

Abstract
The human pancreas expresses two major trypsinogen isoforms, cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2). Mutations in PRSS1 cause hereditary pancreatitis by altering cleavage of regulatory nick sites by chymotrypsin C (CTRC) resulting in reduced trypsinogen degradation and increased autoactivation. Despite 90% identity with PRSS1 and a strong propensity for autoactivation, mutations in PRSS2 are not found in hereditary pancreatitis suggesting that activation of this isoform is more tightly regulated. Here, we demonstrated that CTRC promoted degradation and thereby markedly suppressed autoactivation of human anionic trypsinogen more effectively than previously observed with cationic trypsinogen. Increased sensitivity of anionic trypsinogen to CTRC-mediated degradation was due to an additional cleavage site at Leu-148 in the autolysis loop and the lack of the conserved Cys-139–Cys-206 disulfide bond. Significant stabilization of anionic trypsinogen against degradation was achieved by simultaneous mutations of CTRC cleavage sites Leu-81 and Leu-148, autolytic cleavage site Arg-122, and restoration of the missing disulfide bridge. This stands in stark contrast to cationic trypsinogen where single mutations of either Leu-81 or Arg-122 resulted in almost complete resistance to CTRC-mediated degradation. Finally, processing of the trypsinogen activation peptide at Phe-18 by CTRC inhibited autoactivation of anionic trypsinogen, although cationic trypsinogen was strongly stimulated. Taken together, the observations indicate that human anionic trypsinogen is controlled by CTRC in a manner that individual natural mutations are unlikely to increase stability enough to promote intra-pancreatic activation. This unique biochemical property of anionic trypsinogen explains the lack of association of PRSS2 mutations with hereditary pancreatitis.
Keywords: chymotrypsin, chymotrypsin C, pancreas, serine protease, trypsin, trypsinogen
Introduction
The digestive proenzyme trypsinogen is typically expressed in multiple isoforms in the pancreas. The human pancreas secretes three isoforms as follows: cationic trypsinogen, anionic trypsinogen, and mesotrypsinogen, which are encoded by the PRSS1, PRSS2, and PRSS3 genes, respectively (1). Cationic and anionic trypsinogen constitute more than 95% of total trypsinogen in the pancreatic juice, with a slight preponderance of the cationic isoform (2–4). Interestingly, expression of anionic trypsinogen is increased in inflammatory and malignant diseases of the pancreas and in chronic alcoholism resulting in a reversal of the relative isoform amounts (4–6). Trypsinogen is activated to trypsin in the duodenum by the serine protease enteropeptidase, and active trypsin serves not only as the most abundant digestive protease but also as an activator of other proteolytic zymogens (7). Trypsinogen can undergo autoactivation, a self-amplifying bimolecular reaction in which trypsin activates trypsinogen to yield two trypsin molecules. Autoactivation plays an important role in ectopic activation of trypsinogen inside the pancreas, which can lead to pancreatitis. Hereditary pancreatitis is an autosomal dominant disorder caused by mutations in PRSS1 that result in increased intra-pancreatic trypsinogen activation (8, 9). The mutations alter cleavage of regulatory nick sites by chymotrypsin C (CTRC)2 and trypsin that control trypsinogen degradation and autoactivation (10). The digestive enzyme CTRC cleaves at Leu-81 in the calcium binding loop, which together with an autolytic cleavage at Arg-122 by trypsin results in trypsinogen degradation (Fig. 1A) (10, 11). A secondary CTRC-mediated cleavage at Leu-41 was also observed. Active cationic trypsin is also degraded by this mechanism but at a much slower rate due to unusual thermodynamic stability of the regulatory nick sites (11, 12). CTRC-mediated degradation effectively curbs autoactivation of trypsinogen, resulting in diminished trypsin levels. Importantly, a single mutation of either Leu-81 or Arg-122 is enough to stabilize cationic trypsinogen and allow robust autoactivation even in the presence of CTRC (10).
FIGURE 1.

Regulation of human cationic (A) and anionic (B) trypsinogen isoforms by CTRC. Hereditary pancreatitis-associated PRSS1 mutations that alter activation and/or degradation of cationic trypsinogen are indicated. CTRC cleavage sites are highlighted in red, trypsin cleavage sites in green, the activation peptide in yellow, and the position of the missing disulfide bridge in anionic trypsinogen in blue. See text for further details.
In addition to promoting degradation, CTRC also stimulates autoactivation of cationic trypsinogen by processing the trypsinogen activation peptide at Phe-18 to a shorter form (Fig. 1A) (10, 13). PRSS1 mutations associated with hereditary pancreatitis can decrease or eliminate cleavage at Leu-81 (N29I, N29T, V39A, R122C, and R122H) and Arg-122 (V39A, R122C, and R122H) or increase cleavage of the activation peptide at Phe-18 (A16V and N29I) (Fig. 1A). As a result, cationic trypsinogen mutants autoactivate to high trypsin levels in the presence of CTRC (10).
The discovery of the association of PRSS1 mutations and hereditary pancreatitis stimulated a number of investigations into the potential role of PRSS2 in chronic pancreatitis (14–16). PRSS2 is abundantly expressed; it shares 90% identity with PRSS1 at the amino acid level, and it also readily autoactivates (17–19) suggesting that it might initiate or contribute to pathological intra-pancreatic trypsinogen activation. However, genetic studies failed to identify PRSS2 mutations in hereditary pancreatitis families or in association with sporadic chronic pancreatitis (14–16). Interestingly, a polymorphic variant, G191R, was found with higher frequency in healthy controls (3.4%) versus pancreatitis patients (1.3%), indicating an ∼2.7-fold protective effect, as judged by the odds ratio in the largest cohort studied (14). Biochemical analysis revealed that variant G191R rendered anionic trypsinogen susceptible to autodegradation by introducing a new trypsin-sensitive cleavage site. Although the findings confirmed that anionic trypsinogen does contribute to intra-pancreatic trypsinogen activation, the small protective effect pointed to a limited role. Taken together with the lack of pathogenic PRSS2 mutations, the observations to date led us to hypothesize that anionic trypsinogen may be more tightly regulated by CTRC, possibly preventing natural mutations from causing significantly increased activation of this isoform.
Experimental Procedures
Materials
Recombinant human CTRC was expressed and purified as described previously (10). PageRuler prestained protein ladder was obtained from Thermo Scientific (catalog no. 26617, lot 00340439).
Nomenclature
Amino acid residues in human trypsinogen isoforms were numbered starting with the initiator methionine of the primary translation product, according to the recommendations of the Human Genome Variation Society.
Plasmid Construction and Mutagenesis
The pTrapT7 intein-PRSS1 expression plasmid containing the coding sequence of human cationic trypsinogen N-terminally fused to a self-splicing mini-intein was constructed previously (20). The analogous pTrapT7 intein-PRSS2 expression plasmid was constructed by replacing the EcoRI-SacI fragment in the pTrapT7 intein-PRSS1 plasmid containing the N29I mutation with the PRSS2 coding sequence from the pTrapT7 PRSS2 plasmid (19). Mutations were generated by overlap extension PCR mutagenesis and cloned into the expression plasmids.
Expression and Purification of Recombinant Trypsinogen
Wild-type and mutant intein-trypsinogen fusion constructs were expressed in the aminopeptidase P-deficient LG-3 Escherichia coli strain (20, 21). Upon self-splicing of the mini-intein moiety intracellularly, the liberated trypsinogen contains a homogeneous intact N terminus. In vitro refolding and purification of trypsinogen on immobilized ecotin was performed as described previously (21, 22). Concentrations of trypsinogen preparations were determined from the UV absorbance at 280 nm using extinction coefficients 38,890 m−1 cm−1 (PRSS2) and 37,525 m−1 cm−1 (PRSS1).
Trypsinogen Autoactivation
Wild-type and mutant trypsinogens (1 μm) were incubated at 37 °C in the absence or presence of 10, 25, 50, and 100 nm human CTRC and 10 nm initial trypsin in 0.1 m Tris-HCl (pH 8.0), the indicated concentrations of NaCl (typically 25 mm for anionic trypsinogen and no salt added for cationic trypsinogen), 1 mm CaCl2, and 0.05% Tween 20 (final concentrations). At the indicated times, 10-μl aliquots were withdrawn and mixed with 40 μl of assay buffer containing 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20. Trypsin activity was measured by adding 150 μl of 200 μm N-succinyl-Ala-Ala-Pro-Lys-p-nitroanilide substrate (catalog no. L1725, Bachem, Torrance, CA) and following the release of the yellow p-nitroaniline at 405 nm in a SpectraMax Plus384 microplate reader (Molecular Devices, Sunnyvale, CA) for 1 min. Reaction rates were calculated from fits to the initial linear portions of the curves. The substrate was dissolved in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20. Trypsin activity was expressed as percent of maximal activity, which was determined either by autoactivation in 10 mm CaCl2 or by activation with 28.2 ng/ml human enteropeptidase (catalog no. 1585-SE, R&D Systems, Minneapolis, MN).
Experimental Uncertainty and Reproducibility
Autoactivation experiments were performed in three technical replicates from two independent enzyme preparations. Representative time courses are shown. For clarity, error bars were omitted in most figures. All replicates were submitted for review. SDS-PAGE analyses were carried out in duplicates; both gels were reviewed, but only representative gels are shown.
Results
Autoactivation of Human Anionic Trypsinogen
Autoactivation of human cationic trypsinogen has been extensively studied as the main pathogenic mechanism of trypsin generation in hereditary pancreatitis (10, 23–25). Although prior studies demonstrated that human anionic trypsinogen also undergoes autoactivation (17–19), purity of native trypsinogen preparations was uncertain, and recombinant preparations differed from the native proenzyme in their N termini due to cloning considerations and unwanted processing by bacterial aminopeptidases. Here, we utilized self-splicing mini-intein fusion technology to generate human trypsinogens with homogeneous authentic N-terminal sequences (see “Experimental Procedures”) (20, 21). When autoactivation was measured at pH 8.0 in 1 mm calcium, conditions chosen to approximate the physiological milieu in the pancreatic juice, both human trypsinogens were rapidly converted to active trypsin (Fig. 2, A and C). On SDS-PAGE, the activation was visualized as a characteristic small shift in mobility (Fig. 2, B and D). Autoactivation of anionic trypsinogen proceeded about 2-fold faster than its cationic counterpart; however, the trypsin plateau reached was lower (∼70%), indicating some degradation at this calcium concentration (Fig. 2C). SDS-PAGE analysis and N-terminal sequencing revealed that both trypsinogens were cleaved at the Arg-122–Val-123 peptide bond by trypsin, but anionic trypsinogen was also autolyzed at the Lys-193–Asp-194 peptide bond (Fig. 2, B and D, see also Fig. 1). Although cleavage at Arg-122 does not alter trypsin activity (26–28), cleavage at Lys-193 inactivates the enzyme (29, 30). Increasing NaCl concentrations progressively reduced the rate of autoactivation with no significant difference in salt sensitivity between the two isoforms (Fig. 2, A and C). For subsequent studies, we selected experimental conditions under which rates of autoactivation of cationic and anionic trypsinogen were comparable, and the entire time course could be measured within an hour. Therefore, cationic trypsinogen was assayed in the absence of added salt, whereas anionic trypsinogen was tested in the presence of 25 mm NaCl.
FIGURE 2.
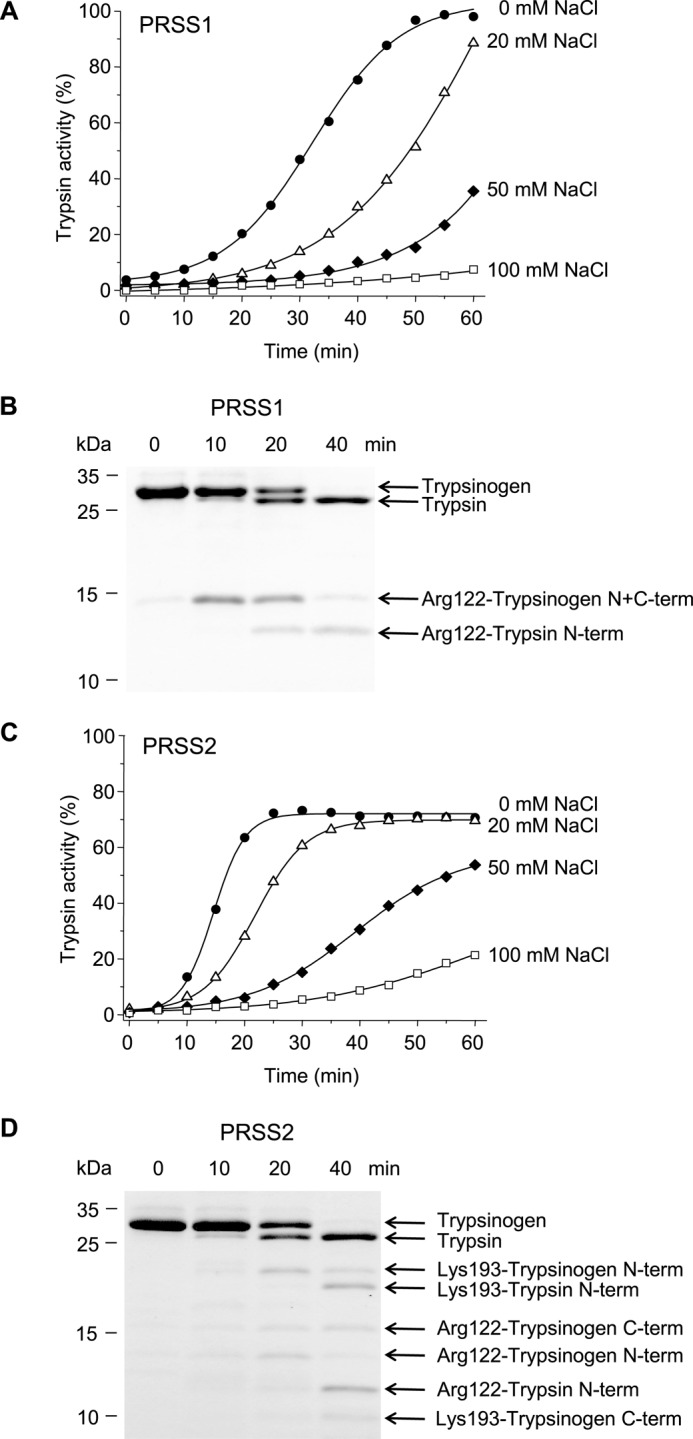
Autoactivation of human cationic (PRSS1, A and B) and anionic trypsinogen (PRSS2, C and D). Trypsinogen (1 μm) was incubated at 37 °C with 10 nm initial trypsin in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, 0–100 mm NaCl, and 0.05% Tween 20 (final concentrations). A and C, at the indicated times aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” B and D, trypsinogen (150 μl) was precipitated with 10% trichloroacetic acid (final concentration) and analyzed by reducing SDS-PAGE and Coomassie Blue staining. Gels shown correspond to activity curves measured in the absence of NaCl for PRSS1 and in 20 mm NaCl for PRSS2. Autolytic bands resulting from cleavage of the Arg-122–Val-123 and Lys-193–Asp-194 peptide bonds are indicated. N-term, N-terminal fragment; C-term, C-terminal fragment. Cleavage sites were determined by N-terminal sequencing after transferring the bands to PVDF membranes. See “Experimental Procedures” for statement on experimental uncertainty and reproducibility.
CTRC Reduces Activation of Human Cationic Trypsinogen
Previously, we presented data demonstrating that autoactivation of human cationic trypsinogen was suppressed by human CTRC (10). To provide a framework for this study, we repeated these experiments using a somewhat higher concentration range (10–100 nm) of CTRC (Fig. 3). As expected, at pH 8.0, in 1 mm calcium, CTRC slightly increased the initial rate of autoactivation and markedly reduced final trypsin levels, although complete suppression was not achieved even at 100 nm CTRC concentration (Fig. 3A). Notably, degradation of active trypsin was negligible during the time course studied. Individual mutations of either the CTRC cleavage site Leu-81 (L81A) or the trypsin-sensitive autolytic site Arg-122 (R122A) resulted in almost complete stabilization against degradation and the development of high trypsin levels (Fig. 3, B and C). In the absence of degradation, the stimulatory effect of CTRC on the initial rate of autoactivation was more apparent in mutants L81A and R122A.
FIGURE 3.
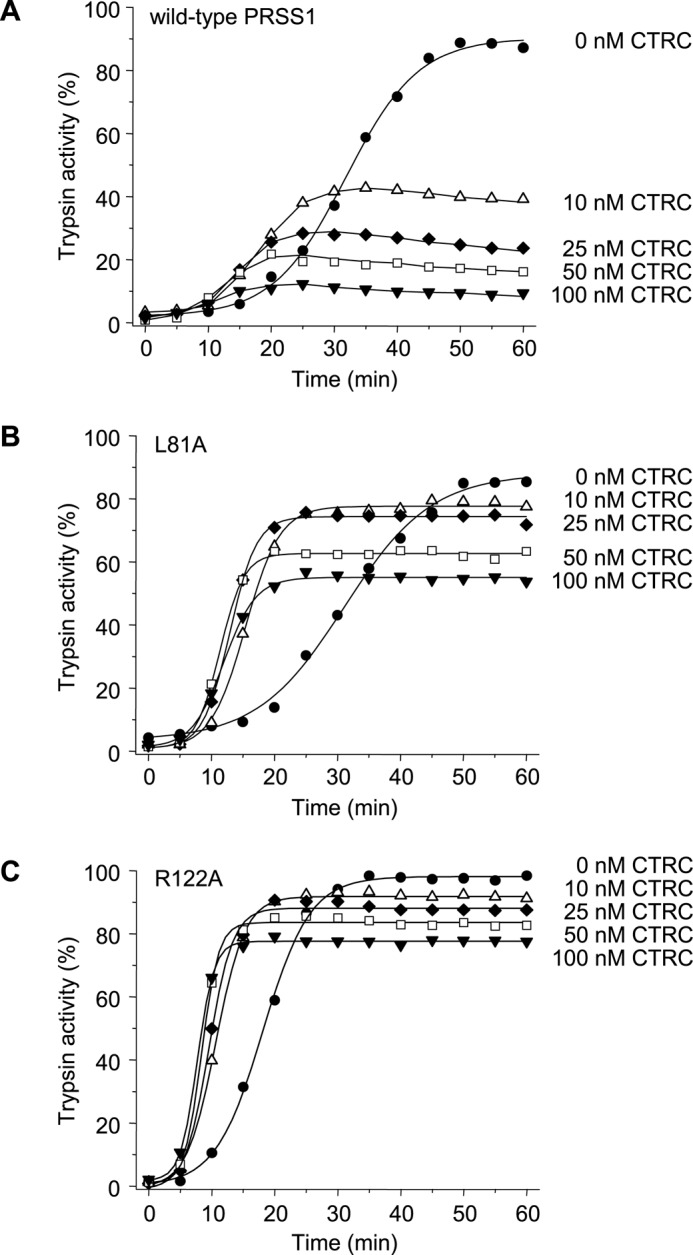
Effect of CTRC on the autoactivation of human cationic trypsinogen (PRSS1). Wild-type (A), L81A (B), and R122A (C) mutant cationic trypsinogen (1 μm) was incubated at 37 °C with 0–100 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20 (final concentrations). At the indicated times aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” Experimental uncertainty and reproducibility are given under “Experimental Procedures.”
CTRC Abolishes Activation of Human Anionic Trypsinogen
When measured under similar conditions, autoactivation of anionic trypsinogen was more strikingly diminished by CTRC, and essentially no trypsin activity was detectable at 50 nm CTRC concentration (Fig. 4A). Mutation of Leu-81 (L81A) or Arg-122 (R122A) had no protective effect whatsoever; in fact, mutant L81A was even more sensitive to CTRC-mediated degradation (Fig. 4, B and C). In contrast to cationic trypsinogen, CTRC did not seem to increase the initial rate of autoactivation of anionic trypsinogen.
FIGURE 4.
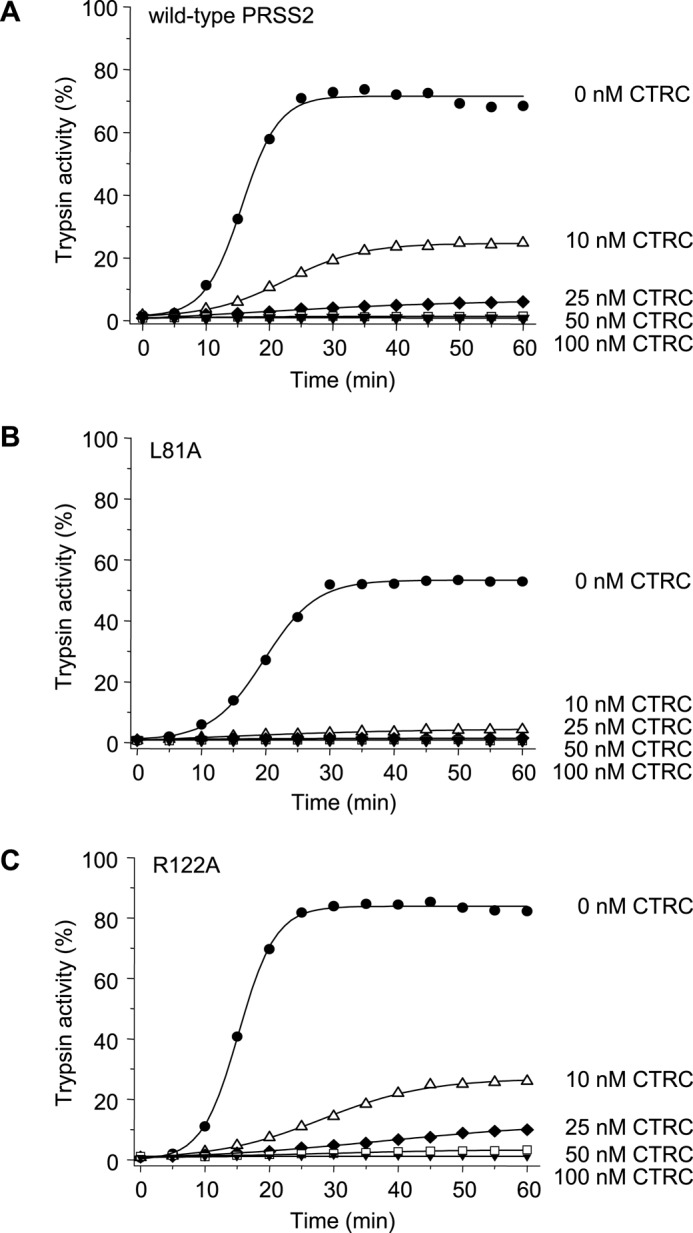
Effect of CTRC on the autoactivation of human anionic trypsinogen (PRSS2). Wild-type (A), L81A (B), and R122A (C) mutant anionic trypsinogen (1 μm) was incubated at 37 °C with 0–100 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, 25 mm NaCl, and 0.05% Tween 20 (final concentrations). At the indicated times, aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” Experimental uncertainty and reproducibility are discussed under “Experimental Procedures.”
CTRC Cleaves at Leu-148 in the Autolysis Loop of Anionic Trypsinogen
SDS-PAGE analysis of anionic trypsinogen treated with 10 nm CTRC for 10 min at pH 8.0 in the absence of calcium revealed that the earliest cleavage generated two bands that migrated in the vicinity of the 15-kDa molecular mass marker (Fig. 5A). The same bands were prominent when the cleavage pattern of mutant L81A was analyzed, indicating that CTRC rapidly cleaved human anionic trypsinogen at a site other than Leu-81 (Fig. 5B). The bands were transferred to PVDF membrane and subjected to Edman degradation, which identified the cleaved peptide bond as Leu-148–Ser-149 located in the so-called autolysis loop (Fig. 5C). Position 148 is Ala in cationic trypsinogen, which explains why this cleavage was not observed there. Mutation of Leu-148 (L148A) in anionic trypsinogen resulted in significant but incomplete stabilization against CTRC-mediated degradation (Fig. 6A, cf. Fig. 4A). Stability of anionic trypsinogen was progressively further increased by the addition of mutations L81A (double mutant L81A,L148A, Fig. 6B) and R122A (triple mutant L81A, R122A,L148A, Fig. 6C); however, degradation by CTRC still proceeded more rapidly than observed with cationic trypsinogen single mutants L81A or R122A (cf. Fig. 3, B and C).
FIGURE 5.
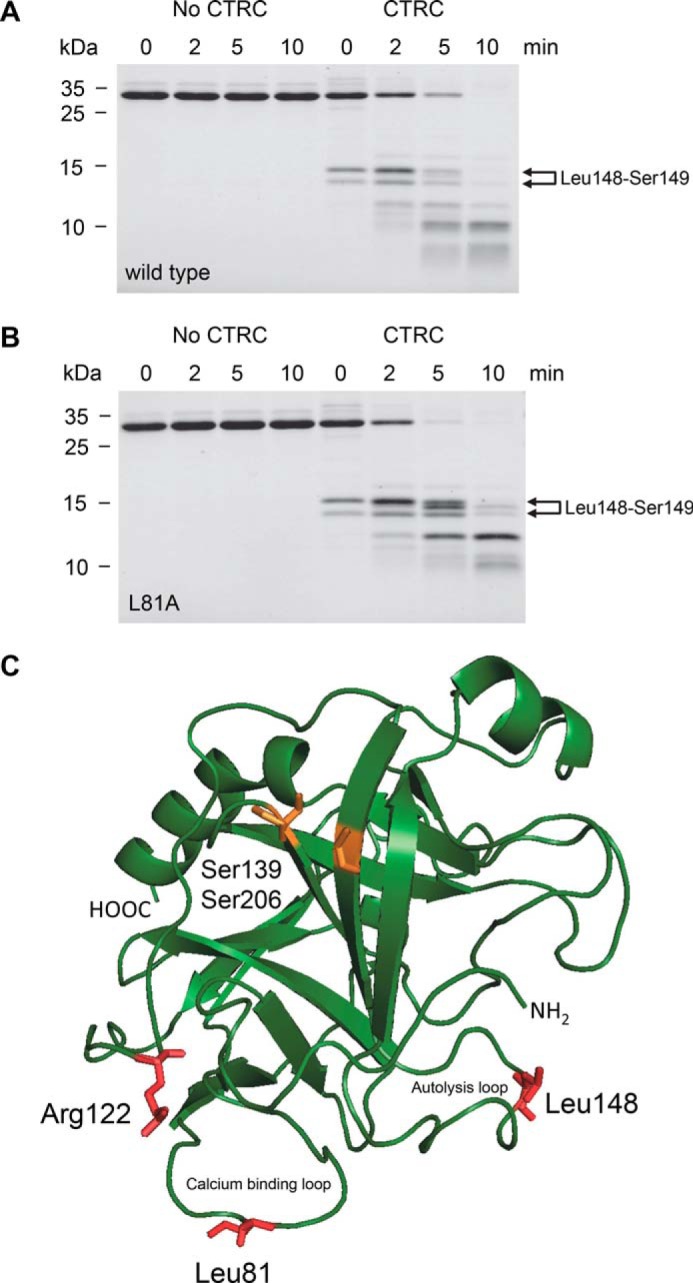
Cleavage of human anionic trypsinogen by CTRC. Wild-type (A) and L81A mutant (B) anionic trypsinogen (1 μm) was incubated at 37 °C with 10 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 25 mm NaCl, and 20 nm human SPINK1 trypsin inhibitor (final concentrations). Trypsin inhibitor was included to prevent autoactivation during the cleavage reaction. At the indicated times, trypsinogen (150 μl) was precipitated with 10% trichloroacetic acid (final concentration) and analyzed by reducing SDS-PAGE and Coomassie Blue staining. The two bands generated by cleavage of the Leu-148–Ser-149 peptide bond are indicated. Note that the N-terminal upper band slightly shifts over time as the activation peptide is processed at Phe-18 by CTRC. See text for details. See “Experimental Procedures” for statement on experimental uncertainty and reproducibility. C, model of human anionic trypsinogen with regulatory cleavage sites indicated in red. The position of the missing disulfide replaced by Ser-139 and Ser-206 is highlighted in orange. Bovine cationic trypsinogen (Protein Data Bank file 1TGN (34)) was used as template to generate a model for anionic trypsinogen using the SWISS-MODEL structure homology-modeling server (35). The image was rendered by PyMOL 3.1.
FIGURE 6.
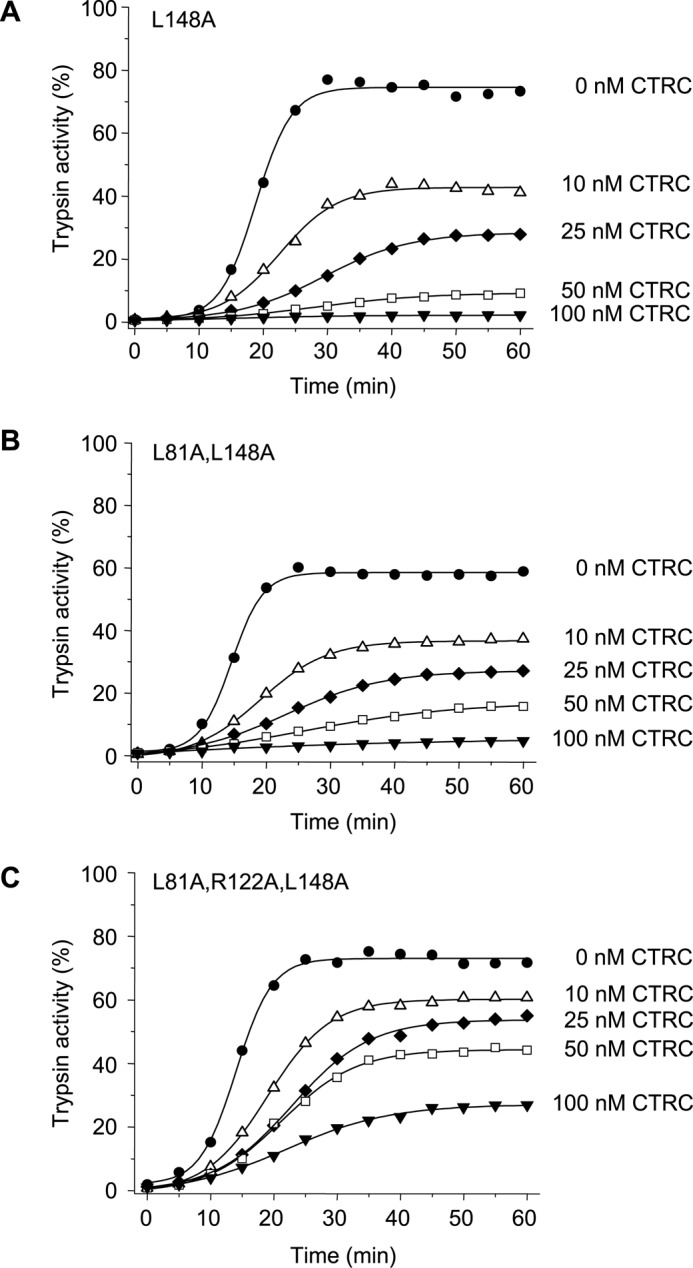
Effect of mutation L148A on the autoactivation of human anionic trypsinogen in the presence of CTRC. Mutant L148A (A), double mutant L81A,L148A (B), and triple mutant L81A,R122A,L148A (C) anionic trypsinogen (1 μm) were incubated at 37 °C with 0–100 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, 25 mm NaCl, and 0.05% Tween 20 (final concentrations). At the indicated times, aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” Experimental uncertainty and reproducibility are given under “Experimental Procedures.”
Absence of a Conserved Disulfide Bridge Contributes to Proteolytic Instability of Human Anionic Trypsinogen
Despite mutations of the primary CTRC cleavage sites Leu-81 and Leu-148 and the autolytic site Arg-122, anionic trypsinogen was still prone to CTRC-mediated degradation, suggesting that additional sites are susceptible to attack. N-terminal sequencing of the smaller bands seen in Fig. 5A identified a number of secondary CTRC cleavage sites, i.e. peptide bonds Tyr-37–Gln-38, Leu-41–Asn-42, Leu-166–Ser-167, and Leu-189–Glu-190 (see Fig. 1B). Although present, with the exception of Leu-41, none of these sites are cleaved in cationic trypsinogen suggesting that increased flexibility of anionic trypsinogen might be responsible for the exposure of potential CTRC-sensitive cleavage sites. Anionic trypsinogen is 90% identical to cationic trypsinogen in its primary amino acid sequence, but a notable difference is the absence of the disulfide bridge Cys-139–Cys-206 (Figs. 1A and 5C). Both Cys residues contributing to this bond in cationic trypsinogen are replaced with Ser in anionic trypsinogen. Overall, human anionic trypsinogen contains only four disulfide bridges, which is unique among vertebrate trypsinogens, the large majority of which harbor six. Restoration of the missing disulfide by mutations S139C and S206C resulted in measurable stabilization against CTRC (Fig. 7A). Remarkably, essentially complete stabilization was achieved with combination of the restored disulfide bridge and elimination of the Leu-81, Arg-122, and Leu-148 cleavage sites (quintuple mutant L81A,R122A,S139C,L148A,S206C) (Fig. 7B). SDS-PAGE analysis revealed no detectable cleavage fragments when the quintuple mutant was treated with 10 nm CTRC for 10 min (Fig. 7C, cf. Fig. 5A).
FIGURE 7.
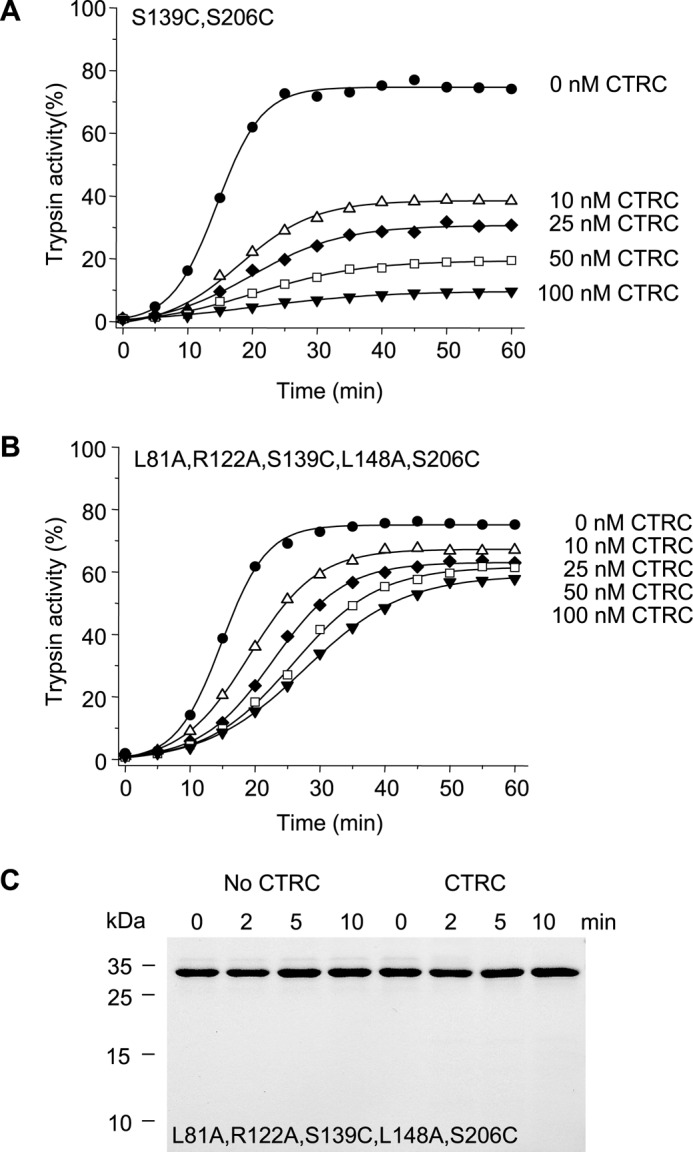
Effect of restoration of the Cys-139–Cys-206 disulfide bond on the autoactivation of human anionic trypsinogen in the presence of CTRC. Double mutant S139C,S206C (A) and quintuple mutant L81A,R122A, S139C,L148A,S206C (B) anionic trypsinogen (1 μm) were incubated at 37 °C with 0–100 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, 25 mm NaCl, and 0.05% Tween 20 (final concentrations). At the indicated times aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” C, cleavage of the quintuple anionic trypsinogen mutant L81A,R122A,S139C,L148A,S206C by CTRC. Trypsinogen (1 μm) was incubated at 37 °C with 10 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 25 mm NaCl, and 20 nm human SPINK1 trypsin inhibitor (final concentrations). At the indicated times, trypsinogen (150 μl) was precipitated with 10% trichloroacetic acid (final concentration) and analyzed by reducing SDS-PAGE and Coomassie Blue staining. See “Experimental Procedures” for statement on experimental uncertainty and reproducibility.
N-terminal Processing of Anionic Trypsinogen Slows Autoactivation
Cleavage of the activation peptide close to its N terminus at Phe-18 by CTRC accelerates autoactivation of human cationic trypsinogen (10, 13). Mutation A16V increases N-terminal processing by CTRC and causes hereditary or sporadic chronic pancreatitis (see Fig. 1A) (10, 13). Selective study of CTRC-mediated cleavage at Phe-18 in wild-type anionic trypsinogen was hampered by the multitude of other cleavages; therefore, we used the stable quintuple mutant to estimate cleavage rates. Anionic trypsinogen was processed at Phe-18 by CTRC 5-fold faster than cationic trypsinogen (Fig. 8A). Interestingly, however, we did not observe acceleration of autoactivation in the presence of CTRC in any of the experiments involving anionic trypsinogen (see Figs. 4, 6, and 7), not even with the quintuple mutant where degradation is unlikely to mask a stimulatory effect. To allow for a better comparison of how CTRC-mediated cleavage at Phe-18 affects anionic and cationic trypsinogen, we generated constructs with shortened activation peptides, starting with Asp-19 instead of Ala-16 (Ala-Pro-Phe deleted, APF-del). When autoactivation of N-terminally truncated (APF-del) cationic and anionic trypsinogen was compared, the two isoforms exhibited contrasting effects; activation of cationic trypsinogen was markedly increased and that of anionic trypsinogen was reduced, relative to wild type (Fig. 8B).
FIGURE 8.
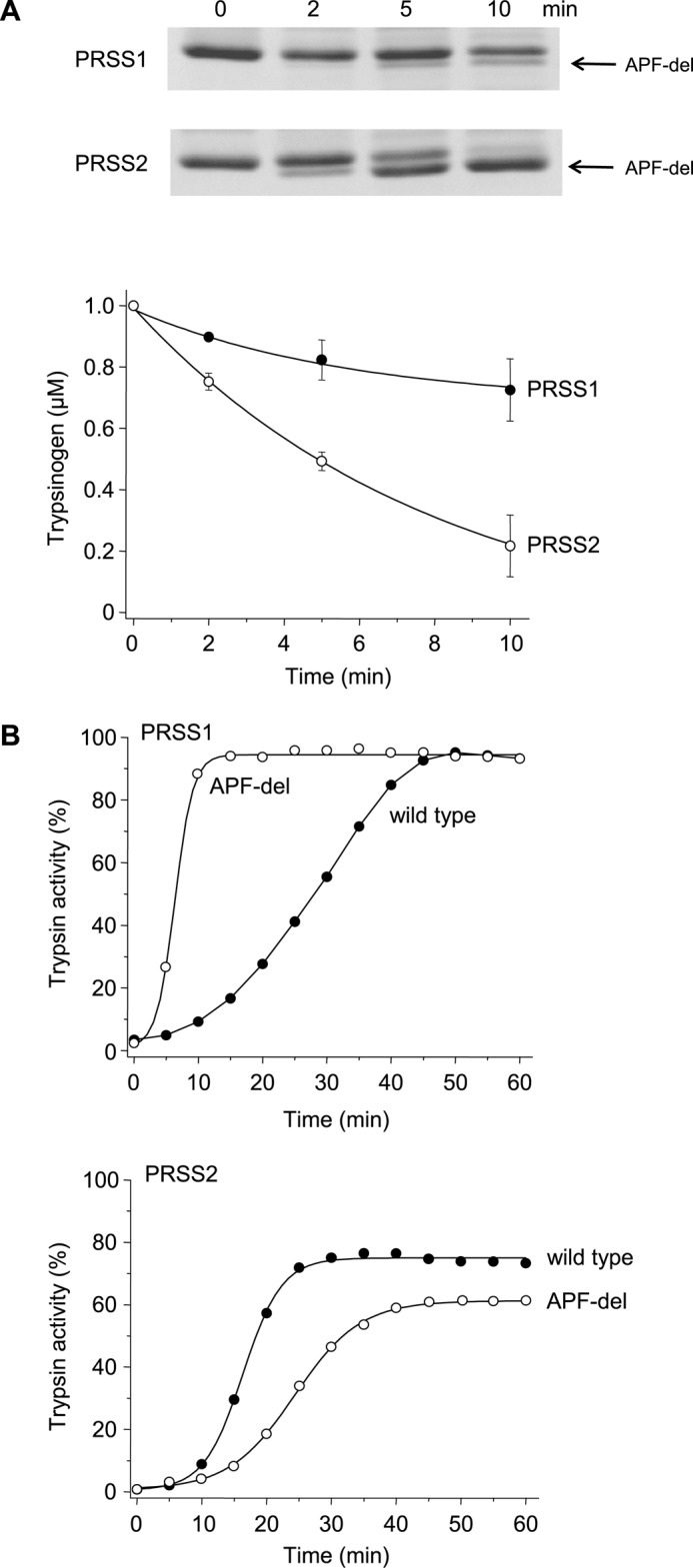
Cleavage of the trypsinogen activation peptide by CTRC and its effect on autoactivation of human cationic (PRSS1) and anionic (PRSS2) trypsinogen. A, wild-type human cationic and anionic trypsinogen (1 μm) were incubated at 37 °C with 25 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 20 nm human SPINK1 trypsin inhibitor (final concentrations). To remain consistent with the autoactivation experiments, reactions with anionic trypsinogen also contained 25 mm NaCl. At the indicated times trypsinogen (150 μl) was precipitated with 10% trichloroacetic acid (final concentration) and analyzed by SDS-PAGE under non-reducing conditions followed by Coomassie Blue staining. APF-del indicates the band cleaved at Phe-18 by CTRC. The graph shows densitometric quantitation of the trypsinogen band from three experiments (mean ± S.E.). The calculated rates of cleavage were 30.9 nm/min for PRSS1 and 152.8 nm/min for PRSS2. B, wild-type and N-terminally truncated (APF-del) mutant trypsinogen (1 μm) were incubated at 37 °C in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 25 mm NaCl (PRSS2) or no salt added (PRSS1). At the indicated times aliquots (10 μl) were removed, and trypsin activity was determined and expressed as described under “Experimental Procedures.” Experimental uncertainty and reproducibility are discussed under “Experimental Procedures.”
Discussion
The causative role of cationic trypsinogen mutations in hereditary pancreatitis was discovered in 1996 (8); however, a mechanistic understanding of how these mutations trigger the disease was not achieved until 2012 (10). Although increased trypsinogen activation as an intuitive model was suggested early on, biochemical studies demonstrated only a slightly increased propensity for autoactivation of mutant trypsinogens, which seemed insufficient to explain pathogenesis (23, 24). A breakthrough in our research came with the realization that a lesser known digestive enzyme, CTRC, controls activation of human cationic trypsinogen through proteolytic cleavage of regulatory sites (see Fig. 1A) (10, 11). The most common hereditary pancreatitis-associated mutations alter these cleavages resulting in more robust activation and higher trypsin levels (10). Even in the absence of trypsinogen mutations, the protective effect of CTRC against pancreatitis can be compromised by loss-of-function mutations in CTRC, which increase risk for sporadic chronic pancreatitis (31). As our mechanistic insight into how PRSS1 mutations cause hereditary pancreatitis advanced, the nagging question why PRSS2 mutations are not associated with the disease has remained unanswered. Anionic trypsinogen is expressed to high levels and is capable of rapid autoactivation (17–19), suggesting that the lack of a pathogenic role may be due to stronger control by protective mechanisms in the pancreas. The findings of this study confirm this notion as they demonstrated that CTRC promotes degradation of anionic trypsinogen more effectively than it does with cationic trypsinogen. Furthermore, in stark contrast with cationic trypsinogen, CTRC does not stimulate autoactivation of anionic trypsinogen through processing of the activation peptide; in fact, a slight inhibitory effect was observed.
We found that robust degradation of anionic trypsinogen by CTRC was explained by the presence of the additional Leu-148 cleavage site in the autolysis loop and the absence of the Cys-139–Cys-206 disulfide bridge, which seems to increase flexibility and exposure of secondary CTRC cleavage sites such as Tyr-37, Leu-41, Leu-166, and Leu-189 (see Fig. 1B). Complete stabilization of anionic trypsinogen against CTRC-mediated degradation during autoactivation required elimination of the Leu-81, Leu-148, and Arg-122 cleavage sites and restoration of the missing disulfide bridge. This stood in dramatic contrast to cationic trypsinogen where single mutations of Leu-81 or Arg-122 afforded essentially complete protection (see Fig. 3) (10). Although data are not shown, introduction of the Leu-148 cleavage site into cationic trypsinogen (A148L mutation) resulted in efficient degradation by CTRC even when Leu-81 was mutated. Furthermore, elimination of the Cys-139–Cys-206 disulfide bond from cationic trypsinogen (C139S,C206S double mutation) caused significant autolytic degradation indicating markedly increased proteolytic instability (data not shown). Taken together, the observations demonstrate that because of specific evolutionary differences between the otherwise highly similar human trypsinogen isoforms, anionic trypsinogen is more susceptible to CTRC-dependent degradation. Consequently, natural mutations are less likely to induce significant stabilization that would increase intra-pancreatic activation to pathogenic levels as seen with cationic trypsinogen mutations in hereditary pancreatitis.
A seemingly paradoxic effect of CTRC on cationic trypsinogen is the stimulation of autoactivation by cleaving the activation peptide at Phe-18 (10, 13). Relative to trypsinogen degradation, this stimulatory effect is almost negligible in 1 mm calcium (see Fig. 3A). However, PRSS1 mutations A16V and N29I stimulate cleavage at Phe-18 by 5.8- and 2.6-fold, respectively, leading to increased autoactivation and chronic pancreatitis (10). The activation peptide sequence of anionic trypsinogen is identical to that of the cationic isoform, and as expected, anionic trypsinogen was also processed by CTRC at Phe-18. The rate of cleavage was 5-fold increased relative to cationic trypsinogen, and this is partly explained by the presence of Ile-29 (versus Asn-29) in anionic trypsinogen, which mimics the effect of the N29I mutation in cationic trypsinogen. Importantly, however, the N-terminal processing of anionic trypsinogen resulted in slight inhibition of autoactivation, indicating that natural mutations that might alter cleavage at Phe-18 in this isoform would not have a pathogenic effect. The opposing effects of CTRC on autoactivation of the two human trypsinogen isoforms are the consequence of Asp-218 in cationic trypsinogen, which is replaced with Tyr in anionic trypsinogen (25). Asp-218 forms part of the S3 subsite and interacts with Asp-21 of the activation peptide resulting in electrostatic repulsion and inhibition of autoactivation (see Fig. 5 in Ref. 25). This inhibitory interaction is partly relieved by CTRC-mediated cleavage of the activation peptide likely due to increased flexibility. Introduction of Asp-218 into anionic trypsinogen reconstitutes the CTRC-mediated stimulation of autoactivation, whereas a Tyr-218 replacement in cationic trypsinogen abolishes the effect (25).
Although PRSS2 mutations may not be causative in hereditary pancreatitis, anionic trypsinogen still contributes to intra-pancreatic trypsinogen activation and pancreatitis risk, as demonstrated by the small protective effect of the self-degrading G191R variant (14–16). As we observed in this study, single mutations may cause limited stabilization of anionic trypsinogen against CTRC, which can result in slightly elevated autoactivation. Such a biochemical change can, in turn, translate to a small but measurable increase in pancreatitis risk. Therefore, it is possible that pancreatitis-associated variants in PRSS2 will be discovered in the future; however, none of these are expected to confer the high penetrance disease risk observed with PRSS1 mutations. Despite this theoretical possibility, genetic investigations to date failed to identify such PRSS2 variants (14–16).
A possible caveat to this study is that native human trypsinogens are sulfated on Tyr-154 (32). Previously, we compared properties of sulfated and non-sulfated anionic trypsinogen and found that sulfation slightly inhibited autoactivation and CTRC-mediated degradation (33). Although these small effects of sulfation may change the quantitative aspects of some of our results, the main conclusions would remain the same.
In summary, we found that intra-pancreatic activation of human anionic trypsinogen is prevented through CTRC-mediated degradation much more effectively than previously demonstrated for cationic trypsinogen. The observations offer a plausible explanation for the conspicuous absence of anionic trypsinogen mutations in hereditary pancreatitis.
Author Contributions
Z. J. performed experiments, analyzed data, prepared figures for publication, and approved the final version of the manuscript. M. S. T. conceived and designed the project, analyzed the data, and wrote the manuscript.
Acknowledgment
András Szabó is gratefully acknowledged for helping with the initiation of the project.
This work was supported by National Institutes of Health Grants R01 DK058088, R01 DK082412, and R01 DK095753. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
- CTRC
- chymotrypsin C.
References
- 1. Chen J.-M., and Ferec C. (2000) Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas 21, 57–62 [DOI] [PubMed] [Google Scholar]
- 2. Guy O., Lombardo D., Bartelt D. C., Amic J., and Figarella C. (1978) Two human trypsinogens. Purification, molecular properties, and N-terminal sequences. Biochemistry 17, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 3. Scheele G., Bartelt D., and Bieger W. (1981) Characterization of human exocrine pancreatic proteins by two-dimensional isoelectric focusing/sodium dodecyl sulfate gel electrophoresis. Gastroenterology 80, 461–473 [PubMed] [Google Scholar]
- 4. Rinderknecht H., Renner I. G., and Carmack C. (1979) Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics and patients with pancreatitis and cancer of the pancreas. Gut 20, 886–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rinderknecht H., Stace N. H., and Renner I. G. (1985) Effects of chronic alcohol abuse on exocrine pancreatic secretion in man. Dig. Dis. Sci. 30, 65–71 [DOI] [PubMed] [Google Scholar]
- 6. Borgström A., and Andrén-Sandberg A. (1995) Elevated serum levels of immunoreactive anionic trypsin (but not cationic trypsin) signals pancreatic disease. Int. J. Pancreatol 18, 221–225 [DOI] [PubMed] [Google Scholar]
- 7. Rinderknecht H. (1986) Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig. Dis. Sci. 31, 314–321 [DOI] [PubMed] [Google Scholar]
- 8. Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K. Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., and Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 [DOI] [PubMed] [Google Scholar]
- 9. Németh B. C., and Sahin-Tóth M. (2014) Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G466–G473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szabó A., and Sahin-Tóth M. (2012) Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 287, 20701–20710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szmola R., and Sahin-Tóth M. (2007) Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szabó A., Radisky E. S., and Sahin-Tóth M. (2014) Zymogen activation confers thermodynamic stability on a key peptide bond and protects human cationic trypsin from degradation. J. Biol. Chem. 289, 4753–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nemoda Z., and Sahin-Tóth M. (2006) Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 281, 11879–11886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witt H., Sahin-Tóth M., Landt O., Chen J. M., Kähne T., Drenth J. P., Kukor Z., Szepessy E., Halangk W., Dahm S., Rohde K., Schulz H. U., Le Maréchal C., Akar N., Ammann R. W., et al. (2006) A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat. Genet. 38, 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santhosh S., Witt H., te Morsche R. H., Nemoda Z., Molnár T., Pap A., Jansen J. B., and Drenth J. P. (2008) A loss of function polymorphism (G191R) of anionic trypsinogen (PRSS2) confers protection against chronic pancreatitis. Pancreas 36, 317–320 [DOI] [PubMed] [Google Scholar]
- 16. Kume K., Masamune A., Takagi Y., Kikuta K., Watanabe T., Satoh K., Satoh A., Hirota M., Hamada S., and Shimosegawa T. (2009) A loss-of-function p.G191R variant in the anionic trypsinogen (PRSS2) gene in Japanese patients with pancreatic disorders. Gut 58, 820–824 [DOI] [PubMed] [Google Scholar]
- 17. Colomb E., and Figarella C. (1979) Comparative studies on the mechanism of activation of the two human trypsinogens. Biochim. Biophys. Acta 571, 343–351 [DOI] [PubMed] [Google Scholar]
- 18. Colomb E., Figarella C., and Guy O. (1979) The two human trypsinogens. Evidence of complex formation with basic pancreatic trypsin inhibitor-proteolytic activity. Biochim. Biophys. Acta 570, 397–405 [DOI] [PubMed] [Google Scholar]
- 19. Kukor Z., Tóth M., and Sahin-Tóth M. (2003) Human anionic trypsinogen. Properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur. J. Biochem. 270, 2047–2058 [DOI] [PubMed] [Google Scholar]
- 20. Király O., Guan L., Szepessy E., Tóth M., Kukor Z., and Sahin-Tóth M. (2006) Expression of human cationic trypsinogen with an authentic N terminus using intein-mediated splicing in aminopeptidase P deficient Escherichia coli. Protein Expr. Purif. 48, 104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Király O., Guan L., and Sahin-Tóth M. (2011) Expression of recombinant proteins with uniform N termini. Methods Mol. Biol. 705, 175–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lengyel Z., Pál G., and Sahin-Tóth M. (1998) Affinity purification of recombinant trypsinogen using immobilized ecotin. Protein Expr. Purif. 12, 291–294 [DOI] [PubMed] [Google Scholar]
- 23. Sahin-Tóth M. (2000) Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J. Biol. Chem. 275, 22750–22755 [DOI] [PubMed] [Google Scholar]
- 24. Sahin-Tóth M., and Tóth M. (2000) Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem. Biophys. Res. Commun. 278, 286–289 [DOI] [PubMed] [Google Scholar]
- 25. Nemoda Z., and Sahin-Tóth M. (2005) The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. J. Biol. Chem. 280, 29645–29652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maroux S., and Desnuelle P. (1969) On some autolyzed derivatives of bovine trypsin. Biochim. Biophys. Acta 181, 59–72 [DOI] [PubMed] [Google Scholar]
- 27. Ru B. G., Du J. Z., Zeng Y. H., Chen L. S., Ni Y. S., Tan G. H., and Zhang L. X. (1980) Active products of porcine trypsin after autolysis. Sci. Sin. 23, 1453–1460 [PubMed] [Google Scholar]
- 28. Kukor Z., Tóth M., Pál G., and Sahin-Tóth M. (2002) Human cationic trypsinogen. Arg117 is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J. Biol. Chem. 277, 6111–6117 [DOI] [PubMed] [Google Scholar]
- 29. Smith R. L., and Shaw E. (1969) Pseudotrypsin. A modified bovine trypsin produced by limited autodigestion. J. Biol. Chem. 244, 4704–4712 [PubMed] [Google Scholar]
- 30. Sahin-Tóth M. (1999) Hereditary pancreatitis-associated mutation Asn21→Ile stabilizes rat trypsinogen in vitro. J. Biol. Chem. 274, 29699–29704 [DOI] [PubMed] [Google Scholar]
- 31. Rosendahl J., Witt H., Szmola R., Bhatia E., Ozsvári B., Landt O., Schulz H. U., Gress T. M., Pfützer R., Löhr M., Kovacs P., Blüher M., Stumvoll M., Choudhuri G., Hegyi P., et al. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sahin-Tóth M., Kukor Z., and Nemoda Z. (2006) Human cationic trypsinogen is sulfated on Tyr-154. FEBS J. 273, 5044–5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rónai Z., Witt H., Rickards O., Destro-Bisol G., Bradbury A. R., and Sahin-Tóth M. (2009) A common African polymorphism abolishes tyrosine sulfation of human anionic trypsinogen (PRSS2). Biochem. J. 418, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kossiakoff A. A., Chambers J. L., Kay L. M., and Stroud R. M. (1977) Structure of bovine trypsinogen at 1.9 Å resolution. Biochemistry 16, 654–664 [DOI] [PubMed] [Google Scholar]
- 35. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]