

Abstract
Valosin-containing protein/p97(VCP) is a hexameric ATPase vital to protein degradation during endoplasmic reticulum stress. It regulates diverse cellular functions including autophagy, chromatin remodeling, and DNA repair. In addition, mutations in VCP cause inclusion body myopathy, Paget disease of the bone, and frontotemporal dementia (IBMPFD), as well as amyotrophic lateral sclerosis. Nevertheless, how the VCP activities were regulated and how the pathogenic mutations affect the function of VCP during stress are not unclear. Here we show that the small ubiquitin-like modifier (SUMO)-ylation of VCP is a normal stress response inhibited by the disease-causing mutations in the N-domain. Under oxidative and endoplasmic reticulum stress conditions, the SUMOylation of VCP facilitates the distribution of VCP to stress granules and nucleus, and promotes the VCP hexamer assembly. In contrast, pathogenic mutations in the VCP N-domain lead to reduced SUMOylation and weakened VCP hexamer formation upon stress. Defective SUMOylation of VCP also causes altered co-factor binding and attenuated endoplasmic reticulum-associated protein degradation. Furthermore, SUMO-defective VCP fails to protect against stress-induced toxicity in Drosophila. Therefore, our results have revealed SUMOylation as a molecular signaling switch to regulate the distribution and functions of VCP during stress response, and suggest that deficiency in VCP SUMOylation caused by pathogenic mutations will render cells vulnerable to stress insults.
Keywords: amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), endoplasmic reticulum-associated protein degradation (ERAD), post-translational modification (PTM), stress granule, stress response, sumoylation, hexamer, valosin-containing protein/p97 (VCP)
Introduction
Valosin-containing protein (VCP/p97, cdc48, Ter-94)2 is a highly conserved member of AAA (ATPase associated with diverse cellular activities) family proteins. It is mainly composed of N-domain and two ATPase domains. By forming a homohexamer and binding various co-factors via the N-domain, VCP helps to remodel, unfold, or degrade protein substrates using the energy derived from ATP hydrolysis (1–3). Due to its cellular abundance and broad interaction with a number of co-factors, key substrates, and regulators of the ubiquitin proteasome system, VCP has emerged as a vital modulator of a large variety of cellular activities, including protein degradation, ER stress response, autophagy/mitophagy, endosomal trafficking, cell cycle, and DNA repair (2, 5, 6). However, the factors that regulate VCP activities are not clear.
The importance of VCP is also demonstrated by its association with cancer and degenerative diseases. VCP is highly expressed in non-small cell lung carcinoma (7), and its expression is correlated with tumor progression and prognosis (8). On the other hand, mutations in VCP have been associated with degenerative diseases, including inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD) and amyotrophic lateral sclerosis (ALS) (6, 9). Most pathogenic mutations of VCP are found in the N-domain, with a few in the ATPase domains (10). The pathology of VCP-associated degenerative diseases features ubiquitin-positive inclusions. Although some of the pathogenic VCP mutations have been shown to affect the ATPase activities (11, 12) or mitochondrial function (13, 14), the effects of these mutations in protein homeostasis, especially in the cellular stress response associated with the development of degenerative diseases, are unclear. As weakened cellular stress response contribute to aging and the development of neurodegenerative diseases (15–17), it will be of great interest to determine whether the pathogenic mutations in VCP also affect cellular stress response.
Protein post-translational modifications (PTMs) likely play important roles in the development of neurodegenerative diseases (18–20). Like epigenetic modifications (21), PTMs could be one of the links between environment input/life experiences and the functional regulation of disease-associated proteins in the decades leading to neurodegeneration. One form of PTMs is protein SUMOylation, which is a lysine modification homologous to ubiquitination, and is facilitated by the E1-activating enzymes SAE1 or SAE2, the solo E2 conjugating enzyme Ubc9 and various E3 ligases (22). By attaching the SUMO groups to certain lysine residues in the substrates, SUMOylation can modulate the stability, localization, and interaction pattern of the target proteins (23). VCP has been shown to be phosphorylated, acetylated, or oxidized mainly at the C terminus (24), and these modifications affect the ATPase activities of VCP. Interestingly, a few recent reports have shown the SUMOylation of VCP-binding co-factors or substrates (25, 26), and suggested the interplay between the SUMO and ubiquitin pathway in the functional modulation of the VCP complexes. Recently, mass spectrometry-based global studies of SUMOylation have identified VCP as a potential SUMO target protein among sometimes thousands of SUMOylated targets (27–30). However, the SUMOylation of VCP has never been further characterized. How SUMOylation affects the functions of VCP, and whether it is relevant to neurodegenerative diseases are unknown.
In this study, using in vitro and in vivo models, as well as primary cells derived from a patient carrying VCP mutation, we characterized the SUMOylation of VCP and identified it as a stress response that affects the activities of VCP. Furthermore, we found that N-domain pathogenic mutations in VCP caused attenuated SUMOylation of VCP, weakened hexamer assembly, and defective stress response.
Results
Pathogenic Mutations at the N-domain Inhibit the SUMOylation of VCP
The human VCP or its yeast homolog cdc48 has been suggested to be a potential SUMO substrate either by SUMO-binding domain prediction or global mass spectrometry analysis (27, 28, 31). It is unclear how the putative SUMOylation of VCP is regulated, and whether SUMOylation affects VCP function. To address these questions, we first set out to biochemically validate VCP as a real SUMO substrate. We co-transfected His-tagged VCP, SUMO conjugating enzyme Ubc9, and CFP-tagged SUMO-1 in HeLa cells, and detected the presence of SUMOylated VCP using denaturing RIPA or 4 m urea buffer (Fig. 1A). Furthermore, by fusing VCP with a functional (WT) or defective (C93S) Ubc9 (the Ubc9 fusion-directed SUMOylation approach) (32), and co-expressed it with SUMO-1 (Fig. 1, B and C), we found the presence of SUMOylated, higher molecular weight VCP species only in cells expressing the functional VCP-Ubc9 fusion protein (Fig. 1C). To determine whether VCP is SUMOylated in vivo, we immunoprecipitated VCP from rat cortical tissues under a denaturing condition, and detected the presence of SUMOylated VCP (Fig. 1D). In addition, the SUMOylation of endogenous VCP was suppressed in cells with knocked down Ubc9 expression (Fig. 1E). Taken together, these results confirmed that VCP is a SUMO substrate.
FIGURE 1.

VCP is modified by SUMO in the N-domain. A, VCP is a SUMOylated protein. HeLa cells co-transfected with His-tagged VCP, CFP-tagged SUMO-1, and SUMO E2 Ubc-9 as indicated were lysed in RIPA buffer (left panel) or 4 m urea buffer (right panel). Lysates were incubated with nickel-nitrilotriacetic acid-agarose beads to pulldown VCP-His, and then immunoblotted with anti-SUMO-1 (top panel) or anti-His antibodies (middle and bottom panels). Longer exposed gel image was used to show the amount of SUMOylated VCP relative to the unmodified VCP. B, diagram of VCP construct with Ubc9 fusion-directed SUMOylation (UFDS). C, UFDS of VCP. HeLa cells were transiently transfected with VCP-Ubc9-His and CFP-SUMO1. Total lysates were analyzed by immunoblotting with anti-His and Tubulin antibodies. D, endogenous VCP is SUMOylated in rat cortex. The cortical tissue from adult rats lysed in RIPA buffer were subjected to immunoprecipitation using anti-VCP antibody and then blotted with antibody to SUMO-1. E, suppressed endogenous VCP SUMOylation in cells with reduced Ubc9. HEK293 cells transfected with indicated siRNAs were cultured for 72 h, followed by analysis of VCP SUMOylation as described in D. Note the decreased global SUMOylation after Ubc9 knockdown. F, schematic representation of full-length and truncation constructs of VCP. G, SUMOylation of the VCP N-terminal truncation constructs. HEK293 cells were co-transfected with the WT or indicated VCP-His deletion constructs, and SUMO-related plasmids. E2, Ubc9; C-S1, CFP-SUMO1. SUMOylated species were determined as described above. Arrows and asterisks indicate unmodified VCP constructs and SUMOylated VCP, respectively. The Tubulin/Actin loading control shows that the identical amount of lysates were used for pulldown.
To identify the SUMO conjugation site within VCP, we first made two His-tagged truncation constructs expressing the N-domain (amino acids 1–208) or C-domain (amino acids 209–806) (Fig. 1F). The N-domain mainly contains the protein binding region, and the C-domain contains the D1 and D2 ATPase domains and the rest of the C-terminal VCP sequences. We found that the N-domain could be SUMOylated as efficiently as the wild-type VCP. In contrast, SUMO modification was completely absent in the C-domain (Fig. 1G). Thus, the N-domain of VCP harbors potential SUMO conjugation sites.
Based on sequence matching, VCP may contain two putative SUMO conjugation sites at lysine residues 190 and 486. We mutated these two lysines to arginines (K190R, K486R), respectively, and tested the SUMOylation capacity of these VCP mutants. Surprisingly, the mutation at neither site had noticeable effect on VCP SUMOylation, whereas the pathogenic mutation R191Q significantly decreased the VCP SUMOylation (Fig. 2A). This observation prompted us to test the effect of 5 other ALS-causing point mutations on VCP SUMOylation. All the N-domain pathogenic mutants we tested, including R155C, R159H, R95G, G97E, and A232E, could reduce VCP SUMOylation (Fig. 2, B and C). In contrast, the D592N mutation at the D2 domain did not affect VCP SUMOylation (Fig. 2B). These results suggest that reduced SUMOylation of VCP may be a common consequence of pathogenic mutations in the VCP N-domain.
FIGURE 2.

Pathogenic mutations in the N-domain of VCP inhibit SUMOylation. A and B, the pathogenic mutation reduces the SUMOylation of VCP. HEK293 cells were co-transfected with the WT or indicated mutant VCP-His constructs together with SUMO-related plasmids, and analyzed for SUMO conjugation as described. C, the quantification of SUMOylation levels of indicated VCP constructs relative to that of the WT VCP in B. Data are shown as mean ± S.E., n = 3 experiments. D, protein sequence of the VCP N-domain with the sites of lysine (K) to arginine (R) mutations indicated. Every Lys in the N-domain was mutated and the position and the number of Lys to Arg mutations in each construct indicated (e.g. K60–3 harbors 3 Lys to Arg mutations starting at the Lys60). E, mutagenesis and SUMOylation analysis indicates 3 likely sites of SUMO conjugation in the N domain of VCP. All the Lys to Arg VCP mutant constructs were analyzed. F, the reduction of VCP SUMOylation by the pathogenic mutant is comparable with that of the SUMO-deficient mutant K60–3. The SUMO-insensitive K148 mutant was included as a control. HEK293 cells were transfected as indicated, followed by VCP SUMOylation analysis.
As VCP was apparently SUMOylated at non-consensus sites, we mutated every lysine residue in the N-domain to characterize the SUMO conjugation sites. These mutants with 1 to 3 lysine residues switched to arginine (Fig. 2D) were tested for their SUMOylation capability. The triple mutations at lysine residues at 60, 62, and 63 (K60–3), single point mutation at Lys136 or Lys164 led to clearly decreased SUMOylation of VCP (Fig. 2E). Furthermore, the reduction of VCP SUMOylation caused by pathogenic mutations and K60–3 were comparable (Fig. 2F). Although it was surprising that our extensive mutagenesis failed to completely abolish VCP SUMOylation, these results suggested that SUMOylation at the N-domain of VCP is a dynamic event, and could occur at several sites subject to active regulation. It is conceivable that a number of lysine residues could act as SUMO conjugation sites to compensate for each other, and could be affected by the conformation changes caused by disease mutations even at non-lysine residues at the N-domain.
SUMOylation of VCP Is a Stress Response Regulating the Translocation of VCP to Stress Granules and Nucleus
Defective cellular responses to oxidative and ER stress contribute to the progression of neurodegenerative diseases (33, 34). As VCP is a key regulator of ER stress, we aimed to test whether the SUMOylation VCP were altered during cellular stress response. We exposed HEK293 cells and primary neuronal cultures (data not shown) to various inducers of oxidative stress (paraquat or arsenite) or ER stress (MG-132 or tunicamycin) and determined the change of global protein SUMOylation. Arsenite and MG132 produced the most consistent induction of protein SUMOylation (Fig. 3A). Next, we found that the SUMOylation of endogenous VCP gradually increased with rising dosage of MG132 or arsenite (Fig. 3B). Similarly, the SUMOylation of VCP increased in mice intraperitoneally injected with arsenite or paraquat (Fig. 3C).
FIGURE 3.
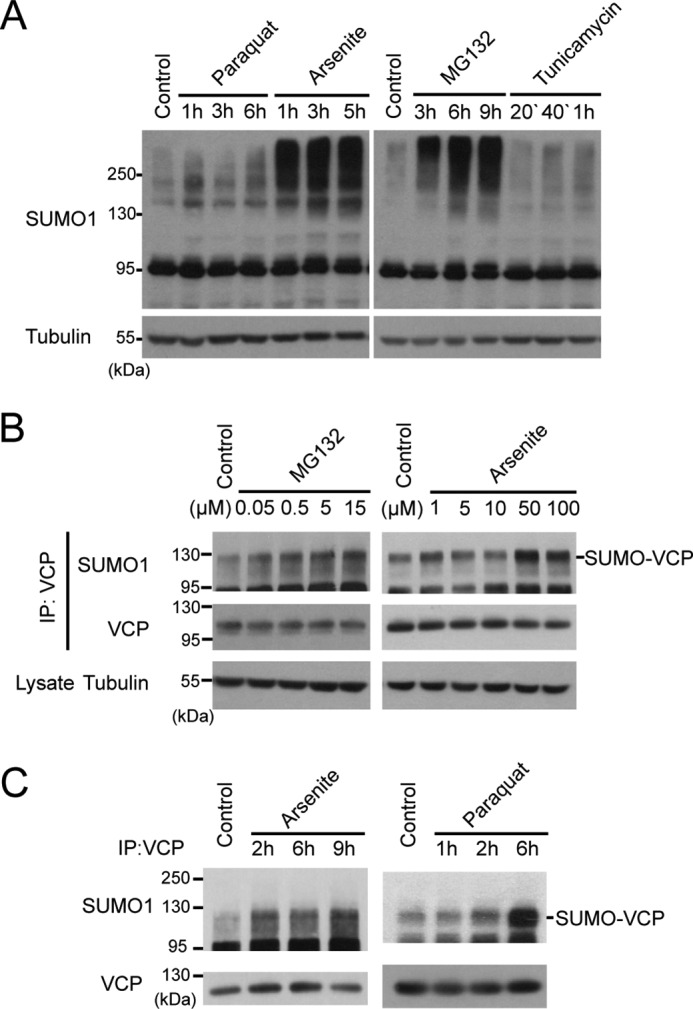
SUMOylation of VCP is a stress response. A, oxidative and ER stress increase global SUMOylation. HEK293 cells were treated with stress inducers and harvested at various time points as indicated. The global SUMO levels were determined by immunoblotting for SUMO-1. The concentration of each stressor is: paraquat (250 μm), sodium arsenite (100 μm), MG132 (15 μm), and tunicamycin (5 μg/ml). B, the SUMOylation of VCP is enhanced by arsenite or MG132 in a dose-dependent manner. HEK293 cells were treated with MG132 for 4 h or arsenite for 2 h at increasing concentrations, and the SUMOylation of endogenous VCP was determined using immunoprecipitation (IP)-immunoblotting as described in the legend to Fig. 1D. C, SUMOylation of VCP increases in vivo during stress. Wild type C57/B6 mice were treated with sodium arsenite or paraquat via intraperitoneal injection for indicated time. SUMOylation of VCP in the liver was analyzed by immunoprecipitation and immunoblotting.
VCP is an abundant protein capable of shuttling between various cellular compartments to regulate a variety of activities, including autophagy, endoplasmic reticulum-associated degradation (ERAD), and DNA repair (35). We assessed whether the localization of endogenous VCP was altered under stress conditions. First, using biochemical methods, we extracted cytosolic proteins with 0.2% Nonidet P-40 buffer, and lysed the remaining Nonidet P-40-insoluble proteins, which mainly contain nuclear fractions, with the RIPA buffer. The purity of the cytosolic fractions was confirmed by the presence of tubulin and the absence of nuclear lamin (Fig. 4A). It is worth noting that the RIPA fractions also contain cytosolic markers for stress granule, PABP and YB1 (Fig. 4A), suggesting that some stress granules were insoluble to Nonidet P-40 yet soluble in RIPA buffer. In cells treated with a higher concentration of MG132 or arsenite, we have observed a clear increase in the abundance of VCP in the RIPA-soluble fractions, and a corresponding slight decrease of cytosolic VCP (Fig. 4, A and B), suggesting increased distribution of VCP to the nucleus and stress granules upon stress. Furthermore, the increased distribution of VCP in the nucleus/stress granules upon stress was also observed in the primary neuronal cultures (Fig. 4C).
FIGURE 4.

Translocation of VCP to the stress granules and nucleus under stress conditions. A, translocation of VCP to Nonidet P-40-insoluble fractions in HEK293 cells exposed to increasing doses of MG132 or arsenite. The total amount of Nonidet P-40-soluble or RIPA-soluble VCP in HEK293 cells treated by MG132 (4 h) or arsenite (2 h) was determined by SDS-PAGE. Note the increase of VCP in the RIPA lysates from cells treated with a high concentration of stress inducers. Tubulin and Lamin A/C were probed to serve as markers for cytosol and nuclear proteins, respectively. YB1 and PABP were markers for stress granules. B, quantification of the data shown in A; data are represented as mean ± S.E.; n = 3 experiments. C, stress-induced translocation of VCP to RIPA-soluble fractions in primary rat cortical culture. Rat cortical cultures were treated with arsenite for 2 h. Results represent mean ± S.E. n = 3 experiments.
To further characterize the change in the subcellular distribution of endogenous VCP during stress response, we triple labeled VCP, nuclear DAPI, and stress granule marker YB-1 for immunofluorescence microscopy (Fig. 5A). Our quantitative analysis of VCP subcellular localization showed a dose-dependent and statistically significant increase of VCP in the nucleus and stress granules when cells were exposed to arsenite (Fig. 5, A–C), confirming the biochemical analysis in Fig. 4A.
FIGURE 5.

The accumulation of endogenous VCP in the nucleus and stress granules under stress condition. A, HeLa cells treated with increasing concentrations of sodium arsenite (2 h) were triple-labeled for VCP, DAPI, and stress granule marker YB-1 for confocal immunofluorescence microscopy analysis. Up arrows indicate nuclear VCP puncta, and arrowheads indicate VCP puncta in stress granules. Scale bar = 10 μm. B, the quantification of the fluorescence intensity of nuclear VCP, the number of cells (≥30) analyzed at each condition was labeled on each bar. C, quantification of the YB-1-positive stress granules (SG) (diameter ≥ 2 μm) per cell in cells exposed to sodium arsenite, with ≥30 cells analyzed at each condition.
Finally, to evaluate the role of SUMOylation in the subcelluar distribution of VCP, we transfected SUMO protease SENP1 to block the global SUMOylation and VCP SUMOylation (Fig. 6A), and then evaluated the presence of endogenous VCP in the RIPA-soluble fractions upon arsenite exposure. Inhibition of SUMOylation almost largely abolished the nuclear and stress granule translocation of VCP (Fig. 6B). To directly assess the effect of VCP SUMOylation on the subcellular distribution upon stress, we determined the abundance of WT and SUMO-defective mutants VCP K60–3, K136–1, or K164–1 in RIPA-soluble fractions in arsenite-treated cells (Fig. 6C). SUMO deficiency in VCP led to a statistically significant decrease in the distribution to the nucleus and stress granules upon stress (note the RIPA-soluble, arsenite + group).
FIGURE 6.

SUMOylation promotes the VCP translocation into the nucleus and stress granules. A, confirmation of decreased global and VCP SUMOylation caused by SENP1 overexpression. B, inhibition of global SUMOylation by SENP1 blocks stress-induced translocation of VCP. HEK293 cells were transfected with or without SUMO protease SENP1, followed by 50 μm arsenite treatment for 2 h. VCP in each fraction was analyzed as described in the legend to Fig. 4A. Data are shown as mean ± S.E., n = 3 experiments. C, SUMO-deficient VCP exhibit decreased translocation upon stress. HEK293 cells transfected with the indicated SUMO-deficient mutants were treated with 50 μm sodium arsenite for 2 h, and biochemically analyzed for distribution as described above. Quantification of the data represents as mean ± S.E. n = 5 experiments. D, pathogenic N-domain mutant VCP shows reduced nuclear distribution in cells exposed to arsenite. HEK293 cells were transfected as indicated. Data are shown as mean ± S.E., n = 3 experiments.
As pathogenic N-domain mutant VCP showed reduced SUMOylation, we assessed their distribution under stress. Like SUMO-defective mutants VCP K60–3, K136–1, or K164–1, the N-domain pathogenic R155C and R159H VCP, but not the SUMOylation-insensitive D592N VCP, showed reduced nuclear distribution in cells exposed to arsenite (Fig. 6D). Taken together, these results suggest that the SUMOylation of VCP is a stress response that facilitates the cellular distribution of VCP to stress granules and nucleus, and the pathogenic mutations that caused SUMO deficiency show defective stress response.
Defective SUMOylation of VCP Caused by Mutations Reduced the Hexamer Assembly under Stress Conditions
As the post-translational modifications may affect protein conformation and protein interaction dynamics, we examined whether VCP SUMOylation might impact its hexamer formation, which is vital for the ATPase and protein degradation activities of VCP (36). First, using native gels, we detected increased VCP hexamer assembly in arsenite or paraquat-treated mice (Fig. 7, A and B) that showed increased VCP SUMOylation (Fig. 3C). Next, to validate the role of SUMOylation directly in a system modeling the cellular environment, we compared the hexamer assembly by the WT and C93S Ubc9-VCP fusion constructs, as the only difference between these two constructs was the SUMO status of VCP (Fig. 1C). VCP-Ubc9-WT showed much enhanced hexamer formation than VCP-Ubc9-C93S or the control VCP-WT (Fig. 7C). We then used the more sensitive gel filtration method to separate the native protein complexes from cells expressing VCP-WT, VCP-Ubc9-WT, or VCP-Ubc9-C93S (Fig. 7, D and E). The main peak of control VCP-WT was eluted in a fraction corresponding to a molecular mass of 670 kDa as indicated by the marker thyroglobulin, consistent with the size of VCP hexamer. Because the two VCP-Ubc9 fusion proteins were larger than the VCP-WT, the main peaks shifted to the left as expected (Fig. 7E). As in native gel (Fig. 7C), the abundance of the hexameric species for the VCP-Ubc9-WT fusion protein was much higher than that of the SUMO-defective VCP-Ubc9-C93S (Fig. 7, D and E). There were multiple high molecular VCP-related bands observed only in the lysates from VCP-Ubc9-WT-expressing cells, likely the SUMOylated species (Fig. 7D). The presence of these bands were also consistent with the high molecular weight protein smears observed in Fig. 6C. Therefore, these results demonstrated that SUMOylation of VCP promotes the VCP hexamer formation in cells.
FIGURE 7.

SUMOylation of VCP promotes its hexamer assembly. A and B, increased VCP hexamer assembly in vivo during stress response. The abundance of the VCP hexamers in the liver of mice treated with sodium arsenite (A) or paraquat (B) were analyzed by PAGE under non-denaturing (−SDS) or denaturing (+SDS) conditions (see “Experimental Procedures”). Quantification of the data represents mean ± S.E. n = 3 experiments. C, Ubc9 fusion-directed SUMOylation (UFDS) of VCP promotes the hexamer formation. HEK293 cells were lysed and analyzed as described above. VCP hexamer formation was assessed in a non-denaturing (−SDS) condition, and the total VCP in each sample was determined by SDS-PAGE, and serve as loading control. D, size exclusion chromatography of His-tagged WT VCP and VCP-Ubc9 fused proteins. HEK293 cells were co-transfected with CFP-SUMO1 and the indicated VCP constructs, and the lysates were fractionated using size exclusion chromatography followed by SDS-PAGE and immunoblotting. E, quantification of the data shown in D. Elution peaks of molecular weight markers are shown. Results represent mean ± S.E. n = 3 experiments.
Previous studies have shown that the VCP hexamer assembly was not affected by pathogenic mutations (11, 37, 38). However, in these studies, the SUMOylation was not a factor to modulate VCP activity. In fact, we have confirmed that in the absence of an increase of global SUMOylation as seen during stress response, there was no difference between the hexamer formed by WT or mutant VCP (data not shown). To test whether N-domain pathogenic mutations affected VCP hexamer formation in the presence of increased cellular SUMO levels, we compared the hexameric VCP formed by the WT, disease-causing mutants (R155C and R159H), and the SUMO-defective K60–3, K164–1, and K136–1 VCP in the presence of SUMO-1 (Fig. 8A). Our results showed reduced hexamer formation by the mutant VCP (Fig. 8A). Furthermore, the level of reduction in hexamer formation by the pathogenic or the SUMO-deficient K60–3 was similar. Although the reduction was only about 30%, the change was very reproducible. To further validate these results in a more physiological condition, we analyzed the VCP hexamer formation in primary cells derived from an IBMPFD patient carrying a heterozygous VCP G97E mutation (39). Like other N-domain pathogenic mutations, the G97E mutation led to deficient SUMOylation (Fig. 2B), and decreased VCP hexamer assembly (Fig. 8C) in primary kidney epithelial cells. These results suggest that decreased SUMOylation caused by N-domain pathogenic mutations in VCP may lead to reduced hexamer formation during stress response.
FIGURE 8.
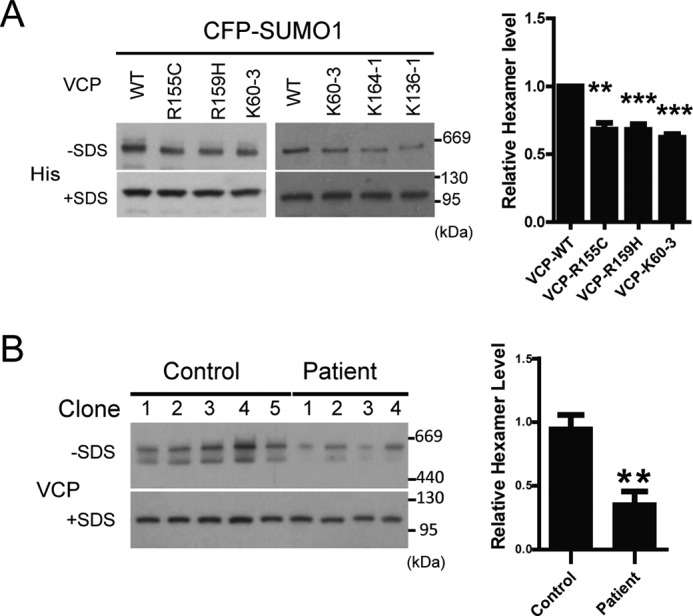
SUMO-defective mutations of VCP impair hexamer assembly. A, pathogenic or SUMO-deficient mutant VCP exerts a comparable inhibitory effect on hexamer formation. HEK293 cells transfected with CFP-SUMO1 together with various VCP constructs were analyzed as described in the legend to Fig. 7A. Values are represented as mean ± S.E. n = 3 experiments. B, reduced VCP hexamer formation in primary kidney epithelial cell cultures from a patient carrying the G97E point mutation in VCP. To minimize the variance among the different clones derived from the initial culturing expansion, 5 clones from the healthy control and 4 from the patient were analyzed for hexamer assembly. The quantification of the data are represented in the adjacent bar graph as mean ± S.E.
Defective SUMOylation of VCP Causes Altered Co-factor Binding, Impaired ERAD, and Increased Vulnerability to Stress Insults
VCP interacts with a large number of co-factors to exhort its diverse cellular functions, with most of the co-factors binding the N-domain of VCP (40). For example, VCP forms a complex with NPL4 and UFD1 to facilitate the degradation of polyubiquitinated protein substrates in ER stress (41). To determine whether deficient SUMOylation of VCP may affect co-factor binding, we assessed the binding between some main N-domain targeting co-factors (NPL4, UFD1, and p47) and the SUMO-capable (Ubc9-WT) or SUMO-defective (Ubc9-C93S) VCP-Ubc9 fusion proteins (Fig. 9A). The SUMO-defective VCP showed clearly increased binding of NPL4 and p47, and mildly increased binding to UFD1 (Fig. 9A). In contrast, the interaction between VCP and the ubiquitin chain-editing factor E4B, another co-factor binding to the VCP N-domain (42), was not affected by the status of VCP SUMOylation (Fig. 9A), suggesting that the effect of VCP SUMOylation on protein binding is selective. Furthermore, pathogenic VCP mutants R155C and R159H, as well as the SUMO-deficient mutant K60–3, showed statistically significant increased binding to NPL4 and p47 in the presence of increase cellular SUMOylation (Fig. 9, B and C). However, in the absence of increased SUMO levels, there was no clear difference in NPL4 binding between the WT VCP and VCP mutants (data not shown). Therefore, defective SUMOylation of VCP leads to increased binding between VCP and co-factors NPL4 and p47.
FIGURE 9.

Defective SUMOylation of VCP causes altered co-factor binding, impaired ERAD, and increased vulnerability to stress insults. A, defective SUMOylation of VCP leads to altered co-factor binding. His-tagged Ubc9 fusion-directed SUMOylation (UFDS) constructs of VCP were transfected in HEK293 cells and the binding to UFD1, NPL4, p47, or E4B were determined. B and C, the pathogenic or SUMO-deficient mutant VCP showed increased binding to co-factor NPL4 and p47. HEK293 cells transfected with CFP-SUMO1 together with various VCP constructs were analyzed as in A. Results represent mean ± S.E. n = 3 experiments. D and E, pathogenic or SUMO-deficient VCP mutants fails to degrade ERAD substrates. HeLa cells stably expressing the ERAD substrate TCRα-GFP were first transfected with siRNA targeting 5′ UTR to knockdown endogenous VCP, and then transiently transfected with the WT or mutant VCP as indicated. The degradation of ERAD substrate TCRα-GFP was determined by antibody against GFP. Results represent mean ± S.E. n = 3 experiments. F, confirmation of decreased SUMOylation of Ter94KK. Drosophila Ter94 and TER94KK were subcloned in mammalian expression vector and transiently expressed in HEK293 cells. The SUMOylation capacity of Ter94 and TER94KK were assessed by immunoprecipitation (IP) using an anti-V5 antibody followed by immunoblotting. Direct analysis of Ter94 SUMOylation in flies was attempted but not successful as the SUMO signals were beyond the detection limit. G, survival curve of male Elav[C155]/+ (black), Elav[C155]>Ter94 (green), and Elav[C155]>Ter94KK (red) after 1 mm arsenite treatment. Ter94KK is K57R,K59R double mutant, homologous to the human VCP K60–3 mutant. There are only two Lys residues in Ter-94 at this region. Each genotype contained at least 4 vials with 20 flies each, and 2 other replicate experiments showed similar results. Data are represented as mean ± S.E.; n ≥ 4 vials, Elav[C155]>Ter94 (green) were compared with Elav[C155]>Ter94KK for statistical analysis. All transgenic flies have normal lifespan without exposure to stress.
We then compared the regulation of ERAD by the pathogenic and SUMO-defective VCP using a well characterized ERAD reporter TCR-α-GFP (43). We first knocked down endogenous VCP using RNAi targeting the 5′-untranslated region, and then co-transfected the RNAi-resistant WT or various VCP mutants in cells stably expressing TCR-α-GFP. Silencing the endogenous VCP significantly blocked the degradation of the TCR-α-GFP. Although the WT VCP could fully restore the degradation of TCR-α-GFP, the pathogenic mutants or the SUMO-deficient K60–3 showed a significant functional impairment (Fig. 9, D and E).
Finally, we assessed the effect of VCP SUMOylation in the protection against stress-induced toxicity in vivo. We made transgenic flies expressing the WT Ter94, the Drosophila VCP homolog, or the one carrying the double Lys to Arg mutations, Ter94KK. The mutations in Ter94KK were comparable with the SUMO-deficient K60–3 mutant in the human VCP. Each transgene was inserted at the same integration site to achieve the same expression level, and the reduced SUMOylation capacity of Ter94KK was confirmed (Fig. 9F). Although the WT Ter94 was able to protect against arsenite-induced toxicity, Ter94KK worsened the arsenite toxicity (Fig. 9G), suggesting that SUMO-deficient Ter94KK has reduced protective capacity. These results suggest that defective SUMOylation may render flies vulnerable to stress insults.
Discussion
In this report, we have described a novel stress response, featuring the SUMOylation of ALS/FTD-associated proteins VCP. Under stress conditions, SUMOylation of VCP promoted VCP translocation to the nucleus and stress granules, increased VCP hexamer assembly, altered co-factor interaction, and facilitated ERAD. In contrast, N-domain pathogenic mutations in VCP caused attenuated SUMOylation of VCP, weakened hexamer assembly, and defective stress response. Therefore, our study highlights SUMOylation as a novel signaling switch to control the function of an essential regulator of misfolded-protein degradation during stress response, and suggests pathogenic mutations interfere with this process and render cells more susceptible to degeneration.
Two recent studies have suggested the emerging roles of VCP in the clearance and assembly of stress granules (44, 45). Besides, the nuclear activities of VCP are crucial in the protection against the neurotoxicity in polyglutamine diseases, or in the degradation of misfolded nuclear proteins during ERAD (46, 47). Mutations in the N-domain also lead to reduced nuclear entry of VCP, although with unclear mechanism (35). Thus, our results suggest that the SUMOylation of VCP could be the upstream signal that promotes the distribution of VCP into the stress granules and nucleus upon stress insults.
Protein SUMOylation is a dynamic process, and it is an efficient means to quickly modulate the activities of the target proteins. Typically, only 1–5% of a SUMO substrate is modified (48). However, this important form of modification is known to change the protein conformation and affect some key properties of the substrates, such as stability and protein interaction pattern (48). For VCP, we have observed reproducible and consistent SUMOylation in the N-domain in cultured cells and in vivo. Moreover, our studies revealed that at least five pathogenic mutations in the N-domain reduced VCP SUMOylation, strongly suggesting defective SUMOylation as an underlying mechanism for abnormal VCP function. Some of the proteomics-based studies have previously identified VCP among thousands of putative SUMO substrates (27, 28, 49, 50). However, there are large discrepancies in the substrates identified among various studies, sometimes even among the repeats performed in the same studies (30). Depending on the sensitivity and methods used, not all the SUMO conjugation sites can be identified for every SUMO substrate. Using mutagenesis, we have attempted to identify the main SUMO conjugation site in VCP. Although we failed to completely abolish the SUMO conjugation, we have identified 3 sites (K60–3, K136, and K164) that can greatly affect VCP SUMOylation. Based on gel patterns, it seems that these SUMO sites may not all be modified at the same time, as most of time we only observed one major SUMO-related band. However, in the presence of high SUMO concentration, poly-SUMOylation can occur. These observations suggest that there might be a few dominant SUMO reception sites; however, when mutated, there are other sites that can be SUMOylated as compensation. This idea is consistent with the study showing that SUMO conjugation could occur at many different lysine residuals, although with different affinity and frequency (28). Thus, although the SUMOylation of VCP is readily detectable, the exact modification appears to be a dynamic process. Regardless, our results have showed the functional significance and disease relevance of the VCP SUMOylation.
Previous studies have failed to detect the effects of pathogenic mutations on VCP hexamer formation or co-factor binding under basal conditions (11, 37, 38, 51, 52). However, our experiments have indicated that increased SUMOylation can promote the VCP hexamer assembly, and highlighted SUMOylation as a signaling switch to efficiently regulate VCP activities during stress response. Meanwhile, SUMO-defective VCP mutants showed increased binding to some co-factors targeting the VCP N-domain. Even though our results strongly suggest that defective SUMOylation causes altered co-factor binding, we could not entirely exclude the possibility that the altered co-factor binding to the VCP mutants may affect SUMOylation. As defective SUMOylation of VCP also lead to reduced hexamer formation during stress, the increased binding to co-factors may reflect increased availability of monomeric VCP. It has been shown that each VCP homohexamer can only bind one NPL4 (53). It is worth noting that although the hexamer assembly and co-factor binding were not dramatically affected by the decreased SUMOylation observed in mutant VCP, the changes were very reproducible. Given the slow progressive nature of neurodegenerative diseases, small deficiency in VCP activities during aging-associated stress will likely accumulate and gradually make a big impact in the long run. It is a common feature that patients carrying pathogenic mutations at birth will not develop neurodegenerative symptoms until adulthood, even for diseases as severe as ALS (54, 55).
Our results also provide new insights into the role of SUMOylation and PTMs in general in neurodegeneration. Genetic predispositions, such as N-domain mutations in VCP, could cause deficiency in PTMs that affect the protein function, especially during cellular stress. The non-genetic factors, such as aging or environmental-related stress, may further amplify the functional deficiency caused by the genetic factors and eventually accelerate the disease progression. As VCP is known to be phosphorylated and acetylated (24), the functions of VCP are likely governed by potential cross-talk mechanisms among various forms of PTMs, especially given VCP itself as a key regulator of the degradation of ubiquitinated proteins. Our current study has provided evidence to demonstrate SUMOylation as an important molecular signal to enhance VCP function during stress response. Given the many functions attributed to VCP in various subcellular compartments, the cross-talk among the PTMs of VCP could be a fast and efficient approach to regulate VCP activities in stress response and aging, and warrants further investigation in the future.
Experimental Procedures
Human Primary Cell Culture
For the analysis of VCP hexamer formation under disease conditions, a 58-year-old male patient with the G97E mutation in VCP (described previously (39)) and age and sex-matched healthy controls were recruited in this study for the culture of primary renal epithelial cells. Informed consent was obtained from all participants, and the study was approved by the ethics committee of Shanghai Jiao Tong University Affiliated People's No. 6 Hospital. Urine samples were collected as the source of the primary epithelial cell culture using the published protocol (56).
Materials
The cDNA for the human VCP was reverse-transcribed and subcloned into the pcDNA3.1 expression vector (Invitrogen) with His6 tag as described (57). Various point mutations or truncation mutations of VCP were generated by PCR mutagenesis. Plasmids for Ubc9, CFP-SUMO1, and SENP1 were previously described (58). The Ubc9 fusion-directed SUMOylation constructs of VCP were generated by fusing the VCP with the WT or C93S Ubc9 as described (32). TCRα-GFP was kindly provided by Dr. Ron R. Kopito (Stanford university) (43). VCP and Ubc9 siRNA were chemically synthesized (GenePharma) with the following sequences: CGGGAGAGGCGCGCGCCAU (VCP) (59); CAAAAAAUCCCGAUGGCAC (Ubc9 SiRNA1) (60); ACCACCAUUAUUUCACCCGAA (Ubc9 SiRNA2) (61). The primary antibodies used in the study were: anti-VCP antibody (MA3-004, Pierce); anti-YB1 antibody (ab76149, Abcam); anti-FLAG tag (DYKDDDDK-Tag) antibody (M20008, Abmart); anti-SUMO1 antibody (33–2400, Invitrogen); anti-His tag antibody (M30111, Abmart); anti-GFP antibody (M20004, Abmart); anti-Lamin A/C antibody (2921, Epitomics); anti-V5 tag antibody (R960–25, Invitrogen); anti-E4B antibody (5550, Epitomics); anti-UFD1 antibody (10615-1-AP, Proteintech); anti-NPL4 antibody (11638-1-AP, Proteintech); anti-p47 antibody (15620-1-AP, Proteintech); and anti-Ubc9 antibody (51018-2-AP, Proteintech).
Cell Culture, Transfection, and Treatment
HeLa and HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum (FBS; Invitrogen) and antibiotics, at 37 °C with 5% CO2. Cells were transfected with plasmids using Lipofectamine 2000 reagent (Invitrogen) and with siRNA by Lipofectamine RNAimax (Invitrogen). Cells were treated with sodium arsenite (S7400, Sigma), MG132 (47490, Merck), paraquat (PS366, Sigma), and tunicamycin (T7765, Sigma) as indicated. Primary cortical neuronal cultures were prepared from neocortical fragments of postnatal day 0 (P0) Sprague-Dawley rat pups as described (62). HeLa cells stably expressing TCRα-GFP were established by transient transfection followed by G418 (800 μg/ml) selection.
Immunoblotting, SUMOylation Analysis, and Affinity Pulldown Experiments
The procedures for Western blotting and immunoprecipitation were described previously (58). For the analysis of SUMOylation of endogenous VCP, one 10-cm plate of HEK293 cells was lysed in denaturing RIPA (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) lysis buffer or 4 m urea buffer (4 m urea, 1% SDS, 10% glycerol, 25 mm Tris-HCl, pH 6.8, 1 mm EDTA, 0.7 m mercaptoethanol) supplemented with protease inhibitor mixture (Roche Applied Science) and 20 mm N-ethylmaleimide (E3876, Sigma). Lysates were incubated with anti-VCP mouse monoclonal antibodies and protein A/G-Sepharose (A10001, Abmart) for 16 h at 4 °C with gentle inversion mixing. The beads were washed four times with lysis buffer, and eluted by 1× SDS-sample buffer followed by SDS-PAGE analysis. For His-tagged VCP affinity pulldown, lysates were incubated with nickel-nitrilotriacetic acid-agarose beads (30230, Qiagen) for 3 h at 4 °C. The amount of lysate input in immunoprecipitation and affinity pulldown experiments was similar in each sample as confirmed by loading control (actin or tubulin) using immunoblots. In addition, equal expression of various VCP constructs was also experimentally confirmed (data not shown). Beads were washed 5 times with washing buffer containing 20 mm imidazole, and eluted with 40 μl of elution buffer containing 250 mm imidazole. Eluted proteins were first probed with anti-SUMO antibody to reveal SUMOylated proteins, or with anti-UFD1, NPL4, p47, or E4B antibody to reveal VCP binding co-factors, and then re-probed with anti-VCP antibody.
Immunofluorescence Microscopy
Z stack images were obtained with an inverted laser scanning confocal microscope (Nikon A1R) with ×60 TIRF oil immersion lens. Fluorescence intensity of VCP was determined using Image-Pro Plus 6.0 software. Nuclear VCP distributions were represented as: (nuclear VCP intensity/total cell VCP intensity) × 100%. The number of stress granules (with a cut off diameter ≥2 μm) in each cell analyzed in Fig. 6E were scored.
Analysis of VCP Hexamer Assembly
The abundance of VCP hexamer was analyzed as described (11, 63, 64) with a minor modification. Cells or tissue samples were first grinded in liquid nitrogen, and then collected in 0.2% Nonidet P-40 lysis buffer (20 mm HEPES, pH 7.2, 0.32 m sucrose, 5 mm MgCl2, 2 mm ATP, 0.2% Nonidet P-40 and protease inhibitors). One part of the supernatants from each sample was mixed with 2× native sample buffer (62.5 mm Tris-HCl, pH 6.8, 40% glycerol, 0.01% bromphenol blue) for the analysis of VCP hexamer assembly by native PAGE (−SDS). To assess the abundance of VCP in the supernatant, an equal amount of the supernatants were analyzed by SDS-PAGE and immunoblotting analysis (+SDS).
For the gel filtration assay, lysates were set to 100 μl and subjected to size exclusion chromatography using Superdex 200 10/300 GL column (GE Healthcare) at a flow rate of 0.4 ml/min. Gel filtration standards (MWGF1000, Sigma) were used as molecular weight markers. The protein containing eluted samples were collected and analyzed by SDS-PAGE and immunoblotting.
Nonidet P-40-soluble/RIPA-soluble Fractionation
To biochemically analyze VCP translocation under stress, cells were first lysed in 0.2% Nonidet P-40 lysis buffer and centrifuged at 10,000 rpm for 10 min at 4 °C. The supernatant was collected as the Nonidet P-40-soluble fraction. The pellet was then lysed using denaturing RIPA buffer as the RIPA-soluble fraction. The purity of the nuclear and cytosolic fractions using this method was comparable with the commercial fractionation kit.
Stress Toxin Treatment in Mice
The treatment procedures were described previously (65). Briefly, wild-type female C57BL/6 mice (purchased from SLAC Laboratory Animal Co.), ages 7 weeks (18–20 g), were treated with sodium arsenite (5 mg/kg body weight) or paraquat (150 mg/Kg) via intraperitoneal injection.
Fly Stocks, Transgenic Fly Generation
Flies were maintained on standard food at 24 °C and 60% humidity in 12-h light/dark cycles. For the generation of the transgenic flies expressing the WT or SUMOylation-deficient Ter94, the V5-tagged Ter94 fragment was subcloned into the pUASTattB vector, with pUAST-Ter94-V5 as a template (a generous gift from Dr. Zhao Yun (4)). PCR mutagenesis was used to alter the lysines at amino acid residues 57 and 59 to arginine to generate the SUMOylation-deficient Ter94 mutant. Finally, the transgenic flies were generated at the Drosophila core facility in Shanghai Institutes for Biological Sciences.
Arsenite Resistance Test in Flies
Male attP-86F8, UAS-Ter94, UAS-Ter94KK were crossed with virgin female C155Elav-Gal4 to obtain the male progeny. 20 Elav/+, Elav/+;Ter94/+, and Elav/+;Ter94KK/+ were placed in each vial, and transferred to vials containing 1 mm sodium arsenite mixed in standard food at the age of 5 days. At least 4 vials were used for each condition. The number of dead flies was scored every 12 h.
Statistics
The band intensity in immunoblots was determined by BIO-RAD Quantity One software. All the data are presented as the mean ± S.E. The statistical significance was analyzed using Student's t test in all the experiments (***, p < 0.001; **, p < 0.01; *, p < 0.05).
Author Contributions
T. W. designed, performed, and analyzed most of the experiments; W. X. designed and performed the experiments related to Drosophila; M. Q., Y. Y., P. B., F. S., and Z. Z. assisted in various experiments and data acquisition; J. X. designed experiments and analyzed data. J. X. and T. W. wrote the manuscript.
Acknowledgments
We thank the donors for the urine collection, and Dr. Kopito and Dr. Yun Zhao for reagents. We also thank Drs. Jianying Xi and Jian Wang at Shanghai Hua Shan Hospital for technical consultation.
This work was supported by the Hundreds of Talents Program, Chinese Academy of Sciences, the State Key Laboratory of Neuroscience, Shanghai Pujiang Talent Program Grant 12PJ1410000, and National Science Foundation of China Grant 81371402 (to J. X.). The authors declare that they have no conflicts of interest with the contents of this article.
- VCP
- valosin-containing protein
- ALS
- amyotrophic lateral sclerosis
- PTM
- post-translational modifications
- SUMO
- small ubiquitin-like modifier
- ER
- endoplasmic reticulum
- ERAD
- endoplasmic reticulum-associated degradation.
References
- 1. Meyer H., Bug M., and Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 2. Meyer H., and Weihl C. C. (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolf D. H., and Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 4. Zhang Z., Lv X., Yin W. C., Zhang X., Feng J., Wu W., Hui C. C., Zhang L., and Zhao Y. (2013) Ter94 ATPase complex targets K11-linked ubiquitinated ci to proteasomes for partial degradation. Dev. Cell 25, 636–644 [DOI] [PubMed] [Google Scholar]
- 5. Chapman E., Fry A. N., and Kang M. (2011) The complexities of p97 function in health and disease. Mol. Biosyst. 7, 700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weihl C. C., Pestronk A., and Kimonis V. E. (2009) Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. 19, 308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valle C. W., Min T., Bodas M., Mazur S., Begum S., Tang D., and Vij N. (2011) Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PloS One 6, e29073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fessart D., Marza E., Taouji S., Delom F., and Chevet E. (2013) P97/CDC-48: proteostasis control in tumor cell biology. Cancer Lett. 337, 26–34 [DOI] [PubMed] [Google Scholar]
- 9. Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., Gibbs J. R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D. G., et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nalbandian A., Donkervoort S., Dec E., Badadani M., Katheria V., Rana P., Nguyen C., Mukherjee J., Caiozzo V., Martin B., Watts G. D., Vesa J., Smith C., and Kimonis V. E. (2011) The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget's disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci. 45, 522–531 [DOI] [PubMed] [Google Scholar]
- 11. Halawani D., LeBlanc A. C., Rouiller I., Michnick S. W., Servant M. J., and Latterich M. (2009) Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol. Cell. Biol. 29, 4484–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manno A., Noguchi M., Fukushi J., Motohashi Y., and Kakizuka A. (2010) Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 15, 911–922 [DOI] [PubMed] [Google Scholar]
- 13. Bartolome F., Wu H. C., Burchell V. S., Preza E., Wray S., Mahoney C. J., Fox N. C., Calvo A., Canosa A., Moglia C., Mandrioli J., Chiò A., Orrell R. W., Houlden H., et al. (2013) Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim N. C., Tresse E., Kolaitis R. M., Molliex A., Thomas R. E., Alami N. H., Wang B., Joshi A., Smith R. B., Ritson G. P., Winborn B. J., Moore J., Lee J. Y., Yao T. P., Pallanck L., Kundu M., and Taylor J. P. (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Austin S., and St-Pierre J. (2012) PGC1alpha and mitochondrial metabolism: emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 125, 4963–4971 [DOI] [PubMed] [Google Scholar]
- 16. Labbadia J., and Morimoto R. I. (2015) The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T., Cho D. H., and Lipton S. A. (2012) Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp. Neurol. 238, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Popovic D., Vucic D., and Dikic I. (2014) Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253 [DOI] [PubMed] [Google Scholar]
- 19. Mathias R. A., Guise A. J., and Cristea I. M. (2015) Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol. Cell. Proteomics 14, 456–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorval V., and Fraser P. E. (2007) SUMO on the road to neurodegeneration. Biochim. Biophys. Acta 1773, 694–706 [DOI] [PubMed] [Google Scholar]
- 21. Landgrave-Gómez J., Mercado-Gómez O., and Guevara-Guzmán R. (2015) Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 9, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 23. Gareau J. R., and Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mori-Konya C., Kato N., Maeda R., Yasuda K., Higashimae N., Noguchi M., Koike M., Kimura Y., Ohizumi H., Hori S., and Kakizuka A. (2009) p97/valosin-containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes Cells 14, 483–497 [DOI] [PubMed] [Google Scholar]
- 25. Nie M., Aslanian A., Prudden J., Heideker J., Vashisht A. A., Wohlschlegel J. A., Yates J. R. 3rd, and Boddy M. N. (2012) Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem. 287, 29610–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bergink S., Ammon T., Kern M., Schermelleh L., Leonhardt H., and Jentsch S. (2013) Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat. Cell. Biol. 15, 526–532 [DOI] [PubMed] [Google Scholar]
- 27. Becker J., Barysch S. V., Karaca S., Dittner C., Hsiao H. H., Berriel Diaz M., Herzig S., Urlaub H., and Melchior F. (2013) Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 20, 525–531 [DOI] [PubMed] [Google Scholar]
- 28. Hendriks I. A., D'Souza R. C., Yang B., Verlaan-de Vries M., Mann M., and Vertegaal A. C. (2014) Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 21, 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuzaki S., Lee L., Knock E., Srikumar T., Sakurai M., Hazrati L. N., Katayama T., Staniszewski A., Raught B., Arancio O., and Fraser P. E. (2015) SUMO1 affects synaptic function, spine density and memory. Sci. Rep. 5, 10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tirard M., Hsiao H. H., Nikolov M., Urlaub H., Melchior F., and Brose N. (2012) In vivo localization and identification of SUMOylated proteins in the brain of His6-HA-SUMO1 knock-in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 21122–21127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hannich J. T., Lewis A., Kroetz M. B., Li S. J., Heide H., Emili A., and Hochstrasser M. (2005) Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J. Biol. Chem. 280, 4102–4110 [DOI] [PubMed] [Google Scholar]
- 32. Jakobs A., Koehnke J., Himstedt F., Funk M., Korn B., Gaestel M., and Niedenthal R. (2007) Ubc9 fusion-directed SUMOylation (UFDS): a method to analyze function of protein SUMOylation. Nat. Methods 4, 245–250 [DOI] [PubMed] [Google Scholar]
- 33. Andersen J. K. (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat. Med. 10, S18–S25 [DOI] [PubMed] [Google Scholar]
- 34. Hetz C., and Mollereau B. (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249 [DOI] [PubMed] [Google Scholar]
- 35. Song C., Wang Q., Song C., Lockett S. J., Colburn N. H., Li C. C., Wang J. M., and Rogers T. J. (2015) Nucleocytoplasmic shuttling of valosin-containing protein (VCP/p97) regulated by its N domain and C-terminal region. Biochim. Biophys. Acta 1853, 222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beuron F., Dreveny I., Yuan X., Pye V. E., McKeown C., Briggs L. C., Cliff M. J., Kaneko Y., Wallis R., Isaacson R. L., Ladbury J. E., Matthews S. J., Kondo H., Zhang X., and Freemont P. S. (2006) Conformational changes in the AAA ATPase p97-p47 adaptor complex. EMBO J. 25, 1967–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang Y. C., Hung W. T., Chang Y. C., Chang H. C., Wu C. L., Chiang A. S., Jackson G. R., and Sang T. K. (2011) Pathogenic VCP/TER94 alleles are dominant actives and contribute to neurodegeneration by altering cellular ATP level in a Drosophila IBMPFD model. PLoS Genet. 7, e1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weihl C. C., Dalal S., Pestronk A., and Hanson P. I. (2006) Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15, 189–199 [DOI] [PubMed] [Google Scholar]
- 39. Gu J. M., Ke Y. H., Yue H., Liu Y. J., Zhang Z., Zhang H., Hu W. W., Wang C., He J. W., Hu Y. Q., Li M., Fu W. Z., and Zhang Z. L. (2013) A novel VCP mutation as the cause of atypical IBMPFD in a Chinese family. Bone 52, 9–16 [DOI] [PubMed] [Google Scholar]
- 40. Buchberger A., Schindelin H., and Hänzelmann P. (2015) Control of p97 function by co-factor binding. FEBS Lett. 589, 2578–2589 [DOI] [PubMed] [Google Scholar]
- 41. Meyer H. H., Shorter J. G., Seemann J., Pappin D., and Warren G. (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19, 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Böhm S., Lamberti G., Fernández-Sáíz V., Stapf C., and Buchberger A. (2011) Cellular functions of Ufd2 and Ufd3 in proteasomal protein degradation depend on Cdc48 binding. Mol. Cell. Biol. 31, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu H., Kaung G., Kobayashi S., and Kopito R. R. (1997) Cytosolic degradation of T-cell receptor α chains by the proteasome. J. Biol. Chem. 272, 20800–20804 [DOI] [PubMed] [Google Scholar]
- 44. Buchan J. R., Kolaitis R. M., Taylor J. P., and Parker R. (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seguin S. J., Morelli F. F., Vinet J., Amore D., De Biasi S., Poletti A., Rubinsztein D. C., and Carra S. (2014) Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 21, 1838–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Foresti O., Rodriguez-Vaello V., Funaya C., and Carvalho P. (2014) Quality control of inner nuclear membrane proteins by the Asi complex. Science 346, 751–755 [DOI] [PubMed] [Google Scholar]
- 47. Fujita K., Nakamura Y., Oka T., Ito H., Tamura T., Tagawa K., Sasabe T., Katsuta A., Motoki K., Shiwaku H., Sone M., Yoshida C., Katsuno M., Eishi Y., Murata M., Taylor J. P., Wanker E. E., Kono K., Tashiro S., Sobue G., La Spada A. R., and Okazawa H. (2013) A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat. Commun. 4, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Geiss-Friedlander R., and Melchior F. (2007) Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 49. Eifler K., and Vertegaal A. C. (2015) Mapping the SUMOylated landscape. FEBS J. 282, 3669–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tammsalu T., Matic I., Jaffray E. G., Ibrahim A. F., Tatham M. H., and Hay R. T. (2014) Proteome-wide identification of SUMO2 modification sites. Sci. Signal. 7, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fernández-Sáiz V., and Buchberger A. (2010) Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep. 11, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hübbers C. U., Clemen C. S., Kesper K., Böddrich A., Hofmann A., Kämäräinen O., Tolksdorf K., Stumpf M., Reichelt J., Roth U., Krause S., Watts G., Kimonis V., Wattjes M. P., Reimann J., et al. (2007) Pathological consequences of VCP mutations on human striated muscle. Brain 130, 381–393 [DOI] [PubMed] [Google Scholar]
- 53. Pye V. E., Beuron F., Keetch C. A., McKeown C., Robinson C. V., Meyer H. H., Zhang X., and Freemont P. S. (2007) Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ling S. C., Polymenidou M., and Cleveland D. W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Renton A. E., Chiò A., and Traynor B. J. (2014) State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou T., Benda C., Dunzinger S., Huang Y., Ho J. C., Yang J., Wang Y., Zhang Y., Zhuang Q., Li Y., Bao X., Tse H. F., Grillari J., Grillari-Voglauer R., Pei D., and Esteban M. A. (2012) Generation of human induced pluripotent stem cells from urine samples. Nat. Protocols 7, 2080–2089 [DOI] [PubMed] [Google Scholar]
- 57. Xu J., Zhong N., Wang H., Elias J. E., Kim C. Y., Woldman I., Pifl C., Gygi S. P., Geula C., and Yankner B. A. (2005) The Parkinson's disease associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum. Mol. Genet. 14, 1231–1241 [DOI] [PubMed] [Google Scholar]
- 58. Zhong N., Kim C. Y., Rizzu P., Geula C., Porter D. R., Pothos E. N., Squitieri F., Heutink P., and Xu J. (2006) DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of PSF. J. Biol. Chem. 281, 20940–20948 [DOI] [PubMed] [Google Scholar]
- 59. Kobayashi T., Manno A., and Kakizuka A. (2007) Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells 12, 889–901 [DOI] [PubMed] [Google Scholar]
- 60. Lin X., Liang M., Liang Y. Y., Brunicardi F. C., and Feng X. H. (2003) SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J. Biol. Chem. 278, 31043–31048 [DOI] [PubMed] [Google Scholar]
- 61. Tong L., Wu Z., Ran M., Chen Y., Yang L., Zhang H., Zhang L., Dong H., and Xiong L. (2015) The role of SUMO-conjugating enzyme Ubc9 in the neuroprotection of isoflurane preconditioning against ischemic neuronal injury. Mol. Neurobiol. 51, 1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peng Y. R., Zeng S. Y., Song H. L., Li M. Y., Yamada M. K., and Yu X. (2010) Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J. Neurosci. 30, 16220–16231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Song C., Wang Q., and Li C. C. (2003) ATPase activity of p97-valosin-containing protein (VCP): D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J. Biol. Chem. 278, 3648–3655 [DOI] [PubMed] [Google Scholar]
- 64. Wang Q., Song C., and Li C. C. (2003) Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem. Biophys. Res. Commun. 300, 253–260 [DOI] [PubMed] [Google Scholar]
- 65. Sahin U., Ferhi O., Jeanne M., Benhenda S., Berthier C., Jollivet F., Niwa-Kawakita M., Faklaris O., Setterblad N., de Thé H., and Lallemand-Breitenbach V. (2014) Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J. Cell Biol. 204, 931–945 [DOI] [PMC free article] [PubMed] [Google Scholar]