

Abstract
Ezrin is a member of the ERM (ezrin/radixin/moesin) family of proteins that links cortical cytoskeleton to the plasma membrane. High expression of ezrin correlates with poor prognosis and metastasis in osteosarcoma. In this study, to uncover specific cellular responses evoked by ezrin inhibition that can be used as a specific pharmacodynamic marker(s), we profiled global gene expression in osteosarcoma cells after treatment with small molecule ezrin inhibitors, NSC305787 and NSC668394. We identified and validated several up-regulated integrated stress response genes including PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 as novel ezrin-regulated transcripts. Analysis of transcriptional response in skin and peripheral blood mononuclear cells from NSC305787-treated mice compared with a control group revealed that, among those genes, the stress gene DDIT4/REDD1 may be used as a surrogate pharmacodynamic marker of ezrin inhibitor compound activity. In addition, we validated the anti-metastatic effects of NSC305787 in reducing the incidence of lung metastasis in a genetically engineered mouse model of osteosarcoma and evaluated the pharmacokinetics of NSC305787 and NSC668394 in mice. In conclusion, our findings suggest that cytoplasmic ezrin, previously considered a dormant and inactive protein, has important functions in regulating gene expression that may result in down-regulation of stress response genes.
Keywords: ezrin, gene expression, metastasis, osteosarcoma, small molecule, transgenic mice
Introduction
Ezrin is a prototype member of the ERM (ezrin/radixin/moesin) protein family of membrane-cytoskeleton linkers (1). Ezrin has been implicated in the regulation of many essential cellular functions including cell adhesion, motility, regulation of ion channels, maintenance and determination of cell shape, morphogenesis, cell proliferation, and apoptosis (2). A growing body of evidence suggests that high ezrin expression correlates with metastatic behavior and poor clinical outcome in a variety of tumors including melanoma, colorectal cancer, endometrioid carcinoma, astrocytic tumors, soft tissue sarcomas, and adenocarcinomas of lung and breast (3–11). Ezrin has also been directly implicated in tumor progression and metastatic process in pediatric solid tumors of mesenchymal origin including osteosarcoma and rhabdomyosarcoma (12–14).
Osteosarcoma is the most common and highly invasive primary malignant tumor of bone. Osteosarcoma can affect people of any age but occurs typically in adolescents and young adults and accounts for ∼5% of all childhood tumors (15). Advances in induction and adjuvant chemotherapy protocols in addition to surgery have largely improved 5-year survival rates of patients with only localized disease to 60–70% (16). Most patients diagnosed with localized osteosarcoma are considered to have micrometastases undetectable by the current procedures at the time of diagnosis (17). Osteosarcoma recurs in ∼30–40% of those patients initially diagnosed with “non-metastatic” disease, which results in a poor prognosis with a 5-year survival of less than 20%. The development of metastasis, commonly to the lungs, remains the major cause of death in osteosarcoma patients (18). A high level of ezrin is associated with a poor prognosis in pediatric osteosarcoma patients and in spontaneous canine osteosarcomas (12, 19). Ezrin directly increases the metastatic capability of osteosarcoma cells by providing an early survival advantage for cancer cells reaching the lung in a mouse model (12, 19). Therefore, development of novel targeted therapeutics that effectively inhibit ezrin to prevent pulmonary metastasis is urgently needed.
The current understanding of ezrin function is based on its dynamic behavior involving a conformational change switching the protein from a closed/inactive state to an open/active state through intramolecular interactions between the N-terminal domain (FERM) and F-actin binding site in the C-terminal region. In its closed state, self-association of the ezrin masks the respective protein binding sites on N-terminal site and renders the protein inactive in the cytoplasm. Unfolding and thus activation of ezrin occurs in response to binding to membrane phospholipid PIP2 through its N-terminal FERM domain and subsequent phosphorylation of a conserved threonine residue within the C-terminal domain (2, 20). This form of ezrin is regarded as fully functional; it can bind various membrane-associated proteins and the underlying cortical F-actin via its N-terminal and C-terminal domains, respectively. Recent data from our lab has demonstrated that the function of ezrin extends beyond its classical role as a cross-linker protein at the membrane-cortex interface to bind with DDX3 as a modulator of translation (21, 22). Thus, our prior findings suggested that ezrin has a role in integrating cellular stress response with the regulation of mRNA translation, thereby possibly rendering the cells more resistant to oncogenic stress encountered during metastatic progression (21).
In our earlier studies, we have discovered two small molecule inhibitors of ezrin, NSC305787 and NSC668394, which bind directly to ezrin and inhibit its activity in mediating the metastatic progression of osteosarcoma cells in multiple in vitro and in vivo experimental models (23). In the present study, we expand on our previous findings by demonstrating that treatment with NSC305787 significantly reduces pulmonary metastasis in a transgenic mouse model of osteosarcoma. We additionally studied the pharmacokinetics of NSC305787 and NSC668394 in mice and used a genomic approach to identify key ezrin-mediated biological pathways in osteosarcoma cells modulated by anti-ezrin compounds that can be used as pharmacodynamic marker(s) of compound treatment. Finally, our analysis of gene expression in NSC305787-treated mice compared with a control group revealed that among the set of compound-up-regulated specific target genes, the stress gene DDIT4/REDD1 may be used as a surrogate pharmacodynamic marker of response to ezrin inhibition.
Experimental Procedures
Cell Lines and Culturing
Human MG63.3 osteosarcoma, mouse K7M2 osteosarcoma, and canine MCKOS, SKKOS, and CSKOS osteosarcoma cells were maintained in DMEM supplemented with 10% FBS in a humidified atmosphere of 5% CO2 at 37 °C. The canine osteosarcoma cells were kindly provided by Dr. D. H. Thamm (Colorado State University, Fort Collins, CO). The human MG63.3 and mouse K7M2 cell lines were kindly provided from Dr. C. Khanna (NCI, National Institutes of Health, Bethesda, MD). K7M2 cells were derived from the clonally related K12 cell line through in vivo selection by repeated cycling of cells from pulmonary metastases into the orthotopic site (24). K7M2 cells express higher levels of ezrin protein, which leads to a greater potential to metastasize to the lungs than K12 cells (25). MG63.3 cells were derived from MG63.2 using in vivo passage by a process of experimental metastasis (25, 26).
Gene Silencing with siRNA
The prevalidated siRNA sequence targeting human ezrin (catalog no. s14796; Invitrogen) or nontargeting control (siGENOME nontargeting siRNA control pools/pool 2 D-001206-14-50; Dharmacon, Lafayette, CO) were transfected using X-tremeGene siRNA transfection reagent (Roche) according to the manufacturer's instructions. The cells were analyzed for ezrin expression after 72 h by immunoblotting.
Quantitative RT-PCR
Changes in transcript expression levels of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were determined by real time quantitative RT-PCR. Total RNA from osteosarcoma cells and mouse peripheral blood mononuclear cells (PBMCs)2 were extracted using the RNeasy Mini Kit (Qiagen; catalog no. 74104). Total RNA from mouse skin was extracted using the TRIzol reagent (Ambion/Thermo Fisher Scientific; catalog no. 15596018). Total RNA was reverse transcribed using a transcriptor first-strand cDNA synthesis kit (Roche) according to the manufacturer's protocol. A portion of the total cDNA was amplified by real time PCR on a LightCycler 480 II system using SYBR green mix (Sigma-Aldrich). The reactions were performed in a 20-μl volume (10 μl of 2× master mix, 1.0 μl of 10 μmol/liter forward and reverse primer mix, and 2.0 μl of cDNA) as triplicates on a 96-multiwell plate. The PCR cycling conditions were as follows: preincubation at 95 °C for 10 min and 40 amplification cycles, including denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s. Finally, a melting curve analysis was performed by a stepwise temperature increase from 65 to 97 °C to check primer specificity. The relative target gene expression was quantified by the ΔΔCt method using either 18S rRNA or GAPDH for normalization. The sequences of human, mouse, and canine primer pairs are shown in Table 1.
TABLE 1.
Sequences of primers used for real time qPCR experiments in this study
| Gene symbol | Protein | Forward primer | Reverse primer |
|---|---|---|---|
| 18S rRNA | CTTAGAGGGACAAGTGGCG | ACGCTGAGCCAGTCAGTGTA | |
| Human | |||
| PTGS2 | Prostaglandin G/H synthase 2 | CTCAGCCATACAGCAAATCCT | CCGGGTACAATCGCACTTAT |
| ATF3 | Activating transcription factor 3 | ATCGGCCCACGTGTATTG | TAGCTCTGCAATGTTCCTTCTT |
| DDIT3 | DNA damage-inducible transcript 3 | CAAGAGGTCCTGTCTTCAGATG | GGGTCAAGAGTGGTGAAGATT |
| DDIT4 | DNA damage-inducible transcript 4 | TGTTTAGCTCCGCCAACTC | TTCTTGATGACTCGGAAGCC |
| TRIB3 | Tribbles homolog 3 (Drosophila) | AGCTGTGTCGCTTTGTCTT | CTTGTCCCACAGGGAATCATC |
| ATF4 | Activating transcription factor 4 | CCCTTCACCTTCTTACAACCTC | TTCACTGCCCAGCTCTAAAC |
| Mouse | |||
| PTGS2 | Prostaglandin G/H synthase 2 | CGGACTGGATTCTATGGTGAAA | CTTGAAGTGGGTCAGGATGTAG |
| ATF3 | Activating transcription factor 3 | CTCCTGGGTCACTGGTATTTG | CCGATGGCAGAGGTGTTTAT |
| DDIT3 | DNA damage-inducible transcript 3 | TCACACGCACATCCCAAA | CCTAGTTCTTCCTTGCTCTTCC |
| DDIT4 | DNA damage-inducible transcript 4 | TTAAGTTCTGCCAACTCTTCCT | CGGAGCTGTAGAGTTTCTTCTT |
| TRIB3 | Tribbles homolog 3 (Drosophila) | AACCTTCAGAGCGACTTGTGGC | ATCTCCCTTCGGTCAGACTGTG |
| ATF4 | Activating transcription factor 4 | CCTGACTCTGCTGCTTACATTA | AGGACTCTGGGCTCATACA |
| PSMD7 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 7 | GAGCATGTGACTAGCGAGATT | CCCACTGTAGTGTCCTTGATG |
| Canine | |||
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | TGTCCCCACCCCCAATGTATC | CTCCGATGCCTGCTTCACTACCTT |
| PTGS2 | Prostaglandin G/H synthase 2 | CGAAGAACTTACAGGAGAGAAGG | TACCAAGAGGGCAGGATACA |
| ATF3 | Activating transcription factor 3 | CGGCGGGAAAGAAATAAGATTG | GTTCACACTCTCCAGCTTCTC |
| DDIT3 | DNA damage-inducible transcript 3 | AAAGACCTAGGAAACGGAAACA | CCACTTTCCTCTCGTTCTCTTG |
| DDIT4 | DNA damage-inducible transcript 4 | CGAGGATGAGCACTTGTGTG | AGCTCTTGCCTTGCTCTACG |
| TRIB3 | Tribbles homolog 3 (Drosophila) | GCGGTTCAAGTTGGACAATGA | GGAGGATGTAGGGCCCAAG |
| ATF4 | Activating transcription factor 4 | CTTTACCTTTCTGCAACCACTTC | ACTTCACTGCCTAGCTCTAAAC |
| PSMD7 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 7 | CAGGCCCTAAACTACACAAGAA | AGGTCCTTTGGCTTCACATC |
Immunoblotting
To verify ezrin knockdown, MG63.3 human osteosarcoma cells were lysed on ice for 30 min in phospholysis buffer (50 mmol/liter HEPES, pH 7.9, 100 mmol/liter NaCl, 4.0 mmol/liter sodium pyrophosphate, 10 mmol/liter EDTA, 10 mmol/liter sodium fluoride, and 1% Triton X-100) containing 2.0 mmol/liter sodium vanadate, 1.0 mmol/liter PMSF, 4.0 μg/ml aprotinin, 4.0 μg/ml leupeptin, and 1.0 μg/ml calyculin A. Protein concentrations of cell lysates were determined by the BCA assay and using bovine serum albumin as a standard (Pierce). Immunoblotting was performed as previously described (21). Proteins were resolved by SDS-PAGE and then transferred to an Immobilon-P membrane (Millipore, Billerica, MA). Proteins were probed using following primary antibodies: antiezrin, 1:2,500 (Sigma-Aldrich; E8897); and antiactin-HRP, 1:5,000 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-1615). The blots were developed using Millipore Immobilon Western chemiluminescent HRP substrate according to the manufacturer's instructions (Millipore, Billerica, MA). Chemiluminescence was detected using a Fujifilm LAS-3000 imaging system.
Gene Expression Profiling
For microarray analysis, 2 × 105 cells were plated on a 100-mm dish and grown until they reached at 60–70% confluency. The cells were starved in serum-free DMEM for 2 h prior to treatment. The cells were treated with 3.0 μmol/liter of NSC305787, 3.0 μmol/liter of NSC668394, or vehicle control (DMSO) for 6 h at 37 °C. Gene expression analysis was performed on quintuple cultures for each condition. Total RNA was isolated using QIAshredder and RNeasy mini kit (Qiagen). Total RNA concentration and quality control was assessed using NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). RNA integrity was assessed using the Agilent 2100 Bioanalyzer system (Agilent Technologies, Palo Alto, CA). Samples with RNA integrity number >7 were used in subsequent RNA amplification and hybridization steps. Transcriptome-wide expression profiling was performed using Illumina HumanHT-12 v4 Expression BeadChip Arrays (Illumina, San Diego, CA). Briefly, the raw data were preprocessed by Illumina GenomeStudio, and the preprocessed data were loaded into Partek Genomics Suite 6.6 for normalization and following statistical analysis. ANOVA model was employed to calculate overall and pair-wise p values, and a p value lower than 0.05 was used in combination with a fold change of 1.5 as cutoff thresholds to identify differentially expressed genes.
Transgenic Mouse Model of Osteosarcoma
All animal studies were conducted with the approval of Georgetown University's Institutional Animal Care and Use Committee in accordance with the requirements of the NIH. For all animal experiments, NSC305787 and NSC668394 were solubilized in DMSO, and the dosing solution of each compound was prepared at a concentration of 0.1 mmol/liter in 1% (v/v) DMSO prepared in sterile PBS.
The Osx-Cre+p53fl/flpRBfl/fl transgenic mouse model of osteosarcoma was generously provided by Dr. S. H. Orkin and Dr. K. A. Janeway (Harvard Medical School, Boston, MA) and has been previously described (27). Female mice homozygous for the p53lox and pRBlox genes (p53fl/flpRBfl/fl) on a mixed background were crossed with male mice hemizygous for a Osx-Cre transgene (Osx-Cre+p53fl/flpRBfl/fl; bearing only a single conditional allele of Cre transgene) to generate Osx-Cre+p53fl/flpRBfl/fl animals. The resulting offspring were genotyped by PCR analysis of genomic DNA isolated from tail biopsies of weanlings using the following primer pairs specific to Cre Recombinase transgene: Cre1 (5′-GACCAGGTTCGTTCACTCATGG-3′) and Cre2 (5′-AGGCTAAGTGCCTTCTCTACAC-3′). Female mice were fed with doxycycline-containing food (2,000 mg/kg diet; Harlan Laboratories) throughout the pregnancy until the weaning of the offspring at 21 days to repress the expression of Cre recombinase. All of the animals were observed daily, and tumors were measured at least once a week using a slide caliper. The tumor volumes were calculated using the following formula: tumor volume = (π/6) × length2 × width. Once tumors were detected, mice were randomly assigned to control (DMSO), NSC305787 treatment, and NSC668394 treatment groups. Mice were treated with 240 μg/kg/day NSC305787, 226 μg/kg/day NSC668394, or vehicle (DMSO, 1%) once daily, five times a week by i.p. injection in a volume of 100 μl. At the end of the study, lung and tumor samples were isolated upon necropsy. Half of each sample was flash frozen immediately in liquid nitrogen, and the other half was fixed in 10% formalin for 18–24 h, transferred to 70% ethanol, and stored at room temperature.
Pharmacokinetic Studies in Mice
Female 4–6-week-old Balb/c mice used for pharmacokinetics studies were purchased from Harlan Sprague-Dawley, Inc. (Indianapolis, IN) or Taconic Farm, Inc. (Germantown, NY). Mice received either NSC305787 or NSC668394 at a dose of 240 μg/kg body weight and 226 μg/kg body weight, respectively, via a single i.v. or i.p. injection in a volume of 100 μl. For each time point, three mice were used. Blood was collected at indicated time points by cardiac puncture using a 1-ml syringe under anesthesia and transferred to 1.3-ml EDTA tubes (Sarstedt, Nuermbrecht, Germany). Blood samples were centrifuged at 1,000 × g for 10 min at 4 °C to obtain plasma. Plasma samples were stored at −80 °C until further analysis.
NSC305787 or NSC668394 (50 μl of plasma) was extracted with 200 μl of acetonitrile containing 50 ng/ml of the internal standard NSC668394 or NSC305787, respectively. After centrifugation, the supernatant was injected into the LC/MS/MS system. Separation was achieved with an Agilent Zorbax XDB-C18 (4.6 × 50 mm, 5 μm) column at room temperature at 0.5 ml/min for 3 min. A gradient was implemented using mobile phases A (water, 0.1% formic acid) and B (acetonitrile, 0.1% formic acid): 60% B for 0.0–0.5 min, increased from 60% B to 90% B over 0.5–1.5 min, held at 90% B from 1.5–2.5 min, and then decreased to 60% B for 2.5–3 min for column re-equilibration. The analytes were monitored using an AB Sciex triple quadrapoleTM 5500 mass spectrometric detector (Applied Biosystems, Foster City, CA) using electrospray ionization operating in positive mode. The spectrometer was programmed to allow the [MH+] ions of NSC305787 and NSC668394 at m/z 445.3 and 452.9, respectively, to pass through the first quadrupole (Q1) and into the collision cell (Q2). The daughter ions for NSC305787 (m/z 427.2) and NSC668394 (m/z 189.2) were monitored through the third quadrupole (Q3). Calibration curves for NSC305787 or NSC668394 were computed using the area ratio peak of the analyte to the internal standard by using a quadratic equation with a 1/x2 weighting function over the range of 1–500 ng/ml.
Mean NSC305787 or NSC668394 concentrations were calculated at each time point. Pharmacokinetic parameters were calculated from the mean concentration-time data using noncompartmental methods as analyzed in Pheonix WinNonlin version 6.3 (Pharsight A Certara Company, Cary, NC).
Pharmacodynamic Studies
CD-1 male mice used for gene expression analysis of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 in PBMCs and skin were purchased from Harlan Sprague-Dawley, Inc. The mice were dosed with either vehicle (DMSO, 1%) or NSC305787 (240 μg/kg/day) through i.p. injection once daily for 3 consecutive days in a volume of 100 μl. The animals were euthanized 3 h after the last injection. Blood was collected through cardiac puncture in EDTA tubes (Sarstedt, Nuermbrecht, Germany). PBMCs were then isolated by Histopaque-1077 density gradient cell separation protocol according to the manufacturer's instructions (Sigma-Aldrich; catalog no. 10771). Skin samples from the back were collected as well and snap-frozen in liquid nitrogen immediately upon necropsy.
Osteosarcoma Phenotypes: Histopathological Analysis
An expert pathologist performed the analysis of the biopsies stained with hematoxylin and eosin, with attachment on the criteria established by the World Health Organization (28). Both primary tumors and lungs were completely embedded and included in histopathologic analysis regardless of macroscopical absence of neoplasm.
Statistical Analysis
Statistical analysis of microarray data were performed by ANOVA model, and the unpaired pair-wise comparison was made among groups using Partek Genomic Suite 6.6. The statistical significance of differences between groups in gene expression levels determined by RT-qPCR was evaluated by an unpaired Student's t test using Prism version 6.0c (GraphPad Software, La Jolla, CA). Statistical analysis of differences between the control and NSC305787-treated animal groups in the incidence of lung metastasis and tumor weight was performed by Fisher's exact and Mann-Whitney U tests, respectively (IBM SPSS Statistics, version 21). Although a type I error level of less than 5% was used to infer statistical significance, when accompanied by clinically significant differences, analyses used a p value of less than 0.10 to flag differences that are close to the level of statistical significance.
Results
NSC305787 Inhibits Osteosarcoma Lung Metastasis in a Transgenic Mouse Model
We investigated the in vivo anti-metastatic effects of NSC305787 and NSC668394 in a clinically relevant model of osteosarcoma (27). This genetically engineered mouse model is based on the restricted conditional deletion of Rb and Trp53 genes in osteoblast precursors using an osterix promoter-driven Cre recombinase, which induces spontaneous osteosarcoma formation with 100% penetrance (27). Osx-Cre+p53fl/flpRBfl/fl mice were maintained on a doxycycline diet during the period from conception until weaning (3 weeks of age) to repress the expression of Cre from Osx-Cre. This resulted in delayed osteosarcoma development with higher frequency of metastasis as previously reported (27). These mice developed tumors mostly in the limbs (forelimbs/hindlimbs), followed by the sacrum, spine, and ribs (Table 2).
TABLE 2.
Summary of transgenic mouse with osteosarcoma (Osx-Cre+p53fl/flpRBfl/fl) data for in vivo testing of the anti-metastatic effects of NSC305787
| Gender | Age at diagnosis | Survival since diagnosis | Tumor site |
|---|---|---|---|
| days | days | ||
| Control | |||
| F | 235 | 60 | Sacrum |
| F | 128 | 59 | Forelimb |
| F | 196 | 49 | Hindlimb |
| F | 173 | 31 | Underarm |
| F | 175 | 53 | Forelimb |
| M | 223 | 36 | Hindlimb |
| M | 174 | 53 | Forelimb |
| F | 164 | 42 | Underarm |
| F | 140 | 15 | Forelimb |
| F | 200 | 80 | Sacrum/underarm |
| F | 249 | 39 | Spine |
| M | 193 | 67 | Hindlimb |
| M | 188 | 77 | Hindlimb |
| Mean ± S.D. | 187.5 ± 34.7 | 50.9 ± 18.4 | |
| NSC305787 | |||
| F | 200 | 72 | Ribcage |
| F | 130 | 15 | Jaw |
| F | 176 | 31 | Hindlimb |
| M | 168 | 111 | Hindlimb |
| F | 207 | 53 | Hindlimb |
| F | 236 | 59 | Hindlimb |
| M | 209 | 62 | Sacrum |
| F | 270 | 62 | Sacrum/ribcage |
| F | 223 | 23 | Hindlimb/ribcage |
| F | 217 | 90 | Hindlimb |
| Mean ± S.D. | 203.6 ± 38.8 | 57.8 ± 29.6 | |
The mice were monitored daily for clinical signs of tumorigenesis, such as changes in body condition, attitude, limping, poor feeding, grooming, or weight loss and followed up by x-ray examination (Fig. 1A). All mice demonstrated typical pathological features of osteosarcoma characterized by the presence of variable quantity of osteoid formation within malignant neoplasm of mesenchymal origin composed of cytologically malignant cells. They usually were showing extension into the adjacent muscle and bone tissue, and some cases showed distant metastasis to lungs (Figs. 1B and 2).
FIGURE 1.

Histopathologic analysis of transgenic mice (Osx-Cre+p53fl/flpRBfl/fl) demonstrates typical pathological features of osteosarcoma. A, x-ray imaging of a representative animal demonstrating osteosarcoma of the hindlimb and spine in the coronal plane (left panel) and the sagittal plane (right panel). Arrows indicate osteosarcoma lesions. B, four representative hematoxylin and eosin images of Osx-Cre+p53fl/flpRBfl/fl mice show a histologically classical ostesarcoma morphology similar to that seen in human tumors, with different amounts of osteoid production along with osteoblastic areas. Diverse morphology was seen in different parts of the same tumor, as well as in different tumors from differing mice. In some cases, tumor showed extension and invasion into the surrounding muscle tissues (white arrows). Scale is indicated on each image.
FIGURE 2.
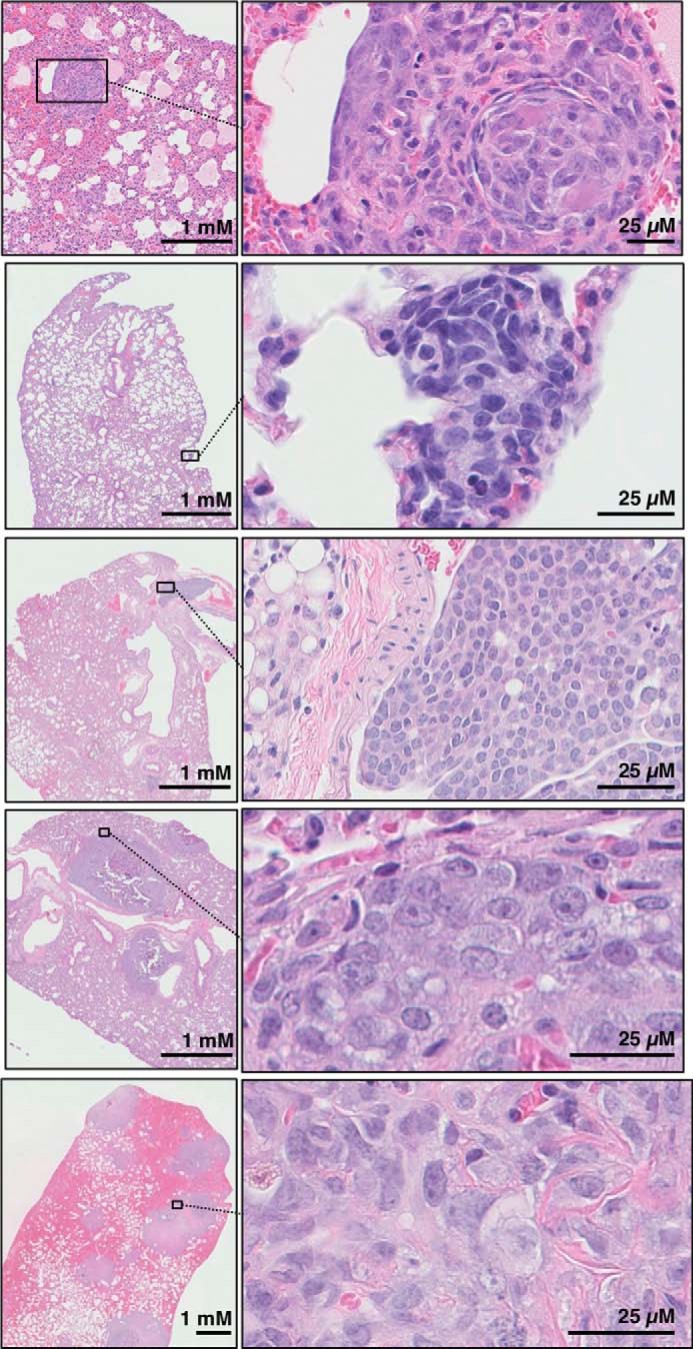
Osx-Cre+p53fl/flpRBfl/fl mice develop lung metastases that are detectable at necropsy. Representative images of lung metastasis with low (left panels) and high (right panels) magnification. Lung metastasis in all animals showed the same histopathological features as the primary tumor in the same subject.
Once a tumor was detected, the mice were randomized to control (DMSO) and treatment groups (i.p. with NSC305787 or NSC668394 at doses of 240 and 226 μg/kg body weight, respectively, once daily, five times a week). The animals were euthanized when one of the following was observed: if primary tumor reached to 2.0 cm3 in size, if the tumor showed signs of ulceration, or if animals showed signs of pain and distress. We started the experiment with 13 mice/group. However, in the NSC305787 treatment group three mice were found dead during the study. Mortalities occurred on weekends that prevented proper tissue recovery. This mortality rate of 23% (3 of 13) may indicate the potential toxicity of long term NSC305787 treatment. Because osteosarcoma commonly metastasizes to the lungs, we assessed the metastatic dissemination of tumor cells in the lung (Fig. 2). We found that 85% (11 of 13) of mice in the control group had lung metastasis at the time of necropsy by histologic evaluation (Fig. 3A). Treatment of mice with NSC305787 significantly suppressed pulmonary metastasis compared with the vehicle-treated control. Lung metastasis was observed only in 40% (4 of 10) of NSC305787-treated animals (p = 0.039, Fisher's exact test) (Fig. 3A). In contrast to NSC305787, NSC668694 treatment did not inhibit metastatic tumor formation in lungs (data not shown).
FIGURE 3.

NSC305787 inhibits pulmonary metastasis in a transgenic mouse model of osteosarcoma (Osx-Cre+p53fl/flpRBfl/fl) and has a more favorable pharmacokinetic profile compared with NSC668394 in the mouse model. A, the incidence of lung metastasis in NSC305787-treated (n = 10; 240 μg/kg i.p. administration once daily, five times per week) and untreated (n = 13) mice were compared (*, p = 0.039, Fisher's exact test). B, no significant difference was observed in primary tumor growth between compound-treated (n = 10) and vehicle control animals (n = 13) (ns, non-significant; p = 0.14, Mann-Whitney U test). C and D, time course of NSC305787 and NSC668394 concentrations in female Balb/c mouse plasma following i.p. (C) and i.v. (D) administration at single doses of 240 and 226 μg/kg, respectively. Mean concentration ± S.D. from three animals was determined at each time point.
To rule out the possibility that the lower incidence of lung metastasis in NSC305787 treatment group might have resulted from smaller primary tumor volumes at the time of euthanasia, we assessed the effect of NSC305787 on primary tumor growth. Although NSC305787 treatment strongly reduced metastasis, it did not reduce primary tumor growth (p = 0.14, Mann-Whitney U test) (Fig. 3B). Because animals were euthanized because of the primary tumor growth, no significant difference was observed in overall survival between two groups (Table 2). Taken together, these findings suggest that NSC305787 treatment inhibits ezrin-dependent lung metastasis of osteosarcoma.
Pharmacokinetic Analysis Demonstrates NSC305787 Has a Substantially Longer Plasma Half-life than NSC668394
We previously published that the anti-ezrin compound NSC668394 did inhibit invasive phenotype in cell culture assays with osteosarcoma cells, and this activity was comparable with NSC305787 treatment (23). However, in that manuscript the potent in vitro activity of NSC668394 did not translate into a robust in vivo activity, because it was found to be less effective than NSC305787 in increasing the overall survival in an experimental mouse metastasis model of osteosarcoma, in which highly metastatic K7M2 osteosarcoma cells were delivered by tail vein injection into mice (23). Consistent with these observations, NSC668394 treatment had also no effect in reducing the incidence of lung metastasis in a transgenic mouse model of osteosarcoma in the present study as well (data not shown). Because the differences in exposures may account for differences in their in vivo activities, we assessed the pharmacokinetics of NSC305787 and NSC668394 in female Balb/c mice. We first determined the plasma concentrations of NSC305787 and NSC668394 over a 6-h time course after a single i.p. injection at doses of 240 and 226 μg/kg, respectively (Fig. 3C). NSC305787 was observed at all the time points collected and was relatively consistent achieving ∼16 ng/ml exposure after 6 h, whereas NSC668394 was undetectable after 1 h (Fig. 3C). We next monitored the plasma concentrations of NSC305787 and NSC668394 over an extended time period after a single i.v. injection at the same doses (Fig. 3D). NSC668394 pharmacokinetics demonstrated a monophasic elimination profile whether administered i.p. or i.v. (Fig. 3, C and D). Similar to i.p. administration, NSC668394 was undetectable after 0.5 h, thus limiting the pharmacokinetic parameters that could be calculated. NSC305787 demonstrated a triphasic elimination profile after i.v. administration (Fig. 3D). The Cmax and AUCinf were calculated to be 195.1 ng/ml and 415.3 ng·h/ml, respectively, following i.v. administration. NSC305787 had an elimination half-life and clearance of 13.6 h and 9.6 ml/min/kg, respectively (Table 3). These findings revealed that NSC305787 had a more favorable pharmacokinetic profile compared with NSC668394, which may be responsible for the lack of anti-metastatic effect of NSC668394 in mice.
TABLE 3.
Plasma pharmacokinetic parameters after i.v. administration of NSC305787 (240 μg/kg) in Balb/c mice
Pharmacokinetic parameters are as follows: Cmax is the maximum plasma concentration; Tmax is the time to reach maximum plasma concentration; AUC is the area under the compound concentration versus time; T½ is the elimination half-life; V is the volume of distribution; and Cl is the clearance.
| Parameter | Value |
|---|---|
| Dose/route | 240 μg/kg IV |
| Tmax | 0.03 h |
| Cmax | 195.1 ng/ml |
| AUC14h | 228.5 ng·h/ml |
| AUCinf | 415.3 ng·h/ml |
| T½ | 13.6 h |
| V | 11.3 liter/kg |
| Cl | 9.6 ml/min/kg |
Inhibition of Ezrin Function in Osteosarcoma Cells Induces Activation of Genes Involved in Integrated Stress Response Signaling
We profiled global gene expression in MG63.3 human osteosarcoma cells after treatment with NSC305787 and NSC668394 to search for a specific pharmacodynamic marker(s) of response to ezrin inhibition. The cells were treated with 3.0 μmol/liter of either compound or vehicle control for 6 h using five biological replicates per condition. Total RNA was isolated, and whole genome expression analysis was performed using Illumina HumanHT-12 v4 Expression BeadChips, which provides coverage for more than 47,000 transcripts and known splice variants across the human transcriptome. The comparison of microarray gene expression data in response to NSC305787 and NSC668394 treatment revealed that 16 genes were commonly up-regulated (p < 0.05, with a fold change cutoff of >1.5) (Fig. 4A and supplemental Tables S1 and S2). It is of note that nearly all genes (15 of 16 genes) commonly induced, except LURAP1L, have been previously reported to be linked to integrated stress response pathway (Table 4), implicating that a common underlying mechanism may be shared by these compounds. Only 2 genes were down-regulated following NSC305787 treatment, whereas 90 transcripts were found to be down-regulated in NSC668394-treated cells (p < 0.05, with a fold change cutoff of >1.5). We did not detect any transcripts commonly down-regulated in response to both compounds (supplemental Tables S1 and S2).
FIGURE 4.

The small molecule inhibitors of ezrin up-regulate the integrated stress response-related genes. A, a Venn diagram showing number of up-regulated genes in response to either NSC305787 or NSC668394 treatment (p < 0.05, with a fold change cutoff of >1.5). B, validation of a set of genes identified from microarray analysis comparing MG63.3 human osteosarcoma cells treated with either NSC305787 or NSC668394 or vehicle control using real time qPCR. The cells were treated with 3.0 μmol/liter of either compound or vehicle control for 6 h, and mRNA levels of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were measured. The results were normalized to 18S rRNA levels and expressed as fold change over the control group. The values are presented as the means and standard deviations of triplicate determinations. (*, p < 0.05: for microarray analysis, statistical significance between treatment versus control group was determined using an ANOVA model (n = 5 biological replicates); for RT-qPCR experiments statistical significance between treatment versus control group (n = 3 technical replicates) was determined by an unpaired Student's t test). C, K7M2 mouse osteosarcoma cells were treated with 3.0 μmol/liter of either compound for 6 h, and expression levels of the selected integrated stress response signature genes were determined using mouse-specific primers as explained in B (*, p < 0.05, compared with control; using an unpaired Student's t test). D, MCKOS, SKKOS, and CSKOS canine osteosarcoma cells were treated with 5.0 μmol/liter of NSC305787 for 6 h, and expression levels of the selected integrated stress response signature genes were determined using canine-specific primers as explained in B except that GAPDH was used for normalization (*, p < 0.05, compared with control; using an unpaired Student's t test; ns, non-significant).
TABLE 4.
List of genes that were commonly up-regulated in response to NSC305787 and NSC668394 treatment
| Probe set | Gene title | Fold changea |
UniGene ID | |
|---|---|---|---|---|
| NSC305787 | NSC668394 | |||
| ILMN_1677511 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) (PTGS2) | 2.66 | 4.12 | Hs.196384 |
| ILMN_2374865 | Activating transcription factor3 (ATF3) | 2.51 | 3.11 | Hs.460 |
| ILMN_1676984 | DNA damage-inducible transcript3 (DDIT3) | 2.48 | 3.13 | Hs.505777 |
| ILMN_1661599 | DNA damage-inducible transcript4 (DDIT4) | 2.38 | 4.94 | Hs.744875 |
| ILMN_2376403 | TSC22 domain family, member3 (TSC22D3) | 1.99 | 3.92 | Hs.522074 |
| ILMN_2374159 | Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member1 (HERPUD1) | 1.92 | 2.46 | Hs.146393 |
| ILMN_1781285 | Dual specificity phosphatase1 (DUSP1) | 1.83 | 1.70 | Hs.171695 |
| ILMN_1800512 | Heme oxygenase (decycling) 1 (HMOX1) | 1.81 | 36.0 | Hs.517581 |
| ILMN_2188862 | Growth differentiation factor 15 (GDF15) | 1.71 | 2.24 | Hs.616962 |
| ILMN_1701361 | Leucine-rich adaptor protein 1-like (chromosome 9 open reading frame 150, C9orf150) (LURAP1L) | 1.63 | 2.18 | Hs.445356 |
| ILMN_1787815 | Tribbles homolog 3 (Drosophila) (TRIB3) | 1.61 | 3.35 | Hs.516826 |
| ILMN_1659936 | Protein phosphatase 1, regulatory subunit 15A (PPP1R15A) | 1.61 | 1.96 | Hs.631593 |
| ILMN_1773742 | DnaJ (Hsp40) homolog, subfamily B, member 9 (DNAJB9) | 1.56 | 2.86 | Hs.741182 |
| ILMN_1679041 | Solute carrier family 3 (activators of dibasic and neutral amino acid transport), member 2 (SLC3A2) | 1.56 | 2.24 | Hs.502769 |
| ILMN_1784602 | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) (CDKN1A) | 1.51 | 1.67 | Hs.370771 |
| ILMN_1713505 | Niemann-Pick disease, type C1 (NPC1) | 1.58 | 1.50 | Hs.715623 |
a p < 0.05. The data represent all commonly up-regulated genes (1.5-fold) from microarray analysis of MG63.3 human osteosarcoma cells after treatment with anti-ezrin compounds NSC305787 and NSC668394.
We further validated the microarray data through extensive testing using real time qPCR. We determined the relative expression of five genes critically involved in integrated stress response, PTGS2, ATF3, DDIT3, DDIT4, and TRIB3 in human MG63.3 osteosarcoma cells and highly metastatic mouse K7M2 osteosarcoma cells. Canine osteosarcoma exhibits many similarities to its human counterpart with respect to tumor histology, biological behavior, transcriptional profiles, response to conventional therapies, early metastasis, and dysregulated expression of ezrin (12). Given the similarities of canine and pediatric osteosarcomas, we included three canine osteosarcoma cells including MCKOS, SKKOS, and CSKOS to examine the effect of ezrin inhibitors on stress response gene expression, which may be used as surrogate pharmacodynamic marker in clinical trials involving canine osteosarcoma cohort.
Even though the microarray data demonstrated that ATF4 was induced 1.43- and 2.24-fold in response to NSC305787 and NSC668394 treatment, respectively, we included this gene in our panel for qPCR validation experiments because of its critical role in integrated stress response and homology to ATF3. As shown in Fig. 4B, the results for the analyzed genes in MG63.3 cells in response to both compounds were consistent with the microarray data. By using mouse- and canine-specific primers, we observed that mouse K7M2 osteosarcoma cells and canine MCKOS, SKKOS, and CSKOS osteosarcoma cells also exhibited the same gene expression pattern as the MG63.3 human osteosarcoma cells (Fig. 4, C and D). We used PSMD7 gene encoding 26S proteasome non-ATPase regulatory subunit 7 protein as a control, because its expression was determined to be unaffected by either compound treatment in microarray gene expression analysis. We observed no change in PSMD7 mRNA levels in response to NSC305787 treatment for all canine osteosarcoma cells, suggesting a specific involvement of this pathway in ezrin function (Fig. 4D). Recently, we have identified a compound, MMV667492, from the MMV400 “Malaria Box” library as a novel small molecule inhibitor of ezrin (29). MMV667492 exhibited potent anti-ezrin activity in blocking cell motility and invasion in vitro, in vivo, and ex vivo experimental models (29). To further verify that up-regulation of these transcripts is a specific response of cells to ezrin inhibition, we determined whether MMV667492 treatment affects the expression of integrated stress response effector genes. Consistent with above findings, treatment of MG63.3 and K7M2 cells with MMV667492 induced activation of genes with known functional significance related to integrated stress response signaling (Fig. 5A).
FIGURE 5.

Pharmacological inhibition of ezrin by NSC305787 and NSC668394 up-regulates the expression of integrated stress response signature genes in a dose-dependent manner. A, human MG63.3 and mouse K7M2 osteosarcoma cells were treated with 20.0 μmol/liter MMV667492 or vehicle control for 6 h, and mRNA levels of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were measured by real time qPCR. The results were normalized to 18S rRNA levels and expressed as fold change over the control group. The values are presented as the means and standard deviations of triplicate determinations (*, p < 0.05, compared with control; using an unpaired Student's t test). B, K7M2 mouse osteosarcoma cells were treated with indicated concentrations of compounds, NSC305787 or NSC668394, or vehicle control for 6 h, and mRNA levels of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were measured by real time qPCR. The results were normalized to 18S rRNA levels and expressed as fold change over the control group. The values are presented as the means and standard deviations of triplicate determinations (*, p < 0.05, compared with control; using an unpaired Student's t test; ns, non-significant).
We further evaluated dose-dependent effects of ezrin inhibitors on the expression of downstream integrated stress response factors in K7M2 osteosarcoma cells. We tested the effects of compounds on stress response gene expression at sublethal concentrations. Because NSC305787 is ∼2-fold more toxic than NSC668394 in K7M2 cells (IC50 for 72 h: 2.9 μm versus 5.9 μm), we chose 1–3 and 3–10 μm dose ranges for NSC305787 and NSC668394, respectively. Both NSC305787 and NSC668394 induced expression of integrated stress response signature genes in a dosage-dependent manner, whereas no change was observed in the expression of PSMD7 (Fig. 5B).
We next examined whether the effect of ezrin inhibitors on the expression of genes involved in integrated stress response signaling can be recapitulated by siRNA-mediated depletion of ezrin in MG63.3 cells. We observed that there was a clear transcriptional response indicating an integrated stress response signature to the reduced levels of ezrin protein (Fig. 6A). The relatively lower induction levels of ATF3, DDIT3, and TRIB3 compared with those in response to either compound treatment could be attributed to residual ezrin protein in cells (Fig. 6B).
FIGURE 6.

Suppression of ezrin expression leads to increased mRNA levels of downstream integrated stress response signaling effector genes and results in loss of transcriptional response to anti-ezrin compound. A, expression of ezrin protein was inhibited by siRNA targeting ezrin in human MG63.3 osteosarcoma cells and mRNA levels of a set of selected integrated stress response signature genes including PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were measured by real time qPCR. The results were normalized to 18S rRNA levels and expressed as fold change over the control. The values are presented as the means and standard deviations of triplicate determinations (*, p < 0.05, compared with siControl; using an unpaired Student's t test). B, Western blot showing ezrin levels from A. The protein levels were detected from total cell lysates at 72 h after transfection of cells with siRNA for ezrin. MW, molecular mass. C, ezrin expression was inhibited by a siRNA in MG63.3 cells, and cells were treated with either 3.0 μmol/liter of NSC668394 or vehicle control for 6 h on the third day of transfection. The mRNA levels of PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 were measured by real time qPCR. The results were normalized to 18S rRNA levels and expressed as fold change over the vehicle control in both control siRNA and ezrin siRNA-transfected cells. The values are presented as the means and standard deviations of triplicate determinations (*, p < 0.05, compared with siControl; using an unpaired Student's t test). D, DDIT4/REDD1 gene may serve as a pharmacodynamic marker of response to ezrin inhibition by NSC305787 in vivo. DDIT4/REDD1 mRNA levels of PBMCs and skin were measured by real time qPCR after a short term i.p. treatment of CD-1 mice with NSC305787 at a dose of 240 μg/kg once daily for 3 consecutive days. The results were normalized to 18S rRNA levels and expressed as 1/ΔCt. Each sample was analyzed in triplicate, and each mouse in treatment and control group is designated by a specific symbol. Horizontal lines indicate mean values of all measurements in each group (*, p < 0.05, compared with control; using an unpaired Student's t test).
To confirm that anti-ezrin compound-mediated changes in target gene expression occur through inhibition of ezrin function, we compared the mRNA expression levels of the same set of genes in wild-type versus ezrin-depleted MG63.3 human osteosarcoma cells treated with the ezrin inhibitor NSC668394. We observed that up-regulation of integrated stress response signature genes was much weaker in cells with reduced ezrin levels compared with wild-type cells, indicating the specificity of the compounds on the ezrin-related biological pathways (Fig. 6C).
Expression Analysis of Integrated Stress Response Signature Genes in Mice Reveals That DDIT4/REDD1 Can Be Used as a Specific Pharmacodynamic Marker of NSC305787 Activity
We next sought to investigate whether NSC305787 treatment could lead to overexpression of integrated stress response signature genes including PTGS2, ATF3, DDIT3, DDIT4, TRIB3, and ATF4 in vivo. CD-1 mice were given a single i.p. injection of 240 μg/kg NSC305787 once daily for 3 consecutive days. Expression analysis of the integrated stress response-related genes in PBMCs and skin of NSC305787-treated mice compared with a control group demonstrated up-regulation of DDIT4/REDD1 (Fig. 6D), but not the other genes analyzed. Thus, these results suggest that the stress gene DDIT4/REDD1 may be used as a surrogate pharmacodynamic marker of response to ezrin inhibition by NSC305787 in future in vivo studies.
Discussion
Current conventional therapeutics targeting primarily cancer growth have shown limited efficacy in successful treatment of patients with metastatic disease, because of the exceedingly complex nature of metastatic process. Translation of biological knowledge into clinical practice remains challenging because there is currently a lack of anti-metastatic compounds specifically targeting metastatic regulators (30, 31). Ezrin has been validated as a key determinant in the development of metastatic osteosarcoma, which supports the development of small molecule ezrin inhibitors (12, 19). Ezrin is involved in all phases of metastasis including invasion, motility, and survival in a new environment, thereby highlighting its importance as a key therapeutic target in cancer (32).
In the present study, we demonstrated that NSC305787 treatment but not NSC668394 inhibited the lung metastasis in Osx-Cre+p53fl/flpRBfl/fl transgenic mice. Nevertheless, NSC 668394 can still be used as a tool for probing the biological mechanisms of action underlying ezrin-mediated metastatic behavior in cell culture assays. It should be noted that the inhibition of lung metastasis in response to NSC305787 treatment did not result in a significant reduction of primary tumor size and therefore did not alter the overall survival, because the animals were euthanized because of the primary tumor growth but not from the development of metastasis. Therefore, we were unable to examine the effect of NSC305787 on the overall survival of mice. Future studies involving the use of human xenograft or mouse allograft model of osteosarcoma, in which cells are injected orthotopically into the tibia of mice, should be able to overcome this obstacle. Amputation of the tumor-bearing limb would be an essential step in those models to avoid that mice becoming moribund because of primary tumor growth. This would allow us to study the likely favorable effect of NSC305787 treatment on overall survival caused by the suppression of metastatic development.
Disseminated metastatic cells encounter various microenvironmental stresses in the circulation or after reaching a new environment where they must adapt and endure these stresses to survive and establish secondary tumors (33, 34). Studies using human and mouse osteosarcoma cells expressing inactive ezrin demonstrated that dysregulation of ezrin function results in a consistent increase in apoptosis of cells early after their arrival in the lung with no effect on the growth of primary tumor (35). In the present study, the comparison of global gene expression changes in response to NSC305787 and NSC668394 treatment indicated a common transcriptional regulation of genes with functional relevance to integrated stress response, suggesting a specific ezrin-mediated response in both human and mouse osteosarcoma cells (Table 4). The integrated stress response is critical to cope with and alleviate physiological stress conditions (36–38), whereas its prolonged activation can signal cell death through the pro-apoptotic DDIT3/CHOP and TRIB3 through a not well understood mechanism (39, 40). Our previous findings through a nano-LC-MS-MS-based proteomic approach and co-IP experiments demonstrated that ezrin interacts with proteins involved in translation initiation process and stress granule dynamics (21). Given that the activation of integrated stress response promotes the formation of stress granules (41), our observations suggest that ezrin may allow the cancer cells subjected to oncogenic/stress conditions to survive by integrating stress response with the regulation of gene expression at both transcriptional and translational levels.
The transcription factor ATF3 has been previously identified to be highly expressed in EC109 esophageal squamous cell carcinoma (ESCC) cells after siRNA-mediated suppression of ezrin (Gene Expression Omnibus accession no. GSE6233) (42, 43). Furthermore, consistent with this microarray data, immunohistochemical analysis of ESCC specimens and normal esophageal mucosae has demonstrated a negative correlation between ezrin levels and the level of ATF3 (42). We compared our findings with the published data obtained using ESCC cells following ezrin knockdown to further test that whether a similar transcriptional signature involving integrated stress response signaling exists in epithelial origin esophageal cancer cells. Similar to our findings, we identified that 11 genes of 16 from Table 4 including PTGS2, ATF3, TRIB3, HERPUD1, TSC22D3, GDF15, PPP1R15A, DUSP1, CDKN1A, NPC1, and ATF4 demonstrated significantly higher expression levels in ESCC cells after ezrin ablation, whereas no change was observed in the expression levels of control gene PSMD7 (Fig. 7). Although we have demonstrated that the expression of DDIT3/CHOP and DDIT4/REDD1 genes were induced after treatment of osteosarcoma cells with either ezrin inhibitors or siRNA targeting ezrin, no such change was observed in ezrin-depleted ESCC cells, suggesting that their expression can be regulated in a context-dependent manner (Fig. 7).
FIGURE 7.
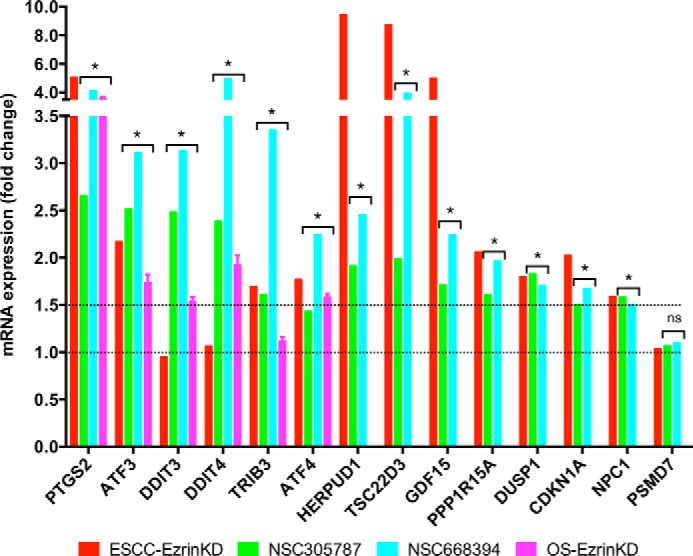
Suppression of ezrin expression and its pharmacological inhibition results in increased mRNA levels of integrated stress response signature genes in both osteosarcoma and ESCC. The published data on mRNA expression levels of integrated stress response signature genes in ESCC cells following ezrin depletion (42, 43) were compared with those generated in this work using specific molecule inhibitors of ezrin and siRNA-mediated silencing of ezrin (*, p < 0.05: for microarray analysis, statistical significance between treatment versus control group was determined using ANOVA model (n = 5 biological replicates); for RT-qPCR experiments statistical significance between treatment versus control group (n = 3 technical replicates) was determined by an unpaired Student's t test; ns, non-significant).
Although studies have linked integrated stress response to autophagy, cell survival and death, differentiation, metastasis, amino acid metabolism, angiogenesis, resistance to oxidative stress, and drug resistance, much remains to be learned about how the integrated stress response is regulated and determines the fate of cancer cells. Suppression of ezrin expression or disruption of its function by expression of a dominant-negative ezrin T567A mutant has been shown to decrease the phosphorylation of both S6K1 and 4E-BP1, indicating the involvement of mTOR/S6K1/4E-BP1 pathway of translational regulation in ezrin-related metastatic behavior (44). Furthermore, rapamycin, an inhibitor of mTORC1, led to decreased cell migration and invasion in K7M2 mouse osteosarcoma cells and significant inhibition of experimental pulmonary metastasis in mice (44). It is interesting that several studies have identified the stress response gene DDIT4/REDD1 as an essential regulator of mTOR activity, which is transcriptionally activated in response to diverse stress conditions and leads to inhibition of mTORC1 pathway (45–48). Thus, our present findings together with those reports suggest that DDIT4/REDD1-mediated mTORC1 inhibition and dependent deregulation of translational machinery caused by inhibition of ezrin function or its reduced expression may lead to inhibition of metastasis. Accordingly, we postulate that the DDIT4/REDD1 gene might serve as a pharmacodynamic marker to monitor the effect of therapeutic inhibition of ezrin function for the inhibition of metastatic phenotype in osteosarcoma. Despite the fact that both the level and pattern of gene expression may change with respect to the dose and duration of inhibitor treatment, our findings nevertheless suggest a potential use for the stress gene DDIT4/REDD1 in patients for pharmacodynamic monitoring of ezrin inhibition. Early clinical trials will have to validate these findings for the clinical applicability of DDIT4/REDD1 as a surrogate pharmacodynamic marker in PBMCs and skin.
The pharmacokinetic analysis demonstrated that plasma concentrations of NSC305787 were in the nanomolar range achieving an average steady-state level of 20.0–35.0 nm within 6–14 h of i.p. or i.v. administration (Fig. 3, C and D), which were much lower than the concentrations used in cell line experiments (usually 1–3 μm). The use of higher doses in in vivo experiments would likely achieve a stronger effect regarding the inhibition of metastasis. Hence, these findings will be helpful in guiding the dose optimization in future animal experiments as well as in clinical trials. Development of NSC305787 derivatives with improved solubility through medicinal biochemistry and evaluating different formulations could also improve the pharmacokinetic profile that can increase effective plasma concentration.
In summary, our data establish anti-ezrin compound NSC305787 as a potent anti-metastatic agent leading to inhibition of metastatic dissemination to the lung in a transgenic model of osteosarcoma and suggest that cytoplasmic ezrin, previously considered a dormant and inactive, may have important functions in regulating gene expression. A potential clinical application of ezrin inhibitors at early stages of disease before there is clinically detectable metastasis in lungs could be a promising therapy against this devastating and aggressive disease.
Author Contributions
H. Ç. and A. Ü conceived and designed the experiments. H. Ç., G. B., J. H., S.-H. H., G. T. P., M. Ö., A. A., and M. A. R. performed the experiments. H. Ç., G. B., J. H., G. T. G., G. T. P., M. H., X. L., M. Ö., A. A., M. A. R., and A. Ü analyzed the data. T. Z. M., E. J. C., and J. A. T. contributed reagents/technical expertise/materials/intellectual discussion/analysis tools. H. Ç. and A. Ü wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the Animal Models Shared Resource, Histopathology & Tissue Shared Resource and the Genomics & Epigenomics Shared Resource at the Lombardi Comprehensive Cancer Center (Georgetown University), which are supported by a grant P30CA51008 from the National Cancer Institute. We thank Dr. Chand Khanna from National Cancer Institute (NCI/NIH, Bethesda, MD) for providing human MG63.3 osteosarcoma and mouse K7M2 osteosarcoma cell lines. We thank Dr. Douglas H. Thamm (Colorado State University, Fort Collins, CO) for providing MCKOS, SKKOS and CSKOS canine osteosarcoma cell lines. We thank Dr. Stuart H. Orkin and Dr. Katherine A. Janeway (Harvard Medical School, Boston, MA) for providing mice to generate the Osx-Cre+p53fl/flpRBfl/fl transgenic mouse model of osteosarcoma and helpful discussion. We thank the Brandon Carrington Lee Foundation and Children's Cancer Foundation for continuous support of our research program.
This work was supported by Department of Defense Grant W81XWH-10-1-0137 and by funds from the Brandon Carrington Lee Foundation and the Children's Cancer Foundation (to A. Ü.). This work was also supported by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins through National Institutes of Health Grants P30CA006973 and UL1TR001079 (from the National Center for Advancing Translational Sciences, a component of the National Institutes of Health, and the National Institutes of Health Roadmap for Medical Research) and Shared Instrument Grant S10RR026824. Georgetown University has filed a patent application for using NSC305787 and related compounds in cancer therapy, where G. B., J. A. T., and A.Ü. are listed as inventors. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2.
- PBMC
- peripheral blood mononuclear cell
- qPCR
- quantitative PCR
- ANOVA
- analysis of variance
- ESCC
- esophageal squamous cell carcinoma.
References
- 1. Bretscher A., Edwards K., and Fehon R. G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599 [DOI] [PubMed] [Google Scholar]
- 2. Fehon R. G., McClatchey A. I., and Bretscher A. (2010) Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Federici C., Brambilla D., Lozupone F., Matarrese P., de Milito A., Lugini L., Iessi E., Cecchetti S., Marino M., Perdicchio M., Logozzi M., Spada M., Malorni W., and Fais S. (2009) Pleiotropic function of ezrin in human metastatic melanomas. Int. J. Cancer 124, 2804–2812 [DOI] [PubMed] [Google Scholar]
- 4. Leiphrakpam P. D., Rajput A., Mathiesen M., Agarwal E., Lazenby A. J., Are C., Brattain M. G., and Chowdhury S. (2014) Ezrin expression and cell survival regulation in colorectal cancer. Cell. Signal. 26, 868–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Köbel M., Langhammer T., Hüttelmaier S., Schmitt W. D., Kriese K., Dittmer J., Strauss H. G., Thomssen C., and Hauptmann S. (2006) Ezrin expression is related to poor prognosis in FIGO stage I endometrioid carcinomas. Mod. Pathol. 19, 581–587 [DOI] [PubMed] [Google Scholar]
- 6. Geiger K. D., Stoldt P., Schlote W., and Derouiche A. (2000) Ezrin immunoreactivity is associated with increasing malignancy of astrocytic tumors but is absent in oligodendrogliomas. Am. J. Pathol. 157, 1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weng W. H., Ahlén J., Aström K., Lui W. O., and Larsson C. (2005) Prognostic impact of immunohistochemical expression of ezrin in highly malignant soft tissue sarcomas. Clin. Cancer Res. 11, 6198–6204 [DOI] [PubMed] [Google Scholar]
- 8. Li Q., Gao H., Xu H., Wang X., Pan Y., Hao F., Qiu X., Stoecker M., Wang E., and Wang E. (2012) Expression of ezrin correlates with malignant phenotype of lung cancer, and in vitro knockdown of ezrin reverses the aggressive biological behavior of lung cancer cells. Tumour Biol. 33, 1493–1504 [DOI] [PubMed] [Google Scholar]
- 9. Chen Q. Y., Xu W., Jiao D. M., Wu L. J., Song J., Yan J., and Shi J. G. (2013) Silence of ezrin modifies migration and actin cytoskeleton rearrangements and enhances chemosensitivity of lung cancer cells in vitro. Mol. Cell. Biochem. 377, 207–218 [DOI] [PubMed] [Google Scholar]
- 10. Elliott B. E., Meens J. A., SenGupta S. K., Louvard D., and Arpin M. (2005) The membrane cytoskeletal crosslinker ezrin is required for metastasis of breast carcinoma cells. Breast Cancer Res. 7, R365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J., Wei K., Yu H., Jin D., Wang G., and Yu B. (2015) Prognostic value of ezrin in various cancers: a systematic review and updated meta-analysis. Sci. Rep. 5, 17903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khanna C., Wan X., Bose S., Cassaday R., Olomu O., Mendoza A., Yeung C., Gorlick R., Hewitt S. M., and Helman L. J. (2004) The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med. 10, 182–186 [DOI] [PubMed] [Google Scholar]
- 13. Yu Y., Khan J., Khanna C., Helman L., Meltzer P. S., and Merlino G. (2004) Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat. Med. 10, 175–181 [DOI] [PubMed] [Google Scholar]
- 14. Bruce B., Khanna G., Ren L., Landberg G., Jirström K., Powell C., Borczuk A., Keller E. T., Wojno K. J., Meltzer P., Baird K., McClatchey A., Bretscher A., Hewitt S. M., and Khanna C. (2007) Expression of the cytoskeleton linker protein ezrin in human cancers. Clin. Exp. Metastasis 24, 69–78 [DOI] [PubMed] [Google Scholar]
- 15. Kansara M., Teng M. W., Smyth M. J., and Thomas D. M. (2014) Translational biology of osteosarcoma. Nat. Rev. Cancer 14, 722–735 [DOI] [PubMed] [Google Scholar]
- 16. Isakoff M. S., Bielack S. S., Meltzer P., and Gorlick R. (2015) Osteosarcoma: current treatment and a collaborative pathway to success. J. Clin. Oncol. 33, 3029–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gill J., Ahluwalia M. K., Geller D., and Gorlick R. (2013) New targets and approaches in osteosarcoma. Pharmacol. Ther. 137, 89–99 [DOI] [PubMed] [Google Scholar]
- 18. Daw N. C., Chou A. J., Jaffe N., Rao B. N., Billups C. A., Rodriguez-Galindo C., Meyers P. A., and Huh W. W. (2015) Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br. J. Cancer 112, 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ren L., and Khanna C. (2014) Role of ezrin in osteosarcoma metastasis. Adv. Exp. Med. Biol. 804, 181–201 [DOI] [PubMed] [Google Scholar]
- 20. McClatchey A. I. (2014) ERM proteins at a glance. J. Cell Sci. 127, 3199–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Çelik H., Sajwan K. P., Selvanathan S. P., Marsh B. J., Pai A. V., Kont Y. S., Han J., Minas T. Z., Rahim S., Erkizan H. V., Toretsky J. A., and Üren A. (2015) Ezrin binds to DEAD-box RNA helicase DDX3 and regulates its function and protein level. Mol. Cell. Biol. 35, 3145–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Briggs J. W., Ren L., Nguyen R., Chakrabarti K., Cassavaugh J., Rahim S., Bulut G., Zhou M., Veenstra T. D., Chen Q., Wei J. S., Khan J., Uren A., and Khanna C. (2012) The ezrin metastatic phenotype is associated with the initiation of protein translation. Neoplasia 14, 297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bulut G., Hong S. H., Chen K., Beauchamp E. M., Rahim S., Kosturko G. W., Glasgow E., Dakshanamurthy S., Lee H. S., Daar I., Toretsky J. A., Khanna C., and Uren A. (2012) Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 31, 269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khanna C., Khan J., Nguyen P., Prehn J., Caylor J., Yeung C., Trepel J., Meltzer P., and Helman L. (2001) Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res. 61, 3750–3759 [PubMed] [Google Scholar]
- 25. Khanna C., Prehn J., Yeung C., Caylor J., Tsokos M., and Helman L. (2000) An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin. Exp. Metastasis 18, 261–271 [DOI] [PubMed] [Google Scholar]
- 26. Ren L., Mendoza A., Zhu J., Briggs J. W., Halsey C., Hong E. S., Burkett S. S., Morrow J., Lizardo M. M., Osborne T., Li S. Q., Luu H. H., Meltzer P., and Khanna C. (2015) Characterization of the metastatic phenotype of a panel of established osteosarcoma cells. Oncotarget 6, 29469–29481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walkley C. R., Qudsi R., Sankaran V. G., Perry J. A., Gostissa M., Roth S. I., Rodda S. J., Snay E., Dunning P., Fahey F. H., Alt F. W., McMahon A. P., and Orkin S. H. (2008) Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 22, 1662–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fletcher C. D. M., World Health Organization., and International Agency for Research on Cancer. (2013) WHO Classification of Tumours of Soft Tissue and Bone, 4th Ed., IARC Press, Lyon, France [Google Scholar]
- 29. Çelik H., Hong S. H., Colón-López D. D., Han J., Kont Y. S., Minas T. Z., Swift M., Paige M., Glasgow E., Toretsky J. A., Bosch J., and Üren A. (2015) Identification of novel ezrin inhibitors targeting metastatic osteosarcoma by screening open access malaria box. Mol. Cancer Ther. 14, 2497–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wan L., Pantel K., and Kang Y. (2013) Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 19, 1450–1464 [DOI] [PubMed] [Google Scholar]
- 31. Iiizumi M., Liu W., Pai S. K., Furuta E., and Watabe K. (2008) Drug development against metastasis-related genes and their pathways: a rationale for cancer therapy. Biochim. Biophys. Acta 1786, 87–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brambilla D., and Fais S. (2009) The Janus-faced role of ezrin in “linking” cells to either normal or metastatic phenotype. Int. J. Cancer 125, 2239–2245 [DOI] [PubMed] [Google Scholar]
- 33. Mehlen P., and Puisieux A. (2006) Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458 [DOI] [PubMed] [Google Scholar]
- 34. Nguyen D. X., and Massagué J. (2007) Genetic determinants of cancer metastasis. Nat. Rev. Genet. 8, 341–352 [DOI] [PubMed] [Google Scholar]
- 35. Ren L., Hong S. H., Chen Q. R., Briggs J., Cassavaugh J., Srinivasan S., Lizardo M. M., Mendoza A., Xia A. Y., Avadhani N., Khan J., and Khanna C. (2012) Dysregulation of ezrin phosphorylation prevents metastasis and alters cellular metabolism in osteosarcoma. Cancer Res. 72, 1001–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., and Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 [DOI] [PubMed] [Google Scholar]
- 37. Marciniak S. J., and Ron D. (2006) Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 86, 1133–1149 [DOI] [PubMed] [Google Scholar]
- 38. Baird T. D., and Wek R. C. (2012) Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 3, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., and Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohoka N., Yoshii S., Hattori T., Onozaki K., and Hayashi H. (2005) TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson P., and Kedersha N. (2008) Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150 [DOI] [PubMed] [Google Scholar]
- 42. Xie J. J., Xu L. Y., Xie Y. M., Zhang H. H., Cai W. J., Zhou F., Shen Z. Y., and Li E. M. (2009) Roles of ezrin in the growth and invasiveness of esophageal squamous carcinoma cells. Int. J. Cancer 124, 2549–2558 [DOI] [PubMed] [Google Scholar]
- 43. Wu B., Xie J., Du Z., Wu J., Zhang P., Xu L., and Li E. (2014) PPI network analysis of mRNA expression profile of ezrin knockdown in esophageal squamous cell carcinoma. BioMed Res. Int. 2014, 651954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wan X., Mendoza A., Khanna C., and Helman L. J. (2005) Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res. 65, 2406–2411 [DOI] [PubMed] [Google Scholar]
- 45. Brugarolas J., Lei K., Hurley R. L., Manning B. D., Reiling J. H., Hafen E., Witters L. A., Ellisen L. W., and Kaelin W. G. Jr. (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sofer A., Lei K., Johannessen C. M., and Ellisen L. W. (2005) Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 25, 5834–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ellisen L. W. (2005) Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle 4, 1500–1502 [DOI] [PubMed] [Google Scholar]
- 48. Kimball S. R., Do A. N., Kutzler L., Cavener D. R., and Jefferson L. S. (2008) Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J. Biol. Chem. 283, 3465–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.