

Abstract
Crystallographic studies of insulin bound to receptor domains have defined the primary hormone-receptor interface. We investigated the role of TyrB26, a conserved aromatic residue at this interface. To probe the evolutionary basis for such conservation, we constructed 18 variants at B26. Surprisingly, non-aromatic polar or charged side chains (such as Glu, Ser, or ornithine (Orn)) conferred high activity, whereas the weakest-binding analogs contained Val, Ile, and Leu substitutions. Modeling of variant complexes suggested that the B26 side chains pack within a shallow depression at the solvent-exposed periphery of the interface. This interface would disfavor large aliphatic side chains. The analogs with highest activity exhibited reduced thermodynamic stability and heightened susceptibility to fibrillation. Perturbed self-assembly was also demonstrated in studies of the charged variants (Orn and Glu); indeed, the GluB26 analog exhibited aberrant aggregation in either the presence or absence of zinc ions. Thus, although TyrB26 is part of insulin's receptor-binding surface, our results suggest that its conservation has been enjoined by the aromatic ring's contributions to native stability and self-assembly. We envisage that such classical structural relationships reflect the implicit threat of toxic misfolding (rather than hormonal function at the receptor level) as a general evolutionary determinant of extant protein sequences.
Keywords: diabetes, hormone, non-standard mutagenesis, protein structure, receptor-tyrosine kinase
Introduction
Insulin, a small globular protein critical to the maintenance of metabolic homeostasis (1), provides a classical model for studies of protein structure and self-assembly (2) with long-standing application to human therapeutics (3). Insulin contains two polypeptide chains, designated A and B, that are linked by two disulfide bridges (cystines A7-B7 and A20-B19); the A chain is further stabilized by an intrachain bridge (cystine A6-A11). In pancreatic β-cells, insulin is stored within glucose-regulated secretory granules as zinc-coordinated hexamers (Fig. 1A). The hormone binds to a receptor-tyrosine kinase, designated the insulin receptor (IR).5 The product of a single gene, the IR precursor is processed in the trans-Golgi network into its final disulfide-linked (αβ)2 homodimer conformation. The extracellular α subunit contains hormone-binding elements, whereas the transmembrane β subunit contains the intracellular tyrosine kinase domain (4). Alternative splicing leads to two receptor isoforms, IR-A and IR-B (5). The three-dimensional structure of the holoreceptor has been visualized only at low resolution (>20 Å), but these findings have been inconclusive (for review, see Ref. 6). How extracellular binding of insulin alters the structure of the receptor, leading in turn to activation of the intracellular tyrosine kinase domains, is a major unsolved problem (7).
FIGURE 1.

Structure of insulin and the ectodomain of the IR. A, assembly of the zinc insulin coordinated WT hexamer. The insulin monomer (A and B chains) forms zinc-free dimers via anti-parallel association of B-chain α-helices and C-terminal β-strands (brown); two zinc ions then mediate assembly of three dimers to form a classical hexamer (T6). The A chain is shown in yellow (ribbon representation), and the B chain is in beige (B1-B19) or brown (B20-B30). The conserved aromatic residues of PheB24 and PheB25 are shown as black sticks, whereas TyrB26 is red. B, inverted V-shaped assembly of IR ectodomain homodimer. One monomer is in ribbon representation (labeled), the second is in surface representation. Domains are labeled as follows: L1, first Leu-rich repeat domain; CR, Cys-rich domain; L2, second Leu-rich repeat domain. αCT, α-chain C-terminal segment. C, model of WT insulin in its receptor-free conformation overlaid onto the structure of the insulin-bound μIR (4). L1 and part of CR are shown in powder blue; αCT is shown in purple. Residues PheB24, PheB25, and TyrB26 are as in panel A. The B chain of μIR-bound insulin is shown in black (B6-B19); the brown tube indicates classical location within the overlay of residues B20-B30 of insulin in its receptor-free conformation, highlighting steric clash of B26-B30 with αCT. Coordinates were obtained from PDB entries 4INS, 4ZXB, and 3W11.
Dissection of the IR into discrete domains has enabled crystallographic analysis of its parts. The relevant structural biology is as follows. (i) Structures of the intracellular tyrosine kinase domains at 1.9 Å resolution have been obtained, and their mode of interaction has been determined (8, 9). (ii) The structure of the N-terminal three domains of the α subunit (Leu-rich domain 1, Cys-rich domain, and Leu-rich domain 2) has been determined at 2.3 Å (10) and is similar to the homologous fragment of the type 1 insulin-like growth factor receptor (11). (iii) A lower resolution structure (3.8 Å) has been determined of the dimeric ectodomain (containing the entire α subunit and the extracellular portion of the β subunit; (αβΔ)2). This structure (Fig. 1B) enabled the orientation of the Leu-rich domain 1, Cys-rich domain, and Leu-rich domain 2 fragment to be determined relative to fibronectin-III homology domains 1, 2, and 3 (FnIII-1, FnIII-2, FnIII-3). An insert domain within FnIII-2, including the insulin-binding element (αCT) within the C-terminal region of the α subunit, was incompletely traced in the electron-density map. The overall conformation of the (αβΔ)2 ectodomain resembles an inverted-V in which the presumed high-affinity site of insulin binding lay within the crux of the dimer (12). An improved crystallographic model of the ectodomain has recently been described (12).
A recent structural advance exploited domain-minimized models of the α subunit containing the primary insulin-binding elements L1 and αCT. Notably, a co-crystal structure has been determined at 3.5 Å resolution of a ternary complex between insulin, an L1-CR fragment, and a synthetic αCT peptide spanning residues 704–719 of IR-A (4). In this structure (designated the micro-receptor (μIR) complex) the C-terminal segment of the insulin B chain is detached from the hormone's α-helical core; such detachment is incompatible with classical structures of insulin (Fig. 1C) but enables insertion of this segment (including TyrB26; red and asterisk in Fig. 2A) between the conserved surfaces of L1 and the αCT peptide (Fig. 2, B and C). This mode of binding, long anticipated based on studies of anomalous insulin analogs (13, 14) and residue-specific photo-cross-linking (15), has defined the binding surfaces in the μIR for insulin's conserved triplet of aromatic residues, PheB24, PheB25, and TyrB26 (Fig. 2A). Surprisingly, whereas the side chain of PheB24 inserts within a classical nonpolar pocket (anchoring the displaced B-chain β-strand; Ref. 16), the B25 and B26 side chains appear less closely packed (Fig. 2, B–D). The latter side chains, despite their broad conservation (17) and efficient IR photo-cross-linking as photo-activatable derivatives (15, 18, 19), appear to contact the μIR surface only loosely.
FIGURE 2.

Insulin sequence and μIR complex. A, sequence of WT insulin and sites of modification. A and B chains are shown in white and gray, respectively. Conserved aromatic residues PheB24 and PheB25 are highlighted as black circles. The present study focused on substitutions of TyrB26 (red circle); additional substitutions were made at position B29 (Orn; encircled X) to facilitate semi-synthesis. B, stick representation of residues B20-B27 (carbon atoms (green), nitrogen atoms (blue), and oxygen atoms (red) packed between αCT and the L1-β2 strand. B-chain residues B8-B19 are shown as a black ribbon, and the A chain is shown as a yellow ribbon; residues A1-A3 are concealed behind the surface of αCT. Key contact surfaces of αCT with B24-B26 are highlighted in magenta and of L1 with B24-B26 are highlighted in cyan; L1 and αCT surfaces not in interaction with B24-B26 are shown in lighter shades. Insertion of the B20-B27 segment between L1 and αCT is associated with a small rotation of the B20-B23 β-turn and changes in main-chain dihedral angles flanking PheB24 (4). C, orthogonal view to B, showing interaction of the side chain of PheB24 with the nonpolar surface of the L1-β2 sheet. TyrB26 is hidden below the surface of αCT. Engagement of conserved residues A1-A3 against the nonpolar surface of αCT is shown at the top. D, environment of TyrB26 within Site 1 complex (stereo). Neighboring side chains in L1 and αCT are as labeled. Coordinates were obtained from PDB entry 4OGA.
Our interest in TyrB26 was motivated by its broad conservation among vertebrate insulins (17) and insulin-like growth factors (IGF-I and IGF-II) (20, 21). Classical structure-activity relationships at B26 are complex, presumably because of a subtle interplay between direct effects of modifications at the hormone-receptor interface and indirect effects of conformational changes at this interface. Whereas substitution of TyrB26 by Phe impairs IR affinity by 2-fold (22), a finding consistent with a specific (although modest) contribution by the para-OH group, studies of an Ala substitution have given rise to conflicting results (23–25). Although apparently dispensable in a truncated yet active insulin analog lacking residues B26-B30 (des-pentapeptide[B26-B30]-insulin-amide; Refs. 26 and 27), mutations or non-standard modifications at B26 in full-length or truncated insulin analogs may impair or enhance activity (28, 29). Thus, no coherent pharmacophore has been obtained.
To clarify the contribution of TyrB26 to receptor binding, we undertook a systematic mutational survey based on the semi-synthetic preparation of 18 insulin analogs. In accordance with the peripheral B26-related interface in the μIR complex (4), this survey revealed that high IR affinity can be conferred by non-aromatic polar or charged side chains (either acidic or basic). Although well tolerated at the receptor interface, these substitutions were observed to impair the hormone's thermodynamic stability, self-assembly, and susceptibility to fibrillation. Our results thus suggest that, among the 20 natural amino acids, the wild type TyrB26 best meets the simultaneous challenges of biological activity, protein stability, self-assembly, and protection from toxic misfolding (30). Such co-optimization highlights the multidimensional biophysical and biological landscape of protein evolution.
Experimental Procedures
Preparation of Insulin Analogs
Analogs were made by trypsin-catalyzed semi-synthesis using an insulin fragment, des-octapeptide[B23-B30]-insulin and modified octapeptides as described (23). The des-octapeptide[B23-B30]-insulin was generated via cleavage of human insulin with trypsin and purified by reverse-phase high performance liquid chromatography (HPLC); octapeptides were synthesized by solid-phase synthesis (31). The formation of a peptide bond between ArgB22 and a synthetic octapeptide was mediated by trypsin (in a mixed solvent system containing 1,4-butanediol and dimethylacetamide) as previously described (32). Insulin analogs were purified by preparative reverse-phase C4 HPLC (Higgins Analytical Inc., Proto 300 C4 10 μm, 250 × 20 mm), and their purity was assessed by an analytical reverse-phase C4 HPLC (Higgins Analytical Inc., Proto 300 C4 5 μm, 250 × 4.6 mm). Molecular masses of purified analogs were verified using an Applied Biosystems 4700 proteomics analyzer (matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry; MALDI-TOF MS).
Receptor-binding Assays
Affinities for IR-B were measured by a competitive-displacement scintillation proximity assay. This assay employed detergent-solubilized holo receptor with C-terminal streptavidin-binding protein tags purified by sequential wheat germ agglutinin and Strep-Tactin-affinity chromatography from detergent lysates of polyclonal stably transfected 293PEAK cell lines expressing each receptor. A dilution series of human insulin (a generous gift from Novo-Nordisk A/S, Bagsværd, Denmark) or analog (11 dilutions, 5-fold each with a maximum initial concentration of 2 μm) in 100 μl of binding buffer (100 mm HEPES (pH 7.8), 100 mm NaCl, 10 mm MgSO4, 0.025% (v/v) Tween 20, and 0.5% (w/v) bovine serum albumin) was made in a 96-well plate (Costar). The assay was initiated by the addition to the wells of a premixed solution containing (i) wheat germ agglutinin scintillation proximity assay (SPA) beads (PerkinElmer Life Sciences), (ii) solubilized receptor, and (iii) 125I-TyrA14-insulin in binding buffer. The final concentration of 125I-labeled ligand was 7.5 pm, and the amount of receptor added was adjusted so that the extent of labeled ligand binding in the absence of competitor was <15% of the total added counts in order to avoid ligand-depletion artifacts. Plates were incubated with gentle shaking for 24 h at room temperature, centrifuged, and counted for 5 min/well in a 12-detector Trilux scintillation counter (PerkinElmer Life Sciences/Wallac). To obtain analog dissociation constants, competitive binding data were analyzed by non-linear regression by the method of Wang (33), a model that provides an analytical solution for the binding of two ligands to a single receptor.
Receptor-binding Screening Protocol
The ability of insulin analogs to displace bound 125I-TyrA14-insulin from antibody-immobilized wheat germ agglutinin-purified receptor was tested at an analog concentration of 0.75 nm. This concentration corresponded to displacement of 95% of receptor-bound 125I-TyrA14-insulin by the control analog, OrnB29-insulin. The fraction of 125I-TyrA14-insulin displaced by a given analog permitted assignment to the following three categories: <60% (low affinity), 60–80% (intermediate affinity), or >80% (high affinity).
Insulin Self-assembly
Oligomeric states of the insulin analogs were monitored by size-exclusion chromatography (SEC) using HPLC. The exclusion void volume (Vo) was established with apoferritin (443 kDa). Insulin analogs were made 0.6 mm in a buffer consisting of 25 mm Tris-HCl (pH 7.4), 0.65 mg/ml phenol, 1.6 mg/ml meta-cresol, 16 mg/ml glycerol, and ZnCl2 at a ratio of 2 zinc ions per insulin hexamer. For zinc-free conditions, analogs were also made 0.6 mm in phosphate-buffered saline (pH 7.4). Protein samples (volume 10 μl) were applied through a Waters 717 autosampler onto a Zenix-C SEC-150 column (Sepax Technologies, Southborough, MA) with a nominal fractionation range of 0.5–150 kDa. Proteins were fractionated at a flow rate of 1 ml/min using a Waters Binary HPLC system. Protein elution was monitored at 215 and 280 nm using a dual-lambda Waters 2487 absorbance detector. The mobile phase consisted of 10 mm Tris-HCl (pH 7.4) and 140 mm NaCl with or without 0.3 mm ZnCl2 and 50 mm cyclohexanol; the latter provided a non-aromatic replacement for the phenolic compounds often employed as R6-hexamer stabilizing- and anti-microbial agents in pharmaceutical formulations (34–36). Data acquisition and processing utilized Waters HPLC Empower® software. The column was calibrated for apparent molecular mass determination by fractionating standard proteins individually on the column.
TR Transition and R6 Co2+ Kinetic Assay
Visible absorption spectroscopy was used to probe the formation and disassembly of phenol-stabilized R6 Co2+-substituted insulin hexamers. Insulin analogs were made 0.6 mm in a buffer containing 50 mm Tris-HCl (pH 7.4), 50 mm phenol, 0.2 mm CoCl2, and 2 mm NaSCN. Sample pH was readjusted in each case to 7.4, and samples were incubated overnight at room temperature before the studies to ensure that a conformational equilibrium was reached. Spectra (400–750 nm) were obtained to monitor tetrahedral Co2+ coordination with its signature peak absorption band at 574 nm (37). To assess variation in the amplitude and λmax of the d-d transition band, the wild-type (WT) spectrum was obtained in four separate preparations containing three replicates each from solutions that were independently prepared; respective S.D. were ±3.7% and 0.5 nm. To determine the rate of Co2+ release from the hexamers, metal ion sequestration was initiated at 25 °C by the addition of an aliquot of EDTA (50 mm at pH 7.4) to a final concentration of 2 mm; this yielded a molar ratio of EDTA per formal insulin trimer of 6.7. Attenuation of the 574-nm absorption band was monitored on a timescale of seconds to hours. Post-dissociation absorption spectra (400–750 nm) were observed to confirm complete attenuation of the 574-nm absorption band (38). Kinetics data were fit to mono-exponential decay functions to determine dissociation rate and half-life of Co2+-coordinated R6 hexamers. Independent replicates of the EDTA assay of WT insulin (using freshly prepared stock solutions in each case) yielded a S.D. of 15% (±69 s) in the lifetime.
Circular Dichroism
Far-ultraviolet (UV) CD spectra were obtained using an AVIV spectropolarimeter equipped with an automated syringe-driven titration unit. Wild type insulin or insulin analogs were made 50 μm in 10 mm potassium phosphate (pH 7.4) and 50 mm KCl. Spectra were obtained from 190–250 nm as described (39). Thermodynamic stabilities were probed by guanidine hydrochloride-induced denaturation monitored by CD at helix-sensitive wavelength 222 nm. Data were fit by nonlinear least squares to a two-state model (40),
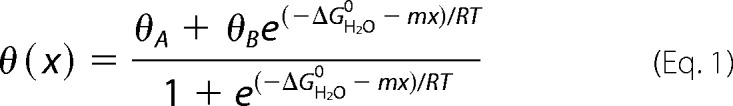 |
where x is the concentration of guanidine hydrochloride, and θA,B represents respective estimates of the baseline ellipticities of the protein in its native and unfolded states as extrapolated to a guanidine concentration of 0 m. Baseline values were approximated via pre- and post-transition lines represented by equations θA(x) = θAH2O + mAx, and θB(x) = θBH2O + mBx. Such simultaneous fitting avoids artifacts of linear plots of ΔG versus concentration of denaturant (41).
Assessment of Fibril Formation
Insulin or insulin analogs were made 60 μm in phosphate-buffered saline (pH 7.4) containing 0.1% sodium azide and gently rocked at 37 °C in glass vials in the presence of a liquid/air interface. Aliquots were taken at regular intervals and frozen for later analysis of thioflavin T fluorescence. The assay was terminated on visual appearance of cloudiness (42). Statistical significance of differences in lag times among analogs was evaluated using Student's t test.
Results
Receptor-binding Studies Defined Three Classes of Analogs
18 insulin analogs containing substitutions at B26 were prepared at small scale (Table 1). To eliminate the B29 tryptic site (and so facilitate semi-synthesis; Ref. 43), the analogs each contained OrnB29 (in place of LysB29). Similarly, to provide a basic side chain at B26, an analog was prepared containing both substitutions OrnB26 and OrnB29. CysB26-OrnB29-insulin was not prepared to avoid possible disulfide interchange and/or formation of covalent dimers.
TABLE 1.
Receptor-binding affinities of insulin analogs
| B26 residuea | Kd | B26 residuea | Kd |
|---|---|---|---|
| nm | nm | ||
| Tyra | 0.042 ± 0.007 | Pro | Ic |
| Gly | Hd | Ser | 0.021 ± 0.003 |
| Ala | 0.042 ± 0.007 | Thr | I |
| Val | 0.12 ± 0.02 | Cys | –e |
| Leu | 1.2 ± 0.2 | Asn | H |
| Ile | 0.52 ± 0.08 | Asp | H |
| Met | I | Gln | 0.043 ± 0.007 |
| Glu | 0.021 ± 0.002 | His | H |
| Phe | 0.10 ± 0.020 | Lys/Arg | –f |
| Trp | H | Orng | 0.038 ± 0.006 |
a Analogs were prepared in a template in which LysB29 was substituted by Orn unless otherwise noted. Assays employed the B isoform of the purified and detergent-solubilized IR as described under “Experimental Procedures.”
b This represents OrnB29-insulin; the dissociation constant of WT insulin under these conditions was 0.040 ± 0.006 nm.
c I represents the intermediate affinity group in the initial coarse screening (Fig. 3B).
d H represents the high affinity group in the initial coarse screening (Fig. 3B).
e Not determined as the CysB26 analog was not prepared.
f Not determined as substitution of TyrB26 by Lys or Arg would have complicated semi-synthesis (see “Experimental Procedures”).
g OrnB26 provided a model of a basic side chain.
A coarse receptor-binding assay (using IR-B) was first undertaken that enabled subgroups of the insulin analogs to be distinguished based on displacement of pre-bound 125I-labeled insulin at a uniform analog concentration of 0.75 nm (Fig. 3A). At this concentration WT insulin displaced 95% of the prebound tracer (125I-TyrA14-insulin; see “Experimental Procedures”). The results defined three classes (Fig. 3B): (i) high affinity (tracer displacement >80%; i.e. similar or greater than that observed on binding of WT insulin); (ii) intermediate affinity (tracer displacement 60–80%), and (iii) low affinity (tracer displacement <60%). The low affinity class contained two aliphatic residues (Ile and Leu). The intermediate affinity class comprised a diverse set of residues, including Phe, Met, Pro, Thr, and Val; the remaining analogs (representing 10 of the 18 analogs tested) were placed in the high-affinity group.
FIGURE 3.
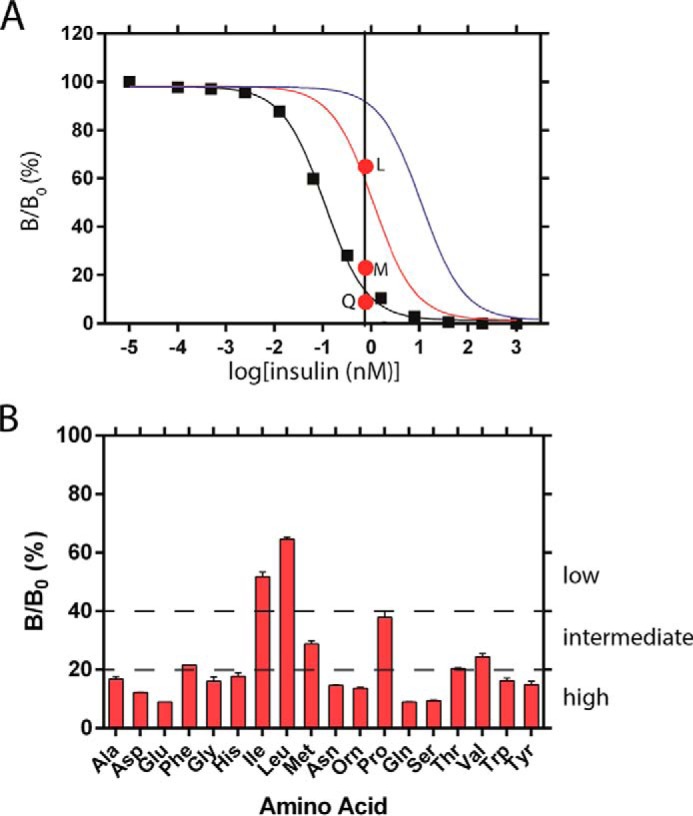
Functional screening of insulin analogs. A, competitive receptor-binding assay of OrnB29-insulin (black squares; the line indicates fitted model); its Kd is estimated to be 0.042 (±0.007) nm. Model curves simulated based on Kd values that are 10-fold (red) or 100-fold (blue) greater than that of OrnB29-insulin are also shown. Red dots indicate binding of insulin analogs LeuB26-OrnB29 (top), MetB26-OrnB29 (middle), and GlnB26-OrnB29 (bottom) at a concentration of 0.75 nm. Vertical axis: B/B0 where B is 125I-TyrA14-insulin bound by receptor at the designated insulin concentration, and B0 is 125I-TyrA14-insulin bound by receptor in the absence of unlabeled insulin. B, coarse screening at analog concentration of 0.75 nm. The analogs were classified as being of low, intermediate, or high affinity depending on the degree of 125I-TyrA14-insulin displacement.
Definitive IR-B-binding assays were then undertaken of selected high, intermediate, and low affinity analogs (Table 1) using a scintillation proximity assay performed with purified detergent-solubilized receptor isoform expressed in the same cell line. Affinities greater than WT insulin were conferred only by SerB26 and GluB26 (Kd ∼ 0.02 nm), whereas the affinities conferred by TyrB26, AlaB26, and OrnB26 were indistinguishable (Kd ∼ 0.04 nm). The high affinities of AlaB26 and GluB26 insulin analogs have previously been reported (3, 25). Also in accordance with past studies (22), substitution of TyrB26 by Phe reduced affinity (between 2- and 3-fold). Whereas a similar reduction was conferred by ValB26, substitution of TyrB26 by Ile or Leu led to more severe impairments (by ∼10-fold and 30-fold, respectively).
Two sets of observations were particularly striking: (a) the high affinity of analogs containing charged side chains (of either sign) or a short polar side chain (Ser) at B26 and (b) the functional incompatibility of aliphatic substitutions larger than Ala at B26. Although this pattern would in general be unexpected on systematic mutagenesis of a conserved aromatic residue in a globular protein (and indeed stands in contrast to results of substitutions at insulin positions TyrB16 and PheB24; Refs. 13, 16, 22), the B26-contacting surface within the μIR complex (4) comprises a shallow solvent-exposed depression at the αCT/L1 junction. Although its structural analysis was limited by low resolution (3.5 Å), this surface contains multiple potential sites for hydrogen bonding and favorable electrostatic interactions, potentially extended by a network of bound water molecules. We speculate that these features of the B26-related surface underlie the enhanced affinities of SerB26 and GluB26 insulin analogs and general exclusion of aliphatic substitutions.
SEC Studies Provided Evidence of Decreased or Aberrant Self-assembly
Competence of the insulin analogs for zinc-free or zinc-stabilized self-assembly was assessed using a calibrated SEC method (Fig. 4). In the absence of zinc ions, WT insulin was predominantly dimeric as expected at a protein concentration of 0.6 mm under these conditions; the parent OrnB29-insulin analog exhibited similar elution behavior with a slight decrease in extent of dimerization (Fig. 4, A–C). A control for a monomeric insulin analog was provided by insulin lispro (KP-insulin; ProB28 → Lys and LysB29 → Pro) (38, 44). The elution time of the OrnB26 analog is similar to that of KP-insulin. The GluB26 analog exhibits two modes of self-association: ∼40% as dimeric (elution time similar to that of the parent analog) and 60% as a higher molecular mass aggregate (asterisk in Fig. 4A). Although the aberrant elution peak is broad, the mean apparent mass is ∼80 kDa, corresponding to 12–14 monomeric units. Perturbed overall dimerization of GluB26-insulin is in accordance with a previous study by equilibrium ultracentrifugation (3).
FIGURE 4.

HPLC size-exclusion chromatography and hormone self-assembly. A, retention times of the various analogs (1–5) are shown; identities are described in C. Elution of ∼ 50% of GluB26-OrnB29 as a large multimer of ∼80 kDa is indicated by an asterisk (*5). B, plot of log molecular weight versus Ve/Vo of different molecular mass standard proteins. Ve is the elution volume of each protein, and Vo is the column's void volume. Calibration: apoferritin (443 kDa, Vo), bovine serum albumin (67 kDa) ovalbumin (45 kDa), carbonic anhydrase (29 kDa), ribonuclease A (13.7 kDa), cytochrome C (12.3 kDa), and synthetic peptides (4.0 and 1.2 kDa). The line represents a linear fit, whereas arrows indicate relative elution of analogs 1–5. C, masses of analogs determined from calibrated standards (fitted line, in panel B). D–F, HPLC SEC in the presence of zinc and cyclohexanol. Proteins were fractionated as in A with inclusion of 50 mm cyclohexanol and 0.3 mm ZnCl2; the column was recalibrated in this buffer. With one exception, the OrnB29-insulin analogs, WT human insulin (HI) and KP each eluted as hexamers as indicated in E and F. Although a small fraction of the GluB26-OrnB29-insulin also eluted as a hexamer (5), most of this protein dissociated on the column with a broad elution profile (*5).
SEC studies were extended to conditions of WT R6 assembly through the addition of zinc ions and phenol (Fig. 4, D–F). To facilitate UV detection of the eluted proteins, cyclohexanol (which is transparent at 215 and 280 nm) was used instead of phenol in the running buffer (see “Experimental Procedures”). Under these conditions WT insulin and the parent OrnB29-insulin each exhibited an apparent mass slightly larger than a hexamer (43 and 37 kDa, respectively), whereas KP-insulin exhibited an apparent mass of one hexamer (formal molecular mass 35 kDa inclusive of two zinc ions; Fig. 4F). The OrnB26 analog was predominantly hexameric with a slight reduction in apparent mass (33 kDa), whereas the GluB26 analog predominantly exhibited a broad distribution of lower molecular mass entities (asterisk in Fig. 4D). A small percentage of the GluB26 analog (ca 5%) eluted as a small bump (41 kDa) on the leading edge of the diffuse profile.
Visible Absorption Co2+ Spectroscopy Provided Evidence of Altered R6 Assembly with Accelerated Disassembly
The structure and stability of the phenol-stabilized R6 insulin hexamer may readily be probed by visible absorption spectroscopy on substitution of the zinc ions by Co2+ (37). This spectrum exhibits a prominent absorption band due to the tetrahedral coordinate of Co2+ in each R3 trimer of the R6 hexamer (or in the R3f trimer of a T3R3f hexamer); no such band exists in the spectrum of an octahedral Co2+ complex as in the T3 trimer of an insulin hexamer (or in an EDTA complex) (38). The magnitude of the d-d band in KP-insulin (gray line in Fig. 5A) was slightly attenuated relative to the WT hexamer (black line), presumed to represent a small shift in the conformational equilibrium from R6 to T3Rf3 (37, 38, 45). The spectrum of the parent OrnB29-insulin (red line in Fig. 5A) was similar to that of WT insulin, whereas the peak signal of the OrnB26 analog (violet) was attenuated by 7 (±1)%. The spectrum of the GluB26 analog (turquoise) was attenuated by 39 (±2)%, suggesting that the substitution partially impairs the TR transition.
FIGURE 5.
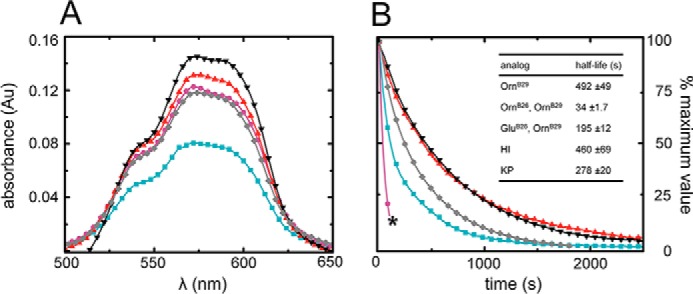
Visible absorption spectra of cobalt-stabilized hexamers and kinetics of metal ion release. A, Co2+ d-d bands of OrnB29-insulin (red), GluB26-OrnB29-insulin (turquoise), and OrnB26-OrnB29 -insulin (violet) near 550 nm provide a signature of the R (or Rf) hexameric state. Amplitudes of both OrnB26 and GluB26 variants were attenuated in relation to OrnB29-insulin. Control spectra were provided by WT insulin (black) and KP-insulin (gray). Whereas attenuation of amplitude of the 550-nm band of the OrnB26 variant may be explained by the decreased hexamer formation of the analog, the marked differences in the GluB26 spectrum may be the result of nonspecific aggregates of the analog forming in aqueous solution, as suggested by gel filtration experiments. Au, absorption units. B, sequestration of divalent cobalt ions from insulin analogs by EDTA; OrnB29-insulin (red), GluB26-OrnB29 (turquoise) insulin, and OrnB26-OrnB29 (violet) analogs are shown in relation to those of WT insulin (black) and KP-insulin (gray). OrnB26-OrnB29-insulin formed aggregates that precipitated from solution at intermediate stages of hexamer dissociation (marked by black asterisk): the initial region of the curve was fitted to a monoexponential equation.
The rate of disassembly of the insulin analog hexamers may likewise be probed through EDTA sequestration of Co2+. This assay's underlying principle exploits transient release of the metal ion when insulin hexamers dissociate and re-assemble within its conformational equilibrium. Because the affinity of EDTA for Co2+ is >108 greater than that of insulin, such transient release results in essentially irreversible sequestration of the metal ion in a colorless complex (38). Whereas the lifetime of the parent hexamer was similar to that of WT insulin (red and black lines in Fig. 5B), the lifetime of the GluB26 hexamer (turquoise) was reduced by 2.6 (±0.2)-fold. Kinetic studies of the OrnB26 analog were limited by its progressive precipitation on the addition of EDTA. Data collected until the appearance of a visible precipitate displayed an even shorter lifetime (reduced by 15 (±3)-fold), but interpretation of this value is unclear given the competing aggregation of unknown structure.
CD Studies of High Affinity Analogs Provided Evidence of Native-like Structure with Decreased Dimerization
Although well tolerated in relation to receptor binding, substitution of TyrB26 by Orn, Glu, or Ser was in each case associated with an altered far-UV CD spectrum at a protein concentration of 50 μm (Fig. 6A). At this concentration WT insulin and OrnB29-insulin are partially dimeric (46) as assessed by size exclusion chromatography (data not shown). The variant spectra exhibited attenuated ellipticity at 222 nm and deepening of the minimum near 208 nm, spectroscopic features associated with partial loss or dynamic destabilization of α-helices (47). In accordance with the above SEC findings, we ascribe these CD changes to decreased dimerization; analogous CD changes were previously found to accompany dilution of WT insulin in the range 5–100 μm (48), presumably due to enhanced flexibility of the monomer (49–51).
FIGURE 6.
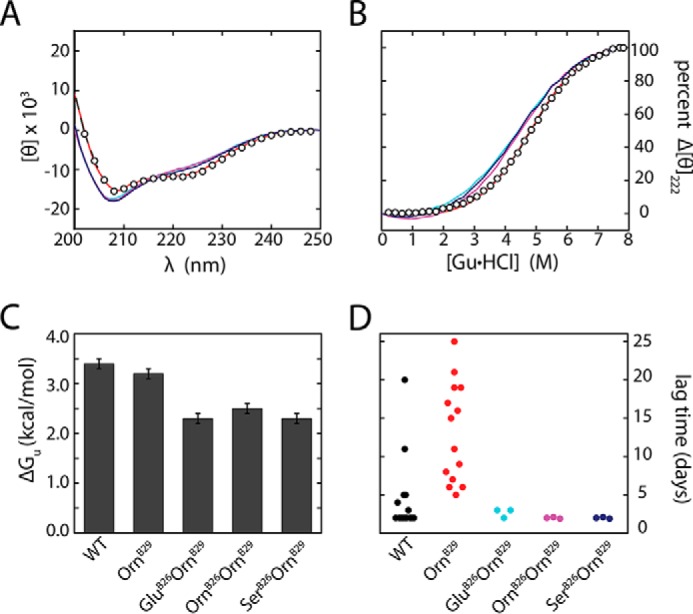
Studies of structure, stability, and fibrillation. A, far-UV CD spectra of OrnB29-insulin (red), GluB26-OrnB29-insulin (cyan), OrnB26-OrnB29-insulin (magenta), and SerB26-OrnB29 insulin (dark blue) relative to WT insulin (open circles; black) at neutral pH 7.4 and 25 °C. Ellipticity was normalized per residue. B, corresponding guanidine-unfolding transitions as monitored at 222 nm. Thermodynamic stabilities were derived using a two-state model (see Table 2). Color code is as in A. C, histogram of ΔGu values in kcal mol−1. Marked changes in stability were evident depending on the identity of the substitution. D, dot plot of lag time (days) to fibril formation of insulin analogs. Onset of fibrillation was defined by a 2-fold enhancement of thioflavin T fluorescence; see the section entitled “Functional Substitutions Led To Accelerated Fibrillation” for statistical analysis and p values.
Functional Substitutions Impair Thermodynamic Stability
Substitutions OrnB26, GluB26, and SerB26 impaired global stability. Estimates of respective free energies of unfolding ΔGu) were obtained at 25 °C based on CD studies of fractional unfolding on chemical denaturation (Fig. 6B and histogram in Fig. 6C). A trend was observed wherein lower concentrations of denaturant (guanidine hydrochloride) were required for 50% unfolding of analogs containing the above substitutions (Fig. 6B and Table 2, column heading Cmid). Application of a two-state model (native and unfolded; Ref. 40) yielded ΔGu values that were in each case at least 0.5 kcal mol−1 lower than that of OrnB29-insulin (baseline stability 3.2 (±0.1) kcal mol−1). Respective decrements in stability (ΔΔGu) for the OrnB26, GluB26, and SerB26 analogs were 0.7 (±0.2), 0.9 (±0.2), and 0.9 (±0.2) kcal mol−1. Such perturbations were thus similar to that reported in studies of an insulin analog containing substitution of PheB24 by Ala (ΔΔGu 0.8 (±0.2) kcal mol−1; Ref. 52).
TABLE 2.
Properties of insulin analogs
| Analog | ΔGua | Cmid | m | Fibrillation lag timeb |
|---|---|---|---|---|
| kcal mol−1 | m | kcal mol−1 m−1 | Days (N) | |
| Wild-type insulin | 3.4 ± 0.1 | 4.8 ± 0.1 | 0.69 ± 0.02 | 4.4 ± 4.9 (15) |
| OrnB29-insulin | 3.2 ± 0.1 | 4.8 ± 0.1 | 0.66 ± 0.01 | 13.1 ± 6.5 (14) |
| GluB26-OrnB29 | 2.3 ± 0.1 | 4.4 ± 0.1 | 0.53 ± 0.02 | 2.7 ± 0.6 (3) |
| OrnB26-OrnB29 | 2.5 ± 0.1 | 4.3 ± 0.2 | 0.59 ± 0.03 | 2.0 ± 0.1c (3) |
| SerB26-OrnB29 | 2.3 ± 0.1 | 4.3 ± 0.3 | 0.53 ± 0.04 | 2.0 ± 0.1c (3) |
a Thermodynamic parameters were inferred from CD-detected guanidine denaturation data by application of a two-state model.
b Fibrillation lag times pertain to zinc-free wild type insulin (in a monomer-dimer equilibrium) and analogs (monomeric); each protein was made 60 μm in phosphate-buffered saline (pH 7.4). A 2-fold increase over baseline in thioflavin T fluorescence provided a criterion for onset of fibrillation.
c All individual samples in this set exhibited the same lag time of 2 days. As the method employed in this study could not distinguish fibril lag times with a resolution given in hours, some variance was added to the data by adding ±0.1 to individual data points to obtain an estimate of the S.D.
The m-values obtained in the fitting (which correlate with extent of solvation of nonpolar surfaces on protein denaturation; Ref. 40) were significantly attenuated relative to OrnB29-insulin or WT insulin (Table 2, column heading m). Such attenuation suggests that in their respective native states, the analogs exhibited less efficient desolvation of nonpolar surfaces. This trend may be due to a direct perturbation of the (A2, A3)-related interchain crevice (2) by OrnB26, GluB26, and SerB26; transmitted structural perturbations cannot be excluded.
Functional Substitutions Led To Accelerated Fibrillation
The reduced stabilities, impaired dimerization, and perturbed m-values of the OrnB26, GluB26, and SerB26 analogs motivated assessment of lag times before onset of fibrillation relative to OrnB29-insulin (Fig. 6D and Table 2). These assays were performed in the absence of zinc ions. Although OrnB29-insulin exhibited a broad range of lag times >5 days (with a mean of 13 days in n = 14 trials), the analogs consistently exhibited lag times <4 days (n = 3). Despite the small sample size, p values were <0.01 for each analog. These trends are, therefore, unrelated to the charge of the B26 side chain and net charge of the protein (53). We speculate that perturbation of the (A2, A3)-related interchain crevice favors local unfolding (42) and non-native conformational excursions, in turn favoring formation of an amyloidogenic nucleus (Fig. 7C) (54).
FIGURE 7.

Evolutionary constraints and insulin fibrillation. A, Venn diagram showing intersection of multiple constraints: function, foldability, misfolding, and assembly. We envisage that TyrB26 is conserved due to its explicit roles in folding and assembly and implicit role in avoiding misfolding. B, surface representation of a T-state monomer (PDB entry 4INS) with residues B23–30 (stick model) within a groove between the A and B chains. The aromatic side chains of PheB24 and PheB25 (both dark gray) and TyrB26 (red) are shown. C, general scheme of insulin fibrillation via a partially unfolded monomer intermediate. The native state is protected by classic self-assembly. Disassembly leads to an equilibrium between native and partially folded monomers. The receptor-bound conformation of insulin (top) may also participate in this equilibrium. This partial fold may unfold completely (bottom) as an off-pathway event or aggregate to form a nucleus en route to a proto-filament (right).
Discussion
Vertebrate insulin sequences exhibit broad conservation (17), including several invariant residues recently shown to pack at the primary hormone-receptor interface (4). Examples are provided by ValA3, ValB12, and PheB24, non-polar side chains of distinctive shape and size whose respective binding sites in the IR are also invariant (7). Such framework contacts are also conserved among IGFs and the cognate type 1 IGF receptor (20, 21). Diverse amino acid substitutions at A3, B12, B24, or B25 impair receptor binding, including mutations as subtle as ValA3 → Leu, ValB12 → Leu, PheB24 → Tyr, or PheB24 → Leu (13, 55–57). Co-evolution of the hormone-receptor interface has presumably led to the strict conservation of such contact sites. Although current co-crystal structures of model insulin-μIR complexes were obtained at low resolution (3.5 Å; Ref. 4), we anticipate that future improvements in resolution may enable quantitative rationalization of such classical structure-activity relationships.
Systematic Mutagenesis of the Insulin Surface
Mutational surveys of key residues in insulin through systematic synthesis of all possible substitutions have provided broad insight into structure-activity relationships (58, 59). Such surveys may complement crystallographic studies of “micro-receptor” complexes (4), especially as to date such fragments lack the FnIII domains in the IR α-subunit and so provide an incomplete description of the hormone-receptor interface. A mutational survey of GluB13, for example, has suggested that this side chain contacts the IR (59) despite its lack of contact to αCT or L1 (4); this inference is in accordance with a proposed Site 2 contact within a FnIII domain (6, 60). Mutational surveys may also provide insight into conserved contacts visualized within the μIR complex. We recently utilized this approach to survey mutations at position B24 (16). The results provided a probe of a deep nonpolar pocket within the hormone-μIR complex (4). At this site only the native side chain of PheB24 among natural amino acids confers high activity. The non-standard aliphatic residue cyclohexanylalanine (ChaB24) was found to be compatible with high activity, demonstrating that aromaticity per se is not required within the B24-related pocket (16).
Structure-Activity Relationships at B26
Because of the low resolution of the present μIR co-crystal structure (4) and enigmatic structure-activity relationships in prior studies (23–29), we undertook a systematic mutagenesis of the B26 position with all standard amino acids, with the exceptions of Cys (to avoid a disulfide exchange) and Lys and Arg (to avoid tryptic cleavage of the octapeptide in the course of semi-synthesis; see “Experimental Procedures”). The latter two residues were represented by the basic amino acid Orn, an analog resistant to tryptic cleavage. The analogs also contained OrnB29 in place of the native Lys for the same reason. Semi-synthesis thus enabled rapid and efficient introduction of diverse substitutions at B26 without encountering barriers to recombinant protein expression (24).
The present B26 survey has demonstrated, in striking contrast to our prior B24 survey (16), that chemically diverse side chains are functional, whereas side chains associated with impaired IR binding were hydrophobic (IleB26, LeuB26, and ProB26). TrpB26 and PheB26 were compatible with near WT affinities, suggesting that their “weakly polar” electronic characteristics or planarity (each feature a consequence of aromaticity (61)) are favorable at the B26-related μIR surface. The displaced conformation of residues B24-B27 in the μIR complex leaves TyrB26 partially exposed at a solvated edge of the L1 domain (Fig. 2B). The reduced activity of ProB26-OrnB29-insulin may reflect not only its non-polarity but also its constrained and distinctive conformation.
All of the tested polar or charged amino acids conferred substantial activity (Asp, Glu, His, Asn, Orn, Gln, Ser, and the native Tyr; with Thr in the intermediate class). Of these, the highest affinities were conferred by Glu, Ser, Orn, and the native Tyr. Indeed GluB26 and SerB26 analogs of OrnB29-insulin exhibited IR affinities 2-fold greater than those of WT insulin or OrnB29-insulin itself; the affinity of OrnB26-OrnB29-insulin was indistinguishable from WT. Yet, Glu, Ser, and Lys (by analogy to Orn) are rarely found in nature as alternatives for TyrB26. To investigate this seeming evolutionary paradox, we measured free energies of unfolding (ΔGu) by chemical denaturation (40). The thermodynamic stabilities of these active variants were each markedly reduced relative to WT insulin or its parent control (ΔΔGu 0.9 (±0.2), 0.9 (±0.2), and 0.7(±0.2) kcal mol−1, respectively.) In SEC studies the GluB26 and OrnB26 analogs exhibited impaired zinc-free dimerization and zinc-stabilized hexamer formation in accordance with the classical packing of TyrB26 at an aromatic-rich dimer interface (2). Whereas decreased dimerization of GluB26-insulin has previously been reported (3), the SEC data also demonstrated aberrant higher order zinc-independent aggregation together with instability of the R6 zinc hexamer. Furthermore, this substitution and OrnB26 perturbed the kinetic stability of the R6 Co2+-substituted insulin hexamer. Finally, we observed that the high-affinity analogs each exhibited (under zinc-free conditions) markedly reduced lag times before fibril formation. Susceptibility to such aggregation-coupled misfolding would be expected to be further magnified by impaired native self-assembly.
The present set of analogs contained the platform substitution LysB29 → Orn, introduced to simplify the protocol of trypsin-mediated semi-synthesis). This non-standard residue (related to Lys by removal of a single methylene moiety from a linear side chain; i.e. [2,5]-diaminopentanoic acid versus [2,6]-diaminohexanoic acid) does not perturb the activity or stability of insulin. Its use as a template was unlikely to have influenced the pattern of activities among B26 analogs. Indeed, to the extent that our survey of substitutions recapitulated prior studies of particular analogs in a native context (such as AlaB26-insulin and GluB26-insulin), similar results were obtained with respect to activity (23) and self-assembly (3). Such template independence is not surprising in this case as residue B29 is peripheral to the hormone's receptor-binding surface (62, 63), does not contribute to classical self-association surfaces (2, 64), and is not conserved among vertebrate insulin sequences (17). Furthermore, in several otherwise high-resolution crystal structures of insulin, the side chain of LysB29 exhibits high thermal B factors (2) and/or lacks continuous electron density (in particular toward its distal end), suggesting dynamic disorder (as exemplified by Protein Data Bank entries 1BEN, 1EV3, 1TRZ; also see PDB entries 1TYL, 1TYM, 1G7A, 1EV3, 1ZNJ, 1EV6, 1BPH, and 1DPH for further examples) (65–67); such flexibility is in accordance with motional narrowing of 1H NMR resonances observed in a variety of NMR studies (49, 68). In pharmacologic applications these considerations have favored B29 as a site of substitution or chemical modification (e.g. ProB29 in Humalog®, GluB29 in Apidra®, and acylation of LysB29 in Levemir® and Tresiba®; Refs. 69–72). Surprisingly, the platform substitution LysB29 → Orn (introduced only to simplify the protocol of trypsin-mediated semi-synthesis) was itself associated with enhanced resistance to fibrillation at neutral pH, a finding that suggests that the mechanism of amyloid formation can be influenced by subtle changes despite its seeming universality among polypeptide sequences (30). It is possible that long-range electrostatic interactions by the δ-amino group of OrnB29 (made more distant and so weakened as compared with the ϵ-amino group of LysB29) may impose kinetic barriers to partial transient unfolding as envisioned in a putative amyloidogenic intermediate (73). Interpretation of relative fibrillation lag times is limited by the lack of structural information pertaining to such non-native intermediates.
Interest in position B26 has recently been renewed by evidence that substitution of TyrB26 by Asn (and possibly other amino acids) can alter the ratio of affinities to isoform IR-A versus IR-B in cellular assays, possibly as a consequence of a B26-related conformational change (74). Although the degree of selectivity was small, such studies motivated elegant use of non-standard protein engineering to create a novel class of constrained analogs (75). The latter studies employed cell lines that respectively expressed either one isoform or the other but in different lineage-specific contexts. Because such membranes may differ in composition or proteome, it would be of future interest to investigate whether such isoform selectivity might be observed in studies of purified receptor isoforms (as distinct from intact cells) and, if so, might extend to receptor fragments (such as the μIR model).
It would be of future interest to investigate the solution structures of selected B26 analogs by NMR spectroscopy. Although such studies would ordinarily require use of a monomeric insulin template (such as insulin lispro; Ref. 76) to circumvent insulin self-association, a confounding issue at the high protein concentrations required for NMR, our SEC studies of OrnB26-OrnB29-insulin and GluB26-OrnB29-insulin suggest that these analogs may be amenable to high-resolution study without further modification. Of particular interest would be the extent to which these B26 substitutions may lead to nonlocal changes in the dynamics of the hormone's α-helical domain (such as in its pattern of conformational broadening; Ref. 77) due to perturbation of the IleA2/ValA3-associated interchain crevice. In light of the displaced conformation of the B23-B27 segment in the μIR complex (4), it would be of further interest to probe whether such B26 substitutions weaken the attachment of this segment to the insulin core. A recent study of the insulin monomer by molecular-dynamics simulations has predicted that long-range interactions by TyrB26 in WT insulin regulate transient detachment and re-attachment of this segment through a series of transient conformational substates (78). Use of heteronuclear NMR methods to uncover such “excited-state” conformations represents a promising frontier of protein science (79).
Toxic Misfolding as an Evolutionary Constraint
Although in vivo formation of insulin fibrils is rare in humans, a hystricomorph rodent in South America (Octodon degus) develops senile diabetes mellitus mostly in association with deposition of an amyloid strictly composed of insulin in its islets of Langerhans (80, 81). The insulin sequence in this rodent contains the unusual substitution HisB10 → Asn, which removes the central Zn2+ coordination site in the classical insulin hexamer. In addition, the variant insulin of O. degus contains substitution TyrB26 → Arg, which would further be expected to impair or block hexamer assembly. Because the self-assembly of Zn2+-insulin hexamers is thought to protect the insulin from partial unfolding and non-native aggregation within the storage granules of pancreatic β-cells (1), the combined adverse effects of AsnB10 and ArgB26 on self-assembly may provide a molecular mechanism for the formation of an insulin-specific amyloid in this species. Such an experiment of nature highlights in the breach the implicit constraint that we propose underlies the broad conservation of Tyr at B26: avoidance of toxic misfolding. We thus envisage that a combination of inefficient or unstable disulfide pairing, perturbed hexamer assembly, and heightened susceptibility to fibrillation might be associated with a risk of toxic protein deposition as an amyloidogenic disease and so impose independent evolutionary constraints (Fig. 7A) (68). The displacement of the C-terminal segment of the B chain from the hormone's α-helical core (Fig. 7B) would otherwise favor a mechanism of fibrillation based on distortion of the monomeric structure with aberrant exposure of nonpolar surfaces (Fig. 7C) (53).
Although residue B26 is broadly conserved as Tyr among vertebrate insulins and as Phe among IGFs (17), such conservation is not strict. In addition to the amyloidogenic O. degus sequence, for example, several hystricomorph insulins contain Arg or Ser at B26 (17). Furthermore, the genome of Poecilia formosa (the Amazon molly fish) encodes a proinsulin/IGF-like protein wherein the B24-B26 segment contains a non-aromatic residue at B26 (PheB24-TyrB26-AsnB26; SwissProt entry A0A096M678_POEFO). Such anomalies are in accordance with the dispensability of TyrB26 in truncated insulin analogs with native activity, as exemplified by des-pentapeptide[B26-B30]-insulin-amide (26). It is possible that the life spans of such animals are shorter than the time scale of toxic protein deposition, and so their reproductive success is unaffected by the variant insulin.
Concluding Remarks
Our results suggest that conservation of TyrB26 is unlikely to be enjoined by the topography of the IR. The reduced stability of alternative high-affinity analogs and their enhanced susceptibility to fibrillation instead suggest that its conservation reflects co-optimization of several factors, including not only activity but also stability and avoidance of toxic misfolding (30). The latter's evolutionary importance is highlighted by the monogenic proinsulin syndrome (82). Indeed, the broad conservation of TyrB26 may reflect the multiple roles played by specific side chains in the course of a complex “conformational life cycle” from nascent folding to receptor binding. It would be of future interest to extend our survey from insulin to proinsulin in relation to foldability in the endoplasmic reticulum and corresponding folding efficiency in vitro.
Author Contributions
V. P. prepared the set of insulin analogs, performed the fibrillation- and guanidine-titration studies, and coordinated the receptor-binding studies. N. B. P. undertook SEC-HPLC studies and contributed to these sections of the manuscript. N. R. and V. P. performed spectroscopic studies of cobalt insulin hexamers. M. C. L. contributed to in silico analysis of the B26-related binding surface of the insulin-microreceptor complex. J. W. oversaw the receptor-binding experiments. M. A. W. coordinated the study and preparation of the manuscript.
Acknowledgments
We thank Drs Q.-X. Hua, W. Jia, S. H. Nakagawa, and Z.-l Wan for advice regarding experimental procedures, L. Whittaker for assistance with receptor-binding assays, J. Racca for assistance with figures, and Profs. P. Arvan, T. L. Blundell, M. Karplus, P. G. Katsoyannis, and M. Liu for general discussion.
This work was supported in part by National Institutes of Health Grants R01 DK040949 and DK079233 (NIDDK; to M. A. W.). This work was also supported by Australian National Health and Medical Research Council (NHMRC) Project Grants 1005896 and 1058233 and the Hazel and Pip Appel Fund (to M. C. L.). M. A. W. has equity in Thermalin Diabetes, LLC (Cleveland, OH), where he serves as Chief Scientific Officer. He has also been a consultant to Merck Research Laboratories and DEKA Research and Development Corp. N. B. P. and J. W. are consultants to Thermalin Diabetes, LLC. Part of M. C. L.'s research was funded by Sanofi (Germany). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We dedicate this article to the memory of the late Prof. Donald F. Steiner (University of Chicago).
- IR
- insulin receptor
- αCT
- α-chain C-terminal segment
- CR
- Cys-rich domain
- FnIII-1
- first fibronectin type III domain
- FnIII-2
- second fibronectin type III domain
- FnIII-3
- third fibronectin type III domain
- IR-A and IR-B
- A and B isoforms of the IR
- KP
- insulin lispro
- L1
- first Leu-rich repeat domain
- L2
- second Leu-rich repeat domain
- SEC
- size-exclusion chromatography
- Orn
- ornithine.
References
- 1. Dodson G., and Steiner D. (1998) The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8, 189–194 [DOI] [PubMed] [Google Scholar]
- 2. Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. M., Hubbard R. E., Isaacs N. W., and Reynolds C. D. (1988) The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 319, 369–456 [DOI] [PubMed] [Google Scholar]
- 3. Brange J., Ribel U., Hansen J. F., Dodson G., Hansen M. T., Havelund S., Melberg S. G., Norris F., Norris K., and Snel L. (1988) Monomeric insulins obtained by protein engineering and their medical implications. Nature 333, 679–682 [DOI] [PubMed] [Google Scholar]
- 4. Menting J. G., Yang Y., Chan S. J., Phillips N. B., Smith B. J., Whittaker J., Wickramasinghe N. P., Whittaker L. J., Pandyarajan V., Wan Z. L., Yadav S. P., Carroll J. M., Strokes N., Roberts C. T. Jr., Ismail-Beigi F., Milewski W., Steiner D. F., Chauhan V. S., Ward C. W., Weiss M. A., and Lawrence M. C. (2014) A structural hinge in insulin enables its receptor engagement. Proc. Natl. Acad. Sci. U.S.A. 111, E3395–E3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moller D. E., Yokota A., Caro J. F., and Flier J. S. (1989) Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 3, 1263–1269 [DOI] [PubMed] [Google Scholar]
- 6. De Meyts P., and Whittaker J. (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1, 769–783 [DOI] [PubMed] [Google Scholar]
- 7. Ward C. W., Menting J. G., and Lawrence M. C. (2013) The insulin receptor changes conformation in unforeseen ways on ligand binding: sharpening the picture of insulin receptor activation. Bioessays 35, 945–954 [DOI] [PubMed] [Google Scholar]
- 8. Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cabail M. Z., Li S., Lemmon E., Bowen M. E., Hubbard S. R., and Miller W.T. (2015) The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 6, 6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lou M., Garrett T. P., McKern N. M., Hoyne P. A., Epa V. C., Bentley J. D., Lovrecz G. O., Cosgrove L. J., Frenkel M. J., and Ward C. W. (2006) The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. U.S.A. 103, 12429–12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garrett T. P., McKern N. M., Lou M., Frenkel M. J., Bentley J. D., Lovrecz G. O., Elleman T. C., Cosgrove L. J., and Ward C. W. (1998) Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 394, 395–399 [DOI] [PubMed] [Google Scholar]
- 12. Croll T. I., Smith B. J., Margetts M. B., Whittaker J., Weiss M. A., Ward C. W., and Lawrence M. C. (2016) Higher-resolution structure of the human insulin receptor ectodomain: multi-modal inclusion of the insert domain. Structure 24, 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mirmira R. G., Nakagawa S. H., and Tager H. S. (1991) Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J. Biol. Chem. 266, 1428–1436 [PubMed] [Google Scholar]
- 14. Hua Q. X., Shoelson S. E., Kochoyan M., and Weiss M. A. (1991) Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354, 238–241 [DOI] [PubMed] [Google Scholar]
- 15. Xu B., Huang K., Chu Y. C., Hu S. Q., Nakagawa S., Wang S., Wang R. Y., Whittaker J., Katsoyannis P. G., and Weiss M. A. (2009) Decoding the cryptic active conformation of a protein by synthetic photoscanning: insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 284, 14597–14608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pandyarajan V., Smith B. J., Phillips N. B., Whittaker L., Cox G. P., Wickramasinghe N., Menting J. G., Wan Z.-l., Whittaker J., Ismail-Beigi F., Lawrence M. C., and Weiss M. A. (2014) Aromatic anchor at an invariant hormone-receptor interface function of insulin residue B24 with application to protein design. J. Biol. Chem. 289, 34709–34727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conlon J. M. (2001) Evolution of the insulin molecule: insights into structure-activity and phylogenetic relationships. Peptides 22, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 18. Shoelson S. E., Lee J., Lynch C. S., Backer J. M., and Pilch P. F. (1993) BpaB25 insulins: photoactivatable analogues that quantitatively cross-link, radiolabel, and activate the insulin receptor. J. Biol. Chem. 268, 4085–4091 [PubMed] [Google Scholar]
- 19. Kurose T., Pashmforoush M., Yoshimasa Y., Carroll R., Schwartz G. P., Burke G. T., Katsoyannis P. G., and Steiner D. F. (1994) Cross-linking of a B25 azidophenylalanine insulin derivative to the carboxyl-terminal region of the α-subunit of the insulin receptor: identification of a new insulin-binding domain in the insulin receptor. J. Biol. Chem. 269, 29190–29197 [PubMed] [Google Scholar]
- 20. Rinderknecht E., and Humbel R. E. (1978) The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 253, 2769–2776 [PubMed] [Google Scholar]
- 21. Rinderknecht E., and Humbel R. E. (1978) Primary structure of human insulin-like growth factor II. FEBS Lett. 89, 283–286 [DOI] [PubMed] [Google Scholar]
- 22. Hu S.-Q., Burke G. T., and Katsoyannis P. G. (1993) Contribution of the B16 and B26 tyrosine residues to the biological activity of insulin. J. Protein Chem. 12, 741–747 [DOI] [PubMed] [Google Scholar]
- 23. Inouye K., Watanabe K., Tochino Y., Kobayashi M., and Shigeta Y. (1981) Semisynthesis and properties of some insulin analogs. Biopolymers 20, 1845–1858 [DOI] [PubMed] [Google Scholar]
- 24. Kristensen C., Kjeldsen T., Wiberg F. C., Schäffer L., Hach M., Havelund S., Bass J., Steiner D. F., and Andersen A. S. (1997) Alanine scanning mutagenesis of insulin. J. Biol. Chem. 272, 12978–12983 [DOI] [PubMed] [Google Scholar]
- 25. Chen H., Shi M., Guo Z. Y., Tang Y. H., Qiao Z. S., Liang Z. H., and Feng Y. M. (2000) Four new monomeric insulins obtained by alanine scanning the dimer-forming surface of the insulin molecule. Protein Eng. 13, 779–782 [DOI] [PubMed] [Google Scholar]
- 26. Cosmatos A., Ferderigos N., and Katsoyannis P. G. (1979) Chemical synthesis of [des(tetrapeptide B27–30), Tyr(NH2)26-B], and [des(pentapeptide B26–30), Phe(NH2)25-B] bovine insulins. Int. J. Pept. Protein Res. 14, 457–471 [DOI] [PubMed] [Google Scholar]
- 27. Fischer W. H., Saunders D., Brandenburg D., Wollmer A., and Zahn H. (1985) A shortened insulin with full in vitro potency. Biol. Chem. Hoppe-Seyler 366, 521–525 [DOI] [PubMed] [Google Scholar]
- 28. Záková L., Kazdová L., Hanclová I., Protivínská E., Sanda M., Budesínský M., and Jirácek J. (2008) Insulin analogues with modifications at position B26. Divergence of binding affinity and biological activity. Biochemistry 47, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 29. Jirácek J., Záková L., Antolíková E., Watson C. J., Turkenburg J. P., Dodson G. G., and Brzozowski A. M. (2010) Implications for the active form of human insulin based on the structural convergence of highly active hormone analogues. Proc. Natl. Acad. Sci. U.S.A. 107, 1966–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 31. Barany G., and Merrifield R. B. (1980). in The Peptides (Gross E., and Meienhofer J., eds.) pp. 273–284, Academic Press, New York [Google Scholar]
- 32. Kubiak T., and Cowburn D. (1986) Enzymatic semisynthesis of porcine despentapeptide (B26–30) insulin using unprotected desoctapeptide (B23–30) insulin as a substrate. Int. J. Pept. Protein Res. 27, 514–521 [DOI] [PubMed] [Google Scholar]
- 33. Wang Z. X. (1995) An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 360, 111–114 [DOI] [PubMed] [Google Scholar]
- 34. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Reynolds C. D., Smith G. D., Sparks C., and Swenson D. (1989) Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 338, 594–596 [DOI] [PubMed] [Google Scholar]
- 35. Brange J. (ed) (1987) Galenics of Insulin: The Physico-chemical and Pharmaceutical Aspects of Insulin and Insulin Preparations, pp. 17–74, Springer Berlin Heidelberg, Berlin [Google Scholar]
- 36. Brange J. (2000) Physical stability of proteins. In Pharmaceutical Formulation Development of Peptides and Proteins (Frokjaer S., and Hovgaard L., eds.) pp. 89–109, 1st Ed., Taylor and Francis, Philadelphia, PA [Google Scholar]
- 37. Roy M., Brader M. L., Lee R. W., Kaarsholm N. C., Hansen J. F., and Dunn M. F. (1989) Spectroscopic signatures of the T to R conformational transition in the insulin hexamer. J. Biol. Chem. 264, 19081–19085 [PubMed] [Google Scholar]
- 38. Birnbaum D. T., Kilcomons M. A., DeFelippis M. R., and Beals J. M. (1997) Assembly and dissociation of human insulin and LysB28ProB29-insulin hexamers: a comparison study. Pharm. Res. 14, 25–36 [DOI] [PubMed] [Google Scholar]
- 39. Sreerama N., and Woody R. W. (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209, 32–44 [DOI] [PubMed] [Google Scholar]
- 40. Sosnick T. R., Fang X., and Shelton V. M. (2000) Application of circular dichroism to study RNA folding transitions. Methods Enzymol. 317, 393–409 [DOI] [PubMed] [Google Scholar]
- 41. Pace C. N., and Shaw K. L. (2000) Linear extrapolation method of analyzing solvent denaturation curves. Proteins 4, 1–7 [DOI] [PubMed] [Google Scholar]
- 42. Yang Y., Petkova A., Huang K., Xu B., Hua Q. X., Ye I. J., Chu Y. C., Hu S. Q., Phillips N. B., Whittaker J., Ismail-Beigi F., Mackin R. B., Katsoyannis P. G., Tycko R., and Weiss M. A. (2010) An Achilles' heel in an amyloidogenic protein and its repair: insulin dynamics, misfolding, and therapeutic design. J. Biol. Chem. 285, 10806–10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Izumiya N., Okazaki H., Matsumoto I., and Takiguchi H. (1959) Action of trypsin and papain on derivatives of diaminobutyric acid, ornithine and lysine. J. Biochem. 46, 1347–1354 [Google Scholar]
- 44. Campbell R. K., Campbell L. K., and White J. R. (1996) Insulin lispro: its role in the treatment of diabetes mellitus. Ann. Pharmacother. 30, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 45. Ciszak E., Beals J. M., Frank B. H., Baker J. C., Carter N. D., and Smith G. D. (1995) Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure 3, 615–622 [DOI] [PubMed] [Google Scholar]
- 46. Attri A. K., Fernández C., and Minton A. P. (2010) pH-dependent self-association of zinc-free insulin characterized by concentration-gradient static light scattering. Biophys. Chem. 148, 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang J. T., Wu C. S., and Martinez H. M. (1986) Calculation of protein conformation from circular dichroism. Methods Enzymol. 130, 208–269 [DOI] [PubMed] [Google Scholar]
- 48. Pocker Y., and Biswas S. B. (1980) Conformational dynamics of insulin in solution: circular dichroic studies. Biochemistry 19, 5043–5049 [DOI] [PubMed] [Google Scholar]
- 49. Jacoby E., Hua Q. X., Stern A. S., Frank B. H., and Weiss M. A. (1996) Structure and dynamics of a protein assembly. 1H NMR studies of the 36-kDa R6 insulin hexamer. J. Mol. Biol. 258, 136–157 [DOI] [PubMed] [Google Scholar]
- 50. Hua Q. X., Ladbury J. E., and Weiss M. A. (1993) Dynamics of a monomeric insulin analogue: testing the molten-globule hypothesis. Biochemistry 32, 1433–1442 [DOI] [PubMed] [Google Scholar]
- 51. Zoete V., Meuwly M., and Karplus M. (2004) A comparison of dynamic behavior of monomeric and dimeric insulin shows structural rearrangements in the active monomer. J. Mol. Biol. 342, 913–929 [DOI] [PubMed] [Google Scholar]
- 52. Hua Q. X., Xu B., Huang K., Hu S. Q., Nakagawa S., Jia W., Wang S., Whittaker J., Katsoyannis P. G., and Weiss M. A. (2009) Enhancing the activity of insulin by stereospecific unfolding: conformational life cycle of insulin and its evolutionary origins. J. Biol. Chem. 284, 14586–14596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nielsen L., Frokjaer S., Brange J., Uversky V. N., and Fink A. L. (2001) Probing the mechanism of insulin fibril formation with insulin mutants. Biochemistry 40, 8397–8409 [DOI] [PubMed] [Google Scholar]
- 54. Jiménez J. L., Nettleton E. J., Bouchard M., Robinson C. V., Dobson C. M., and Saibil H. R. (2002) The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kobayashi M., Ohgaku S., Iwasaki M., Maegawa H., Shigeta Y., and Inouye K. (1982) Characterization of [LeuB24]- and [LeuB25]-insulin analogues: receptor binding and biological activity. Biochem. J. 206, 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kobayashi M., Takata Y., Ishibashi O., Sasaoka T., Iwasaki T. M., Shigeta Y., and Inouye K. (1986) Receptor binding and negative cooperativity of a mutant insulin, [LeuA3]-insulin. Biochem. Biophys. Res. Commun. 137, 250–257 [DOI] [PubMed] [Google Scholar]
- 57. Hu S. Q., Burke G. T., Schwartz G. P., Ferderigos N., Ross J. B., and Katsoyannis P. G. (1993) Steric requirements at position B12 for high biological activity in insulin. Biochemistry 32, 2631–2635 [DOI] [PubMed] [Google Scholar]
- 58. Weiss M. A., Hua Q. X., Jia W., Nakagawa S. H., Chu Y. C., and Katsoyannis P. G. (2002) Activities of insulin analogues at position 8 are uncorrelated with thermodynamic stability. In Insulin and Related Proteins: Structure to Function and Pharmacology (Dieken M. L., Federwisch M., and De Meyts P., eds.) pp. 103–119, Kluwar Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 59. Glendorf T., Sørensen A. R., Nishimura E., Pettersson I., and Kjeldsen T. (2008) Importance of the solvent-exposed residues of the insulin B chain α-helix for receptor binding. Biochemistry 47, 4743–4751 [DOI] [PubMed] [Google Scholar]
- 60. De Meyts P. (1994) The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signaling. Diabetologia. 37, S135–S148 [DOI] [PubMed] [Google Scholar]
- 61. Burley S. K., and Petsko G. A. (1988) Weakly polar interaction in proteins. Adv. Protein Chem. 39, 125–189 [DOI] [PubMed] [Google Scholar]
- 62. De Meyts P., and Whittaker J. (2002) Structure-function relationships of insulin and insulin-like growth factor-I receptor binding. In Insulin and Other Proteins: From Structure to Function and Pharmacology (De Meyts P., Dieken M. L., and Federwisch M. eds.) pp. 131–149, Kluwer Publishing, Dordrecht, the Netherlands [Google Scholar]
- 63. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., Ward C. W., and Lawrence M. C. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. C., Mercola D. A., and Vijayan M. (1971) Atomic positions in rhombohedral 2-zinc insulin crystals. Nature 231, 506–511 [DOI] [PubMed] [Google Scholar]
- 65. Smith G. D., Ciszak E., and Pangborn W. (1996) A novel complex of a phenolic derivative with insulin: Structural features related to the T→R transition. Protein Sci. 5, 1502–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith G. D., Ciszak E., Magrum L. A., Pangborn W. A., and Blessing R. H. (2000) R6 hexameric insulin complexed with m-cresol or resorcinol. Acta Crystallogr. D Biol. Crystallogr. 56, 1541–1548 [DOI] [PubMed] [Google Scholar]
- 67. Ciszak E., and Smith G. D. (1994) Crystallographic evidence for dual coordination around zinc in the T3R3 human insulin hexamer. Biochemistry 33, 1512–1517 [DOI] [PubMed] [Google Scholar]
- 68. Jørgensen A. M., Kristensen S. M., Led J. J., and Balschmidt P. (1992) Three-dimensional solution structure of an insulin dimer: a study of the B9(Asp) mutant of human insulin using nuclear magnetic resonance, distance geometry and restrained molecular dynamics. J. Mol. Biol. 227, 1146–1163 [DOI] [PubMed] [Google Scholar]
- 69. Hirsch I. B. (2005) Insulin analogues. N. Engl. J. Med. 352, 174–183 [DOI] [PubMed] [Google Scholar]
- 70. Garg S. K., Ellis S. L., and Ulrich H. (2005) Insulin glulisine: a new rapid-acting insulin analogue for the treatment of diabetes. Expert. Opin. Pharmacother. 6, 643–651 [DOI] [PubMed] [Google Scholar]
- 71. Goldman-Levine J. D., and Lee K. W. (2005) Insulin detemir: a new basal insulin analog. Ann. Pharmacother. 39, 502–507 [DOI] [PubMed] [Google Scholar]
- 72. Keating G. M. (2013) Insulin degludec and insulin degludec/insulin aspart: a review of their use in the management of diabetes mellitus. Drugs 73, 575–593 [DOI] [PubMed] [Google Scholar]
- 73. Fink A. L. (2005) Natively unfolded proteins. Curr. Opin. Struct. Biol. 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 74. Záková L., Kletvíková E., Lepšík M., Collinsová M., Watson C. J., Turkenburg J. P., Jiráček J., and Brzozowski A. M. (2014) Human insulin analogues modified at the B26 site reveal a hormone conformation that is undetected in the receptor complex. Acta Crystallogr. D Biol. Crystallogr. 70, 2765–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Viková J., Collinsová M., Kletvíková E., Buděšínský M., Kaplan V., Žáková L., Veverka V., Hexnerová R., Aviñó R. J., Straková J., Selicharová I., Vaněk V., Wright D. W., Watson C. J., Turkenburg J. P., Brzozowski A. M., and Jiráček J. (2016) Rational steering of insulin binding specificity by intrachain chemical cross-linking. Sci. Rep. 6, 19431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hua Q. X., Jia W., and Weiss M. A. (2011) Conformational dynamics of insulin. Front Endocrinol. (Lausanne) 2, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hua Q. X., Hu S. Q., Frank B. H., Jia W., Chu Y. C., Wang S. H., Burke G. T., Katsoyannis P. G., and Weiss M. A. (1996) Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J. Mol. Biol. 264, 390–403 [DOI] [PubMed] [Google Scholar]
- 78. Papaioannou A., Kuyucak S., and Kuncic Z. (2015) Molecular dynamics simulations of insulin: elucidating the conformational changes that enable its binding. PLoS ONE 10, e0144058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mulder F. A., Mittermaier A., Hon B., Dahlquist F. W., and Kay L. E. (2001) Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 8, 932–935 [DOI] [PubMed] [Google Scholar]
- 80. Nishi M., and Steiner D. F. (1990) Cloning of complementary DNAs encoding islet amyloid polypeptide, insulin, and glucagon precursors from a New World rodent, the degu, Octodon degus. Mol. Endocrinol. 4, 1192–1198 [DOI] [PubMed] [Google Scholar]
- 81. Hellman U., Wernstedt C., Westermark P., O'Brien T. D., Rathbun W. B., and Johnson K. H. (1990) Amino acid sequence from degu islet amyloid-derived insulin shows unique sequence characteristics. Biochem. Biophys. Res. Commun. 169, 571–577 [DOI] [PubMed] [Google Scholar]
- 82. Weiss M. A. (2013) Diabetes mellitus due to the toxic misfolding of proinsulin variants. FEBS Lett. 587, 1942–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]