

Abstract
We present the results of solid state nuclear magnetic resonance (NMR) experiments on HIV-1 capsid protein (CA) assemblies with three different morphologies, namely wild-type CA (WT-CA) tubes with 35–60 nm diameters, planar sheets formed by the Arg18-Leu mutant (R18L-CA), and R18L-CA spheres with 20–100 nm diameters. The experiments are intended to elucidate molecular structural variations that underlie these variations in CA assembly morphology. We find that multidimensional solid state NMR spectra of 15N,13C-labeled CA assemblies are remarkably similar for the three morphologies, with only small differences in 15N and 13C chemical shifts, no significant differences in NMR line widths, and few differences in the number of detectable NMR cross-peaks. Thus, the pronounced differences in morphology do not involve major differences in the conformations and identities of structurally ordered protein segments. Instead, morphological variations are attributable to variations in conformational distributions within disordered segments, which do not contribute to the solid state NMR spectra. Variations in solid state NMR signals from certain amino acid side chains are also observed, suggesting differences in the intermolecular dimerization interface between curved and planar CA lattices, as well as possible differences in intramolecular helix-helix packing.
Keywords: human immunodeficiency virus (HIV), molecular dynamics, protein assembly, protein structure, solid state NMR
Introduction
The mature capsid of human immunodeficiency virus type 1 (HIV-1), which encloses the RNA genome, is a purely proteinaceous shell formed by roughly 1500 copies of a single 231-residue capsid protein (CA)3 (1). The capsid shell is comprised primarily of a triangular lattice of CA hexamers (2–6). According to the influential “fullerene model” (7), closure of the capsid shell may result from the replacement of precisely 12 CA hexamers at specific locations by CA pentamers (8). However, unlike the icosahedral capsids of certain retroviruses (9), HIV-1 capsids are asymmetric. Although capsids within mature HIV-1 often have an approximately conical morphology, large morphological variations have been reported (10), as well as variations in apparent closure (11). Studies of pure CA performed in vitro have also found a wide variety of morphologies for CA assemblies, including tubes with 40–60 nm diameters (3, 4, 12–15), spheres with minimum diameters of 20 nm and maximum diameters exceeding 10 μm (2, 13, 16), planar sheets (2), and assemblies with asymmetric morphologies resembling those within mature HIV-1 (7, 13, 16).
Essential information about CA molecular structure and lattice structure has been obtained from a large number of investigations by electron microscopy (2–4, 7), x-ray crystallography (5, 6, 8, 17–20), and multidimensional nuclear magnetic resonance (NMR) spectroscopy of soluble monomeric and dimeric constructs (21–29). Results from these investigations show that CA is primarily α-helical, with independently folding N-terminal (helices 1–7) and C-terminal (helices 8–11) domains (NTD and CTD), connected by a short linker that allows the relative orientations of NTD and CTD to vary widely in the unassembled, soluble state. In tubular and planar CA lattices, hexamers form through intermolecular NTD-NTD and NTD-CTD interactions around local 6-fold symmetry sites and are linked to one another by CTD-CTD interactions at local 2-fold symmetry sites. Simulations using highly simplified representations of CA show that these interactions, together with the overall shapes of NTD and CTD, are sufficient to explain triangular lattice formation (30, 31). Additional interactions among groups of three CTD units at local 3-fold symmetry sites also contribute to CA lattice stability (26, 32). In an ideal planar lattice, all CA monomers can be structurally equivalent, as in the planar lattices characterized by electron diffraction and x-ray crystallography (2, 5). In a non-planar assembly, differences in lattice curvature in different directions necessarily lower the symmetry, placing different monomers in different structural environments and allowing differences in intermolecular interactions and monomer conformations.
The major variations in CA assembly morphology observed both in vitro and in mature HIV-1 suggest that certain aspects of the molecular and/or supramolecular structures of CA lattices may also be highly variable. In the case of closed CA assemblies, morphological variations may arise in part from variations in the locations of pentamers, or possibly other types of defects (11, 30, 33, 34). However, pentamer locations alone cannot explain the variations in surface curvature over an entire closed assembly, and cannot explain the differences in surface curvature parallel and perpendicular to the long axes of tubular assemblies (which in principle may contain pentamers at their ends, but not elsewhere).
At least three structural factors, in addition to the presence of pentamers, may contribute to variations in CA assembly morphology and lattice curvature. First, flexibility or conformational heterogeneity in the NTD-CTD linker may permit variations in the relative orientation between NTD and CTD subunits of individual monomers. Second, the identities and geometries of intermolecular interactions may vary within hexamers and/or between hexamers at local 2- and 3-fold symmetry sites. Third, conformational features within individual NTD or CTD subunits, such as helix-helix contacts and angles, may vary.
An all-atom model for CA tubes developed by Zhao et al. (4), based on data from cryo-electron microscopy (cryo-EM; PDB file 3J34), amply illustrates these three factors. Within one hexamer in this model, the relative orientations of NTD and CTD subunits in individual monomers vary by more than 10°; intermolecular angles between helix 9 pairs at local 2-fold symmetric sites vary by roughly 20°; intermolecular distances between helix 10 pairs at local 3-fold symmetry sites vary between 7.5 and 19.1 Å (based on α-carbon sites of Thr200); backbone α-carbon root mean square deviation (r.m.s. deviation) values between pairs of NTD subunits (residues 14–140) within one hexamer exceed 3.5 Å; backbone α-carbon r.m.s. deviation values between pairs of CTD subunits (residues 150–219) within one hexamer exceed 1.5 Å. In contrast, in high-resolution crystal structures of engineered CA hexamers and pentamers (PDB files 3MGE and 3P05) (6, 8), backbone r.m.s. deviation values between NTD subunits in hexamers and NTD subunits in pentamers are less than 0.4 Å; backbone r.m.s. deviation values between CTD subunits in hexamers and CTD subunits in pentamers are less than 0.5 Å. In light of these observations, it remains uncertain which structural factors contribute most to variations in CA assembly morphology and lattice curvature.
It should also be noted that the HIV-1 Gag polyprotein, which contains CA linked covalently to the HIV-1 matrix and nucleocapsid proteins, forms a curved lattice within immature virions (35, 36). According to recent cryo-EM studies (37), this immature lattice is also a triangular lattice of CA hexamers, but with quite different intermolecular interactions and with quite different relative orientations between NTD and CTD subunits of individual monomers.
Publications since 2010 from our laboratory (14, 15) and from Polenova and co-workers (16, 38–40) demonstrate that HIV-1 CA assemblies are amenable to solid state NMR experiments. Solid state NMR measurements can provide high-resolution, site-specific information about protein structure and dynamics in large non-crystalline assemblies (41, 42), thus providing data that are complementary to data from x-ray crystallography, electron microscopy, and liquid state NMR. Recently, Bayro et al. (15) reported extensive solid state NMR experiments on CA tubes, including identification of structurally ordered and disordered protein segments, nearly complete 15N and 13C chemical shift assignments from two- and three-dimensional solid state NMR spectra of samples that were prepared with several different isotopic labeling schemes, and identification of site-specific conformational changes that accompany CA self-assembly.
One of the important findings from the work of Bayro et al. (15) is that 177 of the 231 residues in wild-type CA (WT-CA) produce strong, resolvable signals in multidimensional solid state NMR spectra. Definite site-specific signal assignments were obtained for 159 residues, including residues in helices 9 and 10 that are involved in intermolecular interactions at local 2- and 3-fold symmetry sites, respectively, and including signals from amino acid side chains of these helical segments. (Additional assignments for four residues in WT-CA tubes were obtained subsequently, namely Gly8, Met68, Ala174, and Val181.) This finding implies that the conformations of structurally ordered segments in NTD and CTD and the geometries of intermolecular interactions do not vary greatly within CA tubes, despite the lowering of symmetry discussed above, and despite the fact that the solid state NMR samples contain CA tubes with a variety of diameters and helical pitches. Bayro et al. (15) also identified 29 residues that are not detectable in multidimensional solid state NMR spectra under standard measurement conditions, including portions of the N-terminal “hairpin” segment (specifically, residues 7 and 9–13), the cyclophilin A binding loop (residues 96–97), the NTD-CTD linker (residues 141–146), and the C-terminal tail (residues 220–231). Thus, structural variations within WT-CA tubes may be confined to the protein segments that are invisible to solid state NMR. Conformational distributions within these segments may vary from monomer to monomer. Bayro et al. (15) provided evidence that the conformational disorder within these segments is largely dynamic (15). Similar conclusions were reached by Polenova and co-workers (40) in a recent study of site-specific dynamics in WT-CA tubes.
In this paper, we report the results of solid state NMR measurements on assemblies formed in vitro by the Arg18-Leu mutant of CA (R18L-CA). As demonstrated in earlier studies by Ganser-Pornillos et al. (2), R18L-CA forms spherical assemblies with diameters as small as 20 nm under buffer conditions where WT-CA forms tubes. Under somewhat different buffer conditions, R18L-CA forms two-dimensional crystals (i.e. planar sheets), which were used by Ganser-Pornillos et al. (2) in their characterization of the CA lattice structure by electron diffraction. Although R18L-CA is not a naturally occurring variant, it allows us to make direct comparisons among morphologically distinct CA assemblies. The comparisons of solid state NMR data for WT-CA tubes, R18L-CA spheres, and R18L-CA sheets reported below provide additional insights into structural and dynamical variations that underlie major variations in the morphology of CA assemblies.
Experimental Procedures
Protein Expression and Purification
WT-CA and R18L-CA in pET-11a vector were expressed in Escherichia coli BL21(DE3) cells following the protocol described by Bayro et al. (15) Purification was also performed according to the published procedure, with some modifications for R18L. After lysis of the cells from 1 liter of M9 medium and centrifugation at 9000 × g for 30 min, the supernatant was collected and mixed slowly with saturated (NH4)2SO4 solution until the (NH4)2SO4 solution reached 25% of the final volume. The mixture was kept on ice for 2 h. The protein precipitate was collected and redissolved in 20 ml of 20 mm Tris buffer (pH 8.0) with 5 mm β-mercaptoethanol. The protein solution was dialyzed against 20 mm Tris buffer (pH 8.0, 5 mm β-mercaptoethanol) for 24–48 h, with buffer changes every 8–16 h, to remove residual (NH4)2SO4. After another centrifugation to remove the insoluble precipitate, the protein solution was then dialyzed against buffer A (25 mm K-MOPS at pH 6.9 (WT-CA) or 6.5 (R18L-CA) with 5 mm β-mercaptoethanol) for 24–48 h. R18L-CA was purified using Sepharose resin (HiLoad 16/10 SP Sepharose High Performance, GE Healthcare) with a linear gradient of buffer B (25 mm K-MOPS, 1.0 m NaCl at pH 6.9 (WT-CA) or 6.5 (R18L-A) with 5 mm β-mercaptoethanol) in 20 column volumes. WT-CA eluted around 15% buffer B, whereas R18L-CA eluted around 9% buffer B.
Uniformly 13C,15N-labeled samples (U-WT-CA and U-R18L-CA) were prepared by using d-[13C6]glucose (2.5 g/liter) and 15N-labeled NH4Cl (1.5 g/liter) in the M9 medium. Partially 13C-labeled samples (2-Glyc-WT-CA, Met-WT-CA, and 2-Glyc,Met-R18L-CA) were prepared in a similar way, with [2-13C]glycerol (3.0 g/liter) and/or uniformly 13C,15N-labeled methionine (240 mg/liter) in the growth medium in place of d-[13C6]glucose. Samples and NMR measurements discussed in this paper are listed in Table 1.
TABLE 1.
Summary of NMR measurements and HIV-1 CA samples
All samples are uniformly 15N labeled. U-WT-CA and U-R18L-CA are also uniformly 13C labeled. 2-Glyc-WT-CA is partially 13C labeled by using [2-13C]glycerol (Glyc) as the carbon source in the protein expression medium. Met-WT-CA is uniformly 15N,13C labeled only at methionine residues. 2-Glyc,Met-R18L-CA is partially 13C labeled with [2-13C]glycerol and simultaneously uniformly 15N,13C labeled at methionine residues.
| NMR measurement | Tubes | Spheres | Sheets |
|---|---|---|---|
| Two-dimensional 13C-13C DARR (Figs. 3, 7, 8, and supplemental Fig. S5) | U-WT-CAa | U-R18L-CA | U-R18L-CA |
| 2-Glyc-WT-CAa | 2-Glyc,Met-R18L-CA | 2-Glyc,Met-R18L-CA | |
| Met-WT-CA | |||
| Two-dimensional 15N-13C NCACX (supplemental Figs. S1 and S4) | U-WT-CAa | U-R18L-CA | U-R18L-CA |
| Two-dimensional 15N-13C NCACX (Fig. 6) | 2-Glyc-WT-CA | 2-Glyc,Met-R18L-CA | 2-Glyc,Met-R18L-CA |
| Two-dimensional 15N-13C NCOCX (supplemental Fig. S2) | U-WT-CAa | U-R18L-CA | U-R18L-CA |
| Two-dimensional 15N-13C NCOCX (Fig. 8) | 2-Glyc-WT-CA | 2-Glyc,Met-R18L-CA | 2-Glyc,Met-R18L-CA |
| Two-dimensional 15N-13C NCAROM (Fig. 7) | 2-Glyc-WT-CA | 2-Glyc,Met-R18L-CA | 2-Glyc,Met-R18L-CA |
| Three-dimensional 15N-13C NCACX (Figs. 4 and supplemental Fig. S3) | U-WT-CAa | U-R18L-CA | U-R18L-CA |
| Three-dimensional 15N-13C NCOCX (supplemental Fig. S3) | U-WT-CAa | U-R18L-CA | |
| Two-dimensional 13C-13C INEPT-TOBSY (Fig. 9) | U-WT-CAa | U-R18L-CA | U-R18L-CA |
a Data reported previously by Bayro et al. (15).
Preparation of Tubular, Spherical, and Planar Assemblies
WT-CA tubes were prepared as described by Bayro et al. (15). R18L-CA spheres were prepared by concentrating purified R18L-CA to 60–90 mg/ml in pH 6.5 elution buffer (25 mm K-MOPS with 90 mm NaCl) and mixing with 1.5 m NaCl in 20 mm Tris buffer (pH 8.5), in a 1:2 volume ratio. The final R18L-CA concentration was 20–30 mg/ml, the final NaCl concentration was 1.0 m, and the final pH was 8.0. R18L-CA sheets were prepared as described by Ganser-Pornillos et al. (2). Purified R18L-CA was concentrated to 100–130 mg/ml in elution buffer. The concentrated R18L-CA solution was then diluted to 25–32.5 mg/ml in assembly buffer (18% (w/v) polyethylene glycol (PEG) 20,000, 50 mm sodium cacodylate, and 100 mm calcium acetate) by adding appropriate volumes of 30% (w/v) PEG 20,000, 1.0 m sodium cacodylate (pH 6.5), and 1.0 m calcium acetate (pH 7.0).
WT-CA and R18L-CA solutions were incubated at 37 °C for 60 min to produce the tubes, spheres, and sheets. Solutions were then centrifuged at 80,000 × g for 10 min and the supernatant was removed. For solid state NMR measurements, ∼10 mg of pelleted protein was transferred from the microcentrifuge tube to a 3.2-mm diameter thin-wall magic-angle spinning (MAS) rotor by centrifugation at 50,000 × g for 30–60 min, using a sample packing device made in our laboratory.
Transmission Electron Microscopy and Atomic Force Microscopy
Transmission electron microscopy (TEM) with negative staining was used to confirm assembly of WT-CA and R18L-CA into the desired morphologies. TEM images of WT-CA tubes were obtained as previously described (14, 15). For TEM imaging of R18L-CA spheres, an aliquot of an incubated R18L-CA solution (before centrifugation to pellet the assemblies) was diluted 5-fold using 1.0 m NaCl. A 5-μl aliquot of the diluted solution was applied immediately to the glow-discharged TEM grid (Quantifoil part number S 7/2, 2 nm-carbon film thickness), blotted away with filter paper after a 40-s adsorption period, rinsed twice with 5 μl of water followed by blotting, stained for 40 s with 5 μl of 3% uranyl acetate, blotted again, and dried in air. For TEM imaging of R18L-CA sheets, a 5-μl aliquot of incubated R18L-CA solution was applied to the TEM grid without dilution, allowed to adsorb for 40 s, blotted, and immediately washed using ∼6 ml of 0.1 m KCl solution, applied drop-by-drop to remove excess PEG from the grid surface. The grid was then stained for 40 s with 5 μl of 3% uranyl acetate, blotted, and dried in air. Images were recorded with an FEI Morgagni microscope, operating at 80 keV.
Atomic force microscope (AFM) images of R18L-CA sheets were also recorded, using a Veeco MultiMode AFM and Nanoscope IV controller, equipped with a fluid cell and operating in tapping mode with a silicon nitride probe (0.32 N/m spring constant, triangular cantilever). For AFM imaging, an incubated R18L-CA solution was diluted 10-fold in a dilution buffer that contained 330 mm NaCl, 6% (w/v) PEG 20,000, 17 mm sodium cacodylate (pH 6.5), and 33 mm calcium acetate. A 5-μl aliquot of the diluted solution was applied immediately to a freshly cleaved mica disk, which was placed on the AFM scanner. The fluid cell was filled with 100 μl of dilution buffer, and the AFM head (containing the fluid cell) was placed on top of the scanner. Images were recorded at 9.35 μm/s scan rates. Although stable AFM images of R18L-CA sheets were readily obtained, we did not succeed in obtaining AFM images of R18L-CA spheres under similar conditions, presumably because the spheres were more easily dislodged from the mica surface by the rastering AFM probe.
Solid State NMR
Solid state NMR measurements were performed at 14.1 tesla on a Varian InfinityPlus spectrometer operating at 1H NMR frequency of 599.2 MHz, using a 3.2-mm Varian BioMAS probe. Sample temperatures were maintained at 20 °C with cold nitrogen (FTS XR AirJet chiller), as monitored by the temperature-dependent 1H NMR chemical shift of water in the capsid protein samples. Two- and three-dimensional spectra were recorded at 12.0–12.5 kHz MAS frequency. Typical 1H-13C cross-polarization conditions used a 62 kHz radiofrequency (rf) field for 1H and a ramped 13C rf field centered around 50 kHz, with a 0.75-ms contact time. Typical 1H-15N cross-polarization conditions used a 44 kHz rf field for 1H and a ramped 15N rf field centered around 32 kHz, with a 1.00-ms contact time. 1H decoupling fields were typically 70 kHz, with two-pulse phase modulation (43). Recycle delays were 2.0–2.2 s for all measurements.
Two-dimensional 13C-13C spectra were recorded with 20–500 ms dipolar-assisted rotational resonance (DARR) mixing periods (44, 45). Two- and three-dimensional NCACX and NCOCX experiments were carried out with 3.0–4.0 to 4.0 ms frequency-selective 15N-13C cross-polarization, using rf fields of 30 kHz on 15N and 17.5 kHz on 13C for NCACX spectra (13C rf carrier centered in the 13Cα region), and 30 kHz on 13C and 17.5 kHz on 15N for NCOCX spectra (13C rf carrier centered in the 13CO region). 13C-13C mixing in NCACX and NCOCX experiments was achieved using 25–500 ms DARR periods. For WT-CA tubes and R18L-CA sheets, two-dimensional 15N-13C spectra were also obtained with the 13C rf carrier frequency centered in the Trp aromatic region during 15N-13C cross-polarization (two-dimensional NCAROM), and without an additional 13C-13C polarization transfer period.
Two-dimensional 13C-13C total through-bond correlation (TOBSY) spectra (46) were recorded with preparation of initial 13C polarization by refocused 1H-13C insensitive-nuclei enhanced polarization transfer (INEPT) (47) followed by a 5.04-ms 13C-13C TOBSY mixing period. The MAS frequency was set to 5.952 kHz to allow for a TOBSY matching condition with the 13C rf field of 71.4 kHz. The 1H decoupling field was 64 kHz.
Two- and three-dimensional spectra were processed and analyzed with NMRPipe (48) and Sparky software. Gaussian apodization of 50–60 Hz was applied in all dimensions of two-dimensional and three-dimensional spectra, without artificial resolution enhancement. For two-dimensional INEPT-TOBSY spectra, Gaussian apodization was 30 Hz and 15 Hz in t1 and t2 dimensions, respectively. Additional details of NMR measurement conditions are given in the figure captions.
Results
Morphologies of WT-CA and R18L-CA Assemblies
Fig. 1A shows a typical TEM image of negatively stained WT-CA tubes, formed at pH 8.0 in 1.0 m NaCl. As previously described (14, 15), WT-CA tubes prepared under these conditions have diameters in the 35–60 nm range, indicating a mixture of helical symmetries (3, 4), and lengths that exceed 500 nm. Some of the WT-CA tubes are likely to be multilayered assemblies (14).
FIGURE 1.

TEM images of negatively stained HIV-1 capsid protein assemblies. A, WT-CA tubes. B and C, R18L-CA spheres. D, planar R18L-CA sheet. Non-planar material within ellipses may result from disruption or dissolution of R18L-CA sheets during the TEM grid preparation process. E, Fourier transform of the region of panel D enclosed by a square. The pattern of spots with 6-fold symmetry indicates two-dimensional crystallinity of R18L-CA sheets.
Fig. 1, B and C, show TEM images of negatively stained R18L-CA spheres, also formed at pH 8.0 and 1.0 m NaCl. The apparent diameters of the spheres vary from 20 to 100 nm, consistent with previous observations (2). The smallest diameter is roughly equal to that of a truncated icosahedron, or “soccer ball,” that could be formed by 12 R18L-CA pentamers and 20 R18L-CA hexamers. For an ideal icosahedron with equal distances, d, between neighboring vertices, the distance between diametrically opposed vertices is as shown.
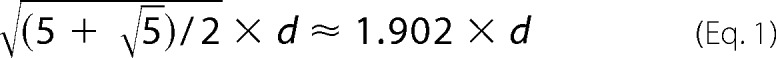 |
In the two-dimensional R18L-CA lattice characterized by electron diffraction (2), the distance between the centers of hexamers is 9.27 nm ≈ d, suggesting a minimum diameter of ∼18 nm for R18L-CA spheres.
At pH 6.5 and in the presence of PEG 20,000 and sodium cacodylate, R18L-CA forms patches of planar sheets (2), which appear to be single layered or multilayered in TEM images with negative staining. Fourier transforms of selected regions show faint but discernible “diffraction” spots with 6-fold symmetry, indicating that the planar sheets contain a nearly crystalline two-dimensional lattice (Fig. 1, D and E). Some objects with undefined structure are also observed in the TEM images. These objects may result from folding of the sheets on the TEM grid or partial dissociation of planar assemblies during preparation of the TEM grids.
As shown in Fig. 2, R18L-CA sheets can also be visualized by AFM, performed with the sheets adsorbed to a mica surface and submerged in a pH 6.5 buffer containing PEG 20,000 and sodium cacodylate. Single sheets have uniform heights of 6.0 ± 0.2 nm, in excellent agreement with the 5.9 nm thickness of the two-dimensional R18L-CA lattice structure identified by electron diffraction (2) and the 5.7-nm interlayer spacing in a recent x-ray crystal structure of WT-CA hexamers (5). Features with 12-nm heights are also seen in the AFM images, indicating the presence of double-layered sheets.
FIGURE 2.
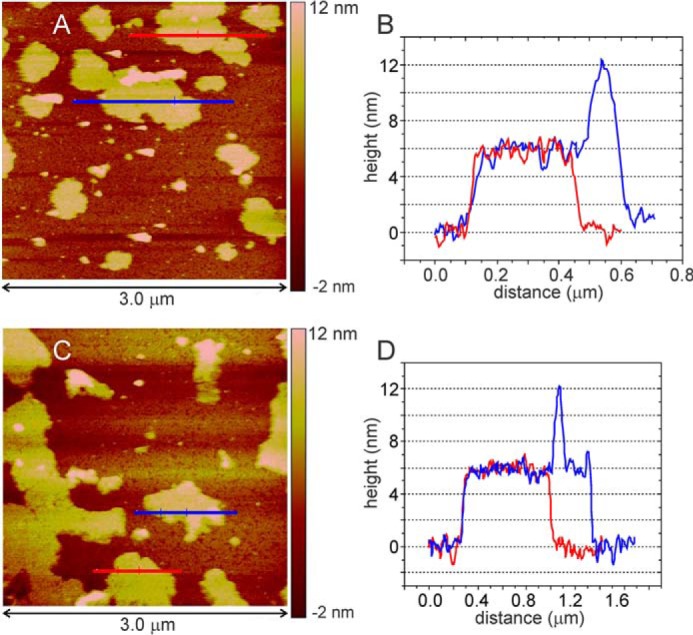
A and C, AFM images of R18L-CA sheets. Images were recorded in tapping mode on mica substrates, immersed in assembly buffer within the fluid cell of the AFM instrument. B and D, height profiles along blue and red lines in panels A and C, showing the uniform 6-nm thickness of single R18L-CA sheets. Features with 12 nm height are attributable to double layers.
Similarity of the Underlying Molecular Structures
Despite the heterogeneous appearance of CA assemblies (e.g. variations in diameters, mixtures of single layer, and multilayer assemblies), solid state NMR spectra of the three samples are comparable in resolution to spectra of other homogenous, non-crystalline protein samples obtained under similar experimental conditions (41, 42). The three types of CA assemblies were also found to be stable under solid state NMR measurement conditions, with moderate MAS frequencies (≤12.5 kHz) and sample temperatures in the 10°−25 °C range. No noticeable changes in the spectra were observed after several weeks of experiments.
Fig. 3 shows two-dimensional 13C-13C correlation spectra of the three uniformly labeled samples, with representative one-dimensional slices to illustrate the resolution and signal-to-noise. Although one might expect U-R18L-CA sheets to show sharper solid state NMR lines than U-R18L-CA spheres or U-WT-CA tubes, due to the absence of lattice curvature in the sheets, in fact the 13C line widths (full width at half-maximum) are typically 0.7 ppm in all three samples. One might also speculate that two-dimensional spectra of U-R18L-CA sheets would show a larger number of detectable cross-peaks than two-dimensional spectra of U-R18L-CA spheres or U-WT-CA tubes (because of a lower degree of conformational disorder in the U-R18L-CA sheets), or conversely that two-dimensional spectra of U-R18L-CA sheets would show a smaller number of detectable cross-peaks (because of the higher symmetry in U-R18L-CA sheets). In fact, two-dimensional spectra of the three types of assemblies show nearly the same numbers of detectable cross-peaks. Differences in cross-peak positions among the three two-dimensional 13C-13C correlation spectra in Fig. 3 are also minor, despite the large differences in morphology. These results indicate that the underlying molecular conformations of WT-CA and R18L-CA are quite similar in the three samples, and that the number of structurally ordered residues is nearly the same in the three samples. Differences in cross-peak intensities are observed in various positions in the spectra, attributable to site-specific differences in conformational rigidity.
FIGURE 3.

Two-dimensional 13C-13C solid state NMR spectra of uniformly 15N,13C-labeled HIV-1 capsid protein assemblies. A, U-WT-CA tubes. B, U-R18L-CA spheres. C, U-R18L-CA sheets. D, one-dimensional slices at 29.3, 51.5, and 65.8 ppm (dashed lines in two-dimensional spectra). The one-dimensional slices are color-coded to match the two-dimensional spectra. DARR mixing periods between t1 and t2 periods were 100 ms for U-WT-CA tubes and 25 ms for U-R18L-CA spheres and sheets. Maximum t1 periods were 5.0–8.9 ms. Total measurement times were ∼18 h. Contour levels increase by factors of 1.5.
Two-dimensional NCACX and NCOCX spectra were also obtained for the three types of assemblies, using U-WT-CA and U-R18L-CA. These two-dimensional spectra are compared in supplemental Fig. S1 (NCACX) and supplemental Fig. S2 (NCOCX). 15N line widths are typically 1.0 ppm for all three samples. As with the two-dimensional 13C-13C spectra, the numbers of cross-peaks and the cross-peak positions are nearly the same in two-dimensional NCACX and in two-dimensional NCOCX spectra of the three samples.
13C and 15N chemical shift assignments for R18L-CA spheres and R18L-CA sheets were obtained from three-dimensional NCACX and NCOCX spectra of the uniformly labeled samples, along with the two-dimensional spectra described above. Examples of two-dimensional planes from the three-dimensional spectra are shown in Figs. 4 and supplemental Fig. S3. Chemical shift differences between R18L-CA spheres and R18L-CA sheets are more obvious in the three-dimensional spectra, due to the higher resolution and dimensionality. Assignments were obtained by matching cross-peaks in two- and three-dimensional spectra of U-R18L-CA assemblies with cross-peaks in spectra of U-WT-CA tubes, for which chemical shift assignments were determined previously by Bayro et al. (41). Assignments in U-R18L-CA assemblies were confirmed by checking connections to signals of neighboring residues, using the NCOCX spectra.
FIGURE 4.
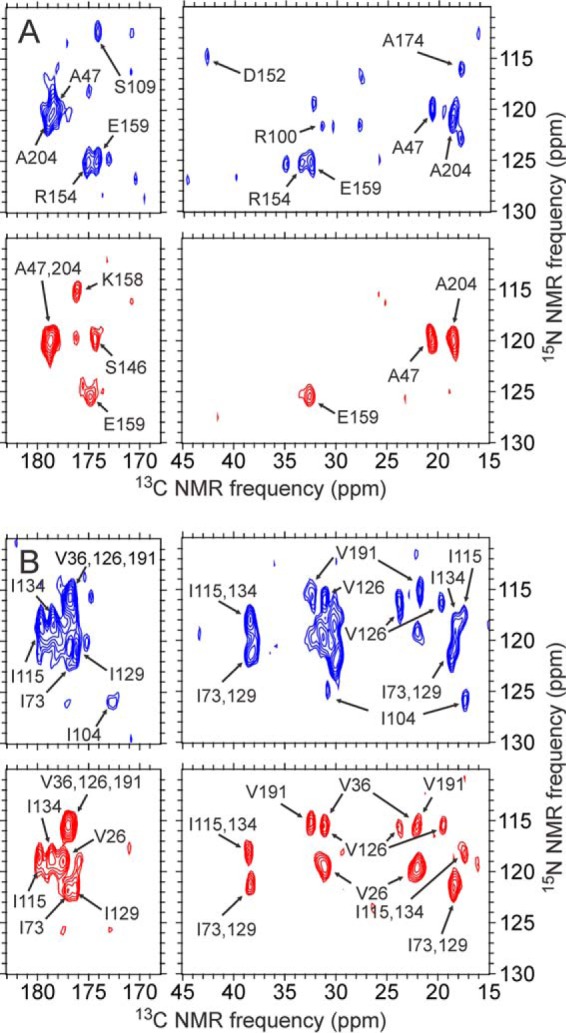
Examples of two-dimensional slices from three-dimensional NCACX spectra of uniformly 15N,13C-labeled R18L-CA spheres (blue) and sheets (red). A, slices at 53.4 ppm in the t2 dimension (i.e. CA dimension). B, slices at 65.5 ppm in the t2 dimension. Cross-peak assignments are based on analyses of two- and three-dimensional spectra and on previously reported assignments for WT-CA tubes (15). Spectra were recorded with 20 ms DARR mixing periods between t2 and t3 periods. Maximum t1 and t2 periods were 5.2–5.9 and 6.0–6.7 ms, respectively. Total measurement times were ∼12 days. Contour levels increase by factors of 1.2.
Of the total of 231 residues, chemical shifts were assigned for 152 residues in R18L-CA spheres; chemical shifts were assigned for 174 residues in R18L-CA sheets. Assignments are listed in supplemental Tables S1 and S2. For R18L-CA sheets, most residues that lack chemical shift assignments are located at in the N-terminal segment (residues 3–7 and 9–13), the C-terminal tail (residues 220–231), the NTD-CTD linker (residues 141–145), and the segment between helices 8 and 9 (residues 170–173, 175–176, and 179–180). For R18L-CA spheres, additional unassigned residues include 28–30, 61–62, 178, and 181–182. The segments that contain unassigned residues in R18L-CA sheets and spheres are essentially the same as the segments that contain unassigned residues in WT-CA tubes, consistent with essentially the same distributions of structurally ordered and disordered segments in the three types of assemblies. Signals assigned to Pro1 and Ser2 were observed in all three types of assemblies, indicating that the very N terminus of CA is structurally ordered, presumably through salt bridge interactions with Asp51 (21, 23). The fact that WT-CA tubes have fewer unassigned residues is attributable to the more extensive set of solid state NMR spectra for WT-CA tubes recorded by Bayro et al. (15).
Residue-specific root mean square differences between solid state NMR chemical shifts in R18L-CA assemblies and chemical shifts in WT-CA tubes are displayed on CA hexamers and monomers in a color-coded manner in Fig. 5. Backbone structures in Fig. 5 are taken from PDB file 3J34. For a given residue, the r.m.s. chemical shift difference is defined as,
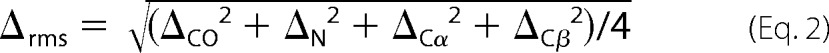 |
where ΔCO, ΔN, ΔCα, and ΔCβ are differences for backbone carbonyl, amide nitrogen, α-carbon, and β-carbon sites. When one or more of these chemical shifts are not available, the denominator in the definition of Δrms is adjusted accordingly. Δrms values are also given in supplemental Tables S1 and S2. For most residues for which chemical shift assignments are available, Δrms < 0.4 ppm (yellow in Fig. 5), indicating a difference that is within the estimated uncertainty in our chemical shift values due to limitations imposed by line widths and signal-to-noise. Residues with Δrms > 0.4 ppm (orange in Fig. 5) are not localized in specific protein segments or at specific regions of intermolecular interactions. These results indicate that conformational differences are relatively minor and distributed over the molecular structures.
FIGURE 5.

Summary of differences in solid state NMR chemical shifts between R18L-CA assemblies and WT-CA tubes. Residue-specific values of chemical shift differences Δrms (defined in the main text) are represented by colors on a schematic representation of a CA hexamer (A, B, D, and E) or monomer (C and F). Hexamer and monomer structures come from PDB file 3J34. Chemical shifts of R18L-CA spheres and WT-CA tubes are compared in panels A–C. Chemical shifts of R18L-CA sheets and WT-CA tubes are compared in panels D–F. Yellow and orange residues have 0.0 ppm < Δrms < 0.4 ppm and Δrms ≥ 0.4 ppm, respectively. Blue residues do not have assigned chemical shifts. The hexamer is viewed from “above” in panels A and D, and “below” in panels B and E. Helical segments are numbered in panels C and F, and the R18L mutation site is shown.
15N and 13C chemical shift values were also used as input for secondary structure predictions by the TALOS+ program (49). Predictions are given in supplemental Tables S1 and S2. The 11 expected α-helical segments are identified by TALOS+. A short helix at the N-terminal end of CTD (residues 150–152) is predicted by TALOS+ for R18L-CA sheets, consistent with the 310-helix seen in some crystalline forms of CTD (PDB files 1A43 and 2XT1), in crystalline forms of full-length CA (PDB files 3MGE, 3P05, and 4XFX), but not in other crystalline or soluble forms of CTD (PDB files 1A8O and 2KOD). The same segment is not predicted to be helical in R18L-CA spheres or WT-CA tubes, although the solid state NMR chemical shifts are similar in all three samples and the backbone φ and ψ torsion angles predicted by TALOS+ are also similar. The difference in secondary structure predictions for this segment is therefore unlikely to indicate real structural differences.
Absence of Detectable Signals Attributable to CA Pentamers
To reduce the complexity of the solid state NMR spectra, samples of WT-CA and R18L-CA were prepared with partial 13C labeling, using [2-13C]glycerol as the carbon source in the protein expression medium (50, 51). For R18L-CA, methionine residues were also fully 13C labeled. Regions of the two-dimensional NCACX spectra of 2-Glyc-WT-CA tubes, 2-Glyc,Met-R18L-CA spheres, and 2-Glyc,Met-R18L-CA sheets are shown in Figs. 6 and supplemental Fig. S4. Two-dimensional 13C-13C spectra are shown in supplemental Fig. S5. Overall, the spectra of the three samples are very similar, with few significant differences in cross-peak positions and widths. Relative intensities of certain cross-peaks vary, most likely due to differences in local dynamics. For example, a cross-peak from Ile201 at 64.7 ppm/123.2 ppm is weaker in the two-dimensional NCACX spectrum of 2-Glyc,Met-R18L-CA spheres than in the corresponding spectra of 2-Glyc-WT-CA tubes and 2-Glyc,Met-R18L-CA sheets. A cross-peak from Thr188 at 63.8 ppm/108.7 ppm is stronger in the two-dimensional NCACX spectrum of 2-Glyc-WT-CA tubes than in the corresponding spectra of 2-Glyc,Met-R18L-CA spheres or sheets. A cross-peak from Phe161 at 62.8 ppm/127.4 ppm in the two-dimensional NCACX spectra of 2-Glyc,Met-R18L-CA spheres and sheets is weaker (and shifted) in the corresponding spectrum of 2-Glyc-WT-CA tubes.
FIGURE 6.

Two-dimensional NCACX spectra of HIV-1 capsid protein assemblies that are uniformly 15N-labeled and partially 13C-labeled. A, 2-Glyc-WT-CA tubes. B, 2-Glyc,Met-R18L-CA spheres. C, 2-Glyc,Met-R18L-CA sheets. D, color-coded one-dimensional slices at 15N chemical shifts of 114.8, 123.2, 124.3, and 128.0 ppm (dashed lines in two-dimensional spectra). DARR mixing periods between t1 and t2 periods were 25 ms. Maximum t1 periods were 7.2 ms. Total measurement times were ∼48 h. Contour levels increase by factors of 1.5.
As discussed above, the smallest R18L-CA spheres may contain both R18L-CA pentamers and R18L-CA hexamers, in a 3:5 ratio. One might expect molecules in pentamers to have significantly different chemical shifts, especially for residues that are involved in intermolecular interactions that differ in pentamers and hexamers (6, 8). However, two-dimensional spectra of R18L-CA spheres in Figs. 6, supplemental Figs. S4 and S5 do not show additional cross-peak signals that can be attributed to pentamers. Because the proportion of pentamers is expected to be smaller in larger spheres, this result may reflect the fact that spheres with minimal diameter represent less than half of the R18L-CA molecules in our samples.
Differences in Trp and Met Side Chain Signals
CA contains five tryptophan residues. Trp23, Trp117, and Trp133 are buried within NTD, whereas Trp80 is partly exposed. Trp184 is located in the intermolecular dimerization interface between CTD pairs (Fig. 7A) and is a critical determinant of CTD dimerization, both in solution and in capsid assemblies (20, 24, 52). Fig. 8 shows regions of two-dimensional 13C-13C and 15N-13C spectra in which tryptophan side chain signals appear. The two-dimensional spectra of R18L-CA spheres or R18L-CA sheets are superimposed on spectra of WT-CA tubes. Cross-peak positions for Trp23 and Trp117 are nearly identical in all three samples. Significant changes in side chain chemical shifts for Trp80 and Trp133 are observed. Interestingly, these two side chains are in close proximity, with a distance of 4.3 ± 0.1 Å between Cϵ3 of Trp80 and Cζ2 of Trp133 in PDB files 4XFX, 1M9C, 3P05, and 3MGE. The observed differences in chemical shifts suggest a minor difference in structure between helices 4 and 7 of NTD in R18L-CA assemblies, compared with WT-CA tubes.
FIGURE 7.
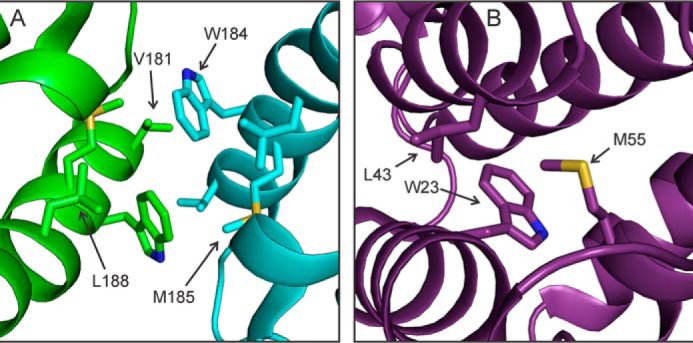
Intermolecular and intramolecular side chain-side chains interactions in HIV-1 capsid protein assemblies. A, intermolecular dimerization interface, involving helix 9 segments of interacting CTD domains from different CA molecules (green and cyan) around local 2-fold symmetry axes. B, intramolecular contacts of helices 1, 2, and 3 within a single NTD domain (purple). Structures in panels A and B are present in crystalline WT-CA (PDB file 4XFX), and may be somewhat different in WT-CA tubes, R18L-CA spheres, and R18L-CA sheets.
FIGURE 8.

Aromatic regions of two-dimensional 13C-13C and two-dimensional 15N-13C spectra of HIV-1 capsid protein assemblies that are uniformly 15N labeled and partially 13C labeled. A and C, two-dimensional spectra of R18L-CA spheres (blue), overlaid on two-dimensional spectra of WT-CA tubes (black). Assignments of cross-peaks from tryptophan residues and one tyrosine residue are shown. B and D, two-dimensional spectra of R18L-CA sheets (red), overlaid on two-dimensional spectra of WT-CA tubes (black). Spectra in panels A and B were obtained with U-R18L-CA and U-WT-CA. Spectra in panels C and D were obtained with 2-Glyc,Met-R18L-CA and 2-Glyc-WT-CA. DARR mixing periods were 500 ms in two-dimensional 13C-13C spectra and 140 ms in two-dimensional 15N-13C spectra. Maximum t1 periods were 6.9 ms in two-dimensional 13C-13C spectra and 6.4 ms in two-dimensional 15N-13C spectra. The 13C carrier frequency during 15N-13C cross-polarization was set to 127 ppm in the two-dimensional 15N-13C spectrum of 2-Glyc-WT-CA tubes and 174 ppm in two-dimensional 15N-13C spectra of 2-Glyc,Met-R18L-CA assemblies, accounting for differences in signals below 115 ppm in these spectra. Contour levels increase by factors of 1.2.
Chemical shifts for Trp184 are nearly identical in WT-CA tubes and R18L-CA spheres. In R18L-CA sheets, a side chain Cδ1/Nϵ1 cross-peak is not observed for Trp184, whereas Cδ1/Nϵ1 cross-peaks are observed for all other tryptophan residues. This observation suggests that the Trp184 side chain has greater disorder in R18L-CA sheets than in other assemblies.
The side chain of Met185 is also located in the CTD-CTD dimerization interface (Fig. 7A). Signals from methionine residues are seen in the regions of two-dimensional 13C-13C spectra shown in Fig. 9. Although cross-peaks from Met185 are clearly seen in spectra of Met-WT-CA tubes and 2-Glyc,Met-R18L-CA spheres, cross-peaks from Met185 are absent from the two-dimensional spectrum of 2-Glyc,Met-R18L-CA sheets. This observation provides further support for greater disorder in the dimerization interface in R18L-CA sheets, as the Met185 side chain also participates in intermolecular interactions in the dimerization interface (5, 19, 26).
FIGURE 9.

Selected regions of two-dimensional 13C-13C spectra of HIV-1 capsid protein assemblies in which methionine residues are uniformly 15N,13C labeled. A, Met-WT-CA tubes. B, 2-Glyc,Met-R18L-CA spheres. C, 2-Glyc,Met-R18L-CA sheets. Orange lines indicate intra-residue correlations to methionine methyl signals. The dashed line in panel C indicates the absence of signals from Met185. Inter-residue cross-peaks in two-dimensional spectra of 2-Glyc,Met-R18L-CA assemblies, which were also partially 13C labeled at non-methionine residues by expression with [2-13C]glycerol as the carbon source, are indicated by purple labels and arrows. D, color-coded one-dimensional slices at 31.8 and 59.9 ppm (dashed lines in two-dimensional spectra). Spectra were recorded with 500-ms DARR mixing periods and maximum t1 periods of 6.9 ms. Contour levels increase by factors of 1.2.
Cross-peaks from Met39 are also clearly seen in the spectrum of Met-WT-CA tubes (Fig. 9A), but are absent or unresolved in the spectra of 2-Glyc,Met-R18L-CA assemblies (Fig. 9, B and C). In crystal structures of CA hexamers (5, 6) and pentamers (8), the Met39 side chain participates in a hydrophobic pocket that presumably stabilizes NTD-NTD contacts, through intermolecular interactions with Leu20 and Val24. Mutation of Met39 to alanine prevents formation of WT-CA tubes (13). Differences in Met39 signals shown in Fig. 9 suggest differences in the intermolecular NTD-NTD interface that involves helices 1 and 2 in capsid protein assemblies with different morphologies.
Two-dimensional 13C-13C spectra of 2-Glyc,Met-R18L-CA assemblies in Fig. 9, which were obtained with 500 ms DARR mixing periods, also show inter-residue cross-peaks that connect Ala22, Trp23, Leu43, and Ser44 with Met55. (Inter-residue cross-peaks are not seen in the spectrum of WT-CA tubes in Fig. 9 because only methionine residues were 13C labeled in the Met-WT-CA sample.) As shown in Fig. 7B, Trp23 is in helix 1 of NTD, Leu43 and Ser44 are in helix 2, and Met55 is in helix 3. The observation of inter-residue cross-peaks indicates inter-residue 13C-13C distances of ∼7 Å or less. The fact that the relative intensities of the inter-residue cross-peaks are somewhat different in the two-dimensional spectra of 2-Glyc,Met-R18L-CA spheres and 2-Glyc,Met-R18L-CA sheets suggests that subtle differences in intramolecular helix-helix distances and/or orientations may exist. Structural differences of this type may contribute to differences in the curvature and morphology of the capsid protein assemblies.
Differences in Dynamics of Disordered Segments
Highly mobile segments of WT-CA and R18L-CA assemblies were probed by two-dimensional 13C-13C INEPT-TOBSY measurements. The two-dimensional INEPT-TOBSY spectra were recorded with conditions resembling those in solution NMR measurements, i.e. 1H-13C and 13C-13C spin polarization transfers mediated by scalar (rather than dipole-dipole) couplings, low-power proton decoupling, and relatively slow MAS. Under these conditions, cross-peak signals can only be observed from residues that undergo rapid (submicrosecond) and nearly isotropic orientational motion. In general, one expects residues that have signal assignments in solid state NMR spectra not to contribute to the two-dimensional INEPT-TOBSY spectra, because such residues should not have sufficient mobility.
The two-dimensional INEPT-TOBSY spectrum of U-WT-CA tubes (Fig. 10A) shows a greater number of strong cross-peaks than the corresponding spectra of U-R18L-CA spheres or sheets (Fig. 10, B and C), indicating a greater number of highly mobile residues. Differences in the numbers of detectable cross-peaks are not simply due to differences in overall signal-to-noise, as shown by the one-dimensional slices in Fig. 10.
FIGURE 10.

Two-dimensional 13C-13C INEPT-TOBSY spectra of uniformly 15N,13C-labeled HIV-1 capsid assemblies, showing signals from protein segments that remain highly dynamic in the assemblies. Two-dimensional spectra are shown for U-WT-CA tubes (A), U-R18L-CA spheres (B), and U-R18L-CA sheets (C), with color-coded one-dimensional slices (D) at 3.18, 42.3, and 53.4 ppm, corresponding to the dashed lines in the two-dimensional spectra. Residue-type assignments, based on random-coil chemical shifts, are shown in panel A. Maximum t1 periods were 7.2–8.9 ms. Total measurement times were ∼24 h. Contour levels increase by factors of 1.4.
Assignment of two-dimensional INEPT-TOBSY cross-peaks to residue types is based on 13C chemical shifts, with the reasonable assumption that chemical shifts of highly mobile residues are approximately equal to random-coil values (53). For U-WT-CA tubes, signals from Ala, Asp/Asn, Glu, Gly, Ile, Lys, Leu, Met, Pro, Arg, Ser, Thr, and Val residues are observed. Signals from Ala, Glu, Gly, Ile, Lys, Leu, Met, Pro, Arg, and Val residues can explained by high mobility in the C-terminal tail (residues 220–231), the NTD-CTD linker (residues 141–145), and the segment between helices 8 and 9 (residues 172–182). Signals from Ser and Thr residues may arise from Ser146 and Thr171, which have tentative assignments in solid state NMR spectra of WT-CA tubes, but not definitive assignments (15). Signals from Asp or Asn residues are problematic, because all Asp and Asn residues have assignments in solid state NMR spectra. A likely explanation for the Asp/Asn cross-peaks in the two-dimensional INEPT-TOBSY spectrum of U-WT-CA tubes (which may also apply to the Ser and Thr cross-peaks) is that the WT-CA tubes contain dynamic heterogeneity, meaning that certain residues are relatively rigid in some WT-CA molecules within the tubes but highly mobile in other WT-CA molecules.
The two-dimensional INEPT-TOBSY spectrum of U-R18L-CA sheets (Fig. 10C) shows cross-peaks from the same residue types as the two-dimensional INEPT-TOBSY spectrum of WT-CA tubes, except that cross-peaks from Asp/Asn residues are absent. Relative intensities of various cross-peaks are clearly different in spectra of U-R18L-CA sheets and U-WT-CA tubes, indicating differences in mobilities. For U-R18L-CA spheres (Fig. 10B), strong cross-peaks are only observed for Lys and Leu residues, possibly arising from Lys227 and Leu231 in the C-terminal tail. Weaker cross-peaks from Arg and Val residues are observed, possibly arising from Arg229 and Val230. No diagonal signals from Gly α-carbons are observed, despite the presence of Gly at residues 220, 222, 223, and 225. The motions of disordered residues in R18L-CA spheres appear to be restricted or slowed, even in the C-terminal tail.
Discussion
The ability of the HIV-1 capsid protein to self-assemble spontaneously into supramolecular structures with several distinct morphologies, all based on the same lattice and nearly the same intermolecular contacts, is an intriguing and biologically relevant phenomenon that must depend on variations in local molecular structure and/or dynamics. Data presented above provide new insights into the nature of these variations. The main result is that solid state NMR spectra of tubular WT-CA assemblies, spherical R18L-CA assemblies, and planar R18L-CA assemblies are remarkably similar. 13C and 15N chemical shifts for the structurally ordered and relatively rigid residues that contribute to solid state NMR spectra vary by less than 0.5 ppm for backbone CO, Cα, Cβ, and backbone N sites of most residues (Fig. 5, supplemental Tables S1 and S2). Two-dimensional 13C-13C and 15N-13C spectra are nearly indistinguishable (Figs. 3, 6, supplemental Figs. S1, S2, and S5). There are no major differences in the backbone conformations or the identities of structurally ordered segments in the tubular, spherical, and planar assemblies.
It is useful to compare these observations with solid state NMR data for the 56-residue B1 domain of streptococcal immunoglobulin-binding protein G (GB1). Rienstra and co-workers (54, 55) have determined chemical shift assignments for GB1 microcrystals prepared under various conditions that result in different crystal forms. The backbone structures are essentially identical in all cases. Comparisons of chemical shifts deposited in the Biological Magnetic Resonance Data Bank (BMRB codes 17810, 15380 formulation D, 15380 formulation E, and 18397) lead to values of Δrms that range from 0.02 to 0.98 ppm, with mean value and standard deviation equal to 0.20 and 0.12 ppm. Chemical shift variations for HIV-1 capsid protein assemblies reported in supplemental Tables S1 and S2 are larger by a factor of about 2.0, with a significant part of the larger variation being attributable to the broader solid state NMR lines and more highly congested spectra of the capsid protein assemblies. Thus, conformational variations in structurally ordered segments of capsid protein assemblies are not much larger than conformational variations of GB1 in different crystal forms.
As an additional assessment of the significance of the observed Δrms values, Fig. 11 shows the results of chemical shift predictions for reported CA structures. Values of Δrms between engineered CA mutants that form crystalline hexamers and pentamers (6, 8) (PDB files 3MGE and 3P05), calculated from predictions generated from atomic coordinates by the SHIFTS (56) and ShiftX (57) programs, are substantially larger than our experimental values of Δrms between WT-CA tubes and R18L-CA sheets (Fig. 11A). Predicted Δrms values between unequivalent molecules within a cryo-EM-based model for WT-CA tubes (4) (PDB file 3J34) are also substantially larger (Fig. 11C). These calculations support the idea that conformational differences among WT-CA tubes, R18L-CA sheets, and R18L-CA tubes are smaller than the differences among these structural models (Fig. 11, B and D).
FIGURE 11.

Predicted chemical shift differences between HIV-1 CA structural models. A, values of Δrms between engineered hexameric (PDB code 3MGE) and pentameric (PDB code 3P05) CA mutants, based on 15N and 13C chemical shift predictions from SHIFTS (red triangles) and ShiftX (blue squares). For comparison, experimental values of Δrms between CA tubes and R18L-CA sheets are shown as solid black circles. B, superposition of Cα traces for the hexameric (magenta) and pentameric (green) CA mutants, for which the r.m.s. deviation between Cα coordinates in residues 10–140 and 150–200 is 1.72 Å. C, values of Δrms between CA chains m and A from a cryo-EM-based model for CA tubes (PDB 3J34), based on chemical shift predication from SHIFTS (red triangles) and ShiftX (blue squares). D, superposition of Cα traces for chains A (magenta) and m (green), for which the r.m.s. deviation between Cα coordinates in residues 10–140 and 150–200 is 2.53 Å.
The r.m.s. deviation for α-carbon coordinates between hexamer-forming and pentamer-forming CA mutants (Fig. 11, A and B) is 1.73 Å. When only residues 150–200 are superimposed, the α-carbon r.m.s. deviation for these residues is only 0.79 Å. Yet predicted chemical shift differences (quantified by Δrms values) are greater than 1.5 ppm for many residues, larger than the chemical shift differences observed in our experiments on WT-CA and R18L-CA assemblies.
For residues 151–173, differences in backbone φ and ψ torsion angles between the crystallographic structures of hexamer-forming and pentamer-forming mutants are less than 20°. Predicted Δrms values for these residues are 0.6 ppm or less using SHIFTX (but 1.8 ppm or less using SHIFTS, which generally predicts stronger dependences of chemical shifts on local protein conformation). Experimental Δrms values for residues 151–173 are less than 0.65 ppm when experimental chemical shifts of WT-CA tubes are compared with those of either R18L-CA sheets or R18L-CA spheres. Thus, the observed chemical shift differences in solid state NMR spectra of the three types of CA assemblies indicate backbone torsion angle differences in structurally ordered segments that are less than 20°.
As discussed above, certain protein segments do not contribute signals to solid state NMR spectra of WT-CA tubes (15). The observation that multidimensional solid state NMR spectra of R18L-CA sheets show essentially the same number of cross-peaks and the same NMR line widths as spectra of WT-CA tubes indicates that the absence of signals from certain segments is not due to static conformational disorder associated with the lower symmetry of the tubular assemblies. Apparently, the curvature of the lattice in WT-CA tubes does not lead to a significant increase in conformational variations in the rigid segments of CA molecules, relative to a planar lattice. Segments that are invisible in solid state NMR measurements on WT-CA tubes remain invisible in measurements on R18L-CA sheets. Most likely, the invisible segments are dynamically disordered (15, 40) in tubular, planar, and spherical assemblies. The precise ranges of conformations explored by the dynamically disordered segments may vary among different types of assemblies (and among symmetry-unequivalent molecules within WT-CA tubes), allowing for the observed variations in lattice curvature. Related suggestions regarding the origin of lattice curvature have been made previously by Bailey et al. (58) and Pornillos et al. (8)
Although the backbone structure is essentially unchanged in the three types of capsid protein assemblies, differences in solid state NMR signals are observed for certain amino acid side chains (Figs. 8 and 9), including side chains of Trp184 and Met185. These side chains are critical for CTD-CTD dimerization, both in unassembled CA (24, 52) and in the CA lattice (5, 6), and are essential for infectivity of HIV-1 virions (59). In particular, R18L-CA sheets show altered or absent signals from Trp184 and Met185 side chains, which are strong and well resolved in spectra of both WT-CA tubes and R18L-CA spheres. Thus, it appears that lattice curvature involves some level of variation in the intermolecular packing of critical side chains in the dimerization interface.
Side chain signals of Trp80 and Trp133 also exhibit variations that suggest differences in intramolecular helix-helix packing within different types of assemblies. Variations in the intensities of inter-residue cross-peaks involving Met55 also suggest differences in intramolecular helix-helix packing. The observed variations in solid state NMR signals from these intramolecularly buried side chains are unexpected, as the corresponding differences between x-ray crystal structures of engineered CA hexamers and pentamers are quite small (6, 8). For all atoms in Trp80 and Trp133 within one CA molecule, the r.m.s. deviation value between PDB files 3P05 and 3MGE is only 0.12 Å; for all atoms in Trp23, Leu43, and Met55, the r.m.s. deviation value is 0.39 Å. Quantification of the structural variations suggested by data in Figs. 8 and 9 may be feasible with additional solid state NMR measurements on selectively labeled samples.
Differences in the dynamics of flexible protein segments are apparent in Fig. 10. Surprisingly, the two-dimensional 13C-13C INEPT-TOBSY spectra indicate fewer highly dynamic residues in R18L-CA spheres, compared with R18L-CA sheets or WT-CA tubes. Differences in these spectra may result from differences in the time scales of molecular motions, rather than differences in the amplitudes of motions.
In conclusion, we have shown that the identities and conformations of structurally ordered, rigid segments of the HIV-1 capsid protein (i.e. segments that contribute to solid state NMR spectra) are nearly identical in tubular, spherical, and planar assemblies. Conformational distributions in disordered segments may vary, accounting for variations in lattice curvature among different types of assemblies. Variations in solid state NMR signals from certain amino acid side chains are also observed, suggesting that the structure of the intermolecular CTD dimerization interface may be different in curved and planar lattices and intramolecular helix-helix packing within the NTD may be somewhat variable.
Author Contributions
J. X. L. designed, performed, and analyzed all experiments on R18L-CA assemblies. M. J. B. designed, performed, and analyzed all experiments on WT-CA assemblies and assisted with the design of experiments on R18L-CA assemblies. R. T. assisted with experimental design and data analysis. J. X. L. and R. T. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Barbie Ganser-Pornillos and Dr. Mark J. Yeager for helpful discussions and advice regarding the preparation of R18L-CA assemblies.
This work was supported by the Intramural Research Program of the NIDDK, National Institutes of Health, under project DK075032, and by the Intramural AIDS Targeted Antiviral Program of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S5 and Tables S1 and S2.
- CA
- capsid protein
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- PDB
- Protein Data Bank
- r.m.s.
- root mean square
- MAS
- magic-angle spinning
- TEM
- transmission electron microscopy
- AFM
- atomic force microscope
- DARR
- dipolar-assisted rotational resonance
- TOBSY
- total through-bond correlation
- INEPT
- insensitive-nuclei enhanced polarization transfer
- Glyc
- glycerol.
References
- 1. Ganser-Pornillos B. K., Yeager M., and Pornillos O. (2012) Assembly and architecture of HIV. Adv. Exp. Med. Biol. 726, 441–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ganser-Pornillos B. K., Cheng A., and Yeager M. (2007) Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131, 70–79 [DOI] [PubMed] [Google Scholar]
- 3. Li S., Hill C. P., Sundquist W. I., and Finch J. T. (2000) Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413 [DOI] [PubMed] [Google Scholar]
- 4. Zhao G., Perilla J. R., Yufenyuy E. L., Meng X., Chen B., Ning J., Ahn J., Gronenborn A. M., Schulten K., Aiken C., and Zhang P. (2013) Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gres A. T., Kirby K. A., KewalRamani V. N., Tanner J. J., Pornillos O., and Sarafianos S. G. (2015) X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pornillos O., Ganser-Pornillos B. K., Kelly B. N., Hua Y., Whitby F. G., Stout C. D., Sundquist W. I., Hill C. P., and Yeager M. (2009) X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ganser B. K., Li S., Klishko V. Y., Finch J. T., and Sundquist W. I. (1999) Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83 [DOI] [PubMed] [Google Scholar]
- 8. Pornillos O., Ganser-Pornillos B. K., and Yeager M. (2011) Atomic-level modelling of the HIV capsid. Nature 469, 424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cardone G., Purdy J. G., Cheng N., Craven R. C., and Steven A. C. (2009) Visualization of a missing link in retrovirus capsid assembly. Nature 457, 694–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Briggs J. A., Wilk T., Welker R., Kräusslich H. G., and Fuller S. D. (2003) Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu Z., Dobro M. J., Woodward C. L., Levandovsky A., Danielson C. M., Sandrin V., Shi J., Aiken C., Zandi R., Hope T. J., and Jensen G. J. (2013) Unclosed HIV-1 capsids suggest a curled sheet model of assembly. J. Mol. Biol. 425, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ehrlich L. S., Agresta B. E., and Carter C. A. (1992) Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66, 4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ganser-Pornillos B. K., von Schwedler U. K., Stray K. M., Aiken C., and Sundquist W. I. (2004) Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78, 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen B., and Tycko R. (2010) Structural and dynamical characterization of tubular HIV-1 capsid protein assemblies by solid state nuclear magnetic resonance and electron microscopy. Protein Sci. 19, 716–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bayro M. J., Chen B., Yau W. M., and Tycko R. (2014) Site-specific structural variations accompanying tubular assembly of the HIV-1 capsid protein. J. Mol. Biol. 426, 1109–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han Y., Ahn J., Concel J., Byeon I. J., Gronenborn A. M., Yang J., and Polenova T. (2010) Solid state NMR studies of HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 132, 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gamble T. R., Vajdos F. F., Yoo S., Worthylake D. K., Houseweart M., Sundquist W. I., and Hill C. P. (1996) Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 18. Momany C., Kovari L. C., Prongay A. J., Keller W., Gitti R. K., Lee B. M., Gorbalenya A. E., Tong L., McClure J., Ehrlich L. S., Summers M. F., Carter C., and Rossmann M. G. (1996) Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 3, 763–770 [DOI] [PubMed] [Google Scholar]
- 19. Worthylake D. K., Wang H., Yoo S., Sundquist W. I., and Hill C. P. (1999) Structures of the HIV-1 capsid protein dimerization domain at 2.6-Å resolution. Acta Crystallogr. D Biol. Crystallogr. 55, 85–92 [DOI] [PubMed] [Google Scholar]
- 20. Pornillos O., Ganser-Pornillos B. K., Banumathi S., Hua Y., and Yeager M. (2010) Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J. Mol. Biol. 401, 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gitti R. K., Lee B. M., Walker J., Summers M. F., Yoo S., and Sundquist W. I. (1996) Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273, 231–235 [DOI] [PubMed] [Google Scholar]
- 22. Campos-Olivas R., and Summers M. F. (1999) Backbone dynamics of the N-terminal domain of the HIV-1 capsid protein and comparison with the G94D mutant conferring cyclosporin resistance/dependence. Biochemistry 38, 10262–10271 [DOI] [PubMed] [Google Scholar]
- 23. Tang C., Ndassa Y., and Summers M. F. (2002) Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 9, 537–543 [DOI] [PubMed] [Google Scholar]
- 24. Wong H. C., Shin R., and Krishna N. R. (2008) Solution structure of a double mutant of the carboxy-terminal dimerization domain of the HIV-1 capsid proteint. Biochemistry 47, 2289–2297 [DOI] [PubMed] [Google Scholar]
- 25. Shin R., Tzou Y. M., and Krishna N. R. (2011) Structure of a monomeric mutant of the HIV-1 capsid protein. Biochemistry 50, 9457–9467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Byeon I. J., Meng X., Jung J., Zhao G., Yang R., Ahn J., Shi J., Concel J., Aiken C., Zhang P., and Gronenborn A. M. (2009) Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139, 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deshmukh L., Schwieters C. D., Grishaev A., Ghirlando R., Baber J. L., and Clore G. M. (2013) Structure and dynamics of full-length HIV-1 capsid protein in solution. J. Am. Chem. Soc. 135, 16133–16147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deshmukh L., Ghirlando R., and Clore G. M. (2014) Investigation of the structure and dynamics of the capsid-spacer peptide 1-nucleocapsid fragment of the HIV-1 Gag polyprotein by solution NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 53, 1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deshmukh L., Ghirlando R., and Clore G. M. (2015) Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 3374–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen B., and Tycko R. (2011) Simulated self-assembly of the HIV-1 capsid: protein shape and native contacts are sufficient for two-dimensional lattice formation. Biophys. J. 100, 3035–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grime J. M., and Voth G. A. (2012) Early stages of the HIV-1 capsid protein lattice formation. Biophys. J. 103, 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meng X., Zhao G., Yufenyuy E., Ke D., Ning J., Delucia M., Ahn J., Gronenborn A. M., Aiken C., and Zhang P. (2012) Protease cleavage leads to formation of mature trimer interface in HIV-1 capsid. PLoS Pathog. 8, e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiao X., Jeon J., Weber J., Zhu F., and Chen B. (2015) Mechanism of polymorphism and curvature of HIV capsid assemblies probed by 3D simulations with a novel coarse grain model. Biochim. Biophys. Acta 1850, 2353–2367 [DOI] [PubMed] [Google Scholar]
- 34. Frank G. A., Narayan K., Bess J. W. Jr., Del Prete G. Q., Wu X., Moran A., Hartnell L. M., Earl L. A., Lifson J. D., and Subramaniam S. (2015) Maturation of the HIV-1 core by a non-diffusional phase transition. Nat. Commun. 6, 5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wright E. R., Schooler J. B., Ding H. J., Kieffer C., Fillmore C., Sundquist W. I., and Jensen G. J. (2007) Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26, 2218–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Briggs J. A., Riches J. D., Glass B., Bartonova V., Zanetti G., and Kräusslich H. G. (2009) Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U.S.A. 106, 11090–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schur F. K., Hagen W. J., Rumlová M., Ruml T., Müller B., Kräusslich H. G., and Briggs J. A. (2015) Structure of the immature HIV-1 capsid in intact virus particles at 8.8-Å resolution. Nature 517, 505–508 [DOI] [PubMed] [Google Scholar]
- 38. Byeon I. J., Hou G., Han Y., Suiter C. L., Ahn J., Jung J., Byeon C. H., Gronenborn A. M., and Polenova T. (2012) Motions on the millisecond time scale and multiple conformations of HIV-1 capsid protein: implications for structural polymorphism of CA assemblies. J. Am. Chem. Soc. 134, 6455–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han Y., Hou G., Suiter C. L., Ahn J., Byeon I. J., Lipton A. S., Burton S., Hung I., Gor'kov P. L., Gan Z., Brey W., Rice D., Gronenborn A. M., and Polenova T. (2013) Magic angle spinning NMR reveals sequence-dependent structural plasticity, dynamics, and the spacer peptide 1 conformation in HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 135, 17793–17803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu M., Hou G., Zhang H., Suiter C. L., Ahn J., Byeon I. J., Perilla J. R., Langmead C. J., Hung I., Gor'kov P. L., Gan Z., Brey W., Aiken C., Zhang P., Schulten K., Gronenborn A. M., and Polenova T. (2015) Dynamic allostery governs cyclophilin A-HIV capsid interplay. Proc. Natl. Acad. Sci. U.S.A. 112, 14617–14622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bayro M. J., Daviso E., Belenky M., Griffin R. G., and Herzfeld J. (2012) An amyloid organelle, solid state NMR evidence for cross-β assembly of gas vesicles. J. Biol. Chem. 287, 3479–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu J.-X., Qiang W., Yau W.-M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bennett A. E., Rienstra C. M., Auger M., Lakshmi K. V., and Griffin R. G. (1995) Heteronuclear decoupling in rotating solids. J. Chem. Phys. 103, 6951–6958 [Google Scholar]
- 44. Takegoshi K., Nakamura S., and Terao T. (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 [Google Scholar]
- 45. Morcombe C. R., Gaponenko V., Byrd R. A., and Zilm K. W. (2004) Diluting abundant spins by isotope edited radio frequency field assisted diffusion. J. Am. Chem. Soc. 126, 7196–7197 [DOI] [PubMed] [Google Scholar]
- 46. Hardy E. H., Verel R., and Meier B. H. (2001) Fast MAS total through-bond correlation spectroscopy. J. Magn. Reson. 148, 459–464 [DOI] [PubMed] [Google Scholar]
- 47. Morris G. A., and Freeman R. (1979) Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 101, 760–772 [Google Scholar]
- 48. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRpipe: a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 49. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hong M. (1999) Determination of multiple φ-torsion angles in proteins by selective and extensive 13C labeling and two-dimensional solid state NMR. J. Magn. Reson. 139, 389–401 [DOI] [PubMed] [Google Scholar]
- 51. Higman V. A., Flinders J., Hiller M., Jehle S., Markovic S., Fiedler S., van Rossum B. J., and Oschkinat H. (2009) Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labeled proteins. J. Biomol. NMR 44, 245–260 [DOI] [PubMed] [Google Scholar]
- 52. Alcaraz L. A., del Alamo M., Barrera F. N., Mateu M. G., and Neira J. L. (2007) Flexibility in HIV-1 assembly subunits: solution structure of the monomeric C-terminal domain of the capsid protein. Biophys. J. 93, 1264–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., and Sykes B. D. (1995) 1H, 13C, and 15N random coil NMR chemical shifts of the common amino acids: 1. investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81 [DOI] [PubMed] [Google Scholar]
- 54. Franks W. T., Zhou D. H., Wylie B. J., Money B. G., Graesser D. T., Frericks H. L., Sahota G., and Rienstra C. M. (2005) Magic-angle spinning solid state NMR spectroscopy of the β-1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J. Am. Chem. Soc. 127, 12291–12305 [DOI] [PubMed] [Google Scholar]
- 55. Schmidt H. L., Sperling L. J., Gao Y. G., Wylie B. J., Boettcher J. M., Wilson S. R., and Rienstra C. M. (2007) Crystal polymorphism of protein GB1 examined by solid state NMR spectroscopy and x-ray diffraction. J. Phys. Chem. B 111, 14362–14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu X. P., and Case D. A. (2001) Automated prediction of 15N, 13Ca, 13Cb, and 13C′ chemical shifts in proteins using a density functional database. J. Biomol. NMR 21, 321–333 [DOI] [PubMed] [Google Scholar]
- 57. Neal S., Nip A. M., Zhang H., and Wishart D. S. (2003) Rapid and accurate calculation of protein 1H, 13C, and 15N chemical shifts. J. Biomol. NMR 26, 215–240 [DOI] [PubMed] [Google Scholar]
- 58. Bailey G. D., Hyun J. K., Mitra A. K., and Kingston R. L. (2012) A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J. Mol. Biol. 417, 212–223 [DOI] [PubMed] [Google Scholar]
- 59. von Schwedler U. K., Stray K. M., Garrus J. E., and Sundquist W. I. (2003) Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77, 5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.