

Abstract
Bacterial catabolism of aromatic compounds from various sources including phenylpropanoids and flavonoids that are abundant in soil plays an important role in the recycling of carbon in the ecosystem. We have determined the crystal structures of apo-HcaR from Acinetobacter sp. ADP1, a MarR/SlyA transcription factor, in complexes with hydroxycinnamates and a specific DNA operator. The protein regulates the expression of the hca catabolic operon in Acinetobacter and related bacterial strains, allowing utilization of hydroxycinnamates as sole sources of carbon. HcaR binds multiple ligands, and as a result the transcription of genes encoding several catabolic enzymes is increased. The 1.9–2.4 Å resolution structures presented here explain how HcaR recognizes four ligands (ferulate, 3,4-dihydroxybenzoate, p-coumarate, and vanillin) using the same binding site. The ligand promiscuity appears to be an adaptation to match a broad specificity of hydroxycinnamate catabolic enzymes while responding to toxic thioester intermediates. Structures of apo-HcaR and in complex with a specific DNA hca operator when combined with binding studies of hydroxycinnamates show how aromatic ligands render HcaR unproductive in recognizing a specific DNA target. The current study contributes to a better understanding of the hca catabolic operon regulation mechanism by the transcription factor HcaR.
Keywords: bacterial transcription, DNA-binding protein, ligand-binding protein, metabolic regulation, protein conformation, DNA binding, human gut bacteria, NUDIX family, arabinose utilization, ligand-induced conformational change
Introduction
The multiple antibiotic resistance regulator (MarR)2/SlyA family of prokaryotic transcriptional factors has been shown to be crucial for intracellular survival and/or replication of both enteroinvasive Escherichia coli and Salmonella enterica serovar Typhimurium in phagocytic host cells (1) by up-regulating expression of molecular chaperones, proteins involved in the response to antibiotic and oxidative stresses, acid resistance, and production of virulence factors as well as down-regulating some metabolic pathways (2–4) and up-regulating the catabolism of aromatic compounds (5–7). MarR/SlyA members are found primarily in bacteria and in some archaea but are not present in eukaryotes (8) (Clusters of Orthologous Groups (COG) database (www.ncbi.nlm.nih.gov/COG/)).
MarR/SlyA modules are also present in larger proteins (representing over 60 different architectures) such as clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein Csa3 (Protein Data Bank code 2WTE) (9) and in papilloma virus helicase (Protein Data Bank code 2GXA) (10). These MarR/SlyA transcription factors are typically dimers and bind to palindromic DNA operators located within gene promoters with winged helix-turn-helix (wHTH) DNA-binding motifs. These protein-DNA interactions involve specific conformational changes in both transcription factors and DNA. The majority of these transcription factors respond to small ligands such as metal ions (zinc-binding AdcR) (11, 12); aromatic compounds such as uric acid (HucR) (13), 2-hydroxybenzoic acid (MarR), p-coumaroyl coenzyme A (CouR) (14), 3-chlorobenzoate (CbaR) (15), 3-hydroxybenzoate (MobR) (16, 17), lignin-derived hydroxycinnamates (HcaR) (5, 18), and protocatechuate/p-hydroxybenzoate (SCO6704 (5) and BadR from Rhodopseudomonas palustris (20)); and other natural and synthetic products (for example, ethidium bromide and 4′,6′-diamidino-2-phenylindole) (21). In most cases, in the absence of ligand, apo-MarR/SlyA proteins bind to specific DNA operators, and upon ligand binding, they show diminished DNA affinity.
In multiple antibiotic resistance, a nonspecific resistance system in bacteria, a single MarR transcription regulator appears to be capable of responding to a large number of compounds, such as 2,4-dinitrophenol, menadione, plumbagin, and salicylate, that can block MarR DNA binding and induce transcription of the mar operon (22). These data suggest that MarR/SlyA-like transcription factors are promiscuous and can accommodate a variety of ligands. These proteins show preference for binding aromatic compounds, although the affinities are not very high, which is consistent with relaxed specificity (23). The following question remains open: what is the mechanism of reducing DNA affinity? At present, there is inadequate and inconsistent structural information available about the interaction of aromatic ligands with MarR/SlyA transcription factors (24). It was suggested that the MarR family uses the unique mechanism where the DNA- and ligand-binding sites overlap, and the reported crystal structures of MarR/SlyA proteins with DNA imply that this family uses indirect DNA recognition to bind to a specific operator (26–29). It was also proposed that ligands may change distances between the two wHTH motifs, leading to steric clashes with the DNA backbone, and as a result abolish DNA binding. The interaction with ligand may also reorient helices of the wHTH motif, reducing or obliterating the target DNA binding. However, alternative mechanisms have also been proposed (6, 30).
Acinetobacter are important Gram-negative soil gammaproteobacteria that contribute to the mineralization of aromatic compounds and together with the Pseudomonas genus seem to prefer organic acids as carbon sources (31). In the Acinetobacter sp. ADP1 genome, the catabolism of 4-hydroxycinnamic acid derivatives is encoded by the hca operon that is located in an island of catabolic diversity (19). The organization of this operon involves transcripts in both directions (hcaABCDEFG and hcaKR) (Fig. 1). The operon codes for the following activities: enoyl-coenzyme A (CoA) hydratase/lyase (hcaA), hydroxybenzaldehyde dehydrogenase (hcaB), coenzyme A ligase (hcaC), acyl-coenzyme A dehydrogenase (hcaD), outer membrane porin of OprD superfamily (hcaE), chlorogenate esterase (hcaG), and a gene product of unknown function (hcaF). The two genes transcribed in opposite directions code for the transporter of hydroxycinnamates (hcaK) and the transcription regulator (hcaR). Three proteins, HcaA, HcaB, and HcaC, carry out four enzymatic activities on 4-hydroxycinnamic acid derivatives (hydratase/lyase/dehydrogenase/ligase) and process caffeate, p-coumarate, and ferulate to protocatechuate, p-hydroxybenzoate, and vanillate, respectively (31). HcaK belongs to the major facilitator superfamily of a large, diverse, and broadly distributed group of transmembrane transporters, which in bacteria are used mainly for nutrient uptake. The Acinetobacter sp. HcaR (AcHcaR) protein is a predicted member of the MarR/SlyA family of transcription regulators. AcHcaR was shown to control the level of transcription of the hca operon, and derepression is hydroxycinnamate-dependent (18). It was proposed that, in the absence of ligands, AcHcaR binds a specific DNA operator located within the −10 to −35 promoter region and blocks transcription. The precise binding region and the mechanism of DNA interaction remained unclear. In the presence of hydroxycinnamates, AcHcaR fails to bind to DNA, and operon transcription can proceed. It is believed that hydroxycinnamates enter the cell through the HcaK transporter, although it is known that ATP-binding cassette transporters are also involved in the transport of aromatic compounds (32). The HcaC CoA ligase initiates metabolism by converting these compounds to corresponding hydroxycinnamoyl-CoA thioesters. These hydroxycinnamates and thioesters are toxic to the cell as Parke and Ornston (18) have shown using mutation analysis on hcaC, hcaA, and hcaR in Acinetobacter sp. strain ADP1. When the hcaA mutation was combined with a mutation in the repressor HcaR, the exposure to caffeate, p-coumarate, or ferulate totally inhibited the growth of cells. However, an active transcription of the hca operon was maintained in the presence of all these compounds with native AcHcaR being relieved from its repression activity as it is able to respond not only to multiple hydroxycinnamates and the thioester intermediates but also to products (vanillin) (18).
FIGURE 1.
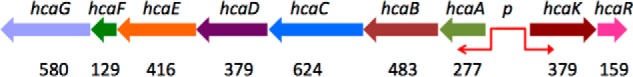
Organization of the hca operon in Acinetobacter sp. ADP1 (18). Genes are drawn with approximate length; the exact protein length in amino acids is shown below. The bidirectional promoter separates the hcaABCDEFG and hcaKR gene clusters.
Other hca operons have been reported, including one in E. coli (hcaA1A2CBD/hcaRT) and related species, that are responsible for the catabolism of phenylpropionic acid and are regulated by a protein also named HcaR (33); however, it belongs to the LysR family of transcription factors. Some other transcription factors, for example E. coli MhpR, involved in regulation of the catabolism of aromatic compounds belong to the IclR family (34). Therefore, in bacteria, the use of widely available aromatic compounds as a source of carbon is important. Multiple families of transcription factors control a variety of catabolic gene clusters and have evolved to respond to the presence of these compounds.
Here we report several crystal structures of AcHcaR: the apo form and in a complex with several substrates for the enzymes of the hca catabolic operon including ferulic acid, 3,4-dihydroxybenzoic acid (DHBA), and p-coumaric acid as well as with vanillin, which is a product of ferulic acid processing by hcaABC gene products. The structures of the apo form of HcaR and its complex with specific 23-bp (24-mer) DNA from the hca promoter region were also determined. The HcaR·DNA complex reveals details of interactions with bases and the sugar-phosphate backbone as well as conformational changes upon binding in both protein and DNA required for specific DNA recognition. The structures with ligands provide molecular details of the interaction between AcHcaR and aromatic compounds and show how ligand binding may interfere with binding to the DNA operator and derepress gene transcription. Biophysical and binding measurements suggest that these ligands stabilize the apo form of the AcHcaR protein. Therefore, our results are consistent with the mechanism of HcaR derepression based on stabilization of protein conformation that is unproductive in recognizing and binding a specific DNA target.
Experimental Procedures
Materials
All DNA used for co-crystallization with HcaR was purchased from Integrated DNA Technologies, Inc. p-Coumaric acid, ferulic acid, vanillin, and DHBA were purchased from Sigma-Aldrich.
Protein Cloning, Expression, and Purification
The gene encoding AcHcaR from Acinetobacter sp. ADP1 was cloned into the pMCSG19 vector using a modified ligation-independent cloning protocol as described earlier (35, 36). AcHcaR was produced as a maltose-binding protein fusion that was cleaved off in vivo in E. coli BL21(DE3) carrying plasmid pRK1037. pRK1037 produces tobacco vein mottling virus protease, which results in AcHcaR being purified as an N-terminal His6-tagged protein (35). The transformed cells were grown at 37 °C in M9 medium supplemented with 0.4% (w/v) glucose, 8.5 m NaCl, 0.1 mm CaCl2, 2 mm MgSO4, and 1% (w/v) thiamine. At a UV absorbance (A595) of 1.0–1.5, 0.01% (w/v) each of l-leucine, l-isoleucine, l-lysine, l-phenylalanine, l-threonine, and l-valine were added to inhibit the metabolic pathway of methionine synthesis and encourage l-selenomethionine (SeMet) incorporation. SeMet (90 mg) was added to 1 liter of culture, and protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were incubated at 18 °C overnight. The harvested cells containing SeMet AcHcaR were resuspended in lysis buffer (500 mm NaCl, 5% (w/v) glycerol, 50 mm HEPES, pH 8.0, 20 mm imidazole, 10 mm 2-mercaptoethanol, and protease inhibitor (one tablet/50 ml of extract, Roche Diagnostics)) and stored at −80 °C. SeMet HcaR from Acinetobacter sp. ADP1 was purified using the procedure described earlier (37, 38). The harvested cells were thawed, and 1 mg/ml lysozyme was added. This mixture was kept on ice for 20 min with gentle shaking and then sonicated. The lysate was clarified by centrifugation at 36,000 × g for 1 h and filtered through a 0.45-μm membrane. The clarified lysate was applied to a 5-ml nickel HisTrap HP column (GE Healthcare) on an ÄKTAxpress system (GE Healthcare). The His6-tagged protein was released with elution buffer (500 mm NaCl, 5% glycerol, 50 mm HEPES, pH 8.0, 250 mm imidazole, and 10 mm 2-mercaptoethanol), and the fusion tag was removed by treatment with recombinant His7-tagged tobacco etch virus protease. Nickel affinity chromatography was used to remove the His6 tag, uncut protein, and His7-tagged tobacco etch virus protease (35). The AcHcaR protein was dialyzed against crystallization buffer containing 250 mm NaCl, 20 mm HEPES, pH 8.0, and 2 mm dithiothreitol (DTT) and then concentrated to 12 mg/ml using an Amicon Ultra centrifugal filter device with a 3,000 molecular weight cutoff (Millipore), flash cooled in liquid nitrogen, and stored at −80 °C. The AcHcaR protein concentration was determined spectrophotometrically by measuring absorbance at 280 nm on a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). The concentration was calculated using the extinction coefficient (8480 m−1cm−1) computed from its amino acid sequence.
Crystallization of Apo-AcHcaR and Complexes with Small Ligands
The AcHcaR protein was crystallized using sitting drop vapor diffusion at 16 °C in a CrystalQuick 96-well round bottom plate (Grainer Bio-One North America, Inc.). A 400-nl droplet of the protein (12 mg/ml) was mixed with a 200-nl droplet of crystallization reagent and allowed to equilibrate over 135 μl of crystallization reagent. The nanopipetting was performed using the Mosquito nanoliter liquid handling system (TTP LabTech). The plate was then incubated at 16 °C within a RoboIncubator automated plate storage system (Rigaku). Automated crystal visualization (Minstrel III, Rigaku) was utilized to locate several crystals, which were cryoprotected and flash cooled in liquid nitrogen. The best crystal of SeMet-labeled AcHcaR was obtained from 1.4 m sodium malonate, pH 7.0, and 0.1 m Bistris propane, pH 7.0. The orthorhombic (P21212) crystals diffracted to 2.35 Å. AcHcaR was also co-crystallized with several ligands. Crystals were obtained for complexes with 5 mm ferulic acid, 5 mm vanillin, or 5 mm DHBA from 1.4 m sodium malonate, pH 7.0, and 0.1 m Bistris propane, pH 7.0, at 16 °C and for AcHcaR complexed with 5 mm p-coumaric acid from 1.4 m sodium malonate, pH 7.0, and 0.1 m Bistris propane, pH 6.8, at 16 °C. The properties of crystals of the AcHcaR complexes with ferulic acid, p-coumaric acid, vanillin, and DHBA are listed in Table 1.
TABLE 1.
Crystallographic statistics of AcHcaR and its complexes with ligands and the cognate DNA
Mol, molecules; AU, asymmetric unit; FOM, figure of merit; COU, coumaric acid; FLA, ferulic acid; VLN, vanillin.
| Apo-AcHcaR | AcHcaR + coumaric acid | AcHcaR + ferulic acid | AcHcaR + vanillin | AcHcaR + DHBA | AcHcaR-DNA peak | AcHcaR-DNA inflection | AcHcaR-DNA high remote | |
|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||
| Space group | P21212 | P21212 | P21212 | P21212 | P21212 | P61 | P61 | P61 |
| Unit cell (Å) | 63.26, 80.54, 63.06 | 64.02, 83.63, 63.06 | 63.67, 82.81, 63.17 | 63.68, 82.82, 63.03 | 63.58, 83.10, 63.05 | a = b = 161.97, 73.83 | a = b = 162.29, 73.89 | a = b = 161.28, 74.09 |
| Molecular mass (Da) (residues) | 33,058 (287)a | 33,058 (287)a | 33,058 (287)a | 33,058 (287)a | 33,058 (287)a | 33,058 (287)a/7,450 (24)b | ||
| Mol (AU) | 2 | 2 | 2 | 2 | 2 | 2 complexesc | ||
| Wavelength (Å) | 0.9794 | 0.9794 | 0.9794 | 0.9794 | 0.9794 | 0.9792 | 0.9794 | 0.9716 |
| Resolution (Å) | 49.8–2.35 | 39.6–2.30 | 29.5–1.89 | 39.4–2.30 | 34.8–2.1 | 50–3.05 (3.05–3.10) | 50–3.00 (3.00–3.05) | 50–3.10 (3.10–3.15) |
| Number of reflections | 13,890/673d | 15,489/747e | 26,574/1,121f | 15,395/738g | 20,059/983h | 20,971/1,710i | 22,204/1,843i | 19,822/1,625i |
| Redundancy | 6.1/5.6 | 7.9/8.1 | 7.3/4.8 | 7.9/8.1 | 7.9/7.7 | 6.0/5.7i | 6.6/6.2i | 7.1/6.4i |
| Completeness (%) | 99.8/100 | 99.7/100 | 97.3/83.0 | 99.9/100 | 99.9/99.8 | 99.3/99.4i | 99.4/99.4i | 99.6/99.9i |
| Rmerge | 0.107/0.674 | 0.090/0.499 | 0.063/0.337 | 0.098/0.674 | 0.098/0.638 | 0.071/0.607i | 0.056/0.736i | 0.110/0.583i |
| I/σ(I) | 9.2/3.3 | 14.1/5.1 | 15.7/4.4 | 10.8/3.7 | 3.5 | 21.1/3.0i | 24.6/2.4i | 12.1/4.0i |
| Phasing | SAD | SAD | SAD | SAD | SAD | MAD | ||
| Phasing power | 1.32 | 2.00 | 1.56 | 1.67 | 1.73 | 1.32/1.14j | ||
| FOM | 0.22 | 0.36 | 0.31 | 0.30 | 0.33 | 0.776 | ||
| Refinement | ||||||||
| Resolution (Å) | 49.8–2.35 | 39.6–2.30 | 29.5–1.89 | 39.4–2.30 | 34.8–2.10 | 38.9–3.00 | ||
| Reflections (work/test) | 25,693/1,274k | 15,439/767 | 26,262/1,325 | 15,337/766 | 19,862/1,014 | 20,010/992 | ||
| Rcrystal/Rfree | 0.187/0.253 | 0.174/0.213 | 0.176/0.206 | 0.167/0.225 | 0.173/0.215 | 0.255/0.294 | ||
| r.m.s.d. bond (Å)/angle (°) | 0.013/1.27 | 0.002/0.618 | 0.006/0.947 | 0.007/1.00 | 0.007/0.920 | 0.004/0.696 | ||
| Protein residues | 12–150, 10–153 | 11–150, 11–153 | 11–153, 11–154 | 11–150, 11–153 | 11–149, 11–152 | 12–150 × 4 | ||
| Solvent/ligand | 67/1 SO4 | 117/2 COU | 166/2 FLA | 86/2 VLN | 183/2 DHBA | −12–11 × 4l | ||
| Wilson B/mean B (Å2) | 47.5/67.2 | 44.1/49.5 | 32.7/47.9 | 44.3/47.5 | 35.1/42.7 | 90.18/173.4 | ||
| Ramachandran plot | ||||||||
| Most favored/disallowed (%) | 91.7/0.0 | 99.6/0.0 | 100.0/0.0 | 98.18/0.0 | 99.64/0.0 | 97.3/0.18 | ||
a Not including cloning artifact.
b DNA.
c Protein-DNA complex.
d Last resolution bin, 2.39–2.35 Å.
e Last resolution bin, 2.34–2.30 Å.
f Last resolution bin, 1.93–1.89 Å.
g Last resolution bin, 2.34–2.30 Å.
h Last resolution bin, 2.14–2.10 Å.
i Highest resolution bin.
j Anomalous/dispersive.
k Including Bijvoet pairs.
l DNA residues.
Co-crystallization of AcHcaR with DNA
We used a specific DNA 20-base pair palindromic sequence recognized by AcHcaR to design the target for crystallization. Self-complementary and semipalindromic DNA duplexes of different lengths (from 19 to 32 bp) were prepared (Integrated DNA Technologies, Inc.) with various modifications on the 5′-end. The synthetic oligonucleotides for co-crystallization were synthesized in 1-μmol scales and prepared by ethanol precipitation followed by a slow annealing step according to a protocol described earlier (39). The DNA was resuspended in 100 μl of 10 mm Tris/HCl, pH 7.5, and 5 mm MgCl2. The concentration was determined spectrophotometrically by measuring absorbance at 260 nm on a Nanodrop ND-1000 spectrophotometer. The DNA concentration was calculated using the extinction coefficient calculated for each DNA sequence. For crystallization, the DNA duplex in a molar ratio of 1.4 or 1.5 was added to the protein dimer.
The HcaR protein was co-crystallized with several different DNA duplexes using sitting drop vapor diffusion at 16 °C in a CrystalQuick 96-well round bottom plate. A 400-nl droplet of the protein·DNA complex was mixed with a 200-nl droplet of crystallization reagent and allowed to equilibrate over 135 μl of crystallization reagent. The nanopipetting was performed using the Mosquito nanoliter liquid handling. The crystallization plate was then incubated at 16 °C in a RoboIncubator automated plate storage system. Automated crystal visualization was utilized to locate several crystals, which were cryoprotected and flash cooled in liquid nitrogen. Macroscopic crystals were obtained for several oligonucleotides. The x-ray quality crystals of the complex were obtained with the 24-base-long (forming 23-bp duplex with 5′ single base overhang) DNA (5′-CGAATATCAGTTAAACTGATATTC) at a concentration of 0.47 mm and HcaR at 0.31 mm from 20 mm MgCl2, 40 mm sodium cacodylate, pH 5.5, and 40% (v/v) 2-methyl-2,4-pentanediol (E4 of Natrix screen from Hampton Research). The properties of crystals are listed in Table 1.
Data Collection and Structure Refinement
Diffraction data of the crystals of apo-AcHcaR and the complexes were collected at 100 K either at the Structural Biology Center 19-BM beamline with an Area Detector Systems Corp. Q210r charge-coupled device detector or the 19-ID beamline with an Area Detector Systems Corp. Q315r charge-coupled device detector (40) at the Advanced Photon Source at Argonne National Laboratory. The crystals were exposed for 3–5 s per 1.0° rotation of ω with a crystal to detector distance of 290–430 mm on 19-ID for the AcHcaR complex with coumaric acid, apo-AcHcaR, the complex with DHBA, the complex with vanillin, and the complex of AcHcaR with DNA. For the complex with ferulic acid, the crystal diffraction data were obtained similarly with a crystal to detector distance of 200 mm on 19-BM. The single wavelength anomalous dispersion (SAD) data sets near the selenium absorption peak (0.9794 Å) for all crystals except for the AcHcaR·DNA complex were recorded scanning 200° on ω. The data collection details for each data set are shown in Table 1. The structures of AcHcaR, apo form, and the complexes with the ligands were determined by SAD phasing using HKL-3000 (41) with SHELXC (42), SHELXD (42), SHELXE (42, 43), MLPHARE (44), dm (45, 46), and SOLVE/RESOLVE (47) as well as Buccaneer (48). For the refinement, phenix.refine (49) and Coot (50) were used for computation and manual adjustment, respectively. The refinement statistics are listed in Table 1. The stereochemistry of the structures was checked with a Ramachandran plot and MolProbity (51). The structure of the AcHcaR·24-mer DNA complex was phased by MAD with the data sets collected from three wavelengths, peak (0.9792 Å), inflection (0.9794 Å), and high remote (0.9716 Å), using HKL-3000. The experimental electron density obtained through MAD phasing for the AcHcaR·DNA complex showed well defined secondary structures of the protein, DNA bases, and sugar-phosphate backbone. MolRep was used to put the apo-AcHcaR dimer in the experimental map, and then the Coot (50)-generated B-form 24-mer (23-bp) DNA duplex was fit manually into the DNA density and adjusted. There are two protein·DNA complexes in the asymmetric unit. For one complex, the electron density was well defined for the protein dimer and DNA duplex. However, for the second complex, only about half of the protein dimer and a third of the DNA duplex could be modeled into initial experimental electron density. Superposing the first complex to the partially built second complex generated the complete second complex. The refinement was carried out with phenix.refine. Occupancies of atoms in the disordered regions in the second complex were adjusted to less than 1.0 during the refinement. Multiple trials of rephasing and remodeling were performed. Several additional manual adjustments using Coot and phenix.refine refinement cycles were required to reach the final structure, resulting in an excellent model for the first complex and reasonable model of the second. The refinement statistics are listed in Table 1. The stereochemistry of each structure was checked with a Ramachandran plot and MolProbity (51). The six structures (apo form and complexes with coumaric acid, vanillin, ferulic acid, DHBA, and 24-mer DNA) have been deposited to the Protein Data Bank with access codes 3K0L, 4RGR, 4RGS, 4RGU, 4RGX, and 5BMZ, respectively.
Electrophoretic Mobility Shift Assay (EMSA)
To confirm DNA binding by AcHcaR, polyacrylamide gel electrophoresis was carried out (Fig. 2) with the following DNA constructs: 1) 43-mer (TTGGATTTAATTTAATATCAGTTAAACTTACATTCAAGTGTTT) containing the potential binding site; 2) 43-mer (GATATTTTATGTGTGTTCTTTGAACATTGACAATAAAAACGTA) not containing the binding site, both located in the intervening sequence between the hcaA and hcaK genes; and 3) the palindromic 24-mer used for co-crystallization trials in the absence and presence of small ligands p-coumaric acid and 3,4-dihydroxybenzoic acid. Each assay of a 10-μl mixture contained 150 mm KCl, 0.1 mm DTT, 0.1 mm EDTA, 10 mm Tris, pH 7.4, 0.86 μm oligonucleotide, and 4.2 μm AcHcaR dimer or small ligands. Samples were incubated at 37 °C for 30 min in a water bath. At the end of the incubation, to each 10-μl mixture, 2 μl of 6× EMSA loading solution (Invitrogen) was added. Samples were electrophoresed on a 6% native polyacrylamide gel in TBE buffer (90 mm Tris borate and 2 mm EDTA, pH 8.3) at 100 V for 50 min at 4 °C. The gel was stained for nucleic acids with SYBR Green (Invitrogen) and incubated at 20 °C with continuous agitation at 50 rpm for 30 min and protection from light. After washing three times in 150 ml of distilled H2O, the stained nucleic acids were visualized using either a Bio-Rad or Syngene gel imaging and analysis system for fluorescence. To confirm the presence of protein in the retarded DNA bands, the gel was washed and stained with SYPRO Ruby for 3 h in the dark. The gel was washed three times in 150 ml of H2O for 10 s. Next the gel was distained in 10% methanol and 7% acetic acid for 60 min. The gel was washed again three times in 150 ml of H2O for 10 s and visualized with either a Bio-Rad or Syngene system for fluorescence.
FIGURE 2.
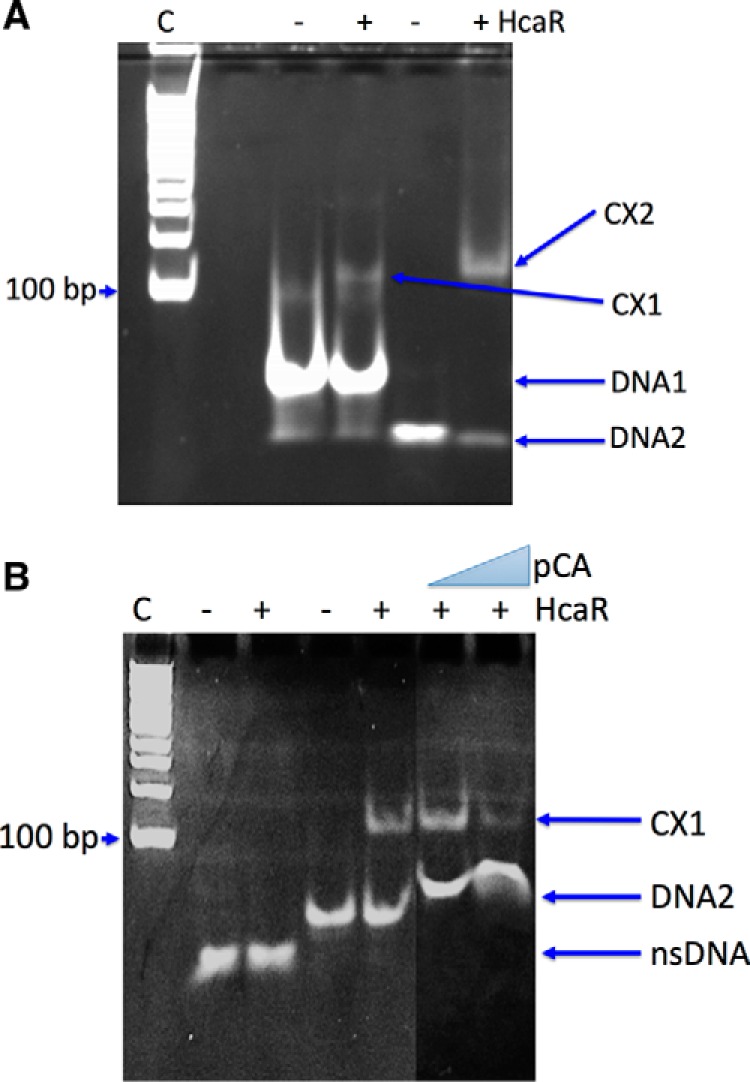
EMSA analysis of HcaR·DNA complex. A, HcaR complexed with DNA1 (42-mer) (labeled as CX1) and DNA2 (24-mer; used for crystallization) (labeled as CX2). B, HcaR interaction with DNA2 in the presence and absence of p-coumaric acid (pCA). Lane C, size markers nsDNA, nonspecific DNA.
Thermal Melting Assay Using Dynamic Light Scattering (DLS)
The thermal melting assay using DLS was performed using a DynaPro Plate Reader Plus (Wyatt Technologies, Inc.) in a 384-microwell plate to measure stabilization effects by hydroxycinnamate derivatives on AcHcaR in the temperature range of 25–55 °C. For all assays, the samples were at a concentration of 1 mg/ml and filtered with a 0.1-μm-cutoff membrane or spun down in 20-μl reactions. The final ligand concentration in initial experiments was with 10-fold molar excess of small molecule to protein.
Oligomeric State Determination in Solution Using Size Exclusion Chromatography
The molecular weight of native AcHcaR protein in solution was determined by HPLC size exclusion chromatography using an SRT SEC-150 (7.8 × 250-mm) column (Sepax Technologies, Inc.) in a buffer containing 20 mm HEPES/NaOH, pH 8.0, 250 mm NaCl, and 2 mm DTT. The column was equilibrated and calibrated using standard proteins from the High Molecular Weight Gel Filtration Calibration kit (GE Healthcare). The chromatography was carried out at 22 °C at a flow rate of 1.2 ml/min. The following proteins were prepared in running buffer at a concentration of 5 mg/ml: aprotinin (6.5 kDa), ribonuclease A (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), conalbumin (75 kDa), aldolase (158 kDa), and thyroglobulin (669 kDa) (GE Healthcare) to determine a calibration profile. The calibration curve of Kav versus log molecular weight was prepared using the equation Kav = (Ve − V0)/(Vt − V0) where Ve is the elution volume for the protein, V0 is the column void volume, and Vt is the total bed volume. Elution volumes were noted, and a linear regression analysis was applied to the standards. The AcHcaR protein (∼5 mg/ml) was resuspended in the running buffer and analyzed under the same conditions as the standards.
Results and Discussion
Overall Structure of AcHcaR
HcaR from Acinetobacter sp. ADP1 belongs to the PF01047 MarR family of transcription factors. The protein's closest sequence relatives are MarR-like proteins from other soil-dwelling bacteria, Acinetobacter, Burkholderia, and Ralstonia. We have cloned, purified, crystallized, and determined the crystal structures of apo-AcHcaR, four complexes with ligands (ferulic acid, DHBA, p-coumaric acid, and vanillin, a product), and an apoprotein complex with a specific DNA target.
The highest resolution structure was obtained with ferulic acid (at 1.89 Å; Table 1), and this structure is used throughout the text to describe the AcHcaR structural details. The 159-residue α/β protein monomer is composed of six helices, one 310 helix, and one β-hairpin, the wing (Fig. 3). The 10 N-terminal, six C-terminal residues, and two residues (96/97) in the β-hairpin loop in molecule B are disordered and are missing in the final model. However, when all six structures are compared, a continuous model covering residues 10–154 can be constructed.
FIGURE 3.
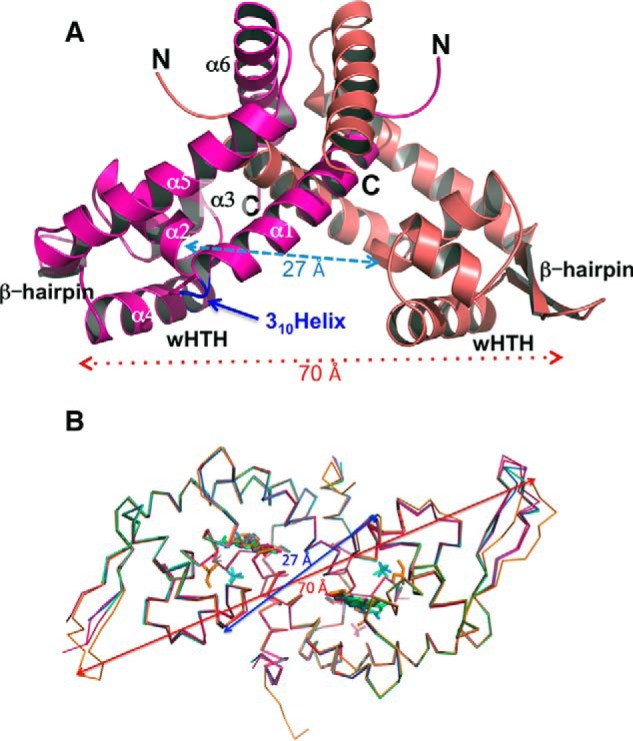
Overall structures of AcHcaR dimers. A, ribbon diagrams of the AcHcaR dimer in stick representation. N and C termini and wHTH motifs are labeled. B, comparison of main chain atom conformation in AcHcaR structures. The main chain structure of apo-AcHcaR (red) is compared with AcHcaR complexes with ligands (AcHcaR·ferulic acid (orange), HcaR·DHBA (pink), HcaR·vanillin (blue), and HcaR·p-coumaric acid (green)). Ligands are shown as sticks using the same color scheme. In addition to aromatic compounds, glycerol and phosphate anions are found in some protein structures. Structures were superimposed with Coot (50). The arrows show distances between Cβ atoms of His91A-His91B (red) and Gln71A-Gln71B (blue). These residues are part of the wHTH DNA-binding motif.
Analysis of the elution profiles of the AcHcaR protein suggests that it is a tetramer in solution (Fig. 4A), in all six crystal structures it is also shown to be a tetramer, a dimer of dimers (Fig. 4B), and each dimer binds separate duplex DNA in the protein·DNA complex (Fig. 4C). Two monomers in a dimer interlock with α-helices forming a 2-fold symmetrical dimer with a triangular shape showing two classic HTH DNA-binding motifs (helices α3 and α4) separated by ∼27 Å and the wings (β-hairpin) separated by ∼70 Å (wHTH) (Fig. 3). The distance and orientation of the wHTH motifs are virtually identical in all six structures with the shortest being the unliganded form (26.9 Å) and the longest for the complex with ferulate (27.3 Å) (Fig. 3). The overall AcHcaR structure is similar to other reported structures of MarR/SlyA-like proteins despite low sequence similarity.
FIGURE 4.
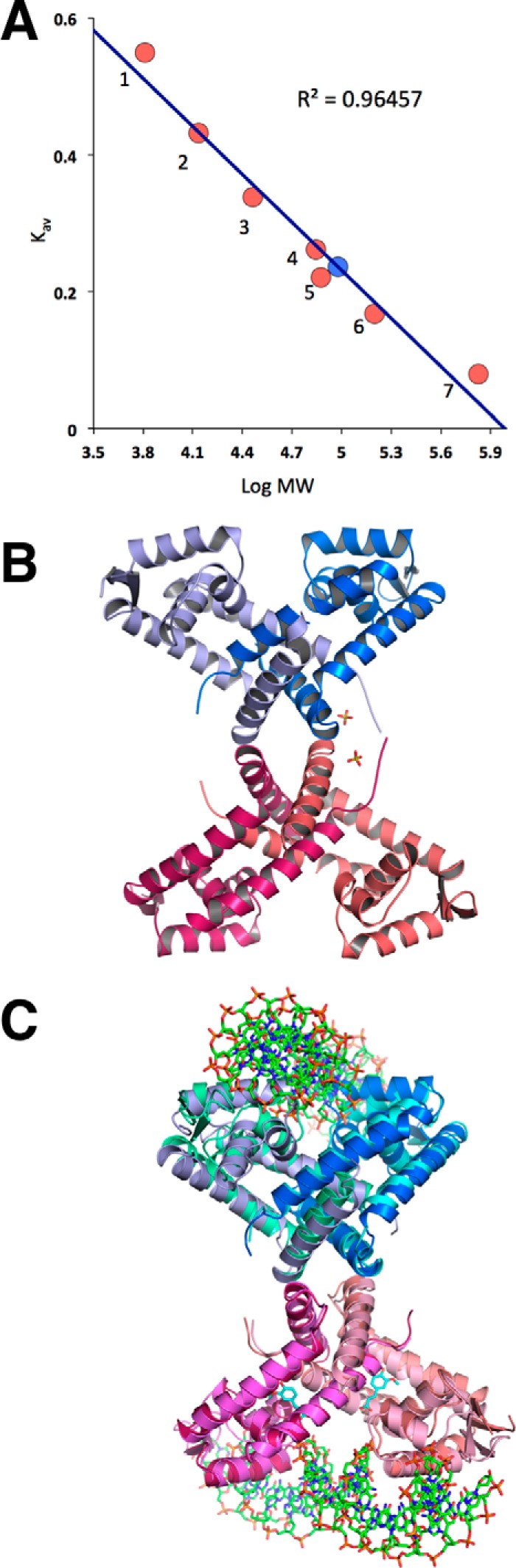
Biochemical property of HcaR. A, size exclusion chromatography of AcHcaR protein. The absorbance at 280 nm is plotted in absorbance units versus retention volume in milliliters for AcHcaR. The inset is the plot of Kav coefficient versus logarithm of molecular weight. Red circles correspond to standard proteins: 1, aprotinin (6.5 kDa); 2, ribonuclease A (13.7 kDa); 3, carbonic anhydrase (29 kDa); 4, ovalbumin (43 kDa); 5, conalbumin (75 kDa); 6, aldolase (158 kDa); and 7, thyroglobulin (669 kDa). The blue circle is AcHcaR (corresponding to 95 kDa). A single peak corresponding to a tetramer is observed. B, the structure of the AcHcaR tetramer as predicted by the PISA server. C, a similar AcHcaR tetramer is maintained in the AcHcaR·DNA complex.
Sequences of a number of transcription regulators governing aromatic catabolism found in soil bacteria are compared in Fig. 5. HcaR sequence similarity to seven MarRs known to respond to aromatic compounds ranges from 44% to no significant identity at all. In these proteins, there are only three conserved residues, Gly85, Thr104, and Gly107 (using HcaR numbering) (Fig. 5A). These residues are at the beginning of the wing motif (Gly85 and Thr104) and in helix 5 (Gly107), and they interact with each other. The hydroxyl of Thr104 makes hydrogen bounds to the main chain carbonyl and amide of Gly85 and Gly107, respectively. Thr104 and Gly107 are also highly conserved in MarR from soil bacteria (Fig. 5B). These MarRs show a different set of highly conserved residues with prominent His95-Gly96-Arg97 sequence in the loop of the β-hairpin. The Arg97 (or Lys97) is also quite conserved in MarRs known to respond to aromatic compounds. This residue is important for DNA binding in the minor groove (see below).
FIGURE 5.
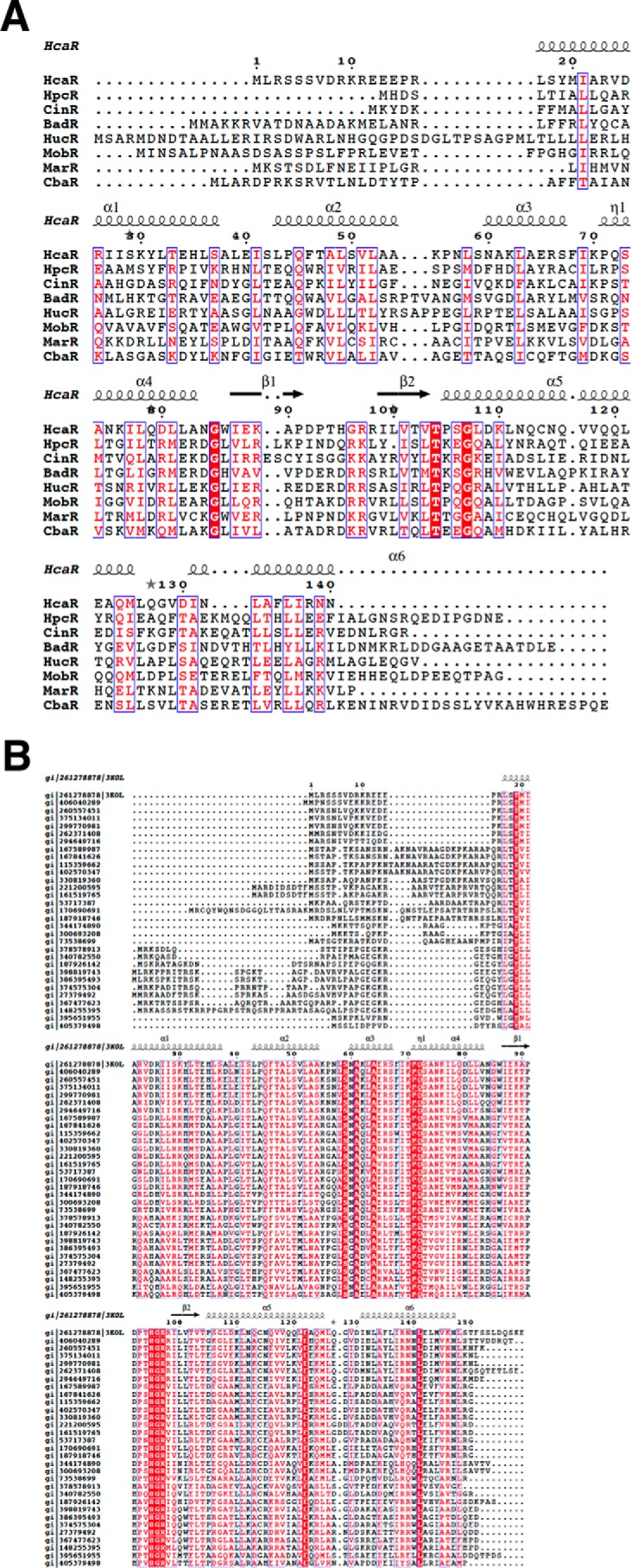
Sequence alignment of HcaR homologs. A, structure-based sequence alignment of select HcaR homologues. HcaR, repressor of 4-hydroxycinnamic acid catabolism in Acinetobacter sp. ADP1; CinR, repressor of cinnamoyl ester from Butyrivibrio fibrisolvens E14; HpcR, regulator of homoprotocatechuate catabolism from E. coli K12; BadR, benzoate anaerobic catabolism regulator from R. palustris CGA009; CbaR, regulator chlorobenzoate catabolism from Conidiobolus coronatus BR60; HucR, regulator of uric acid catabolism from Deinococcus radiodurans; MarR, regulator of 2-hydroxybenzoic acid catabolism from E. coli; MobR, regulator of 3-hydroxybenzoic acid catabolism from Comamonas testosteroni KH122. B, multiple sequence alignment of AcHcaR homologues from soil bacteria using ClustalX (58).
A structural homology search revealed that the nearest AcHcaR homologue is a MarR-like transcription regulator from Silicibacter pomeroyi DSS (Protein Data Bank code 3E6M; Z-score, 9.0; r.m.s.d., 1.74 Å over 132 residues; sequence identity, 20%) and an unannotated protein from Sulfolobus tokodaii (Protein Data Bank code 2YR2; Z-score, 8.2; r.m.s.d., 2.02 Å over 132 residues; sequence identity, 22%). Many other members of the MarR family show relatively high structural homology. However, there are some small but significant differences such as the relative orientation of the secondary structure elements and the subunits. Overall, the crystal structures of AcHcaR with aromatic ligands show only small structural variations (Fig. 3). The r.m.s.d. of Cα atoms for structures of apo and liganded forms ranges from 0.33 to 0.76 Å. The largest differences are observed in the β-hairpin (r.m.s.d., 1.6–2.9 Å; or is disordered), and the C and N termini. In addition to the β-hairpin, small changes are observed in the HTH motif and the recognition helix (see below).
AcHcaR Interaction with Aromatic Ligands
The AcHcaR dimer has two symmetrically disposed deep solvent-accessible cavities (Fig. 6, A and B). These pockets are predominantly lined up with the hydrophobic side chains and are located near the 2-fold axis of the dimer. In each monomer, this cavity is formed by residues from helices α1, α2, and α5 and occupied by an aromatic ligand in all four HcaR·ligand complexes (see below) (Fig. 6B). In addition to aromatic ligands, several other molecules were found associated with the protein on the surface (sulfate and chloride ions and glycerol molecules), and these same small molecules were found in the same sites in different structures.
FIGURE 6.
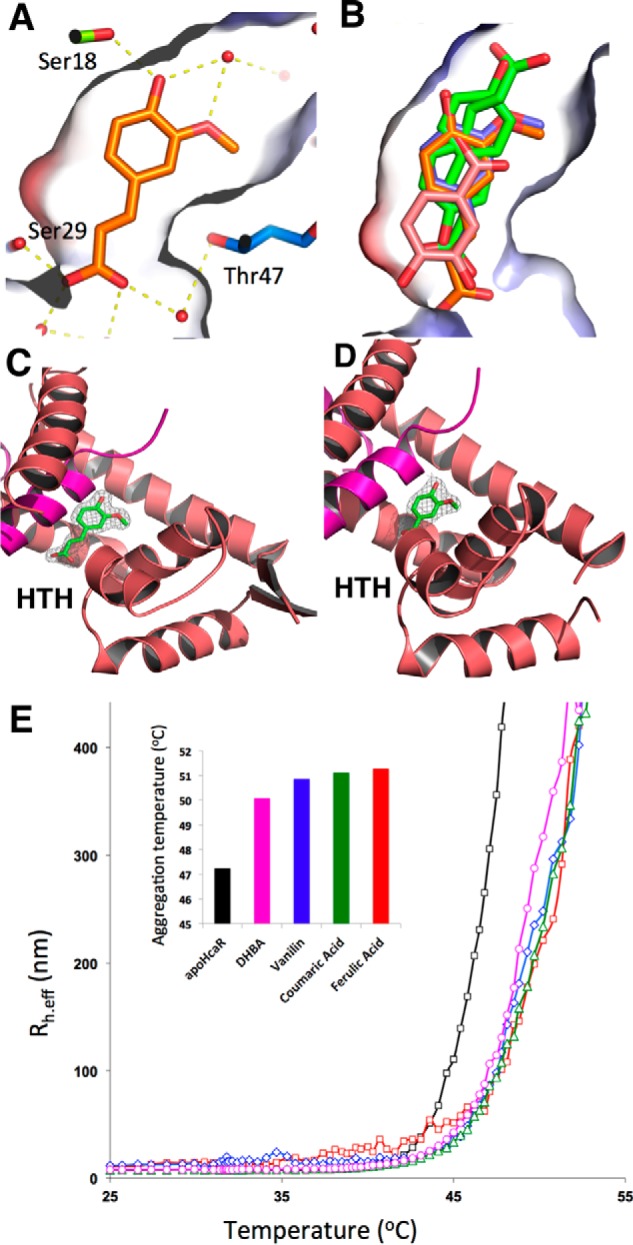
Structures of AcHcaR·ligand complexes. A, binding of ferulic acid in the pocket showing the solvent-accessible surface colored using local electrostatic potential. Hydrogen bonds with protein and solvent are shown in yellow. B, comparison of binding mode for all four ligands (ferulic acid (orange), two conformers of p-coumaric acid (green), vanillin (blue), and DHBA (pink)). Ferulic acid (C) and vanillin (D) caged in the corresponding experimental electron density (from SAD phasing experiment). E, thermal unfolding of AcHcaR monitored using DLS for apo-AcHcaR and complexes with ligands. Effective hydrodynamic radius (Rh.eff) is plotted versus temperature. In the histogram plot, the temperature at which AcHcaR reaches Rh.eff = 250 nm in the absence and presence of ligands is shown. Unfolding is observed at higher temperatures in the presence of ligands.
In all four AcHcaR·ligand complex structures, the electron density for a ligand is of good to excellent quality and can be interpreted with a high degree of confidence (Fig. 6, C and D). Each ligand is bound to the same deep cavity, which is surrounded by a number of electron-rich aromatic/hydrophobic residues (Tyr19, Ile28, Leu32, Phe46, Phe68, and Leu121). Several hydrophilic residues (Ser18, Asp25, Arg26, Ser29, and Thr47) are also in position to make direct or water-mediated hydrogen bonds with the ligand in the dimer interface as a part of the cavity. All of these residues are relatively well conserved in MarR-like proteins from soil bacteria (Fig. 5B). All ligands bind in a similar manner to AcHcaR but show quite distinct interaction patterns.
Binding of ferulic acid and its product vanillin is most similar as expected. Details of the ferulic acid-AcHcaR interaction are depicted in Fig. 6A. All atoms of the ligands are nearly in the plane of the benzene ring. The only direct hydrogen bond (2.7–2.8 Å) is formed for both ligands between 4-hydroxyl and Ser18 at the bottom of the cavity (Fig. 6A). Additional hydrogen bonds are water-mediated, and in the case of ferulic acid, these also connect through two water molecules to a glycerol molecule bound on the surface. At the other end, the carboxylate interacts with Asp25 through water molecules. Interestingly, carboxylate moieties of ferulates from the two subunits are only ∼11 Å apart, and the interaction can be traced through the water molecules. However, with vanillin, this interaction is missing, and instead the aldehyde group forms a hydrogen bond through a water molecule with Arg26. In addition to aforementioned hydrogen bond interactions, both ligands make several van der Waals or hydrophobic interactions with protein using the 3-methoxy group and benzene ring.
p-Coumarate binds to AcHcaR in a similar manner, but the electron density map is consistent with two distinct orientations. In one orientation, it adopts a ferulate-like orientation with the carboxylate facing the solvent, and in the other, the carboxylate points into the pocket. In the first orientation, carboxylate makes a direct hydrogen bond with Ser29 and water-mediated interactions with Asp5 and Arg26 from the opposite subunit. Again, the ligands from the two subunits approach each other within ∼11 Å. In the second orientation, the carboxylate makes a direct hydrogen bond to Ser50 and water-mediated hydrogen bonds to Glu14 as well as to the main chain nitrogen of Arg16.
DHBA shows the most extensive hydrogen bond network; at the same time, it is not able to penetrate the cavity as deeply as the other three ligands. It adopts an orientation similar to one of the p-coumarate poses. Its carboxylate forms a hydrogen bond with Thr47 directly and a hydrogen bond with Ser18 through water. The 4-hydroxyl hydrogen bonds with Ser29, Asp25, and Arg26. The 3-hydroxyl forms a hydrogen bond with Thr47 directly and a hydrogen bond with Ser67 through water. This arrangement allows the ligands to approach each other within ∼10 Å.
There are several structures of MarR/SlyA proteins in complex with salicylic acid available in the Protein Data Bank (codes 3DEU, 1JGS, 3GF2, and 3BPX) (27, 52, 53). In S. enterica serovar Typhimurium, the only two salicylates in SlyA are in a similar location to the ligands in the AcHcaR structures, and the interaction of carboxylate involves a salt bridge with Arg residues (Protein Data Bank code 3DEU). The other ligands are on the protein surface, suggesting a rather nonspecific binding. In the structure of MarR-like protein from S. tokodaii (Protein Data Bank code 3GF2), there are two salicylic acids bound; the carboxylate of the salicylic acid forms hydrogen bonds with two Tyr residues, and the location of binding partly overlaps with the general site of ligands in AcHcaR, but the ligand binds deeper in the pocket. Other structures are inconsistent both in terms of stoichiometry and the location of the binding site. For example, TcaR from Staphylococcus epidermidis binds eight molecules of salicylic acid per dimer, SlyA from S. enterica serovar Typhimurium binds six, and MarR from E. coli binds four.
In AcHcaR, all aromatic ligands bind to the same pocket, establishing it as a functional site. It can be rationalized as an elongated cavity lined mainly with hydrophobic side chains but also containing a hydrophilic side chain and positively charged entry providing limited specificity. In addition, a few residues capable of forming a hydrogen bond at the bottom of the cavity also have solvent-mediated access to the protein surface. More hydrophilic compounds, like DHBA, are captured near the entrance; more hydrophobic and longer compounds can penetrate deeper into the cavity, making a few additional hydrogen bonds that hold a ligand in place, evident by the better defined electron density for the latter compounds. This design of the cavity allows it to accommodate a variety of similar aromatic ligands or to bind the same ligand in multiple poses (Fig. 6B). It would potentially include a number of unbranched benzene-based aromatic natural compounds derived from lignin and other plant material. Perhaps this promiscuity in ligand binding enables AcHcaR, an atypical regulator, to play a role in bacterial catabolic diversity and match well with a broad specificity of hydroxycinnamate catabolic enzymes.
Ligands Stabilize HcaR Conformation
Our data provide new insights into the role of hydroxycinnamate derivatives in DNA binding inhibition. The superpositions of Cα tracings of the apo form AcHcaR and the liganded forms are nearly identical, suggesting that there is no major conformational change in the protein upon aromatic ligand binding. It was suggested that the apo form is predisposed for binding DNA, and the bound ligand may stabilize an inactive form of MarR, preventing the protein from interacting with a specific DNA operator (30, 54). To test the hypothesis that AcHcaR becomes more stable and rigid in the presence of hydroxycinnamates, we performed DLS thermal shift assays. The hydrodynamic radii observed from DLS were not different in the presence and absence of ligands, suggesting no significant physical changes. However, the AcHcaR molecules become significantly more monodispersed in the presence of the ligands. In the presence of ferulic acid, the AcHcaR melting temperature increases by 4 °C (Fig. 6E), and its polydispersity decreases from 21.6 to 9.1%. Similar but smaller effects are observed for DHBA, p-coumaric acid, and vanillin (Fig. 6E). We conclude that, upon binding these aromatic ligands, the protein becomes more stable and is locked in a well defined and compact state. When the cavities are empty or occupied by solvent or other small non-aromatic ligands, the protein is flexible and can adjust to the surface of a specific DNA operator by making necessary conformational changes for maximizing interactions. However, when cavities are occupied by bulky aromatic compounds, the protein loses its flexibility to make necessary changes to bind DNA specifically. Particularly, the interaction of the “wing” hairpins containing three strictly conserved residues (His-Gly-Arg) with the minor groove may be affected (Figs. 3B; 6, C and D; and later on 7B). Therefore these two cavities in the dimer may be directly linked to the control of the DNA binding capacity of a transcription regulator. This seems to be consistent with the suggestion that ligand binding may reduce the flexibility and stabilize an inactive form of AcHcaR that is unable to make a specific interaction with the DNA duplex using the wHTH DNA-binding motif (55). A similar behavior was proposed for the BldR and OhrR regulators (6, 30).
Measurements of AcHcaR properties in solution using size exclusion chromatography and DLS indicate that the protein is a tetramer. The protein migrates on the size exclusion chromatography as a 95-kDa protein (the calculated molecular mass from amino acid sequence for tetramer is 71.54 kDa). This is confirmed by DLS data that show a radius of gyration much larger than expected for a dimer, typically ranging from 75 to 95 kDa. This is observed for apoprotein, complexes with ligands, and the complex with DNA (see below). Analysis of the crystal packing in all six crystal structures, the apo form, and liganded forms using the PISA server also suggests that the protein is a tetramer (dimer of dimers), although the tetramer interface is not very extensive (1484 Å2) (Fig. 4, B and C). It is interesting to note that this same tetramer is also maintained in the AcHcaR·DNA complex (Fig. 4C) and perhaps implicates a possible role of the AcHcaR tetramer in gene regulation. With the tetrameric assembly, AcHcaR would bring together and regulate two distantly located promoters. In addition to the palindromic DNA sequences studied here, HcaR may also recognize less symmetric targets with lower affinity that have not been identified through sequence searches thus far.
Structure of Specific AcHcaR·DNA Complex
The hca promoter of Acinetobacter sp. ADP1 was previously identified in the hca intergenic region through genetic studies (18). Based on analysis of base conservation and the symmetry of the sequence, we have narrowed it down to a 23-bp region between the two open reading frames of hcaK and hcaA that can serve as a DNA-binding site for HcaR. The DNA duplex for co-crystallization was designed using hca operator sequence “CAATATCAGTTAAACTTACATTG” and was symmetrized to “GAATATCAGTTAAACTGATATTC” (modified bases are underlined) to make the 23-bp DNA self-complementary site except for the A:A mismatch at the center of the sequence and the additional C base overhang at the 5′-end. The binding of 42- and 23-bp duplexes containing this specific DNA site by apo-AcHcaR was confirmed using EMSA (Fig. 2A).
The experimental electron density for the AcHcaR·DNA complex showed well defined secondary structures of the AcHcaR and DNA duplex. There are two protein·DNA complexes in the asymmetric unit. The electron density for the first complex (protein chains A and B and DNA chains E and F) was high quality, leading to an excellent model. The second complex (chains C and D of the protein and G and H of the DNA duplex) produced a reasonable model; however, we believe it offers qualitative rather than quantitative information, and for discussion below, we use the first complex. The co-crystal structure of apo-AcHcaR with the hca operator further narrows down the HcaR recognition site to a 15-bp duplex, although the protein interacts with longer DNA through contacts to the sugar-phosphate backbone.
Typically, MarR-type transcription factors bind a specifically deformed B-DNA duplex using their HTH motif in the major groove and a β-hairpin of wHTH in the minor groove. AcHcaR shows a very similar mode of binding. Because we have determined structures including the apo and several liganded forms, it allows us to visualize specific conformational changes upon aromatic ligand and DNA binding. Overall, AcHcaR becomes more ordered, and the dimer is more symmetric in the complex with an aromatic ligand or DNA (Fig. 3B). However, the protein shows more significant and specific conformational changes upon binding to specific DNA. The HTH motifs adjust their orientation in the major groove where Gln72 specifically contacts edges of DNA bases (Fig. 7). Phosphate oxygens of the sugar-phosphate backbone interaction with amino acid side chains and the N-terminal portion (i.e. peptide bond amides of Asn60 and Ala61) of α-helical dipoles contribute to tight binding. A β-hairpin (wing part of wHTH), partly disordered in apo- or liganded HcaR, becomes well ordered and effectively contributes to the interaction with the DNA duplex. The wing is shifted by ∼6 Å (at the tip, Cα His95) toward the minor groove of DNA, making several direct and indirect contacts. Specifically, Arg98 penetrates deeply into the minor groove and contacts DNA bases (Fig. 7C). Because of the extensive interactions with the protein, the DNA duplex deforms, and the ends of the duplex bend toward the protein to make optimized specific contacts. Both the protein and DNA adjust their conformations to enhance binding. The surface correspondence seems to be an important component of indirect recognition, including strong surface electrostatic charge complementarity that allows a close approach (Fig. 8).
FIGURE 7.

Specific DNA binding by AcHcaR. A, overall views of AcHcaR-23-bp plus 5′-C overhang DNA. B, structural comparison of AcHcaR complex with ferulic acid (cyan and red spheres) and apo form bound to DNA (green) show shifts in positions of the HTH motif and the wing. C, specific hydrogen bonds between protein side chains and DNA bases in the major and minor grooves. D, summary of AcHcaR-DNA interaction. Schematic diagram of the 23-bp plus 5′-C overhang DNA sequence used for structure determination is shown. The center of the palindrome is indicated by a 2-fold rotation sign at the A:A base pair in the middle of the diagram. Two chains of HcaR residues are separated by the red diagonally crossing line. Bases are shown as rectangular boxes, large ones for purines and small ones for pyrimidines. Riboses are drawn as pentagons, and phosphates are Ps in small circles. The interactions involved in DNA bases are indicated on the bases in red with interacting protein residue types and numbers. Direct hydrogen bonds are shown in filled squares, and water-mediated hydrogen bonds are in red filled circles. Hydrophobic interactions are depicted as empty red circles. Interactions with phosphates are shown in lines with small pale blue filled circles (water mediated-hydrogen bonds) and squares (direct hydrogen bonds) connected with interacting protein residues (types and numbers). Pro44 in both subunits make interactions with two consecutive phosphates shown as red empty circles between the two P circles. H95* indicates a water-mediated hydrogen bond interaction with the phosphate, but no water molecule is found due to low resolution of the structure. R98*c in red indicates water-mediated interactions with DNA bases in the minor groove.
FIGURE 8.
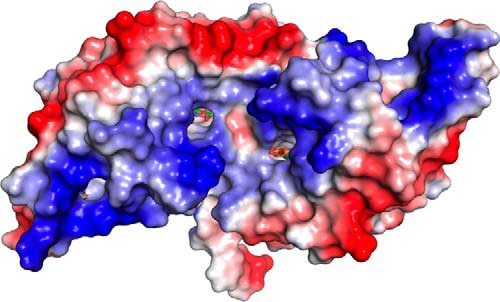
Local electrostatic potential of AcHcaR·p-coumaric acid complex shows the location of the binding sites in the dimer from the direction of the presumed DNA-binding site.
AcHcaR Specific Recognition of DNA
The crystal contains two complexes consisting of a total of four AcHcaR subunits and two duplexes of DNA. The complex involving the A and B subunits is significantly better defined, and all descriptions of the protein-DNA interactions are based on this complex. For the DNA, we number the central base pair A/A “0,” and the subsequent bases are numbered with a “+” sign to the right and a “−” sign to the left on the top strand and with opposite signs on the bottom strand. This numbering reflects the palindromic symmetry and helps to describe protein interactions with bases and phosphate moieties. The interactions of all monomers with DNA are very similar but not identical in details. Here we describe interactions of the A subunit. The sequence-specific interactions of AcHcaR include direct contacts between the protein side chains and the edges of DNA bases in the major and minor grooves. The side chains of Gln72 and Lys76 contact bases in the major groove (Fig. 7C). Lys76 makes a hydrogen bond with N7 of adenine 2, defining purine in this position, and Gln72 contacts guanine 5, cytosine −5, and thymine −6, defining sequence G5/A6 and C−5/T−6 in the opposite strand. Gln72 contacts O6 of guanine 5 with Oϵ1 and N7 through water with Nϵ2. These interactions are possible due to the local conformational changes in DNA (inclination, 3.0/8.5 and 3.9/6.4). In addition, Oγ of Ser73 contacts C5 of cytosine 3 through van der Waals interaction. All of these residues are part of the HTH motif, and their interactions seem to constitute the key determinants of specificity in the protein·DNA complex.
Side chains of several residues directly contact phosphate moieties: Ser42 with P1, Gln79 with P−5, Asn75 to P−6, Lys89 to P−6, Lys70 to P4, and Ser73 to P3 (Fig. 7C). The uniqueness of these interactions is a result of the formation of a hydrogen bond network involving side chains of Asn60/Asn75/Gln79 on the surface of the major groove along with the sugar-phosphate backbone to position aforementioned residues to readily contact DNA bases and phosphates. Some of these residues are at a distance consistent with water-mediated hydrogen bonds to bases or phosphates. However, with the resolution limit at 3.00 Å, the electron density for these waters is not well defined. A partial positive charge at the N-terminal ends of two α-helices of the HTH and the presence of peptide bond amides for hydrogen bonds also contribute to direct contacts to phosphate oxygens: Asn60-Ala61 to P−7, Lys7 to P3, and Ile99 to P−7.
The side chain of Arg98 in the β-hairpin penetrates deep into the minor groove, and the guanidinium nitrogen atoms (Nη1 and Nη2) form direct hydrogen bonds with the O2 atoms of thymines −8 and −9 of the opposite strand (possibly also water-mediated), thereby defining base pairs in the A8T9 steps). With an additional residue (His95) contacting the phosphate moiety of P11 from the minor groove side at both ends of the DNA-binding sites, the DNA is deformed toward the protein and increases the contact surface. Detailed interactions are diagramed in Fig. 7D. The sequence specificity (both through direct and indirect effects) is efficiently achieved by specific conformational changes in both the protein and DNA. As shown in Fig. 7B, a number of protein parts are adjusted to fit in DNA. With all the local deformations, the DNA is deviated from the typical B-form. The DNA geometry was analyzed using the program Curve+ (56). In the complex, the DNA duplex is distorted with the average distance between bases being 3.2 Å (32 Å per turn), which is somewhat shorter than the typical B-DNA (3.4 Å). The total bend is 25–30.4°s over 23 base steps with the majority of local deformations (mostly inclination and X-displacement) concentrated in the T−6C−5/G+5A+6 regions. This distortion results in widening of the major groove, providing the space and surface for the HTH to make specific contacts in the major groove.
There are several MarR-like protein·DNA complex structures (Protein Data Bank codes 1Z9C (BsOhrR), 3Q5F (StSlyA), 3ZPL (ScMarR), 4AIJ and 4AIK (YpRovA), 3GFI (ST1710), 4LLN and 4LLL (SaMepR), 4FX4 (MtMosR), and 4KDP (SeTcaR)) available for this family. Except for the structure of Staphylococcus epidermidis TcaR·single-stranded DNA complex (Protein Data Bank code 4KDP) (25), MarR/SlyA proteins interact with the specifically deformed B-DNA duplex in a quite unique manner. We compared the structures of AcHcaR in the presence and absence of a specific DNA duplex with the aforementioned SlyA/MarR structures. Most secondary structure elements of AcHcaR superposed well despite a low sequence identity (16–26%) among the structures without DNA. However, in the MarR/SlyA·DNA complexes, the wHTH motifs, specifically the α3-α4 helices of the HTH, undergo conformational adjustments to fit into the major groove of the DNA duplex, and particularly the β-hairpin moieties move to embrace DNA and protrude into the minor groove of the DNA with an arginine residue making contact with the bases (Fig. 7C). In each complex, DNA is also distorted from the typical B-form to complement the protein surface.
AcHcaR obeys similar rules. Binding of a specific DNA duplex induces conformational changes in the protein, with the largest occurring in its wing motifs, that allow the protein to reach the DNA bases in the minor groove. The β-hairpins move ∼6 Å (Cα His95) at the tip to allow Arg98 interaction with bases T9 and T−8 in the minor groove. This requires certain flexibility of the protein that may not be possible with an aromatic ligand bound to AcHcaR. The β-hairpins are linked to the aromatic ligand through a series of interactions involving Thr47 and Ser50 from α2 and Asn57 from the loop connecting α2 and α3. In AcHcaR subunit A, Thr47 and Ser50 interact with ferulate through water molecules, and the side chain of Asn57 interacts with the wing motif through the main chain atoms of the Ile101-Leu102-Val103 sequence motif in subunit B. Therefore, information about the bound ligand appears to be transmitted across the subunit interface. Because these residues are only partly conserved in the family (Fig. 5), alternative signaling pathways may be present in other members of the family.
It has been reported that the combination of succinate and acetate can interfere with the derepression effect of p-coumarate in Acinetobacter baylyi (14) and switch bacterial metabolism to process simpler sources of carbon. This suggests that smaller ligands (like succinate or acetate) could bind to the AcHcaR ligand-binding pocket, compete out p-coumarate, and not interfere with conformational changes required for productive DNA binding. This is because they are less bulky and still permit AcHcaR to adopt the DNA binding conformation.
Our model shows that aromatic ligands are too far from the DNA (the closest approach is >5 Å) to interfere, even indirectly, with DNA binding, although there could be some electrostatic repulsion in cases where ligands project the carboxylate group in the direction of the DNA sugar-phosphate backbone. However, this may not be the case in the thioesters (57). These larger ligands, to fit into the cavity, may need to have the thioester group facing out (see ferulic acid conformation in Figs. 6 and 7). In such instance, the CoA moiety would protrude out, likely interfering with DNA binding.
Conclusions
We have investigated AcHcaR, a novel member of the MarR/SlyA family of transcription regulators, and its interactions with small aromatic ligands and a specific DNA operator. We have solved structures of apo-AcHcaR, several complexes with ligands, and the complex of apo-AcHcaR with an idealized 23-bp duplex DNA corresponding to the HcaR operator present the in hca promoter region. These structures show that AcHcaR is promiscuous in accommodating different aromatic compounds in the ligand-binding site. These data when combined with biophysical and biochemical studies suggest how ligand binding may interfere with recognition of DNA. These aromatic compounds relieve AcHcaR repression by binding to the protein, and they appear to stabilize AcHcaR in a state that cannot engage with the specific DNA sites. The protein may not be flexible enough to fit into its DNA-binding site and fails to induce specific changes in DNA necessary for the formation of a productive, high affinity complex with DNA.
With its catabolic diversity, AcHcaR is an atypical regulator and seems to be unique because it is capable of binding substrates, intermediates, and products. As hydroxycinnamates tend to inhibit cell growth at higher concentrations, AcHcaR evolved to possess the dual characteristics of being a repressor and recognizing hydroxycinnamates and their homologues as well as aromatic thioesters to allow expression of enzymes to catabolize these compounds. This property seems to be an adaptation to the apparently broad specificity of hydroxycinnamate catabolic enzymes. Therefore the substrate, intermediate, and product induction of gene expression may help to enhance specificity and reduce toxicity associated with some of these ligands. This function permits the bacterial host to cope with two demands, nutrition and detoxification. It is worth noting that there is a possible link between the location of the operator binding site within the 256-bp hca intergenic region between the genes hcaA and hcaK and the tetrameric (dimer of dimers) assembly observed in the AcHcaR·DNA complex. Parke and Ornston (18) reported a mutational analysis with hcaA, hcaC, and hcaR and found a link between HcaA and HcaR proteins during accumulation of hydroxycinnamate compounds. When mutations in HcaA and HcaR were combined, caffeate, p-coumarate, or ferulate totally inhibited the cell growth at low concentrations (10−6 m). This experiment implicated HcaK, a presumed 12-helix-containing membrane protein, as a possible transporter of hydroxycinnamates and suggested that HcaR regulates HcaK transcription (18). It is plausible that the transcriptional activity of HcaR is regulated by an influx of aromatic compounds resulting from the transporter activity of HcaK. Interestingly, the hcaA and hcaK genes in the hca operon are 256 bp apart and are transcribed in opposite directions from separate promoters and produce two transcripts, hcaABCDEFG and hcaKR. The DNA-binding site hca1 of AcHcaR found in this study (CAATATCAGTTAAACTTACATTG) (underlined bases are involved in interaction with AcHcaR) is located 57 bases upstream of hcaA, which suggests that AcHcaR controls the expression of HcaA. A second less obvious site, hca2 (AAATATTCGAATTGACTATAAAA) (underlined bases are identical with hca1) is located 77 bases upstream of the hcaK gene. Could this sequence serve as a regulatory site for hcaKR genes and be controlled directly by HcaR? Typically, operators overlap with RNA polymerase −15/−35 regions. The hca sites are more distant from the transcription start sites. One possible explanation is the fact that AcHcaR forms tetrameric assemblies (Fig. 4, B and C) and therefore is capable of binding two DNA sites simultaneously. If a similar tetrameric assembly is to be adopted in vivo, perhaps HcaR may control expression of both hcaABCDEFG and hcaKR using hca1 and hca2 within the same intervening sequence: the one dimeric transcription factor binds to one site, and the DNA loops around to have the second site bind to the second dimer of the tetramer. In the 256-bp-long sequence, the two presumed AcHcaR-binding sites are 120 bp apart, and it is conceivable that a tetramer can bind both sites at the same time using a looping mechanism. The resulting structure may be less accessible to RNA polymerase. The second site appears to be less symmetric, and AcHcaR may bind it with lower affinity. Therefore, we hypothesize that the hcaABCDEFG transcript of seven genes may be under tighter control than the hcaKR transcript. The expression of apo-AcHcaR is needed to block transcription, and expression of HcaK is required to transport hydroxycinnamates.
Author Contributions
Y. K., G. B., and A. J. conceived the experiments. G. J., L. B., and Y. K. performed the experiments. Y. K. and A. J. wrote the paper.
Acknowledgments
We thank members of the Structural Biology Center at Argonne National Laboratory for help with data collection at the 19-ID beamline. We thank Gekleng Chhor for proofreading this manuscript.
This work was supported by National Institute of Health Grants GM115586 and GM094585 and by United States Department of Energy, Office of Biological and Environmental Research, Contract DE-AC02-06CH11357. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MarR
- multiple antibiotic resistance regulator
- wHTH
- winged helix-turn-helix
- HTH
- helix-turn-helix
- AcHcaR
- Acinetobacter sp. HcaR
- DHBA
- 3,4-dihydroxybenzoic acid
- SeMet
- l-selenomethionine
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- SAD
- single wavelength anomalous dispersion
- MAD
- multiple wavelength anomalous dispersion
- r.m.s.d.
- root mean square deviation
- DLS
- dynamic light scattering.
References
- 1. Spory A., Bosserhoff A., von Rhein C., Goebel W., and Ludwig A. (2002) Differential regulation of multiple proteins of Escherichia coli and Salmonella enterica serovar Typhimurium by the transcriptional regulator SlyA. J. Bacteriol. 184, 3549–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schumacher M. A., and Brennan R. G. (2002) Structural mechanisms of multidrug recognition and regulation by bacterial multidrug transcription factors. Mol. Microbiol. 45, 885–893 [DOI] [PubMed] [Google Scholar]
- 3. Fiorentino G., Ronca R., Cannio R., Rossi M., and Bartolucci S. (2007) MarR-like transcriptional regulator involved in detoxification of aromatic compounds in Sulfolobus solfataricus. J. Bacteriol. 189, 7351–7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alekshun M. N., and Levy S. B. (1999) The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 7, 410–413 [DOI] [PubMed] [Google Scholar]
- 5. Davis J. R., and Sello J. K. (2010) Regulation of genes in Streptomyces bacteria required for catabolism of lignin-derived aromatic compounds. Appl. Microbiol. Biotechnol. 86, 921–929 [DOI] [PubMed] [Google Scholar]
- 6. Di Fiore A., Fiorentino G., Vitale R. M., Ronca R., Amodeo P., Pedone C., Bartolucci S., and De Simone G. (2009) Structural analysis of BldR from Sulfolobus solfataricus provides insights into the molecular basis of transcriptional activation in Archaea by MarR family proteins. J. Mol. Biol. 388, 559–569 [DOI] [PubMed] [Google Scholar]
- 7. Carmona M., Zamarro M. T., Blázquez B., Durante-Rodríguez G., Juárez J. F., Valderrama J. A., Barragán M. J., García J. L., and Díaz E. (2009) Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol. Mol. Biol. Rev. 73, 71–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tatusov R. L., Koonin E. V., and Lipman D. J. (1997) A genomic perspective on protein families. Science 278, 631–637 [DOI] [PubMed] [Google Scholar]
- 9. Lintner N. G., Frankel K. A., Tsutakawa S. E., Alsbury D. L., Copié V., Young M. J., Tainer J. A., and Lawrence C. M. (2011) The structure of the CRISPR-associated protein Csa3 provides insight into the regulation of the CRISPR/Cas system. J. Mol. Biol. 405, 939–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enemark E. J., and Joshua-Tor L. (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 [DOI] [PubMed] [Google Scholar]
- 11. Guerra A. J., Dann C. E. 3rd, and Giedroc D. P. (2011) Crystal structure of the zinc-dependent MarR family transcriptional regulator AdcR in the Zn(II)-bound state. J. Am. Chem. Soc. 133, 19614–19617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reyes-Caballero H., Guerra A. J., Jacobsen F. E., Kazmierczak K. M., Cowart D., Koppolu U. M., Scott R. A., Winkler M. E., and Giedroc D. P. (2010) The metalloregulatory zinc site in Streptococcus pneumoniae AdcR, a zinc-activated MarR family repressor. J. Mol. Biol. 403, 197–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perera I. C., and Grove A. (2011) MarR homologs with urate-binding signature. Protein Sci. 20, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hirakawa H., Schaefer A. L., Greenberg E. P., and Harwood C. S. (2012) Anaerobic p-coumarate degradation by Rhodopseudomonas palustris and identification of CouR, a MarR repressor protein that binds p-coumaroyl coenzyme A. J. Bacteriol. 194, 1960–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Providenti M. A., and Wyndham R. C. (2001) Identification and functional characterization of CbaR, a MarR-like modulator of the cbaABC-encoded chlorobenzoate catabolism pathway. Appl. Environ. Microbiol. 67, 3530–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hiromoto T., Matsue H., Yoshida M., Tanaka T., Higashibata H., Hosokawa K., Yamaguchi H., and Fujiwara S. (2006) Characterization of MobR, the 3-hydroxybenzoate-responsive transcriptional regulator for the 3-hydroxybenzoate hydroxylase gene of Comamonas testosteroni KH122-3s. J. Mol. Biol. 364, 863–877 [DOI] [PubMed] [Google Scholar]
- 17. Yoshida M., Hiromoto T., Hosokawa K., Yamaguchi H., and Fujiwara S. (2007) Ligand specificity of MobR, a transcriptional regulator for the 3-hydroxybenzoate hydroxylase gene of Comamonas testosteroni KH122-3s. Biochem. Biophys. Res. Commun. 362, 275–280 [DOI] [PubMed] [Google Scholar]
- 18. Parke D., and Ornston L. N. (2003) Hydroxycinnamate (hca) catabolic genes from Acinetobacter sp. strain ADP1 are repressed by HcaR and are induced by hydroxycinnamoyl-coenzyme A thioesters. Appl. Environ. Microbiol. 69, 5398–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dal S., Trautwein G., and Gerischer U. (2005) Transcriptional organization of genes for protocatechuate and quinate degradation from Acinetobacter sp. strain ADP1. Appl. Environ. Microbiol. 71, 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egland P. G., and Harwood C. S. (1999) BadR, a new MarR family member, regulates anaerobic benzoate degradation by Rhodopseudomonas palustris in concert with AadR, an Fnr family member. J. Bacteriol. 181, 2102–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumaraswami M., Schuman J. T., Seo S. M., Kaatz G. W., and Brennan R. G. (2009) Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 37, 1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang Y.-M., Jeng W.-Y., Ko T.-P., Yeh Y.-J., Chen C. K., and Wang A. H. (2010) Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl. Acad. Sci. U.S.A. 107, 8617–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Copley S. D. (2015) An evolutionary biochemist's perspective on promiscuity. Trends Biochem. Sci. 40, 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilkinson S. P., and Grove A. (2006) Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 8, 51–62 [PubMed] [Google Scholar]
- 25. Chang Y.-M., Ho C.-H., Chen C. K., Maestre-Reyna M., Chang-Chien M. W., and Wang A. H. (2014) TcaR-ssDNA complex crystal structure reveals new DNA binding mechanism of the MarR family proteins. Nucleic Acids Res. 42, 5314–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong M., Fuangthong M., Helmann J. D., and Brennan R. G. (2005) Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol. Cell 20, 131–141 [DOI] [PubMed] [Google Scholar]
- 27. Kumarevel T., Tanaka T., Umehara T., and Yokoyama S. (2009) ST1710-DNA complex crystal structure reveals the DNA binding mechanism of the MarR family of regulators. Nucleic Acids Res. 37, 4723–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brugarolas P., Movahedzadeh F., Wang Y., Zhang N., Bartek I. L., Gao Y. N., Voskuil M. I., Franzblau S. G., and He C. (2012) The oxidation-sensing regulator (MosR) is a new redox-dependent transcription factor in Mycobacterium tuberculosis. J. Biol. Chem. 287, 37703–37712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dolan K. T., Duguid E. M., and He C. (2011) Crystal structures of SlyA protein, a master virulence regulator of Salmonella, in free and DNA-bound states. J. Biol. Chem. 286, 22178–22185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh S.-Y., Shin J.-H., and Roe J.-H. (2007) Dual role of OhrR as a repressor and an activator in response to organic hydroperoxides in Streptomyces coelicolor. J. Bacteriol. 189, 6284–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fischer R., Bleichrodt F. S., and Gerischer U. C. (2008) Aromatic degradative pathways in Acinetobacter baylyi underlie carbon catabolite repression. Microbiology 154, 3095–3103 [DOI] [PubMed] [Google Scholar]
- 32. Michalska K., Chang C., Mack J. C., Zerbs S., Joachimiak A., and Collart F. R. (2012) Characterization of transport proteins for aromatic compounds derived from lignin: benzoate derivative binding proteins. J. Mol. Biol. 423, 555–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turlin E., Sismeiro O., Le Caer J. P., Labas V., Danchin A., and Biville F. (2005) 3-Phenylpropionate catabolism and the Escherichia coli oxidative stress response. Res. Microbiol. 156, 312–321 [DOI] [PubMed] [Google Scholar]
- 34. Manso I., Torres B., Andreu J. M., Menéndez M., Rivas G., Alfonso C., Díaz E., García J. L., and Galán B. (2009) 3-Hydroxyphenylpropionate and phenylpropionate are synergistic activators of the MhpR transcriptional regulator from Escherichia coli. J. Biol. Chem. 284, 21218–21228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Donnelly M. I., Zhou M., Millard C. S., Clancy S., Stols L., Eschenfeldt W. H., Collart F. R., and Joachimiak A. (2006) An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr. Purif. 47, 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., and Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 37. Kim Y., Babnigg G., Jedrzejczak R., Eschenfeldt W. H., Li H., Maltseva N., Hatzos-Skintges C., Gu M., Makowska-Grzyska M., Wu R., An H., Chhor G., and Joachimiak A. (2011) High-throughput protein purification and quality assessment for crystallization. Methods 55, 12–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim Y., Dementieva I., Zhou M., Wu R., Lezondra L., Quartey P., Joachimiak G., Korolev O., Li H., and Joachimiak A. (2004) Automation of protein purification for structural genomics. J. Struct. Funct. Genomics 5, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Joachimiak A., and Sigler P. B. (1991) in Methods in Enzymology (Robert T. S., ed) Vol. 208, pp. 82–99, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 40. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., et al. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 42. Schneider T. R., and Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 43. Sheldrick G. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Otwinowski Z. (1991) in Daresbury Study Weekend Proceedings, pp. 80–85, SERC Daresbury Laboratory, Warrington, UK [Google Scholar]
- 45. Cowtan K. (1994) dm: an automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newslett. Protein Crystallogr. 31, 34–38 [Google Scholar]
- 46. Cowtan K. D., and Main P. (1993) Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Crystallogr. D Biol. Crystallogr. 49, 148–157 [DOI] [PubMed] [Google Scholar]
- 47. Terwilliger T. C. (2003) Improving macromolecular atomic models at moderate resolution by automated iterative model building, statistical density modification and refinement. Acta Crystallogr. D Biol. Crystallogr. 59, 1174–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 49. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 51. Davis I. W., Murray L. W., Richardson J. S., and Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alekshun M. N., Levy S. B., Mealy T. R., Seaton B. A., and Head J. F. (2001) The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nat. Struct. Biol. 8, 710–714 [DOI] [PubMed] [Google Scholar]
- 53. Saridakis V., Shahinas D., Xu X., and Christendat D. (2008) Structural insight on the mechanism of regulation of the MarR family of proteins: high-resolution crystal structure of a transcriptional repressor from Methanobacterium thermoautotrophicum. J. Mol. Biol. 377, 655–667 [DOI] [PubMed] [Google Scholar]
- 54. Bordelon T., Wilkinson S. P., Grove A., and Newcomer M. E. (2006) The crystal structure of the transcriptional regulator HucR from Deinococcus radiodurans reveals a repressor preconfigured for DNA binding. J. Mol. Biol. 360, 168–177 [DOI] [PubMed] [Google Scholar]
- 55. Wilkinson S. P., and Grove A. (2005) Negative cooperativity of uric acid binding to the transcriptional regulator HucR from Deinococcus radiodurans. J. Mol. Biol. 350, 617–630 [DOI] [PubMed] [Google Scholar]
- 56. Lavery R., Moakher M., Maddocks J. H., Petkeviciute D., and Zakrzewska K. (2009) Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 37, 5917–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hertz R., Magenheim J., Berman I., and Bar-Tana J. (1998) Fatty acyl-CoA thioesters are ligands of hepatic nuclear factor-4α. Nature 392, 512–516 [DOI] [PubMed] [Google Scholar]
- 58. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]