inhibitor in GBM and demonstrate significant synergistic anti-GBM activity of this inhibitor in combination with PP242. SAR studies identified an improved IRES inhibitor that also demonstrated a significant synergistic antitumor effect in GBM both in vitro and in vivo. Experiments addressing the mechanism of action of the inhibitors demonstrate that the ITAF, hnRNP A1, is the target and that IRES RNA binding is markedly inhibited. Our studies are most consistent with the hypothesis that these inhibitors likely disrupt hnRNP A1-cyclin D1 or -c-MYC IRES binding by binding a small pocket within close proximity to RRM2, altering the conformation of the ITAF to preclude IRES interaction. The improved ability of the SAR-optimized analog IRES-J007 to block cyclin D1 or c-MYC IRES activity may be due to the ability of IRES-J007s to stabilize a conformation of hnRNP A1 that binds IRES RNA less effectively. This notion is supported by the experiments shown in Fig. 5, C and D, in which IRES-J007 appears to more efficiently block IRES activity and IRES RNA binding relative to the parent inhibitor. Although hnRNP A1-mediated exon 5 splicing of Max was unaffected by C11, we cannot rule out the possibility that additional properties of hnRNP A1, which may be affected by inhibitor binding, could contribute to its synergistic anti-GBM effects in combination with mTOR inhibition. Because hnRNP A1 is a nuclear-cytoplasmic shuttling protein (32–35), these inhibitors may have additional effects on the cellular distribution of hnRNP A1, which contributes to their ability to block IRES activity, although in initial experiments with C11, significant nuclear redistribution was not observed in cells following exposure.4
Our inhibitor docking studies suggest that IRES-J007 and its parent compound bind to a small pocket within close proximity to RRM2. The residues in this pocket are well conserved between species, and the pocket appears to have a unique surface structure. We attempted to superimpose this pocket structure on other known binding pocket structures to identify structural similarities and were unable to identify similar pockets searching the Multiple-sketches (PoSSuM) and ProBiS databases (data not shown). This suggests that this surface is distinct and that the C11 and IRES-J007 inhibitors may be less likely to exhibit off-target effects.
Recent crystallographic studies with UP1 bound to the HIV exon splicing silencer 3 stem loop (SL3ESS3) suggest that UP1 binds this RNA via a short, three-nucleotide loop recognition element (36). The structure revealed that RRM1 and the inter-RRM linker fold to form a pocket that sequesters the RNA, whereas RRM2 does not interact with the RNA. Mutagenesis experiments of conserved salt bridge interactions located on the opposite side of the RNA binding surface suggest that RRM1 and RRM2 are conformationally coupled. If UP1 interacts with the cyclin D1 or c-MYC IRESs in a similar manner, then it is conceivable that binding of C11 or IRES-J007 near RRM2 may have widespread conformational effects on RRM1 of UP1 as to inhibit binding to the IRES RNAs.
The plant flavonoid quercetin has been demonstrated to inhibit prostate cancer growth and, more recently, to target hnRNP A1 (37). Quercetin was shown to bind the F peptide region of hnRNP A1, impairing its ability to shuttle between the nucleus and cytoplasm, resulting in cytoplasmic retention. The redistribution of hnRNP A1 correlated with reduced Tnpo1 binding to hnRNP A1 following quercetin treatment, consistent with a defect in shuttling because Tnpo1 is required for hnRNP A1 import into the nucleus (38, 39). The accumulation of cytoplasmic hnRNP A1 was suggested to inhibit IRES-mediated translation of antiapoptotic mRNAs triggering apoptosis. C11 and IRES-J007 appear to act distinctly from quercetin and inhibit hnRNP A1 binding to cyclin D1 and c-MYC IRES RNAs. However, further structural studies are necessary to provide definitive insight into these interactions.
In summary, we have identified inhibitors of cyclin D1 and c-MYC IRES activity that target the ITAF hnRNP A1. We have demonstrated that these inhibitors act in a synergistic manner when combined with PP242, having broad anti-GBM effects in vitro and in xenograft experiments. Ligand docking analyses predict that C11 and IRES-J007 bind to a unique pocket structure within close proximity to RRM2 and interfere with cyclin D1 and c-MYC IRES binding. Our results demonstrate that hnRNP A1 is a compelling target in GBM and can be co-targeted with mTOR in a synthetically lethal manner to achieve significant therapeutic effects.
Author Contributions
B. H., J. L., M. E. J., A. L., and J. G. conceived and designed the experiments. B. H., A. B. S., J. L., K. A. L., and T. B. performed the experiments. B. H., J. L., M. E. J., A. L., and J. G. analyzed the data. B. H., J. L., M. E. J., A. L., and J. G. wrote the paper.
Supplementary Material
Acknowledgments
We thank Ronald Hay, Jacob Fleischmann, Eric Jan, Paul Mischel, and Norimoto Yanagawa for cell lines and reagents. We also thank YiJiang Shi and Cheri Cloninger for technical assistance and Richard Weisbart and Robert Nishimura for comments on the manuscript.
This work was supported by National Institutes of Health Grant R01CA168700 and by Department of Veterans Affairs MERIT Grant I01BX002665. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Experimental Procedures.
B. Holmes and J. Gera, unpublished observations.
- GBM
- glioblastoma
- EGFR
- EGF receptor
- mTOR
- mechanistic target of rapamycin
- IRES
- internal ribosome entry site
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- RRM
- RNA recognition motif
- ITAF
- internal ribosome entry site trans-acting factor
- SAR
- structure-activity relationship
- XTT
- 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt
- CI
- combination index
- C11
- compound 11.
References
- 1. Dunn G. P., Rinne M. L., Wykosky J., Genovese G., Quayle S. N., Dunn I. F., Agarwalla P. K., Chheda M. G., Campos B., Wang A., Brennan C., Ligon K. L., Furnari F., Cavenee W. K., Depinho R. A., et al. (2012) Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 26, 756–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 [DOI] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fan Q. W., and Weiss W. A. (2010) Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr. Top. Microbiol. Immunol. 347, 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cloughesy T. F., Cavenee W. K., and Mischel P. S. (2014) Glioblastoma: from molecular pathology to targeted treatment. Annu. Rev. Pathol. 9, 1–25 [DOI] [PubMed] [Google Scholar]
- 7. Gini B., Zanca C., Guo D., Matsutani T., Masui K., Ikegami S., Yang H., Nathanson D., Villa G. R., Shackelford D., Zhu S., Tanaka K., Babic I., Akhavan D., Lin K., et al. (2013) The mTOR kinase inhibitors, CC214–1 and CC214–2, preferentially block the growth of EGFRvIII-activated glioblastomas. Clin. Cancer Res. 19, 5722–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Betz C., and Hall M. N. (2013) Where is mTOR and what is it doing there? J. Cell Biol. 203, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cloughesy T. F., Yoshimoto K., Nghiemphu P., Brown K., Dang J., Zhu S., Hsueh T., Chen Y., Wang W., Youngkin D., Liau L., Martin N., Becker D., Bergsneider M., Lai A., et al. (2008) Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 5, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang S. M., Wen P., Cloughesy T., Greenberg H., Schiff D., Conrad C., Fink K., Robins H. I., De Angelis L., Raizer J., Hess K., Aldape K., Lamborn K. R., Kuhn J., Dancey J., et al. (2005) Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest. New Drugs 23, 357–361 [DOI] [PubMed] [Google Scholar]
- 12. Wu S. H., Bi J. F., Cloughesy T., Cavenee W. K., and Mischel P. S. (2014) Emerging function of mTORC2 as a core regulator in glioblastoma: metabolic reprogramming and drug resistance. Cancer Biol. Med. 11, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masri J., Bernath A., Martin J., Jo O. D., Vartanian R., Funk A., and Gera J. (2007) mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 67, 11712–11720 [DOI] [PubMed] [Google Scholar]
- 14. Iwanami A., Gini B., Zanca C., Matsutani T., Assuncao A., Nael A., Dang J., Yang H., Zhu S., Kohyama J., Kitabayashi I., Cavenee W. K., Cloughesy T. F., Furnari F. B., Nakamura M., et al. (2013) PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc. Natl. Acad. Sci. U.S.A. 110, 4339–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benavides-Serrato A., Anderson L., Holmes B., Cloninger C., Artinian N., Bashir T., and Gera J. (2014) mTORC2 modulates feedback regulation of p38 MAPK activity via DUSP10/MKP5 to confer differential responses to PP242 in glioblastoma. Genes Cancer 5, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka K., Babic I., Nathanson D., Akhavan D., Guo D., Gini B., Dang J., Zhu S., Yang H., De Jesus J., Amzajerdi A. N., Zhang Y., Dibble C. C., Dan H., Rinkenbaugh A., et al. (2011) Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 1, 524–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi Y., Sharma A., Wu H., Lichtenstein A., and Gera J. (2005) Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J. Biol. Chem. 280, 10964–10973 [DOI] [PubMed] [Google Scholar]
- 18. Martin J., Masri J., Cloninger C., Holmes B., Artinian N., Funk A., Ruegg T., Anderson L., Bashir T., Bernath A., Lichtenstein A., and Gera J. (2011) Phosphomimetic substitution of heterogeneous nuclear ribonucleoprotein A1 at serine 199 abolishes AKT-dependent internal ribosome entry site-transacting factor (ITAF) function via effects on strand annealing and results in mammalian target of rapamycin complex 1 (mTORC1) inhibitor sensitivity. J. Biol. Chem. 286, 16402–16413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jo O. D., Martin J., Bernath A., Masri J., Lichtenstein A., and Gera J. (2008) Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J. Biol. Chem. 283, 23274–23287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cloninger C., Bernath A., Bashir T., Holmes B., Artinian N., Ruegg T., Anderson L., Masri J., Lichtenstein A., and Gera J. (2011) Inhibition of SAPK2/p38 enhances sensitivity to mTORC1 inhibition by blocking IRES-mediated translation initiation in glioblastoma. Mol. Cancer Ther 10, 2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi Y., Yang Y., Hoang B., Bardeleben C., Holmes B., Gera J., and Lichtenstein A. (2016) Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene 35, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Culjkovic B., Topisirovic I., Skrabanek L., Ruiz-Gutierrez M., and Borden K. L. (2005) eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J. Cell Biol. 169, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Babic I., Anderson E. S., Tanaka K., Guo D., Masui K., Li B., Zhu S., Gu Y., Villa G. R., Akhavan D., Nathanson D., Gini B., Mareninov S., Li R., Camacho C. E., et al. (2013) EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab 17, 1000–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonnal S., Pileur F., Orsini C., Parker F., Pujol F., Prats A. C., and Vagner S. (2005) Heterogeneous nuclear ribonucleoprotein A1 is a novel internal ribosome entry site trans-acting factor that modulates alternative initiation of translation of the fibroblast growth factor 2 mRNA. J. Biol. Chem. 280, 4144–4153 [DOI] [PubMed] [Google Scholar]
- 25. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanoh N., Honda K., Simizu S., Muroi M., and Osada H. (2005) Photo-cross-linked small-molecule affinity matrix for facilitating forward and reverse chemical genetics. Angew. Chem. Int. Ed. Engl. 44, 3559–3562 [DOI] [PubMed] [Google Scholar]
- 27. Thompson S. R. (2012) So you want to know if your message has an IRES? Wiley Interdiscip. Rev. RNA 3, 697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., and Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chou T. C. (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 [DOI] [PubMed] [Google Scholar]
- 30. Chou T. C., and Talalay P. (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55 [DOI] [PubMed] [Google Scholar]
- 31. Michlewski G., and Cáceres J. F. (2010) Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 17, 1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dreyfuss G., Kim V. N., and Kataoka N. (2002) Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205 [DOI] [PubMed] [Google Scholar]
- 33. Lewis S. M., and Holcik M. (2008) For IRES trans-acting factors, it is all about location. Oncogene 27, 1033–1035 [DOI] [PubMed] [Google Scholar]
- 34. Lewis S. M., Veyrier A., Hosszu Ungureanu N., Bonnal S., Vagner S., and Holcik M. (2007) Subcellular relocalization of a trans-acting factor regulates XIAP IRES-dependent translation. Mol. Biol. Cell 18, 1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jean-Philippe J., Paz S., and Caputi M. (2013) hnRNP A1: the Swiss army knife of gene expression. Int. J. Mol. Sci. 14, 18999–19024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morgan C. E., Meagher J. L., Levengood J. D., Delproposto J., Rollins C., Stuckey J. A., and Tolbert B. S. (2015) The first crystal structure of the UP1 domain of hnRNP A1 bound to RNA reveals a new look for an Old RNA binding protein. J. Mol. Biol. 427, 3241–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ko C. C., Chen Y. J., Chen C. T., Liu Y. C., Cheng F. C., Hsu K. C., and Chow L. P. (2014) Chemical proteomics identifies heterogeneous nuclear ribonucleoprotein (hnRNP) A1 as the molecular target of quercetin in its anti-cancer effects in PC-3 cells. J. Biol. Chem. 289, 22078–22089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fridell R. A., Truant R., Thorne L., Benson R. E., and Cullen B. R. (1997) Nuclear import of hnRNP A1 is mediated by a novel cellular cofactor related to karyopherin-β. J. Cell Sci. 110, 1325–1331 [DOI] [PubMed] [Google Scholar]
- 39. Rebane A., Aab A., and Steitz J. A. (2004) Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 10, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.