

Abstract
Natural killer (NK) cells induce apoptosis in infected and transformed cells and are important producers of immunoregulatory cytokines. Therefore, they operate under low oxygen conditions (hypoxia) in inflammatory and tumor environments. In vitro studies of NK cells are, however, commonly performed in ambient air (normoxia). We used global gene expression profiling to evaluate changes in transcriptional pathways in primary human NK cells following short term culture under hypoxia compared with normoxia and in response to interleukin 15 (IL-15) priming using a 2 × 2 factorial design. The largest contrasts observed were priming dependences for associations between hypoxia and the hypoxia-inducible factor (Hif) 1 signaling and glycolysis pathways. RT-PCR confirmed positive synergistic hypoxia/IL-15 interactions for genes of key regulatory and metabolic enzymes. IL-15 primes NK cells for effector functions, which were recently demonstrated to depend on glycolytic switching. We did not, however, observe important increases in glycolytic flux through hypoxia and priming alone. Chemical Hif-1α inhibition suggested equal importance of this transcription factor for glycolysis and energy production under normoxia and hypoxia. Hypoxia promoted secretion of CC chemokines Ccl3/4/5 and macrophage migration inhibitory factor. Unexpectedly, hypoxia also stimulated migration of NK cells through the extracellular matrix and shifted amounts of susceptible leukemia target cells toward late apoptosis in a cell killing assay. We conclude that short term hypoxia supports these activities by positively interacting with NK cell priming at the level of glycolytic gene transcription. Hypoxic conditioning of NK cells may thus benefit their use in cell-based immunotherapy of cancer.
Keywords: cell migration, cytokine, glycolysis, hypoxia, interleukin, microarray, natural killer cells (NK cells), cell mediated cytotoxicity
Introduction
Natural killer (NK)3 cells mediate innate immunity by inducing apoptosis in infected and transformed cells and are important producers of immunoregulatory cytokines (1, 2). Therefore, they experience oxygen levels ranging from 5% in venous to 13% in arterial blood and down to 3–2% in healthy tissue and below (hypoxia) in the bone marrow, the lymphatic system, at sites of inflammation, and in the tumor microenvironment (3–9). Tumor hypoxia is thought to support tumor escape from NK cell-mediated cytotoxicity (9, 10). In vitro, promotion of NK cell-mediated cell killing by interleukin-2 (IL-2) was reported to be reduced after 96 h at 1% O2 compared with 20% O2, although antibody-dependent cellular cytotoxicity was not affected (11). Short term hypoxia (14–17 h, 1% O2) similarly reduced target cell killing, but sustaining hypoxia during the 4–4.5-h cell killing assay contributed to this reduction (12, 13). So far, effects of short term hypoxia on NK cell intrinsic activity and on cytolytic susceptibility of target cells, respectively, have not been separated.
NK cell effector functions are controlled by the cytokine milieu and integration of activating and inhibitory signals on potential target cells (14). During an active immune response, the ability of NK cells to respond to activation is substantially enhanced through IL-15, which is produced by monocytes, macrophages, and dendritic cells. IL-15 initiates Janus kinase-signal transducer and activator of transcription (Jak-Stat) signaling that promotes growth of cytotoxic lymphocytes, including NK cells (15, 16). Recently, NK cell treatment with IL-15, in addition to stimulation by cytokines (IL-2, -12, and -18) and binding of ligands for activating NK receptors, was shown to support switching to glycolytic metabolism, which involves nutrient-sensing mechanistic target of rapamycin (mTor) signaling and is a requirement for production of Ifn-γ and cytolytic effector molecules (17–20). Different signaling pathways thus underlie the effectiveness of IL-15 treatment and, consequently, of current ex vivo expansion and stimulation strategies that make use of this cytokine in NK cell-based immunotherapy of cancer (21–24).
Importantly, NK cell treatment with IL-15 also triggers miRNA-27a-5p-mediated down-regulation of the cytotoxic effector molecules granzyme B (Gzmb) and perforin beyond the first 6 h of cytokine addition (25). Here, we thus use the term IL-15 priming to refer to an initial period of IL-15 exposure limited to 6 h. We were interested in transcriptional pathway changes and possible functional differences in NK cells cultured under physiologically low oxygen, namely hypoxia, compared with the commonly used standard condition, i.e. normoxia, in response to IL-15 priming. Hypoxia and priming synergistically drove glycolytic gene transcription, and unexpectedly, hypoxia positively impacted on several NK cell intrinsic activities as follows: secretion of certain cytokines, migration through extracellular matrix (ECM), and progression of target cells to late apoptosis.
The obligatory role of glycolysis for cellular energy (ATP) production under hypoxia precludes interference with it, e.g. by chemical Hif inhibition or addition of glucose analogs, under conditions of low oxygen as a viable experimental strategy in tests of cellular function. Nevertheless, our data emphasize the importance of controlling oxygen levels during the in vitro study of NK cells and suggest that hypoxia can promote NK cell properties desirable for adoptive transfer immunotherapy.
Experimental Procedures
NK Cell Purification and Cell Culture
Ethics approval for this study was obtained from the medical faculty ethics committee. NK cells were prepared from buffy coats obtained through the local Red Cross Blood Donor Service or whole blood of healthy donors after informed consent by negative selection (NK-Cell Isolation Kit, Miltenyi Biotec). Preparations stained ≥93% CD56+CD3− and ≤1% each CD3+, CD14+, CD15+, and CD19+ as judged by flow cytometry. Freshly isolated NK cells were plated at 106/ml in RPMI 1640 medium (Sigma) supplemented with 10% fetal bovine serum (FBS) and 2 mm l-glutamine and were maintained in a standard tissue culture incubator with 5% CO2 resulting in 20% O2 (normoxia, standard condition) or in an oxygen-controlled Galaxy 48R CO2 incubator (New Brunswick) with a nitrogen gas line to establish 1% O2 (hypoxia) at 5% CO2. Human recombinant IL-15 (PeproTech) was used for priming and an equal volume of PBS as control. The Hif-1α inhibitor chetomin (CTM) was prepared as a 1 mm stock solution in dimethyl sulfoxide (DMSO). K562 cells (DSZM accession number 10) were cultured in the same medium and under normoxic standard conditions. At seeding and harvest, NK and K562 cell viabilities by trypan blue staining under all conditions tested were ≥90% (Countess, Invitrogen).
Preparation of Total RNA
We combined use of the mirVana buffer system (Life Technologies, Inc.) and the smaller Pure Link micro kit filter columns and collection tubes (Invitrogen) to obtain higher final RNA concentrations than with the regular mirVana kit procedure. Centrifuge settings were adjusted (10,000 × g, 1 min), and RNA was eluted in 15 μl of nuclease-free water preheated to 95 °C (14,000 × g, 1 min). DNase-treated samples (DNA-free kit, Ambion) were spectrometrically quantified (Tecan Infinite M200 NanoQuant). RNA integrity numbers were ≥8 (Agilent Bioanalyzer 2100).
Microarray and Gene Set Enrichment Analysis (GSEA)
For gene expression profiling, we used Hugene-2_0-st-type arrays (Affymetrix) on an Affymetrix GeneChip platform (Affymetrix Core Facility, Medical Research Center Mannheim), and a Custom CDF Version 17 with Entrez-based gene definitions for array annotation (26). Raw fluorescence intensities were normalized applying quantile normalization and robust multiarray average background correction. One-way analysis of variance was performed to identify differentially expressed genes using JMP10 Genomics version 6 (SAS Institute). A false-positive rate of α = 0.05 with false discovery rate (FDR) correction was considered significant. We performed GSEA to determine whether defined gene sets exhibited statistically significant bias in their distribution within a ranked gene list (27). Gene sets for pathways belonging to various cell functions were obtained from KEGG.
RT-PCR
For relative mRNA quantitation, we reverse-transcribed total RNA using the high capacity cDNA reverse transcription kit and performed TaqMan RT-PCR gene expression analysis on a 7900HT Fast Real Time PCR instrument (Life Technologies, Inc.). Assay IDs are assembled in Table 1. β-Glucuronidase (GUSB) was chosen as endogenous control gene in our experiments among the 16 candidate genes tested by the TaqMan Low Density Array Human Endogenous Control Panel (Life Technologies, Inc.).
TABLE 1.
Specifications of the non-isoform-specific TaqMan assays (Life Technologies, Inc.) used for RT-PCR validation
| Protein | Gene | Assay ID |
|---|---|---|
| β-Glucuronidase | GUSB | Hs00939627a |
| Elongin C | TCEB1 | Hs00855349 |
| Prolyl hydroxylase domain-containing protein 2 (PHD2) | EGLN1 | Hs00254392 |
| 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | PFKFB3 | Hs00998700 |
| Triose-phosphate isomerase | TPI1 | Hs03806547 |
| Phosphoglycerate kinase 1 | PGK1 | Hs00943178 |
| Pyruvate kinase muscle | PKM | Hs00761782 |
| Pyruvate dehydrogenase kinase isozyme 1 | PDK1b | Hs01561850 |
| Transketolase-like 1 | TKTL1 | Hs00202061 |
a This is an endogenous control gene.
b This should not be confused with phosphoinositide-dependent kinase 1 protein.
Protein Secretion
We screened 29 panel analytes selected from MILLIPLEX map kits HCYTOMAG-60K, HCD8MAG-15K, HSP1MAG-63K, and HTH17MAG-14K (Merck Millipore) for secretion by NK cells. Conditioned culture supernatants were stored at −80 °C until analysis on a MAGPIX system (Luminex). Duplicate determinations were averaged and evaluated using MILLIPLEX Analyst software. Human Vegf was determined by ELISA (Quantikine kit, R&D Systems).
Matrigel Invasion and Alamar Blue Assay
The ability of NK cells to migrate through ECM was assessed using Biocoat Matrigel invasion chambers with 8-μm pore size polycarbonate membranes (Corning) based on a published protocol (28). Briefly, 5 × 105 NK cells in 0.3 ml of RPMI 1640 medium (10% FBS, 2 mm l-glutamine), supplemented with IL-15 at a final concentration of 50 ng/ml or 5 μl/ml PBS, were loaded in triplicate into the upper chambers of 24-well plate inserts. Lower chambers were filled with 1.5 ml of likewise supplemented medium, respectively. Assembled chambers were incubated for 48 h at 37 °C and either 20 or 1% O2. From the same NK cell preparation, we set up parallel NK cell cultures under these four conditions for calibration purposes. Four hours before the end of the 48-h incubation period, we added 150 μl of Alamar Blue to the lower chambers. Cells from the parallel cultures were counted, and five serial 1:1 dilutions starting at 5 × 105 cells in final volumes of 1.5 ml were plated in duplicate into free wells on the same plate holding the inserts for the corresponding oxygen concentration, plus two background controls. As to the lower chambers, 150 μl of Alamar Blue was added to standards and controls, and plates were returned to incubators at 20 and 1% O2, correspondingly, for the remaining 4 h. At the end of the 48-h period, inserts were removed. Fluorescence of reduced Alamar Blue was measured as described previously (28). Numbers of migrated NK cells were calculated from calibration curves. The Alamar Blue assay was also applied to assess viabilities of NK cells conditioned for 22 h under normoxia or hypoxia with IL-15, varying concentrations of CTM, and the dye present during the final 6 h of this period. Cells were seeded in 96-well plates at 105 cells in 100 μl per well. The Alamar Blue volume added was 10 μl per well.
Flow Cytometry
NK cell protein expression was assessed using fluorochrome-conjugated monoclonal antibodies (Table 2). For intracellular staining, we used the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences). A total of 105 stained cells were acquired on a FACSCanto II cytometer and analyzed using FACSDiva software (BD Biosciences). Apoptosis in CTM-treated NK cells was detected by staining with annexin V-APC and 7-amino-actinomycin D (7-AAD) (BD Biosciences) and gating on singlets in the forward scatter height versus forward scatter area plot. Duplets were between 3 and 6%.
TABLE 2.
Monoclonal antibody-fluorochrome conjugates used in flow cytometry by provider
| Antigen | Clone | Fluorochrome |
|---|---|---|
| BD Biosciences | ||
| CD56 | NCAM 16.2 | PE |
| CD3 | UCHT1 | FITC |
| CD45 | 2D1 | PerCP |
| CD15 | H198 | APC |
| CD14 | M5E2 | PE-Cy7 |
| CD19 | SJ25C1 | APC-Cy7 |
| CD25 | MA251 | Pe-Cy5 |
| CD69 | FN50 | APC |
| CD107a | H4A3 | APC |
| Ifn-γ | B27 | APC |
| Miltenyi Biotec | ||
| Gzmb | REA162 | APC |
To assess the ability of NK cells to induce apoptosis in target cells, 2.5–5 × 106 K562 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes) at a final concentration of 62.5 nm in 2 ml of PBS for 15 min at 37 °C. Cells were pelleted and resuspended in 4 ml of RPMI 1640 medium (10% FBS, 2 mm l-glutamine), incubated for 30 min at 37 °C, and then washed twice with PBS. Subsequently, CFSE-labeled K562 target cells were resuspended in RPMI 1640 medium (10% FBS, 2 mm l-glutamine) at a concentration of 105 cells/ml and co-incubated with 2.5 × 106 conditioned NK cells in a final volume of 2 ml under standard culture conditions. After 4 h, the killing reaction was stopped by putting the cells on ice and washing twice with PBS followed by addition of annexin V-APC and 7-AAD.
Cellular l-Lactate Dehydrogenase (Ldh) Release and Content
To quantify specific lysis of K562 target cells (T) by effector NK cells (E), release and cellular contents of Ldh were determined enzymatically using the coupled CytoTox 96 non-radioactive cytotoxicity assay (Promega). Fixed numbers of 5 × 104 K562 cells were mixed with conditioned NK cells in volumes of 200 μl at a final FBS concentration of 2% and indicated E/T ratios in triplicate and maintained under standard conditions for 4 h. Ldh activities in lysates of K562 cells and culture supernatants were derived from initial slopes of absorbance versus time progress curves for dye reduction. Cellular Ldh content of NK cells was assessed in lysates of 2 × 105 cells in 100 μl of culture medium using the same lysis procedure as for K562 cells.
Extracellular Flux Analysis
Extracellular acidification rate (ECAR) was measured on a Seahorse XFp analyzer (Seahorse Bioscience) using the Seahorse XF glycolysis stress test kit. Conditioned NK cells were washed twice and resuspended in serum-free unbuffered RPMI 1640 medium with 2 mm l-glutamine and without glucose (Sigma). For comparison of two or three conditions, respectively, cells were plated in triplicate or duplicate into Seahorse miniplates at 2 × 105 cells per well. ECAR was recorded over time with sequential additions of glucose (10 mm), oligomycin (1 μm), and 2-deoxyglucose (50 mm) (Seahorse Bioscience). Data were evaluated using Seahorse Wave software.
Glucose, Lactate, and ATP Measurements
Glucose and lactate levels in culture supernatants from conditioned NK cells were measured enzymatically in single determinations on a Cobas c 311 analyzer using the glucose HK (GLUC2) and lactate (LACT2) assay kits (Roche Applied Science/Hitachi), with a measuring range of 0.11–41.60 and 0.2–15.5 mm, respectively. Cellular ATP levels were assessed in aliquots of 105 conditioned and CTM-treated cells in 100 μl of culture medium in triplicate using the CellTiter Glo® luminescent cell viability assay (Promega).
Statistical Analysis
Differences between experimental groups of paired samples were evaluated using the Wilcoxon signed-rank test (SigmaPlot version 11). p values <0.05 were considered statistically significant.
Results
Transcriptional Up-regulation of Hif-1 Signaling and Glycolytic Pathways by Hypoxia in NK Cells Is Dependent on IL-15 Priming
NK cells conditioned by a 22-h culture under hypoxia or normoxia with or without IL-15 priming for the final 6 h were subjected to global transcriptome profiling. Abundance for 23% of 23,786 genes on the microarray was changed significantly by priming regardless of oxygen concentration. Hypoxia alone only changed it for 1.79% with and for 1.70% without priming. We focused on significantly regulated pathways attributable to NK cells as a single cell type (Fig. 1). Priming was consistently and positively associated with expression of genes involved in protein anabolism, energy production, and immune signaling. Notably, up-regulation of the mitochondrial pathways citric acid (TCA) cycle and oxidative phosphorylation did not depend on oxygen. Hypoxia compared with normoxia did not produce any significant pathway alteration without and only few with priming. Dependences on priming for associations of hypoxia with Hif-1 signaling and with glycolysis/gluconeogenesis (further referred to as glycolysis) were by far the strongest contrasts observed.
FIGURE 1.

Canonical KEGG pathways altered by IL-15 priming and hypoxia. NK cells were isolated from whole blood of five healthy donors, cultured at 20% or 1% O2 for 22 h without or with IL-15 (45 ng/ml) during the final 6 h, and subjected to microarray analysis (Human Gene 2.0 ST array, Affymetrix). GSEA was applied to KEGG pathways. The −log10 of FDR-adjusted probabilities (FDR q-values) of the enrichment of pathways below a 0.05 threshold for at least one of the four comparisons shown are displayed on a color scale with yellow denoting negative and blue positive association. Pathways attributable to NK cells as single cell type are assembled on the left and those involving cells of varying type and tissue origin on the right.
In Fig. 2, significant mean transcriptional changes >1.25-fold for single genes by priming, hypoxia, and/or their combination, each compared with standard conditions, are mapped onto Hif-1 signaling (A) and glycolytic (B) pathways. Priming dominated responses in both. Responses were overall more pronounced when priming was performed under hypoxia, especially for glycolytic genes. Transcriptional changes in response to both stimuli together frequently appeared to display additive to synergistic behavior. Up-regulation by IL-15 of five genes encoding enzymes with pathway regulatory roles appeared to particularly depend on hypoxia, namely elongin C and prolyl hydroxylase domain-containing protein 2 (Phd2) encoded by the transcription elongation factor B polypeptide 1 (TCEB1) and egl-9 family hypoxia-inducible factor 1 (EGLN1) gene, respectively (Hif-1 signaling), and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), triose-phosphate isomerase (TPI1), and pyruvate dehydrogenase kinase isozyme 1 (PDK1) (glycolysis). These were selected for RT-PCR validation. We also selected the phosphoglycerate kinase 1 (PGK1) and pyruvate kinase muscle (PKM) genes for RT-PCR validation because of the important roles of encoded enzymes as ATP producers at low oxygen. In view of strong induction of anabolic processes by priming, we additionally considered the pentose phosphate pathway (PPP) (Fig. 2C). PPP genes from the oxidative branch (NADPH production) were up-regulated by priming, whereas mixed responses were seen for the non-oxidative branch. Here, IL-15 unexpectedly down-regulated transketolase-like 1 (TKTL1) expression opposing its expected up-regulation by hypoxia (29). We thus included TKTL1 in RT-PCR validation. Fig. 3 summarizes transcriptional responses of Hif-1 signaling, glycolytic, and PPP genes and genes of mitochondrial shuttle enzymes.
FIGURE 2.



Mapping of gene expression changes in NK cells by IL-15 priming and hypoxia onto Hif-1 signaling and glycolytic pathways. Microarray analysis was performed on NK cells isolated from whole blood of five healthy donors and cultured at 20% O2 or 1% O2 for 22 h without or with IL-15 (45 ng/ml) during the final 6 h. Total RNA was hybridized onto human genome array chips. Data were subjected to GSEA using KEGG pathways applying an FDR-adjusted probability (FDR q-value) threshold of pathway enrichment of 0.05. By far, the strongest associations for hypoxia were seen with the HIF-1 signaling and glycolysis pathways after priming, although there were no significant associations for hypoxia without priming (Fig. 1). Significant mean transcriptional changes of >1.25-fold relative to standard culture conditions (legend on top) are mapped onto Hif-1 signaling (A) and glycolytic (B) pathways and the PPP (C) using a binary logarithmic color scale with white for no change, green for decreased, and red for increased mean gene expression levels. Glycolytic genes assigned to both KEGG pathways are included in the glycolytic pathway (B), which is further complemented by the SLC16A1/3 and PC genes encoding lactate transporters Mct1/4 and pyruvate carboxylase, respectively. For the display of integer pathways and to avoid gaps, some genes are included even when showing non-significant or below threshold changes (printed in gray). In the Hif-1 signaling pathway (A), mean changes are also given by way of exemption for eponymous HIF1A as well as for PIK3CD and CDKN1A, which both appear to contribute to isoform switches. Names of genes selected for RT-PCR validation (Fig. 4) are printed in bold. Where the gene name is not reminiscent of the protein name, the latter is added in parentheses ((A) Phd2, Hif1B, and p21/27) or replaces the gene name ((B) TIGAR for “TP53-inducible glycolysis and apoptosis regulator” encoded by the C12orf5 gene).
FIGURE 3.

Summary of gene expression profiles for selected pathways regulated by hypoxia and IL-15 in NK cells. Microarray data from NK cells, cultured for 22 h under normoxia (20% O2) or hypoxia (1% O2) and without or with IL-15 (45 ng/ml) during the final 6 h, were subjected to GSEA using KEGG pathways. Gene expression profiles for Hif-1 and glycolytic pathways and the PPP as schematized in Fig. 2 are summarized in a heat map, additionally including glycerol 3-phosphate and malate-aspartate shuttle genes. Tiles are arranged by experimental condition and sample order, and genes are hierarchically clustered. Green indicates minimum and red maximum expression. Genes selected for RT-PCR validation (Fig. 4) are shown with boxes. Pathway assignments are given on the right.
Hypoxia and IL-15 Interact in Transcriptional Control of Key Regulatory and Metabolic Enzymes in NK Cells
Because of the semi-quantitative nature of the microarray method, we quantitated relative changes in expression levels for our genes of interest by RT-PCR (Fig. 4). We did not see important changes in TCEB1 expression by hypoxia, but IL-15 priming consistently induced TCEB1 around 1.7-fold. Compared with standard conditions, EGLN1 was up-regulated 2.4-fold on average by hypoxic priming versus only 1.2- and 1.4-fold, respectively, by priming and hypoxia alone. The expression profile of PFKFB3 was confirmed to be irregular with a decrease by priming and an increase by hypoxia. Highest PFKFB3 transcript levels were confirmed to occur after hypoxic priming. Priming doubled TPI1 transcript levels under normoxia, whereas hypoxia alone raised them very little. Both stimuli concurrently amplified TPI1 expression 3.8-fold. Similar responses were confirmed for PGK1 and PKM. Synergistic induction of PDK1 by hypoxia and priming seen by microarray was confirmed and amounted to 2.2-fold compared with standard conditions. Overall, positive hypoxia/IL-15 interactions in the up-regulation of all genes in our selection but TCEB1 were qualitatively validated by RT-PCR. We confirmed also reverse regulation of TKTL1 with a 24% decrease by IL-15 and a 71% increase by hypoxia and an additive effect by priming under hypoxia.
FIGURE 4.
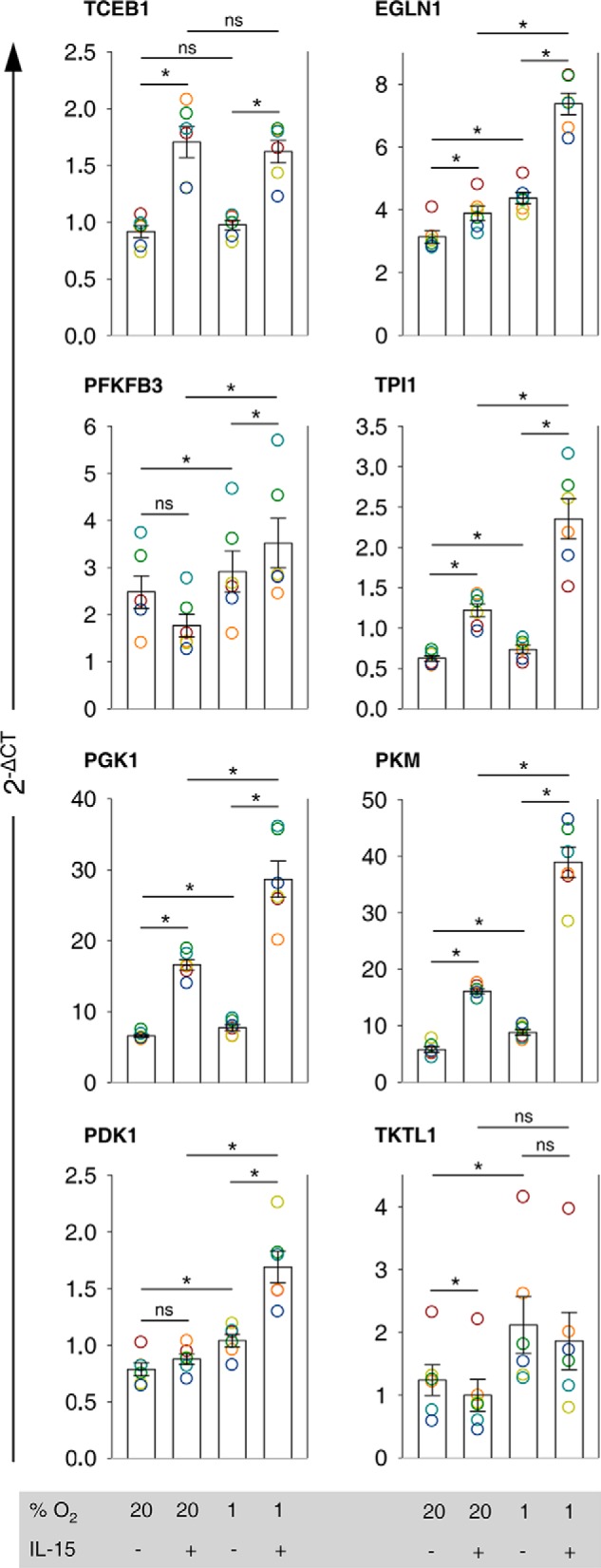
Validation of transcriptional interactions between IL-15 and hypoxia on the level of key regulatory and metabolic enzyme genes. Results from microarray analysis of human NK cells, cultured under normoxia (20% O2) or hypoxia (1% O2) for 22 h without or with IL-15 (45 ng/ml) during the final 6 h, suggested transcriptional interactions between IL-15 and hypoxia in the Hif-1 and glycolytic pathways and the PPP (Figs. 2 and 3). Genes of key regulatory and metabolic enzymes were selected for validation by RT-PCR (TaqMan) in independent NK cell preparations from whole blood of six healthy donors. GUSB was used as endogenous control gene. For each condition, linearized quantities (2−ΔCT) are represented as mean values ± S.E. (bars). Data are displayed as circles in a muted rainbow color scheme to identify values from same NK cell preparations, i.e. same donors. Expression profiles are presented in the order of appearance of the selected genes in the pathway maps shown in Fig. 2. *, p < 0.05; ns, not significant.
Little Changes in Glycolytic Flux by Hypoxia in Quiescent and IL-15-primed NK Cells
In only one of two independent biological samples of conditioned quiescent (S1 and S2) and primed (S3 and S4) NK cells, respectively, hypoxia slightly increased glycolysis as measured by ECAR (Fig. 5A). Under hypoxic conditioning, priming increased glycolysis, glycolytic capacity, and glycolytic reserve slightly in S5 and clearly in S6 (Fig. 5, A, bottom, and B). However, neither hypoxia nor priming stimulated cellular consumption of glucose (Fig. 5C) and did not consistently alter intracellular Ldh enzymatic activity (Fig. 6A) and ATP content (Fig. 6B). Lactate levels in conditioned culture supernatants were barely above the lower limit of detection or undetectable.
FIGURE 5.
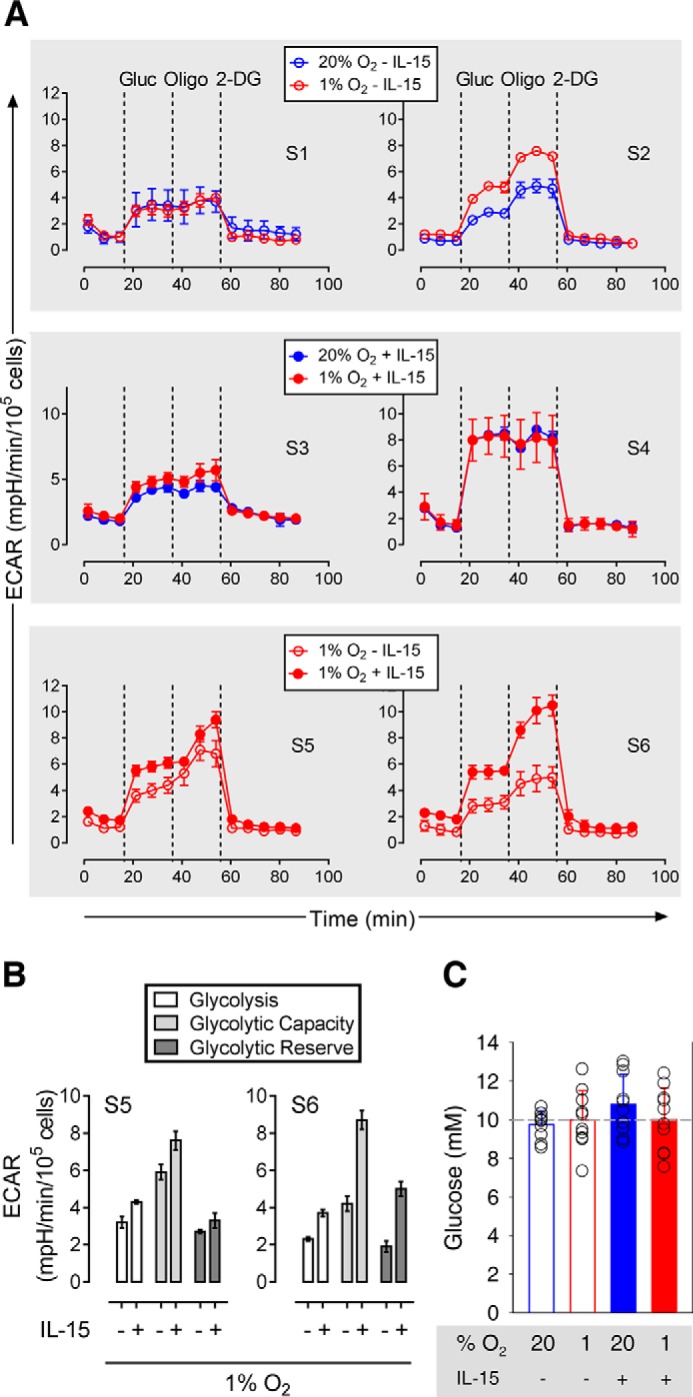
Glycolytic flux in conditioned NK cells. A, ECAR was measured over time for pairwise comparisons. Each data point represents an average of triplicate determinations ± S.D. For three comparisons, results from two independent preparations of human NK cells from healthy volunteers each are shown side by side. Cells were conditioned under normoxia (20% O2) or hypoxia (1% O2) for 22 h without (S1 and S2) or with (S3 and S4) IL-15 (45 ng/ml) during the final 6 h. The third comparison shows the effect of priming under hypoxia (S5 and S6). Dashed lines indicate additions of glucose (Gluc), oligomycin (Oligo), and 2-deoxyglucose (2-DG). For the third comparison, B breaks down glycolysis, glycolytic capacity, and glycolytic reserve as mean values ± S.E. (bars). C compares determinations of residual glucose in conditioned culture supernatants. Mean values ± S.E. (bars) and single values (open circles) for 10 healthy volunteers are shown. The dashed line indicates the level of glucose in fresh supplemented RPMI 1640 medium (10 mm).
FIGURE 6.
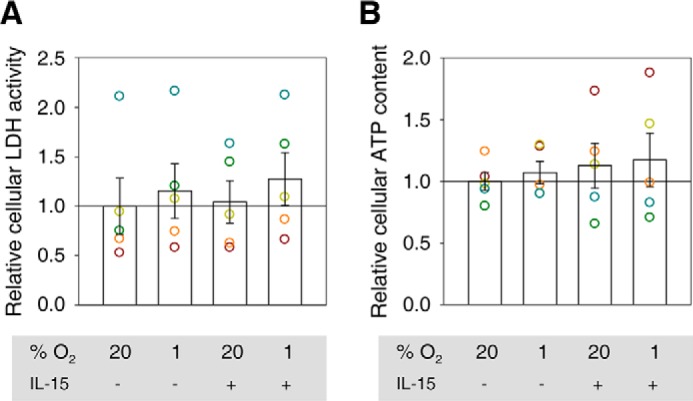
Ldh activity and ATP level in conditioned NK cells. In five independent experiments, human NK cells from buffy coats were cultured under normoxia (20% O2) or hypoxia (1% O2) for 22 h without or with IL-15 (45 ng/ml) during the final 6 h. Using normoxia without priming as a reference (indicated by a horizontal line), Ldh activities (A) and ATP levels (B) from cell lysates are represented as mean values ± S.E. (bars). Data are also displayed as circles in a muted rainbow color scheme to identify values from same NK cell preparations. Differences in this set of samples did not reach statistical significance defined as p < 0.05.
Chemical Inhibition of Hif-1α Transcriptional Activity Reduces Glycolysis and Viability of NK Cells Independent of Oxygen Availability
In two independent biological samples each of IL-15-primed NK cells conditioned either under normoxia (S7 and S8) or hypoxia (S9 and S10), CTM clearly reduced glycolytic activity (Fig. 7A), possibly in a concentration-dependent manner (S9).
FIGURE 7.
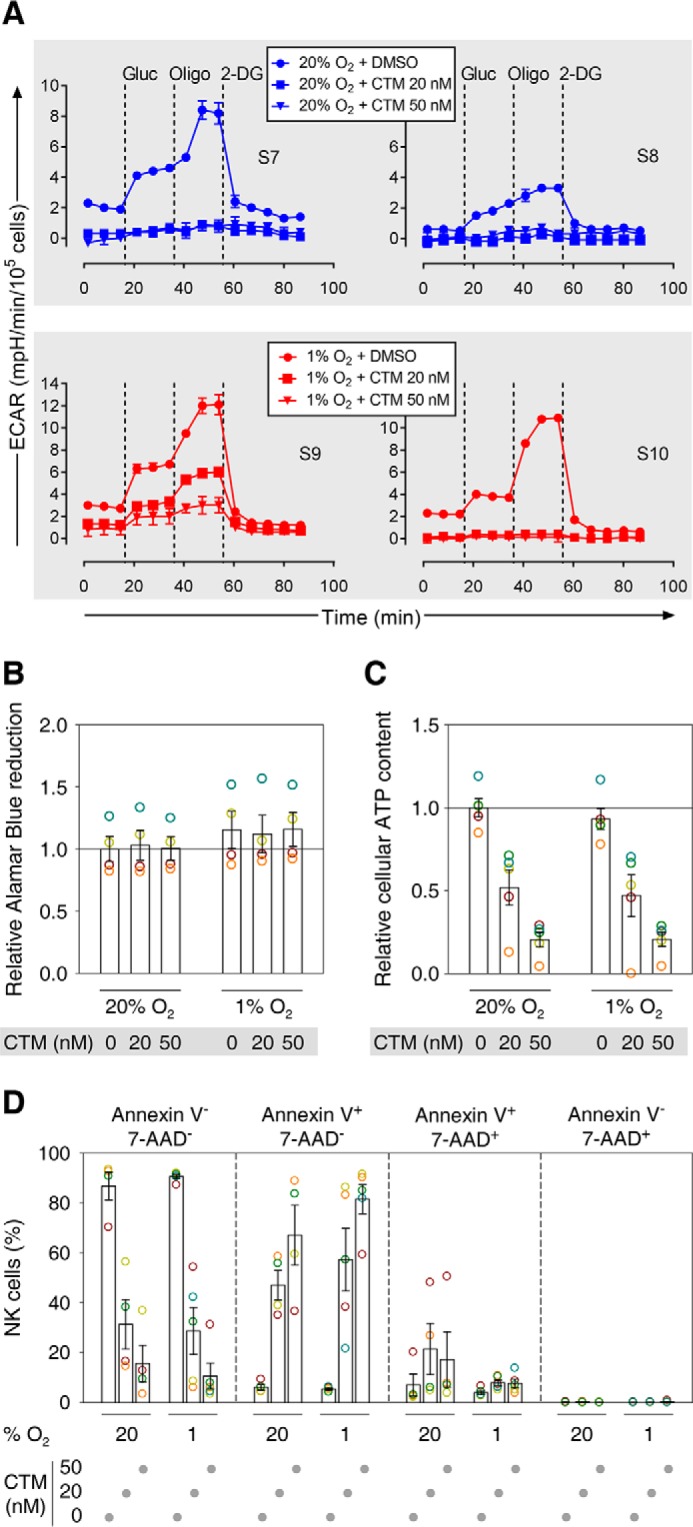
Chemical inhibition of Hif-1α transcriptional activity during IL-15 priming of NK cells reduces glycolysis and viability. A, each data point represents an average of duplicate ECAR determinations ± S.D. For two comparisons, results from two independent preparations of human NK cells from healthy volunteers each are shown side by side. Cells were conditioned under normoxia (20% O2, S7, S8) or hypoxia (1% O2, S9, S10) for 22 h with IL-15 (45 ng/ml) and CTM or DMSO vehicle control during the final 6 h. Dashed lines indicate additions of glucose (Gluc), oligomycin (Oligo), and 2-deoxyglucose (2-DG). B–D show data from four or five independent preparations of NK cells from buffy coats and are displayed as mean values ± S.E. (bars) and also as circles in a muted rainbow color scheme to identify values from the same preparations. Cells were conditioned as in A. B, Alamar Blue was additionally present besides IL-15 and CTM or vehicle during the final 6 h of the conditioning. Alternatively, viability at the end of the conditioning period was evaluated by measuring ATP levels in cell lysates (C) and flow cytometric detection of annexin V-APC/7-AAD staining (D). B and C, normoxia with vehicle served as a reference (indicated by a horizontal line).
Although Alamar Blue reduction was only slightly (on average 12–16%) increased through hypoxia and not changed through CTM (Fig. 7B), the ATP detection-based CellTiter Glo assay indicated stepwise approximate halving of average cell viabilities by 20 and 50 nm CTM both under normoxia and hypoxia. To assess different stages of NK cell apoptosis, we used flow cytometry with fluorescently labeled annexin V and 7-AAD, reporters of early and advanced loss of membrane integrity, respectively. We refer to cells negative for both reporters as viable, annexin V+ 7-AAD− as early apoptotic, annexin V+ 7-AAD+ as late apoptotic, and annexin V− 7-AAD+ as necrotic. This method showed a CTM concentration-dependent reduction in the populations of viable NK cells independent of oxygen concentration (Fig. 7D). This was inversely correlated to an increase in apoptotic cells, mainly at the early apoptotic stage. Necrotic cells were consistently <0.22%.
Increased NK Cell Chemokine Secretion and No Changes in Intracellular and Surface Marker Expression under Hypoxia Compared with Normoxia
Conditioned supernatants from normoxic or hypoxic NK cell cultures with or without IL-15 priming were assayed for secreted proteins mostly known to be produced by NK cells and/or to represent ligands of NK cell-expressed receptors. In no case did we detect Ccl2/Mcp1, Cxcl10/Ip-10, Cxcl8/IL-8, eotaxin, G-Csf, GM-Csf, Gzmb, IL-1ra, IL-1β, IL-1α, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12, β/IL-12 p40, IL-13, IL-17a, IL-22, Ifn-α2, Ifn-γ, Mcp-3, sFasL, Tnf-α, tPai-1, and Vegf.
Ccl-5/Rantes (regulated on activation normal T cell expressed and secreted) was consistently detected at 16–715 pg/ml (Fig. 8A). Ccl3/Mip-1α and Ccl4/Mip-1β required priming to be measurable throughout. Overall mean levels for both were 1 order of magnitude lower than for Ccl5. Despite differences in absolute concentrations between donors for a given condition, respective profile shapes for Ccl3/4/5 were highly consistent. Hypoxia had little effect on secretion by resting NK cells but increased mean levels after priming 1.7-fold (Ccl3/4) and 1.5-fold (Ccl5) (Fig. 8B). Increases in Ccl3/4/5 through priming were stronger under hypoxia than normoxia.
FIGURE 8.

Chemokine secretion and expression of cellular markers by NK cells under hypoxia and IL-15 priming. A and B, NK cells isolated from whole blood were cultured at 20% O2 or 1% O2 for 22 h without or with IL-15 (45 ng/ml) during the final 6 h. Thus conditioned supernatants were screened by multiplex immunoassays for 29 proteins out of which Ccl3/4/5 and Mif were detectable. For supernatants in which no evaluable signal was obtained for a given analyte (A identified by open circles), we used lower limits of detection for comparisons (2.96 pg/ml for Ccl3, 2.86 pg/ml for Ccl4, and 11.32 pg/ml for Mif) thus rather underestimating increases in fold changes (B). A, bars represent averaged double determinations for 10 different donors. B, bars represent mean fold changes ± S.E. of donors 1–10 for hypoxia versus normoxia (open) and IL-15 versus quiescent (gray) comparisons. The y intercept for no change is indicated by a dotted line. C, for flow cytometric analysis of intracellular and surface markers, NK cells were prepared from buffy coats in 3 to 5 independent experiments. Parallel normoxic (blue) and hypoxic (red) cultures were primed with IL-15 (45 ng/ml) 16 h after plating (set to 0 h) and processed consecutively at 6-h intervals. *, p < 0.05; **, p < 0.005; ns, not significant.
Macrophage migration inhibitory factor (Mif) depended on hypoxia to be measurable throughout (Fig. 8A). The hypoxia-dependent average increase in Mif secretion by resting NK cells was 2-fold (Fig. 8B). However, this number resulted from heterogeneous profiles. All-or-nothing responses were observed with four donors where the hypoxia levels of Mif did not exceed 50 pg/ml, although increases were intermediate to small with the other donors for whom Mif levels ranged from ∼50 to 240 pg/ml. Hypoxia response profiles with priming were similar but slightly weaker (1.7-fold versus 2-fold increase). Priming hardly affected Mif concentrations.
We did not observe hypoxia-dependent changes after NK cell priming for intracellular expression of CD107a, Ifn-γ, and Gzmb nor for CD107a, CD25, and CD69 at the cell surface (Fig. 8C). A small increase in Ifn-γ 24 h after priming was lower under hypoxia.
Hypoxia Promotes NK Cell Migration through ECM
The average number of NK cells that migrated through Matrigel under normoxia compared with hypoxia was increased by 45%, equivalent to a 47% raise through IL-15 priming (Fig. 9). Hypoxia also raised the mean of migrated cells under priming conditions, but this effect did not reach statistical significance.
FIGURE 9.
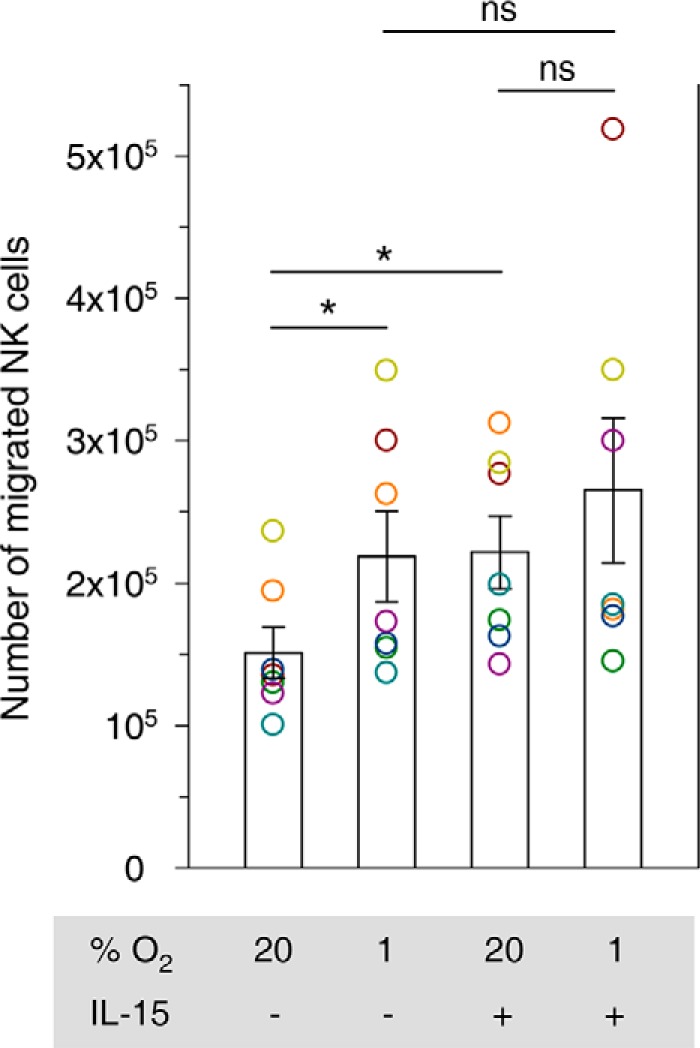
Impact of hypoxia and IL-15 priming on NK cell migration through Matrigel. 5 × 105 NK cells isolated from buffy coats were loaded into Matrigel invasion chambers and cultured for 48 h at 20% O2 or 1% O2 and without or with IL-15 (50 ng/ml) in both lower and upper chambers. Numbers of migrated cells were determined by Alamar Blue reduction and calibration against parallel cultures. Bars represent mean counts ± S.E. of seven independent experiments. Data are displayed as circles in a muted rainbow color scheme to identify values from same NK cell preparations, i.e. same donors. Cells from one donor (brown circle) appeared transmigrated quantitatively under hypoxia in the presence of IL-15. *, p < 0.05; ns, not significant.
Hypoxic Conditioning of NK Cells Promotes Late Apoptosis in Target Cells
Specific lysis of K562 target cells (T) by effector NK cells (E), conditioned by preculture for 22 h under hypoxia or normoxia with or without IL-15 priming for the final 6 h, was determined by Ldh release assay under standard conditions (Fig. 10). Mean lysis increased with the E/T ratio but was independent from preculture oxygen levels. Priming consistently enhanced specific lysis by on average 17–24%. The effect of hypoxia on the cytolytic potential of primed NK cells was a reduction in lysis reaching 4.6% for the 4:1 E/T ratio only.
FIGURE 10.
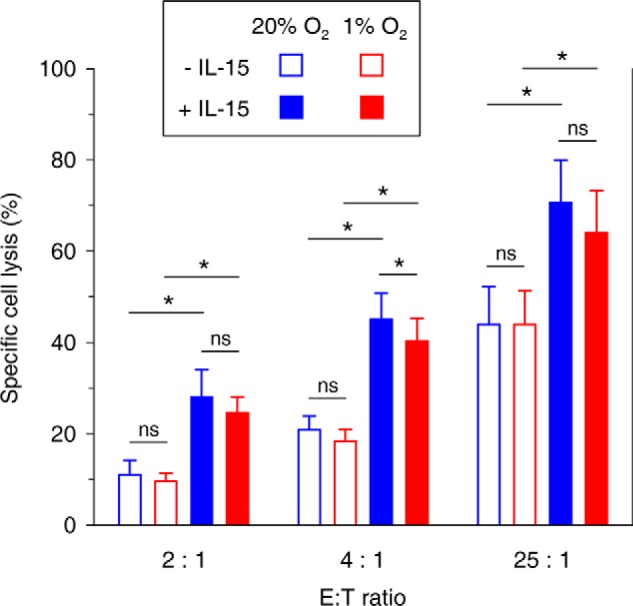
Cytolytic activity of quiescent and primed NK cells conditioned under hypoxia versus normoxia. NK cells isolated from buffy coats were conditioned by preculture at 20% O2 (blue) or 1% O2 (red) for 22 h without (open) or with IL-15 (45 ng/ml, filled) present during the final 6 h. Percentage of specific cell lysis during a subsequent 4-h co-incubation of effector (E) NK cells with K562 target cells (T) at different E/T ratios under normoxic standard culture conditions in triplicate was determined using Ldh release assay applying the following equation to measured released Ldh activities: 100 × (experimental − (E) spontaneous − T spontaneous)/(T maximum − T spontaneous). Bars represent mean values ± S.E. from six independent experiments. *, p < 0.05; ns, not significant.
We assessed different stages of target cell apoptosis by flow cytometric determination of annexin V-APC/7-AAD staining. Conditioned NK cells were co-incubated with CFSE-labeled K562 cells under standard conditions (Fig. 11A, center). Importantly, CFSE was not toxic at the concentration used as annexin V binding and 7-AAD inclusion were equally <5% and <0.2%, respectively, for separately cultured CFSE-labeled and non-labeled K562 cells. After the cell killing reaction, average variations in abundance of CFSE− NK cells at any given stage of apoptosis were <3% of total CFSE− cells (Fig. 11A, top left). Because potential CFSE− debris from apoptotic K562 cells was excluded by forward-scatter thresholding, this indicated that NK cell numbers remained stable during the co-incubations. They were thus used as reference in assessing target cell loss from flow cytometric analysis due to advanced cellular disintegration (Fig. 11A, bottom left). Hypoxic versus normoxic preculture of NK cells did not change the average numbers of captured target cells significantly (86% versus 92%, p = 0.078). In contrast, NK cell priming reduced target cell numbers to 54%, which was partially reversed by hypoxia.
FIGURE 11.

Effects of hypoxia and IL-15 priming during NK cell conditioning on stages of target cell apoptosis. Effector NK cells (E) isolated from buffy coats were conditioned by preculture at 20 or 1% O2 for 22 h without or with IL-15 (45 ng/ml) present during the final 6 h, mixed, and co-incubated with CFSE labeled K562 target cells (T) at an E/T ratio of 25:1 for 4 h under normoxic standard culture conditions. Seven buffy coats were processed in independent experiments out of which one non-primed hypoxia sample was not available for apoptosis analysis. A, representative flow plot (center) shows differentiation of CFSE− effector and CFSE+ target cells. Stages of apoptosis were determined by multicolor flow cytometry in two-factor analysis with annexin V-APC/7-AAD staining as exemplified for target cells (right). The top left diagram summarizes mean proportions of viable (annexin V− 7-AAD−), early apoptotic (annexin V+ 7-AAD−), late apoptotic (annexin V+ 7-AAD+), and necrotic (annexin V− 7-AAD+) effector cells for different preculture conditions. The bottom left diagram displays average CFSE+/CFSE− ratios ± S.E. for each effector cell preculture condition from which average target cell fractions of input cell numbers were calculated on the basis of the E/T ratio of 25:1. B, differential effector cell conditioning caused redistributions of target cells among stages of apoptosis. Changes by hypoxia are highlighted by connecting paired data points, i.e. results from the same buffy coat (left). A stacked bar chart summarizes the distributions (right, cf. A, bottom left). *, p < 0.05; ns, not significant.
To compare amounts of K562 target cells at different stages of apoptosis between NK cell preculture conditions, we transformed relative annexin V-APC/7-AAD staining data from gating on CFSE+ cells (Fig. 11A, center and right) into proportions of original target cell input (Fig. 11B). NK cell priming compared with no priming reduced average amounts of viable K562 cells by 13% (normoxia) and 6.8% (hypoxia) and of early apoptotic K562 cells by 25% (normoxia) and 13% (hypoxia). Without priming, hypoxia reduced K562 cells in early apoptosis by 15%. Conversely, hypoxia increased K562 cells in late apoptosis by 13% (without priming) and 20% (with priming). Necrotic cells were on average below 3.5% for each preculture condition without major changes.
Together, a small negative effect of preculture hypoxia compared with normoxia on the cytolytic activity of IL-15 primed NK cells on K562 cells measured by Ldh release agrees with a smaller loss of CFSE+ target cells when comparing hypoxic to normoxic preculture by flow cytometry. Hypoxia, however, did not change residual amounts of viable target cells significantly but consistently increased amounts of target cells in late apoptosis.
Discussion
In vitro studies of immune cells are commonly performed in ambient air adjusted to 5% CO2. The resultant oxygen concentration of 20% (normoxia) lies well above physiological levels. We used genome-wide transcriptome profiling to gain first insights into the impact of short term hypoxia compared with normoxia on NK cell biology. We found that induction of Hif-1 signaling and glycolytic pathways by hypoxia depended markedly on IL-15 priming. The Hif-1α/Hif-1β dimer is the major transcriptional mediator of hypoxia responses. Hif-1α is stabilized at low oxygen through decreased prolyl hydroxylation by EGLN1-encoded Phd2 (Fig. 2A). This modification otherwise creates a binding site for the von Hippel-Lindau protein that recruits a ubiquitin ligase complex, including TCEB1-encoded elongin C and E3 ubiquitin-protein ligase Rbx1, and leads to proteasomal Hif-1α degradation (30). Despite broad up-regulation of Hif-1α-dependent genes by priming, including glycolytic genes, down-regulation of ARNT, encoding Hif-1β, and up-regulation of RBX1 and TCEB1 (RT-PCR validated, Fig. 4) suggest that a negative feedback loop was operational at 6 h of priming. Validated synergistic up-regulation of EGLN1 by priming under hypoxia additionally indicated increased cellular capacity to degrade Hif-1α and rapid restoration of oxygen sensitivity.
The dependence of glycolytic gene transcription on IL-15 priming appeared especially accentuated and was validated for PFKFB3 and TPI1 (Figs. 2B, 3, and 4). Bifunctional Pfkfb enzymes control intracellular levels of fructose 2,6-bisphosphate, a central allosteric activator of 6-phosphofructo-1-kinase. Contrary to its negative impact under normoxia, priming stimulated the hypoxia-induced expression of Pfkfb3. Pfkfb3 features an exceptionally high kinase/phosphatase activity ratio (31), and its inhibition suppresses T-lymphocytes (32). Pfkfb3 is thus a likely key regulator of glycolysis in primed NK cells also. Tpi1 balances funneling of reducing equivalents into the respiratory chain via the glycerone 3-phosphate shuttle and thereby basal mitochondrial function against salvaging this three-carbon unit for glycolysis. The observed strong stimulation of the priming-induced TPI1 expression by hypoxia indicates tipping the scales toward glycolysis. In addition, RT-PCR revealed positive synergistic transcriptional hypoxia/IL-15 interactions for ATP-producing Pgk1 and Pkm. These effects and overall co-regulation of the linearly arranged core glycolytic genes by hypoxia and IL-15 priming indicate synergistic reinforcement of glycolysis by both stimuli at the transcriptional level. Particularly, apparent gene inductions of LDH and lactate exporter encoding SLC16A1/3, and last but not least of PDK1, for which we also validated a positive hypoxia/IL-15 interaction, are features of anaerobic glycolysis. Hif-1α-regulated Pdk1 inhibits mitochondrial pyruvate dehydrogenase (Pdh), which links glycolysis and the TCA cycle, by reversible phosphorylation on E1. Otherwise, unrestrained Pdh activity in the absence of sufficient oxygen as terminal electron acceptor renders cells sensitive to apoptosis (33, 34).
Our microarray data do not imply stable alterations in NK cells by short term hypoxia and/or IL-15 priming. E1 and E2 of the Pdh complex and TCA cycle and oxidative phosphorylation pathways were transcriptionally up-regulated by IL-15 irrespective of oxygen supply, suggesting that mitochondrial integrity was maintained. Mitochondrial shuttle genes essential to this end also appeared up-regulated by priming (Fig. 3). These observations suggest transitory adaptation to low oxygen, which is in line with rapid restoration of oxygen sensitivity.
Despite the positive interaction between hypoxia and IL-15 priming, there were no clear differences in glycolytic flux through short term hypoxia in both quiescent and primed NK cells (Fig. 5A). This agrees with unchangingly neglectable glucose consumption (Fig. 5C) and no consistent differences in cellular ATP content under all four conditions tested (Fig. 6B). If any, cellular Ldh activity was slightly increased through hypoxia (Fig. 6A). In agreement with published results from murine NK cells primed with IL-15 under normoxia for 4 h (20), the 6-h period of IL-15 exposure under hypoxia used here only slightly raised parameters of glycolytic flux in human NK cells also (Fig. 5, A and B), compared with the reported very prominent enhancements in murine NK cells through IL-2/12 for 18 h (17) and through IL-15 for 18 h (19). Overall, our data, especially the absence of relevant glucose consumption and no measurable lactate production during the 22-h conditioning period throughout, do not support effective glycolytic switching at the metabolic level in NK cells through priming and hypoxia in the absence of additional stimuli. We conclude that glycolytic switching under these conditions mainly affects glycolytic gene transcription (Figs. 2–4).
Contrary to the synergistic effect of hypoxia and priming on glycolytic gene transcription, hypoxia clearly up-regulated and priming slightly down-regulated TKTL1 gene expression (Figs. 2C, 3, and 4). Tktl1 functions in the non-oxidative PPP branch that balances production of glycolytic intermediates and pentose (Fig. 2C) and promotes the Warburg effect (35). The small but negative effect of IL-15 on TKTL1 expression, however, at least questions that hypoxic priming prepares NK cells for switching to a Warburg-type metabolism.
Noteworthy, in central carbon metabolism, the TCA cycle is as an important supplier of anabolic precursors as glycolysis and the PPP (36). Pdk1-mediated inhibition of Pdh activity under hypoxic priming, as suggested by our transcriptional data (Figs. 1–3) and as a protective response to low oxygen (33, 34), limits conversion of pyruvate into acetyl-CoA that condenses with oxaloacetate to form citrate fueling the TCA cycle. This and concomitant removal of TCA cycle intermediates for anabolism would quickly bring the cycle to a standstill. At the same time, transcription of the mitochondrial pyruvate carboxylase (PCK2) gene, encoding an enzyme that forms oxaloacetate from pyruvate and CO2 in an anaplerotic reaction was induced by IL-15 also under hypoxia (Figs. 2B and 3). Together, these changes may reflect a concerted adjustment to reduced oxygen supply in NK cells upon priming to ensure continued generation of anabolic intermediates and thus effector capability under hypoxia. It remains, however, unclear how an activating effect of hypoxia on glycolysis and its limiting effect on carbon entry into the TCA cycle are balanced during the transition of NK cells into hypoxic environments and under the influence of regulatory stimuli, including a changing cytokine milieu. We did not observe any changes in cellular protein mass upon priming, neither under normoxia nor hypoxia, as determined by Bradford assay (data not shown). We thus expect possible changes at the proteome level under these conditions to be rather qualitative than quantitative.
In invading myeloid and expanding lymphoid cells, glycolytic switching, at the transcriptional and functional levels, through to a Warburg effect, not only reflects functional adaptation but also instructs cellular fate through improved meeting increased energetic and anabolic demands by supply of metabolic precursors (36–38). In macrophages, dendritic cells, granulocytes, and T-lymphocytes, such glycolytic switching relies on mTor signaling and involves downstream activation of Hif-1α (37, 39–43). Although glycolytic switching in stimulated NK cells, including IL-15 treatment, also requires mTor signaling, activation of Hif-1α by mTor was not formally investigated in these cells (17–20). It is possible that mTor activates Hif-1α in NK cells as well and that this underlies the synergistic IL-15/hypoxia interactions in glycolytic gene transcription described here. Also, activation of other transcription factors in addition to Hif-1α may account for the different transcriptional profiles (Fig. 4).
Notably, Hif-1α-mediated mTor signaling in vitro in other activated immune cells is functional under normoxia (37, 39–43), including CD8+ T cells (44). We thus probed the importance of Hif-1α transcriptional activity in IL-15-treated NK cells for glycolysis under normoxia compared with hypoxia in the presence of CTM, an inhibitor of its transcriptional activity (45), during priming. Glycolysis was consistently inhibited by CTM (Fig. 7A). Possibly, stabilization of Hif-1α under hypoxia partially counteracted inhibition (S9). We expected strongly reduced glycolysis to be much more detrimental to cell viability under hypoxia than normoxia due to the inherent importance of glycolysis for ATP production at low oxygen. However, slightly higher Alamar Blue dye reduction under hypoxia (Fig. 7B) suggested no reduced viability but favored electron transfer to the dye at limited oxygen as a final electron acceptor. In contrast, with cellular ATP as a measure, viability dropped in a CTM concentration-dependent manner (Fig. 7C), which was expected for hypoxia but not to the same extent for normoxia. Nevertheless, flow cytometric apoptosis detection also indicated equal reduction of viability through CTM under normoxia and hypoxia (Fig. 7D). As in macrophages, dendritic cells, granulocytes, and T-lymphocytes (37, 39–43), aerobic glycolysis in primed NK cells thus appears to rely on Hif-1α, but the existence of an mTor/Hif-1α axis in NK cells remains to be tested. Our results also underscore that our ability to provide direct mechanistic insight into the link between Hif-1α signaling/glycolysis and cellular activities of primed NK cells is seriously hindered by the high importance of glycolysis for ATP production and cell viability. The following possible limitation to these conclusions must be mentioned. Although transcriptional inhibition through CTM has been best documented for Hif-1α target genes, its molecular mechanism is through preventing interaction of the Hif-1α C-terminal activation domain with the cysteine histidine-rich domain 1 (CH1) of its co-activator p300 (45). The p300 CH1 domain also interacts with Stat2 (45), for which, however, no role has yet been established in NK cells.
Experimental proof of the importance of glucose utilization and specific portions of central carbon metabolism for anabolism versus the use of exogenously supplemented metabolic precursors, such as amino acids, for example, will require feeding cells with isotope-labeled glucose followed by detection of label incorporation into anabolic intermediates. But given the demonstrated importance of glycolytic switching for immune function in other leukocyte populations (36–38) on the one hand and the presumably negative effect of hypoxia on effector functions in NK cells on the other hand (9–13), we judged it more relevant to focus our current study on assessing the direct impact of short term hypoxia on several activities of quiescent and IL-15-primed NK cells, typically performed in low oxygen tissue environments.
During invasion of inflammatory sites, NK cells become important producers of cytokines, including chemokines that attract further immune cells (46). Therefore, we tested the effects of hypoxia and IL-15 priming on the release of 29 immune factors, including cytokines from NK cells in conditioned culture supernatants. Hypoxia promoted priming-induced but not resting-state secretion of Ccl3/4/5 (Fig. 8, A and B), chemokines that attract activated T-lymphocytes, monocytes, dendritic cells, eosinophils, and more NK cells (47, 48). Hypoxia and/or Hif-1-dependent expression of individual chemokines has previously been demonstrated in nerve tissue (Ccl3) (49, 50), neutrophils and monocytes (CcL4) (51, 52), and splenocytes (53), endothelial cells (54), adipocytes (55), and breast cancer cells (Ccl5) (56) of human and/or rodent origin. Requirement for priming, as seen here, may augment Ccl3/4/5 release from NK cells selectively in hypoxic inflammatory sites.
We further observed that hypoxia enhanced NK cell migration through reconstituted ECM (Matrigel) (Fig. 9). IL-15 was previously shown to promote human NK cell migration through a fibronectin/gelatin matrix only as a positive gradient (chemotaxis) (57). However, a small chemokinetic component to migration through fibronectin was also seen for NK cells treated with Ccl3, Ccl4, or Ccl5 (58). We propose that IL-15-dependent migration enhancement in our assay, i.e. without gradient but in the presence of IL-15-induced Ccl3/4/5, may have partly resulted from increased random cellular motility through autocrine activities of Ccl3/4/5 and/or through enhanced ECM remodeling activity (59, 60).
Additionally, hypoxia increased secretion of Mif (Fig. 8, A and B), a chemokine-like mediator of innate immunity also produced by monocytes, B- and T-lymphocytes, and endocrine, endothelial, and epithelial cells (61). Hypoxia is known to support Mif secretion by endothelial cells (62) and hepatoma and glioma cell lines (63, 64). Notably, ovarian cancer cell-produced Mif interferes with NK cell cytotoxicity through down-regulation of activating NK receptor NKG2D, which likely contributes to Mif-dependent immune tolerance (65). Recently, hypoxia has been shown to also down-regulate NKG2D on NK cells (11, 13). We propose that this was mediated through autocrine activity of Mif released from NK cells under hypoxia.
Last, but not least, we tested the immediate effect of hypoxic NK cell conditioning on their cell killing activity without interference by hypoxia-activated tumor cell intrinsic resistance mechanisms. As target cells, we used K562 cells that are highly susceptible to perforin/granzyme-induced apoptosis, the main cell death pathway employed by NK cells (66–68). Hypoxic preculture slightly reduced target cell lysis only by primed NK cells (Figs. 10 and 11A, bottom left). However, the amounts of the remaining viable, apoptotic, and necrotic target cells are more informative yardsticks for cell killing activity than total cell lysis. In fact, hypoxic NK cell preculture did not significantly affect the numbers of remaining viable target cells in our cell killing assay (Fig. 11B), indicating unchanged apoptosis triggering, but notably it consistently increased the amounts of target cells in late apoptosis. It is known that interactions between dying and phagocytic cells determine whether apoptosis is tolerogenic or immunogenic, whatever is more beneficial to the host. Specifically, an adaptive immune response is elicited by coordinated exposure of damage-associated molecular patterns by late apoptotic and necrotic cells (69–71). It is not known, however, whether NK cells differentially control this process. Our observation of target cell shifting to late apoptosis through hypoxic NK cell preculture leads us to speculate that oxygen availability during NK cell conditioning can modulate target cell death immunogenicity. This would not withstand hypoxia-mediated immunosuppression through tumor and stroma-derived factors (9, 10). In fact, respiratory hyperoxia leads to NK cell-dependent regression of MCA205 pulmonary tumors in mice (72).
In conclusion, we found that culturing NK cells under hypoxia compared with ambient air (normoxia) and IL-15 priming synergistically augmented glycolytic gene expression without major changes in glycolytic flux and glucose consumption. Hif-1α appeared to be equally important for glycolysis and ATP production under normoxia and hypoxia. Hypoxia synergistically supported priming-induced release of Ccl3/4/5. Unexpectedly, hypoxia alone was able to promote secretion of Mif, migration through ECM, and progression to late apoptosis in target cells. The absence of major changes in glycolytic activity argue against an important anabolic effect through priming. ECM/cell interactions during migration and stress molecules on target cells, however, may provide independent signals for functional glycolytic switching that supported functional changes through improved cellular anabolism. The future investigation of central carbon metabolism under hypoxia is warranted to better understand how cellular anabolism is balanced at low oxygen and regulates NK cell priming and further activation. This may lead to strategies for preparing NK cells for adoptive transfer therapy of cancer that overcome current limitations posed by poor tumor homing and by tumor immune evasion (23, 24).
Author Contributions
S. Y. V., D. K., and H. A. L. conceived the study and designed experiments. S. Y. V., D. K., and J. S. performed experimental work. S. Y. V., D. K., J. S., C. S., and H. A. L. analyzed the data. S. Y. V., D. K., M. T., and H. A. L. interpreted the data. H. A. L. drafted the manuscript. M. T. edited the manuscript. All authors approved the final version of the manuscript.
Acknowledgment
We thank Maria Muciek (Affymetrix Core Facility, Medical Research Center Mannheim) for technical support.
The authors declare that they have no conflicts of interest with the contents of this article.
Raw and normalized microarray data are deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/; accession No. GSE70214).
- NK
- natural killer
- 7-AAD
- 7-amino-actinomycin D
- CTM
- chetomin
- ECAR
- extracellular acidification rate
- ECM
- extracellular matrix
- GZMB
- granzyme B
- HIF-1
- hypoxia inducible factor 1
- MIF
- macrophage migration inhibitory factor
- Pdh
- pyruvate dehydrogenase
- PPP
- pentose phosphate pathway
- STAT
- signal transducer and activator of transcription
- TCA
- tricarboxylic acid or citric acid
- GSEA
- Gene Set Enrichment Analysis
- mTOR
- mechanistic target of rapamycin
- CFSE
- carboxyfluorescein succinimidyl ester
- FDR
- false discovery rate.
References
- 1. Vivier E., Raulet D. H., Moretta A., Caligiuri M. A., Zitvogel L., Lanier L. L., Yokoyama W. M., and Ugolini S. (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smyth M. J., Cretney E., Kelly J. M., Westwood J. A., Street S. E., Yagita H., Takeda K., van Dommelen S. L., Degli-Esposti M. A., and Hayakawa Y. (2005) Activation of NK cell cytotoxicity. Mol. Immunol. 42, 501–510 [DOI] [PubMed] [Google Scholar]
- 3. Bosco M. C., Puppo M., Blengio F., Fraone T., Cappello P., Giovarelli M., and Varesio L. (2008) Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 213, 733–749 [DOI] [PubMed] [Google Scholar]
- 4. Ohta A., Diwanji R., Kini R., Subramanian M., Ohta A., and Sitkovsky M. (2011) In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front. Immunol. 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spencer J. A., Ferraro F., Roussakis E., Klein A., Wu J., Runnels J. M., Zaher W., Mortensen L. J., Alt C., Turcotte R., Yusuf R., Côté D., Vinogradov S. A., Scadden D. T., and Lin C. P. (2014) Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508, 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carreau A., El Hafny-Rahbi B., Matejuk A., Grillon C., and Kieda C. (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 15, 1239–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sitkovsky M., and Lukashev D. (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat. Rev. Immunol. 5, 712–721 [DOI] [PubMed] [Google Scholar]
- 8. Eltzschig H. K., and Carmeliet P. (2011) Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Casazza A., Di Conza G., Wenes M., Finisguerra V., Deschoemaeker S., and Mazzone M. (2014) Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 33, 1743–1754 [DOI] [PubMed] [Google Scholar]
- 10. Baginska J., Viry E., Paggetti J., Medves S., Berchem G., Moussay E., and Janji B. (2013) The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front. Immunol. 4, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balsamo M., Manzini C., Pietra G., Raggi F., Blengio F., Mingari M. C., Varesio L., Moretta L., Bosco M. C., and Vitale M. (2013) Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 43, 2756–2764 [DOI] [PubMed] [Google Scholar]
- 12. Fink T., Ebbesen P., Koppelhus U., and Zachar V. (2003) Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand. J. Immunol. 58, 607–612 [DOI] [PubMed] [Google Scholar]
- 13. Sarkar S., Germeraad W. T., Rouschop K. M., Steeghs E. M., van Gelder M., Bos G. M., and Wieten L. (2013) Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 8, e64835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Long E. O., Kim H. S., Liu D., Peterson M. E., and Rajagopalan S. (2013) Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 31, 227–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mishra A., Sullivan L., and Caligiuri M. A. (2014) Molecular pathways: interleukin-15 signaling in health and in cancer. Clin. Cancer Res. 20, 2044–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van den Bergh J. M., Van Tendeloo V. F., and Smits E. L. (2015) Interleukin-15: new kid on the block for antitumor combination therapy. Cytokine Growth Factor Rev. 26, 15–24 [DOI] [PubMed] [Google Scholar]
- 17. Donnelly R. P., Loftus R. M., Keating S. E., Liou K. T., Biron C. A., Gardiner C. M., and Finlay D. K. (2014) mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 193, 4477–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nandagopal N., Ali A. K., Komal A. K., and Lee S. H. (2014) The critical role of IL-15-PI3K-mTOR pathway in natural killer cell effector functions. Front. Immunol. 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marçais A., Cherfils-Vicini J., Viant C., Degouve S., Viel S., Fenis A., Rabilloud J., Mayol K., Tavares A., Bienvenu J., Gangloff Y. G., Gilson E., Vivier E., and Walzer T. (2014) The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 15, 749–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keppel M. P., Saucier N., Mah A. Y., Vogel T. P., and Cooper M. A. (2015) Activation-specific metabolic requirements for NK Cell IFN-γ production. J. Immunol. 194, 1954–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knorr D. A., Bachanova V., Verneris M. R., and Miller J. S. (2014) Clinical utility of natural killer cells in cancer therapy and transplantation. Semin. Immunol. 26, 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoon S. R., Kim T. D., and Choi I. (2015) Understanding of molecular mechanisms in natural killer cell therapy. Exp. Mol. Med. 47, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Childs R. W., and Carlsten M. (2015) Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat. Rev. Drug Discov. 14, 487–498 [DOI] [PubMed] [Google Scholar]
- 24. Domogala A., Madrigal J. A., and Saudemont A. (2015) Natural killer cell immunotherapy: from bench to bedside. Front. Immunol. 6, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim T. D., Lee S. U., Yun S., Sun H. N., Lee S. H., Kim J. W., Kim H. M., Park S. K., Lee C. W., Yoon S. R., Greenberg P. D., and Choi I. (2011) Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood 118, 5476–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dai M., Wang P., Boyd A. D., Kostov G., Athey B., Jones E. G., Bunney W. E., Myers R. M., Speed T. P., Akil H., Watson S. J., and Meng F. (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edsparr K., Johansson B. R., Goldfarb R. H., Basse P. H., Nannmark U., Speetjens F. M., Kuppen P. J., Lennernäs B., and Albertsson P. (2009) Human NK cell lines migrate differentially in vitro related to matrix interaction and MMP expression. Immunol. Cell Biol. 87, 489–495 [DOI] [PubMed] [Google Scholar]
- 29. Bentz S., Cee A., Endlicher E., Wojtal K. A., Naami A., Pesch T., Lang S., Schubert P., Fried M., Weber A., Coy J. F., Goelder S., Knüchel R., Hausmann M., and Rogler G. (2013) Hypoxia induces the expression of transketolase-like 1 in human colorectal cancer. Digestion 88, 182–192 [DOI] [PubMed] [Google Scholar]
- 30. Semenza G. L. (2009) Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 24, 97–106 [DOI] [PubMed] [Google Scholar]
- 31. Yalcin A., Telang S., Clem B., and Chesney J. (2009) Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 86, 174–179 [DOI] [PubMed] [Google Scholar]
- 32. Telang S., Clem B. F., Klarer A. C., Clem A. L., Trent J. O., Bucala R., and Chesney J. (2012) Small molecule inhibition of 6-phosphofructo-2-kinase suppresses T cell activation. J. Transl. Med. 10, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J. W., Tchernyshyov I., Semenza G. L., and Dang C. V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 [DOI] [PubMed] [Google Scholar]
- 34. Papandreou I., Cairns R. A., Fontana L., Lim A. L., and Denko N. C. (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 [DOI] [PubMed] [Google Scholar]
- 35. Grimm M., Schmitt S., Teriete P., Biegner T., Stenzl A., Hennenlotter J., Muhs H. J., Munz A., Nadtotschi T., König K., Sänger J., Feyen O., Hofmann H., Reinert S., and Coy J. F. (2013) A biomarker based detection and characterization of carcinomas exploiting two fundamental biophysical mechanisms in mammalian cells. BMC Cancer 13, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buck M. D., O'Sullivan D., and Pearce E. L. (2015) T cell metabolism drives immunity. J. Exp. Med. 212, 1345–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palazon A., Goldrath A. W., Nizet V., and Johnson R. S. (2014) HIF transcription factors, inflammation, and immunity. Immunity 41, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pearce E. L., and Pearce E. J. (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ganeshan K., and Chawla A. (2014) Metabolic regulation of immune responses. Annu. Rev. Immunol. 32, 609–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghesquière B., Wong B. W., Kuchnio A., and Carmeliet P. (2014) Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176 [DOI] [PubMed] [Google Scholar]
- 41. Roiniotis J., Dinh H., Masendycz P., Turner A., Elsegood C. L., Scholz G. M., and Hamilton J. A. (2009) Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J. Immunol. 182, 7974–7981 [DOI] [PubMed] [Google Scholar]
- 42. Horng T. (2015) mTOR trains heightened macrophage responses. Trends Immunol. 36, 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pearce E. J., and Everts B. (2015) Dendritic cell metabolism. Nat. Rev. Immunol. 15, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finlay D. K., Rosenzweig E., Sinclair L. V., Feijoo-Carnero C., Hukelmann J. L., Rolf J., Panteleyev A. A., Okkenhaug K., and Cantrell D. A. (2012) PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kung A. L., Zabludoff S. D., France D. S., Freedman S. J., Tanner E. A., Vieira A., Cornell-Kennon S., Lee J., Wang B., Wang J., Memmert K., Naegeli H. U., Petersen F., Eck M. J., Bair K. W., et al. (2004) Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 6, 33–43 [DOI] [PubMed] [Google Scholar]
- 46. Maghazachi A. A. (2010) Role of chemokines in the biology of natural killer cells. Curr. Top. Microbiol. Immunol. 341, 37–58 [DOI] [PubMed] [Google Scholar]
- 47. Maurer M., and von Stebut E. (2004) Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 36, 1882–1886 [DOI] [PubMed] [Google Scholar]
- 48. Grayson M. H., and Holtzman M. J. (2006) Chemokine complexity: the case for CCL5. Am. J. Respir. Cell Mol. Biol. 35, 143–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cowell R. M., Xu H., Galasso J. M., and Silverstein F. S. (2002) Hypoxic-ischemic injury induces macrophage inflammatory protein-1α expression in immature rat brain. Stroke 33, 795–801 [DOI] [PubMed] [Google Scholar]
- 50. Yoshida S., Yoshida A., Ishibashi T., Elner S. G., and Elner V. M. (2003) Role of MCP-1 and MIP-1α in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J. Leukocyte Biol. 73, 137–144 [DOI] [PubMed] [Google Scholar]
- 51. Gonsalves C., and Kalra V. K. (2010) Endothelin-1-induced macrophage inflammatory protein-1β expression in monocytic cells involves hypoxia-inducible factor-1α and AP-1 and is negatively regulated by microRNA-195. J. Immunol. 185, 6253–6264 [DOI] [PubMed] [Google Scholar]
- 52. Walmsley S. R., Print C., Farahi N., Peyssonnaux C., Johnson R. S., Cramer T., Sobolewski A., Condliffe A. M., Cowburn A. S., Johnson N., and Chilvers E. R. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J. Exp. Med. 201, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arnaud C., Beguin P. C., Lantuejoul S., Pepin J. L., Guillermet C., Pelli G., Burger F., Buatois V., Ribuot C., Baguet J. P., Mach F., Levy P., and Dematteis M. (2011) The inflammatory preatherosclerotic remodeling induced by intermittent hypoxia is attenuated by RANTES/CCL5 inhibition. Am. J. Respir. Crit. Care Med. 184, 724–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yeligar S. M., Machida K., Tsukamoto H., and Kalra V. K. (2009) Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-κB, HIF-1α, and AP-1. J. Immunol. 183, 5964–5976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skurk T., Mack I., Kempf K., Kolb H., Hauner H., and Herder C. (2009) Expression and secretion of RANTES (CCL5) in human adipocytes in response to immunological stimuli and hypoxia. Horm. Metab. Res. 41, 183–189 [DOI] [PubMed] [Google Scholar]
- 56. Lin S., Wan S., Sun L., Hu J., Fang D., Zhao R., Yuan S., and Zhang L. (2012) Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci. 103, 904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Allavena P., Giardina G., Bianchi G., and Mantovani A. (1997) IL-15 is chemotactic for natural killer cells and stimulates their adhesion to vascular endothelium. J. Leukocyte Biol. 61, 729–735 [DOI] [PubMed] [Google Scholar]
- 58. Taub D. D., Sayers T. J., Carter C. R., and Ortaldo J. R. (1995) α and β chemokines induce NK cell migration and enhance NK-mediated cytolysis. J. Immunol. 155, 3877–3888 [PubMed] [Google Scholar]
- 59. Edsparr K., Basse P. H., Goldfarb R. H., and Albertsson P. (2011) Matrix metalloproteinases in cytotoxic lymphocytes impact on tumour infiltration and immunomodulation. Cancer Microenviron. 4, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prakash M. D., Munoz M. A., Jain R., Tong P. L., Koskinen A., Regner M., Kleifeld O., Ho B., Olson M., Turner S. J., Mrass P., Weninger W., and Bird P. I. (2014) Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity 41, 960–972 [DOI] [PubMed] [Google Scholar]
- 61. Grieb G., Merk M., Bernhagen J., and Bucala R. (2010) Macrophage migration inhibitory factor (MIF): a promising biomarker. Drug News Perspect. 23, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Simons D., Grieb G., Hristov M., Pallua N., Weber C., Bernhagen J., and Steffens G. (2011) Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J. Cell. Mol. Med. 15, 668–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hira E., Ono T., Dhar D. K., El-Assal O. N., Hishikawa Y., Yamanoi A., and Nagasue N. (2005) Overexpression of macrophage migration inhibitory factor induces angiogenesis and deteriorates prognosis after radical resection for hepatocellular carcinoma. Cancer 103, 588–598 [DOI] [PubMed] [Google Scholar]
- 64. Bacher M., Schrader J., Thompson N., Kuschela K., Gemsa D., Waeber G., and Schlegel J. (2003) Up-regulation of macrophage migration inhibitory factor gene and protein expression in glial tumor cells during hypoxic and hypoglycemic stress indicates a critical role for angiogenesis in glioblastoma multiforme. Am. J. Pathol. 162, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Krockenberger M., Dombrowski Y., Weidler C., Ossadnik M., Hönig A., Häusler S., Voigt H., Becker J. C., Leng L., Steinle A., Weller M., Bucala R., Dietl J., and Wischhusen J. (2008) Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D. J. Immunol. 180, 7338–7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Berthou C., Bourge J. F., Zhang Y., Soulié A., Geromin D., Denizot Y., Sigaux F., and Sasportes M. (2000) Interferon-γ-induced membrane PAF-receptor expression confers tumor cell susceptibility to NK perforin-dependent lysis. Blood 95, 2329–2336 [PubMed] [Google Scholar]
- 67. Kurschus F. C., Bruno R., Fellows E., Falk C. S., and Jenne D. E. (2005) Membrane receptors are not required to deliver granzyme B during killer cell attack. Blood 105, 2049–2058 [DOI] [PubMed] [Google Scholar]
- 68. Martinvalet D., Zhu P., and Lieberman J. (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370 [DOI] [PubMed] [Google Scholar]
- 69. Peter C., Wesselborg S., Herrmann M., and Lauber K. (2010) Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 15, 1007–1028 [DOI] [PubMed] [Google Scholar]
- 70. Kepp O., Galluzzi L., Martins I., Schlemmer F., Adjemian S., Michaud M., Sukkurwala A. Q., Menger L., Zitvogel L., and Kroemer G. (2011) Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 30, 61–69 [DOI] [PubMed] [Google Scholar]
- 71. Bezu L., Gomes-de-Silva L. C., Dewitte H., Breckpot K., Fucikova J., Spisek R., Galluzzi L., Kepp O., and Kroemer G. (2015) Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 6, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hatfield S. M., Kjaergaard J., Lukashev D., Schreiber T. H., Belikoff B., Abbott R., Sethumadhavan S., Philbrook P., Ko K., Cannici R., Thayer M., Rodig S., Kutok J. L., Jackson E. K., Karger B., et al. (2015) Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 7, 277ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]