

Abstract
Filamentation induced by cAMP (Fic) domain proteins have been shown to catalyze the transfer of the AMP moiety from ATP onto a protein target. This type of post-translational modification was recently shown to play a crucial role in pathogenicity mediated by two bacterial virulence factors. Herein we characterize a novel Fic domain protein that we identified from the human pathogen Clostridium difficile. The crystal structure shows that the protein adopts a classical all-helical Fic fold, which belongs to class II of Fic domain proteins characterized by an intrinsic N-terminal autoinhibitory α-helix. A conserved glutamate residue in the inhibitory helix motif was previously shown in other Fic domain proteins to prevent proper binding of the ATP γ-phosphate. However, here we demonstrate that both ATP binding and autoadenylylation activity of the C. difficile Fic domain protein are independent of the inhibitory motif. In support of this, the crystal structure of a mutant of this Fic protein in complex with ATP reveals that the γ-phosphate adopts a conformation unique among Fic domains that seems to override the effect of the inhibitory helix. These results provide important structural insight into the adenylylation reaction mechanism catalyzed by Fic domains. Our findings reveal the presence of a class II Fic domain protein in the human pathogen C. difficile that is not regulated by autoinhibition and challenge the current dogma that all class I-III Fic domain proteins are inhibited by the inhibitory α-helix.
Keywords: mass spectrometry (MS), post-translational modification (PTM), structural biology, x-ray crystallography, x-ray scattering, AMPylation, Clostridium difficile, Fic domain, adenylylation, differential scanning fluorimetry (DSF)
Introduction
Clostridium difficile is a Gram-positive, anaerobic, and spore-forming pathogen recognized as the leading cause of healthcare-associated diarrhea (1–3). The incidence and severity of these infections have been increasing in the past decade, and treatment is difficult and expensive. The virulence is primarily attributed to increased expression of two large toxins, TcdA and TcdB, which have both been shown to inactivate Rho GTPases in host cells by glycosylation of a threonine residue in the switch I region (4, 5). However, there are contrasting reports on the pathological importance of these toxins. In one hamster study, TcdB was found to be essential for C. difficile virulence (6). Another group found that both the A+B− and A−B+ strains are able to cause disease in hamsters (7), whereas a third study concluded that TcdA was the major determinant for virulence (8). Adding to the confusion, a highly virulent strain was recently identified where the pathogenic phenotype could not be attributed to increased toxin production. Instead, this strain was shown to hold additional laterally acquired DNA containing genes of hypothetical function (9). Therefore, increased virulence is also likely associated with accessory virulence factors, which have not yet been described.
Recently, two bacterial virulence factors, IbpA from Histophilus somni and VopS from Vibrio parahaemolyticus, were also discovered to inactivate host cell Rho GTPases by generating a phosphodiester bond with the AMP moiety on either a conserved tyrosine or threonine residue in the switch I loop (10, 11). This modification is known as adenylylation or AMPylation and is catalyzed by the filamentation induced by cAMP (Fic)3 domain. Fic domains have been identified across all domains of life and shown to be responsible for a multitude of functions varying from bacterial virulence effective in host cells to regulation of eukaryotic intracellular proteins (12). SidM/DrrA from Legionella pneumophila was shown to adenylylate Rab1 GTPases and manipulate host membrane trafficking (13). Fic-mediated adenylylation is also involved in eukaryotic cell signaling (14), and very recently the human huntingtin yeast interacting protein E (HYPE) and a homologous Drosophila Fic protein were shown to reversibly adenylylate the molecular chaperone BiP in the endoplasmic reticulum (15, 16). Additionally, the toxin-antitoxin Fic protein complex VbhTA from Bartonella schoenbuchensis was shown to control growth of other bacteria by inactivating DNA gyrase and topoisomerase IV via adenylylation (17).
In the absence of a protein target, Fic domain proteins are known to catalyze autoadenylylation where AMP is transferred to a residue in proximity of the active site (18–22). The biological significance of this self-labeling is unclear but has been proposed to either represent a reaction intermediate in the phosphoryl transfer to a target protein or function in regulation of enzyme activity (22, 23). Some Fic domains are capable of catalyzing post-translational modifications other than adenylylation. The plant pathogen AvrAC from Xanthomonas campestris suppresses the host immune response by transferring UMP from UTP to the host kinases BIK1 and receptor-interacting protein kinase (RIPK) (20), whereas IbpA has affinity for both GTP and ATP (24). In addition, the cofactor specificity is not restricted to triphosphate nucleotides as the Legionella effector AnkX utilizes CDP-choline to target Rab1 GTPase with a phosphocholine modification (25). Finally, the more distantly related bacteriophage toxin Doc inhibits bacterial translation by phosphorylating the translation elongation factor EF-Tu (26).
Fic domains are defined by the highly conserved active site Fic motif HXFX(D/E)(A/G)N(G/K)RXXR but otherwise share very little sequence identity. Also, the Fic domains can be found either as single domain proteins or as individual domains in multidomain proteins. A typical Fic domain consists of a structural core of eight α-helices described as a six-helix up-and-down bundle (α1–α5 and α′1) with helices α6-α7 lying roughly perpendicular to the bundle (18). The highly conserved and essential histidine of the Fic motif is the catalytic residue believed to deprotonate the attacking hydroxyl group of the target protein (19, 21, 27). Fic proteins also include the flap, either a β-hairpin or a loop, bridging helices α2-α3, proposed to mediate substrate binding (18, 19, 23). Recently, it was shown that adenylylation activity of some Fic domains is controlled by a conserved mechanism of ATP binding site obstruction involving an intrinsic inhibitory α-helix (αinh) containing a conserved (S/T)XXXEinh(G/N) inhibitory motif (21). The mechanism of inhibition is divided into three different classes depending on whether αinh is provided intermolecularly by an interacting antitoxin (class I) or positioned intramolecularly in the Fic protein either as an N-terminal (class II) or a C-terminal (class III) α-helix. The conserved glutamate in this inhibitory motif, Gluinh, has been found to cause a steric hindrance of proper positioning of the ATP γ-phosphate (21, 28). This autoinhibition was further supported by substitutions of Gluinh with either alanine or glycine, which resulted in increased levels of autoadenylylation as well as target adenylylation in vitro (15, 16, 21, 28).
We have previously identified a novel Fic domain protein, CdFic, from the Gram-positive human pathogen C. difficile (29). To elucidate the molecular function of CdFic, we have structurally characterized the purified protein both with and without ATP bound in the active site. We show that CdFic forms a stable dimer despite an unusually small interaction interface. Furthermore, ATP binding triggers a transition of the flap from a disordered/open to an ordered/closed conformation. Although the overall structural fold is similar to other Fic domains, we show that CdFic binds ATP as well as catalyzes autoadenylylation independently of the inhibitory motif.
Experimental Procedures
Cloning, Purification, and Crystallization
The gene (CDR20291_0569) from C. difficile strain R20291 (accession number YP_003217073) encoding CdFic and the CdFicSE/AA mutant (containing S31A and Einh35A substitutions in the inhibitory motif) with a C-terminal His6 tag was cloned, expressed, and purified as described previously (29). Codons 37 (AGT), 38 (ACT), 57 (CAT), 163 (GAT), and 200 (AGA) encoding serine, threonine, histidine, and arginine, respectively, were substituted by alanine codons (GCT) using QuikChange site-directed mutagenesis according to the manufacturer's instructions (Agilent Technologies). The Escherichia coli expression plasmids pFVS0015 and pFVS0059, encoding NmFic and the NmFicE186G mutant, respectively, from Neisseria meningitides were kindly provided by Schirmer and co-workers (21) at Biozentrum, University of Basel. After induction in E. coli BL-21 cells, NmFic and NmFicE186G were purified by nickel affinity chromatography using standard procedures. Crystallization experiments were performed using sitting drop vapor diffusion at room temperature. CdFic crystallized in a 2-μl drop with a 1:1 ratio of protein:reservoir solution containing 25% (w/v) PEG-8000, 0.2 m MgCl2, and 0.1 m Hepes, pH 7.5. CdFicSE/AA crystallized in a 4-μl drop with a 3:1 ratio of protein:reservoir solution containing 25% (w/v) PEG-3350, 0.2 m MgCl2, and 0.1 m Hepes, pH 7.5. Finally, CdFicSE/AA-ATP crystals were obtained by gradually soaking drops of CdFicSE/AA crystals with increasing amounts of a 10 mm stock solution of ATP dissolved in reservoir buffer and incubated at room temperature for 20 min. The crystals appeared within 24 h with use of freshly prepared streak seeds from initial smaller crystals using the Seed Bead (Hampton Research). Crystals were flash cooled in liquid N2 after a cryoprotective solution containing reservoir solution and 20% (v/v) glycerol was gradually added to the drop.
Data Collection, Reduction, and Structure Solution
X-ray diffraction data on CdFic crystals were collected to 3.1-Å resolution at beamline ID23 at the European Synchrotron Radiation Facility (λ = 0.979 Å, 100 K). CdFicSE/AA data were collected to 1.8-Å resolution at beamline I911-2 at the MAX-II Laboratory in Lund, Sweden (λ = 1.038 Å, 100 K). Data on the CdFicSE/AA-ATP complex were collected to 2.5-Å resolution at European Synchrotron Radiation Facility beamline ID29 (λ = 0.900 Å, 100 K). All data sets were integrated and scaled by XDS (30). After integration and scaling, all three structures were solved by molecular replacement using the CCP4i module Phaser (31) with the previously solved structure of the signaling protein BtFic from Bacteroides thetaiotaomicron (Protein Data Bank code 3CUC) having 26% sequence identity to CdFic as a search model. Initial rigid body and restrained refinement was carried out using REFMAC5 (32) before iteratively rebuilding the structure using Coot (33) and finally refining it in Phenix (34) including Translation, Liberation, Screw groups (35). For the CdFic and the CdFicSE/AA-ATP structures, NCS restraints were introduced during the refinement steps in Phenix. The atomic coordinates are deposited at the Protein Data Bank. All the structural figures were prepared using PyMOL (36). The buried surface area of the dimer was calculated using PDBePISA (37).
Small Angle X-ray Scattering (SAXS)
All SAXS synchrotron radiation data were collected at beamline I911-4 at the MAX-II Laboratory as 4 × 30-s exposures of a 20–30-μl sample, and scattering profiles were compared to detect radiation damage. The buffer consisted of 20 mm Tris, pH 7.5, 100 mm NaCl2, 5 mm MgCl2, and 5 mm 2-mercaptoethanol. Data were collected at 0.91-Å wavelength at 10 °C using a hybrid pixel Pilatus 1M detector (Dectris). To detect concentration-dependent interparticle effects, measurements were collected at multiple protein concentrations in the range of 0.65–5 mg/ml. The data collected at 2.6 mg/ml were used for further analysis as concentrations above this exhibited minor interparticle effects. Background buffer scattering was subtracted using PRIMUS (38), part of the ATSAS package (39). Pairwise distance distribution function P(r) and the maximum particle dimension Dmax were computed using GNOM (40). The Porod volume was calculated using ATSAS AUTOPOROD (41) and used for molecular weight estimation. Fits between the theoretical SAXS scattering of the crystallographic models and the experimental SAXS data were analyzed using CRYSOL (42) with data below the linear Guinier region removed. The agreement between the theoretical scattering data that were back-calculated from the indicated atomic resolution structures and those of the experimental data were evaluated by means of the χ2 values (42) as well as by visual inspection of the curves.
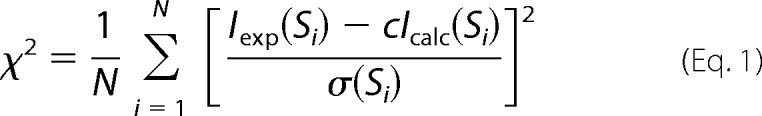 |
where N is the number of experimental points, σ(si) is the experimental errors, and the scaling factor c is as follows.
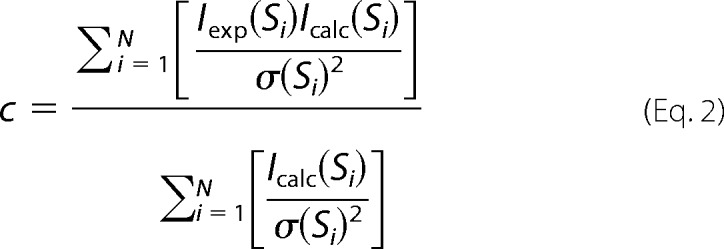 |
Ab initio shape envelope modeling was performed using DAMMIF (43) with both P1 and P2 symmetry constraints. 20 DAMMIF runs were computed, and DAMAVER (44) was used to align and compare the resulting models. The model with the lowest normalized spatial discrepancy was used as a representative, and an envelope was generated based on the DAMMIF bead model using Situs pdb2vol (45). A model for the flexible flap was computed using ensemble optimization method (EOM) (46, 47) and was manually docked into the envelope using USCF Chimera (48).
Differential Scanning Fluorimetry (DSF)
The experiments were performed as described previously (49). A total reaction volume of 25 μl containing either 2 μm CdFic or CdFicSE/AA was mixed with ligand at concentrations ranging from 0 to 17 mm (at ATP 99%, ADP 95%, PPi 99%, GTP 95%, CTP 95%, or UTP 96% purity; Sigma) in a buffer consisting of 50 mm Hepes, pH 7.5, 100 mm NaCl, 5 mm MgCl2, and 5 mm 2-mercaptoethanol and incubated for 20 min at room temperature in 96-well real time PCR plates (triplicates). The ligand concentrations were 7.5, 15, 30, 60, 125, 250, and 500 μm and 1, 2, 4, 6, 8, 10, 12, 15, and 17 mm. SYPRO Orange (Sigma) was added to each well at 5× concentration, the plate was exposed to a temperature gradient ranging from 4 to 95 °C in a 7500 Real Time PCR System machine (Life Technologies), and the fluorescence of the dye was measured at an emission of 569 nm. The gathered data were exported and plotted in GraphPad Prism for analysis.
In Vitro Autoadenylylation
Autoadenylylation activity was assessed in an assay using 200 ng of purified recombinant protein (CdFic, CdFicSE/AA, CdFicS37A, CdFicT38A, CdFicH57A, CdFicH163A, or CdFicR200A) in 30 μl of reaction buffer containing phosphate-buffered saline, pH 7.4, 4 mm EGTA, 5 mm MgCl2, and either 10 μCi of [α-32P]ATP or 10 μCi of [α-32P]CTP. Reactions were incubated at 30 °C for 1 h and stopped by adding 30 μl of 2× Laemmli sample buffer (Sigma), and 20 μl was resolved by SDS-PAGE. Dried gels were exposed to Bio-Rad phosphorimaging screen-K, scanned on a Typhoon 9410 (GE Healthcare), and quantified using the ImageQuant TL program (GE Healthcare). Short exposures are 15 min; long exposures are 2 h. For comparing CdFic and CdFicSE/AA activity with NmFic and NmFicE186G, purified protein was used in the amounts specified in Fig. 6 legend.
FIGURE 6.

Autoadenylylation activity and the acceptor site. A, in vitro autoadenylylation assay. Wild type (WT) CdFic (lane 1), CdFicSE/AA inhibitory motif mutant (lane 2), CdFicH57A dimerization interface mutant (lane 3), and CdFicH163A Fic motif active site mutant as a negative control (lane 4). Proteins were Coomassie-stained (lower panel), dried, exposed on a phosphor screen (top panel), and quantified (right panel). The indicated error bars represent the standard deviation from four independent experiments. B, in vitro autoadenylylation assay as in A but using [α-32P]CTP and longer exposure (exp.) time. C, in vitro autoadenylylation assay with CdFicR200A (lane 2). CdFic is shown (lane 1) along with the CdFicH163A negative control (lane 3). Quantification is shown to the right with the indicated error bars representing the standard deviation from three independent experiments. D, the activity of CdFic and CdFicSE/AA in comparison with the activity of NmFic and NmFicE186G from N. meningitidis. Upper image, equal amounts of NmFic and NmFicE186G (50 ng) exposed for 30 min. Lower image, NmFic (65 ng), NmFicE186G (0.1 ng), and both CdFic and CdFicSE/AA (195 ng) exposed for 43 h. E, in vitro autoadenylylation assay with CdFicS37A and CdFicT38A using [α-32P]ATP. F, structurally based multiple sequence alignment of CdFic, BtFic (B. thetaiotaomicron), HYPE (H. sapiens), VbhA (B. schoenbuchensis), and NmFic. The mapped acceptor sites are colored with green boxes, and the inhibitory residues Glu/Ser of the inhibitory motif are colored in red. The αinh as well as the α′1 helix are both shown in dark yellow color as part of the N-terminal extension to the CdFic core. The fractional conservation is represented below the alignment using bars and sequence conservation with sequence logo. G, superposition of CdFic and NmFic acceptor sites with CdFic colored in dark yellow according to E and NmFic colored in gray. H, the autoadenylylation acceptor site Thr-38 interactions with the dimerization loop colored in black as well as the distance to the ATP α-phosphate shown with a gray dashed line. Hydrogen bonds are shown with yellow dashed lines.
Mass Spectrometry
CdFicSE/AA (4 μg) was incubated with either ATP or the isotope-labeled [13C10,15N5]adenosine 5′-triphosphate derivative at 10 mm (Sigma; purity, >97% and >98%, respectively) in a buffer consisting of 20 mm Tris, pH 7.5, 100 mm NaCl2, 5 mm MgCl2, and 5 mm 2-mercaptoethanol for 1 h and separated by SDS-PAGE. The bands were excised from the gel, washed, in-gel-reduced, S-alkylated with iodoacetamide, and digested with trypsin as described (50). The formed peptides were extracted, dried in a vacuum centrifuge, redissolved in 5% formic acid, and subjected to a desalting/concentration step using StageTips as described (51). Desalted peptides were loaded on an Acclaim PepMap C18 precolumn (300 × 5 × 5 mm; Dionex) and separated using an Acclaim PepMap100 C18 analytical column (75 × 150 × 3 μm, 3-μm particle size; Dionex) by a 75-min gradient controlled by a Dionex Ultimate 3000 system connected to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray source (Proxeon, Odense, Denmark). The flow rate was 200 nl/min; the mobile phases consisted of solvent A (2% (v/v) acetonitrile and 0.1% formic acid) and solvent B (95% (v/v) acetonitrile and 0.1% (v/v) formic acid). The gradient went from 0 to 45% solvent B in 65 min followed by 10 min with 100% solvent B; then data acquisition was stopped, and the column was re-equilibrated with solvent A. MS data were acquired recording full scan spectra (mass/charge (m/z), 250–1,800) in the Orbitrap with 60,000 resolution at 400 m/z. MS/MS data were recorded in parallel in a data-dependent mode, fragmenting the five most abundant ions (charge state, +2 or higher) by collision-induced dissociation in the LTQ ion trap at 35% collision energy. MS/MS spectra were recorded using dynamic exclusion (30 s) to minimize repeated fragmentation of the same peptides and analyzed using MaxQuant version 1.4.1.2 (52) for peptide and protein identification using the built-in Andromeda search engine (53). The following settings were used: His6-CdFicSE/AA sequence (29) with search against common contaminant sequences enabled; variable modifications adenylylation (STY; +329.05 Da), heavy adenylylation (STY; +344.07 Da), oxidation (Met), and acetyl (protein N-terminal); fixed modification, carbamidomethyl (Cys); labels, none; peptide false discovery rate, 1%; protein false discovery rate, 1%; match between runs, 1 min; keep low scoring version of identified peptides, on; all other settings, left at the default settings. After automatic identification of the adenylylated peptides by MaxQuant, the corresponding MS/MS spectra were subjected to manual inspection, and diagnostic ions of 348.1 (AMP) and 363.2 Da ([13C10,15N5]AMP) were observed as described previously (54) and further confirmed the adenylylation of the IAGSTFTTEALALLLDK peptide.
Results
Crystal Structure of CdFic
To investigate the structure and function of the newly identified Fic protein from C. difficile (29), we solved the x-ray crystal structures of CdFic as well as the αinh mutant CdFicSE/AA. This double mutant was chosen hoping that autoinhibition would be released and that αinh would dissociate from the nucleotide binding pocket as seen previously in the NmFicSE/AA mutant structure (21) and thus result in a more active enzyme. Moreover, by soaking CdFicSE/AA crystals with ATP, we also solved the structure of CdFicSE/AA in complex with ATP and Mg2+. The maximum resolution of the diffraction data were 3.1 Å for CdFic, 1.8 Å for CdFicSE/AA, and 2.5 Å for the CdFicSE/AA-ATP complex with final Rfree values of 28.6, 21.8, and 23.3%, respectively. The three structures were solved by molecular replacement using the homologous BtFic from B. thetaiotaomicron as a search model (Protein Data Bank code 3CUC). According to PROCHECK (55), Ramachandran plots for the three structures show that all residues are within the allowed region except for a single outlying residue in the CdFicSE/AA-ATP structure. The details of the data collection and refinement are provided in Table 1. The CdFicSE/AA structure contains two molecules in the asymmetric unit (AU) and belongs to space group P212121, whereas both the CdFic (space group C2221) and the CdFicSE/AA-ATP (space group C2) structures contain four molecules in the AU. In the CdFicSE/AA structure, the ATP binding sites of both molecules in the AU are blocked by a glutamate in the C terminus of a symmetry-related molecule. Luckily, the crystallization condition for CdFicSE/AA also produced another crystal form containing four molecules in the AU of which two of the molecules have an unoccupied binding site. This was the only crystal form in which we were able to soak ATP into the binding site. Similarly, only two of the four molecules in the AU of the CdFic crystal had unoccupied ATP binding sites, but attempts to soak ATP into the binding site of these crystals were unsuccessful.
TABLE 1.
Crystallographic data and refinement statistics
mol, molecules; r.m.s.d., root mean square deviation; ESRF, European Synchrotron Radiation Facility.
| CdFicSE/AA | CdFicSE/AA-ATP | CdFic | |
|---|---|---|---|
| Data collection | |||
| Radiation source | MAX-II I911-2 | ESRF ID29 | ESRF ID23 |
| Wavelength (Å) | 1.038 | 0.900 | 0.873 |
| Data processing | |||
| Space group | P212121 | C2 | C2221 |
| Protein mol/AU | 2 | 4 | 4 |
| Cell dimensions | a = 52.4 Å, b = 67.3 Å, c = 139.6 Å, α = β = γ = 90° | a = 158.1 Å, b = 60.1 Å, c = 124.8 Å, α = γ = 90°, β = 118.6° | a = 57.4, b = 157.4, c = 262.7, α = β = γ = 90° |
| Resolution (Å) | 20.0–1.8 (1.9–1.8)a | 20.0–2.5 (2.6–2.5) | 20.0–3.1 (3.2–3.1) |
| Completeness (%) | 98.6 (97.2) | 99.7 (100) | 99.9 (99.9) |
| CC1/2b | 99.9 (77.9) | 99.9 (57.9) | 99.6 (82.1) |
| Mean I/σ | 19.0 (2.3) | 16.7 (1.3) | 9.7 (1.7) |
| Multiplicity | 6.6 (6.5) | 6.9 (7.2) | 8.0 (8.4) |
| Rmeasc | 7.7 (95.7) | 8.2 (155.2) | 17.3 (114.0) |
| Refinement | |||
| Resolution (Å) | 19.1–1.8 | 19.9–2.5 | 19.9–3.1 |
| Reflectionswork | 45,953 | 35,839 | 20,454 |
| Reflectionstest | 1,378 | 1,076 | 1,026 |
| Rworkd/Rfreee | 18.4/21.8 | 20.48/23.3 | 23.3/28.6 |
| Protein atoms | 3,470 | 6,743 | 6,561 |
| Water atoms | 216 | 14 | |
| Glycerol/PO4/Hepes atoms | 50 | ||
| ATP/Mg2+ atoms | 32 | ||
| Overall B-factor (Å2) | 26.3 | 67.3 | 51.9 |
| r.m.s.d. bond lengths (Å) | 0.008 | 0.010 | 0.003 |
| r.m.s.d. bond angles (°) | 0.999 | 0.921 | 0.779 |
| Ramachandran statistics (%) | |||
| Favored | 99.0 | 98.4 | 98.3 |
| Allowed | 1.0 | 1.5 | 1.7 |
| Outliers | 0 | 0.1 | 0 |
| Protein Data Bank code | 4X2C | 4X2D | 4X2E |
a Values in parenthesis are for the highest resolution shell.
b Percentage of correlation between intensities from random half-data sets (65).
c Rmeas = Σhkl(N(hkl)/[N(hkl) − 1])1/2Σ|Ii(hkl) − 〈/I(hkl)〉|ΣhklΣIi(hkl). Redundancy-independent R factor calculated on intensities (66).
d R = Σ‖Fobs| − |Fcalc‖Σ|Fobs| where |Fobs| and |Fcalc| are observed and calculated structure factor amplitudes, respectively.
e The Rfree value was calculated with a random 3 or 5% subset of all reflections excluded from refinement.
The overall conformation of the three structures is highly isomorphous with a root mean square deviation of 0.49 Å between the A chains of CdFic and CdFicSE/AA as calculated by the SSM Superpose tool in Coot using 197 Cα atoms. The CdFic structure reveals a classical α-helical Fic fold with stacking of three consecutive helix-loop-helix elements (Fig. 1, A and B). In the Fic core element, the Fic motif (residues 163–174) bridges helices α4-α5, whereas the flap bridges helices α2-α3. CdFic contains three additional N-terminal helices as an extension to the Fic core of which the middle helix adopts a location deeply embedded in the Fic core that is analogous to an N-terminal αinh (Fig. 1, A and B). This N-terminal αinh places CdFic among the class II Fic domain proteins (21) and harbors the proposed inhibitory motif residues Ser-31/Gluinh-35, which are situated in the C terminus of the αinh helix. Structural alignment of CdFic with BtFic from B. thetaiotaomicron, the human HYPE, the second Fic domain of Histophilus somni, IbpAFic2, and HpFic from Helicobacter pylori reveals a common arrangement of the Fic core domain consisting of helices α1–α7 (Fig. 1C).
FIGURE 1.

CdFic crystal structures. A, the crystal structure of CdFic. The Fic domain core is shown in green, the N-terminal (N-term.) extension including the αinh is in dark yellow, the C-terminal (C-term.) extension is in violet, the conserved Fic motif loop is in blue, the dimerization loop is in black, and the position of the disordered flap is schematically shown with red spheres. Secondary structures in all other panels are colored accordingly. The αinh residues Ser-31 and Gluinh-35 are represented with sticks and highlighted by arrows. B, topological diagram based on the CdFic structure. C, structural superposition of Fic crystal structures represented with secondary structure and shown as a schematic diagram with each box representing a helix (B. thetaiotaomicron; Protein Data Bank code 3CUC), HYPE (Homo sapiens; Protein Data Bank code 4U07), IbpAFic2 (H. somni; Protein Data Bank code 4ITR), and HpFic (H. pylori; Protein Data Bank code 2F6S). The position of αinh is labeled inh, and TPR is the tetratricopeptide repeat motif of HYPE. D, crystal structure of CdFicSE/AA. The inhibitory motif substitutions S31A and Einh35A are represented with sticks and highlighted by arrows. The electron density for αinh is shown in gray. E, the interaction surface between the αinh helix (dark yellow) and residues in the CdFic core (green) with the αinh surface envelope colored in gray. F, crystal structure of the CdFicSE/AA-ATP complex shown in two orientations related by a 90° rotation about the horizontal axis. ATP is represented with black, blue, red, and yellow sticks, and the ordered/closed flap is colored in red.
Interestingly, in the CdFicSE/AA structure, there is well defined electron density for the αinh helix, and its position is equivalent to the corresponding helix in CdFic (Fig. 1D). Hence, replacing these two residues in the inhibitory motif does not destabilize the αinh helix as seen for the corresponding SEinh/AA mutant of the class III NmFic protein (21). This is not surprising because the interaction surface between αinh and the CdFic core consists mainly of hydrophobic residues (Fig. 1E), which likely stabilize the core of the protein. In support of this, attempts to express and purify a CdFic(Δ1–40) deletion mutant resulted in a highly insoluble protein, indicating that removal of the first two α-helices including αinh dramatically alters the properties of the protein.
In both the CdFic and CdFicSE/AA structures, the major part of the flap is missing from the electron density maps and is supposedly flexible (Fig. 1, A and D). However, in the ATP-bound structure, the entire flap is ordered and almost completely covers the adenine base, leaving only the three phosphates solvent-exposed (Fig. 1F). Only molecule A of the four chains in the AU contains the ATP cofactor in the binding site as symmetry-related molecules block the binding site of molecules C and D. Molecule B does have an unobstructed binding site; however, movements of the flap are possibly prevented by a symmetry-related molecule D, and loop closure by the flap is likely necessary to lock the ATP in the binding site. Conversely, the binding site of molecule A is surrounded by enough space to allow loop closure after ATP binding without disrupting any crystal contacts. Of all 10 molecules within the AUs of the three different CdFic crystal structures, the entire flap is only visible in the molecule with ATP bound. This is unlikely to be a coincidence and strongly suggests that ATP binding triggers a transformation of the flexible flap from a disordered/open to an ordered/closed conformation. As the flaps in other Fic domain proteins structures without ATP are either well defined or only partially disordered (19, 23, 56), the flap in CdFic seems particularly flexible without ATP.
Non-obstructed ATP Conformation
The CdFic structure contains the classical Fic nucleotide binding features consisting of a hydrophobic adenosine binding pocket, the ribose-coordinating arginine, and the GNG anion hole of the Fic motif. In the nucleotide binding pocket of the CdFicSE/AA-ATP structure, there is well defined electron density for an intact ATP molecule coordinating a Mg2+ ion (Fig. 2A). The adenosine is locked into a pocket formed among helix α4, α6, and the flap (Fig. 1F) where Val-122, Ile-124, Leu-161, Leu-203, and Ile-117 form a hydrophobic patch. Only a single hydrogen bond is formed between the surrounding residues (Asn-204) and the adenine base (Fig. 2B). As in other Fic Gluinh/Gly mutant structures in complex with ATP, the ribose is primarily coordinated by hydrogen bonds to the conserved Fic motif Arg-174 and Tyr-199, which help tether the middle of the nucleotide (Fig. 2B) (21, 56). Also, the GNG anion hole and the catalytic Fic motif histidine are situated similarly to other Fic domains. Hence, the Fic motif in the ATP binding pocket is highly conserved and shows no structural features deviating from other ATP-binding Fic domain proteins. Despite this, the ATP phosphates in the CdFicSE/AA-ATP structure are positioned significantly differently in comparison with those of other Fic Gluinh/Gly mutant structures in complex with ATP or ATP analogues (Fig. 2C). In the CdFicSE/AA-ATP structure, the γ-phosphate forms an elaborate network of hydrogen bonds to both GNG backbone amide nitrogens and to Arg-171 of the Fic motif (Fig. 2B). To our knowledge, only one other Fic structure, the VbhT-VbhA-ATP complex (Protein Data Bank code 3ZC7), has a γ-phosphate positioned similarly within the GNG anion hole. However, the substitution of Gluinh with a glycine in VbhA allows the γ-phosphate to move out of the anion hole and into a position overlapping with the Gluinh side chain (Protein Data Bank code 3ZCB). Instead, in all other Einh/G Fic mutant structures in complex with ATP or ATP analogues published to date, the anion hole is responsible for coordinating the α- and β-phosphates with the asparagine in the Fic motif, forming a salt bridge to the α-phosphate (Fig. 2D) (21, 28, 56). In the CdFicSE/AA-ATP structure, however, the α- and β-phosphates are pulled away from the anion hole toward the opposite side of the active side cleft, and the α-phosphate instead forms a salt bridge to Arg-200 positioned in helix α6 of the Fic core (Fig. 2E).
FIGURE 2.

The ATP binding site. A, the CdFicSE/AA-ATP binding pocket. An unbiased Fo − Fc electron density omit map around ATP is contoured at 4 σ and shown in gray. An Fo − Fc electron density omit map around Mg2+ ion is shown in green and contoured at 6.5 σ to emphasize the strong Mg2+ peak. Mg2+ is shown in purple, and the Fic motif and the flap are shown in blue and red, respectively. B, schematic diagram of the CdFicSE/AA-ATP coordination as determined by LIGPLOT (63). C, superposition of ATP from CdFicSE/AA-ATP crystal structure and the structures of HYPEE234G-ATP (H. sapiens; Protein Data Bank code 4U07), NmFicE186G-AMPPNP (N. meningitides; Protein Data Bank code 3ZLM), SoFicE73G-AMPPNP (Shewanella oneidensis; Protein Data Bank code 3ZEC), and BsFic (VbhT-VbhAE24G-ATP) (B. schoenbuchensis; Protein Data Bank code 3ZCB). D, the ATP binding mode of Fic domain structures from C not including CdFicSE/AA. E, the ATP binding mode of CdFicSE/AA highlighting the salt bridge between Arg-200 and α-phosphate shown in identical orientation as D. F, structurally based multiple sequence alignment of Fic domains. Fic domains with the indicated Protein Data Bank accession codes were aligned using the program Strap (64), and the alignment was calculated based on the structural superposition and visualized using the CLC Main Workbench 7.5.1. (Qiagen Bioinformatics). The conservation below 30% identity is not colored, but a progressively darker red color represents conservation in the 30–100% range. The position of Arg-200 residue is shown with green boxes. The fractional conservation is represented below the alignment using bars and sequence conservation with sequence logo. G, superposition of the inhibitory motif residues Ser-31/Gluinh-35 in CdFic (dark yellow) with the CdFicSE/AA-ATP structure (gray). The ATP from the CdFicSE/AA-ATP structure is colored in gray, and a water molecule from the CdFicSE/AA-ATP structure is shown in red color interacting with both the γ-phosphate and the Fic motif Arg-174 and is in an overlapping position with the Gluinh-35 side chain of the CdFic structure.
Structural alignment of known Fic structures shows that CdFic is the only Fic protein containing an arginine at this position as other Fic domain structures predominantly have hydrophobic residues at the structurally equivalent position (Fig. 2, D and F). The Arg-200 residue might therefore explain the unique orientation of the ATP phosphates in CdFic. The carboxylic side chain of Asp-167 in the Fic motif of CdFicSE/AA helps coordinate a Mg2+ ion together with the ATP β- and γ-phosphates (Fig. 2, B and E). This type of coordination is also in contrast to other Fic-ATP structures where the Mg2+ ion is instead situated between the α- and β-phosphates (Fig. 2, C and D). The coordinated Mg2+ ion in CdFic, however, suggests that ATP binds in an adenylylation-competent conformation. Furthermore, this arrangement of ATP phosphates in the CdFicSE/AA-ATP structure implies that Gluinh would not obstruct proper positioning of the γ-phosphate in wild type CdFic. A superposition of the CdFic and CdFicSE/AA-ATP structures shows that Gluinh in CdFic is positioned immediately below the γ-phosphate and that the position of the glutamate side chain is replaced by a water molecule in the CdFicSE/AA-ATP structure that forms hydrogen bonds to both the γ-phosphate and Arg-174 (Fig. 2G).
CdFic Dimerization Interface and Active Site Closure
In all three CdFic structures, two molecules form the same type of dimer (Fig. 3A). The interface is formed by a short loop situated between helices α1 and α′1 (residues 51–59) in the N-terminal extension to the Fic core (Fig. 1B) and buries only ∼6% (642 Å2) of the total surface area (10,755 Å2). However, as we have shown previously by size exclusion chromatography (SEC), the dimer is very stable and elutes as a single monodisperse peak (29). There are six direct hydrogen bond interactions involving only backbone amide nitrogens and carbonyls in the loop, and three water molecules mediate contact between the two monomers (Fig. 3A). The loop conformation is structured mainly by residues Arg-56, His-57, and Asp-61, which tether the loop backbone through hydrogen bond interactions with Ser-37 and Thr-38 in the αinh-α′1 loop. The conformation, but not the sequence, of this minimal dimerization interface is similar to the homologous BtFic (Protein Data Bank code 3CUC) and the human HYPE (56), both likewise situated on a small loop bridging helices α1-α′1 (Fig. 1C). The α1-α′1 loop in BtFic is also stabilized and tethered by hydrogen bonds to residues in the αinh-α′1 loop but also shows no sequence conservation to the structurally equivalent residues in CdFic (Fig. 3A). Interestingly, although CdFic and BtFic share a highly similar relative orientation of the two monomers in the dimer, one monomer of the HYPE dimer is rotated by ∼50° relative to the corresponding CdFic monomer as calculated by DynDom3D (57) (Fig. 3B). This rotation could either be a consequence of the HYPE crystal contacts or a result of the additional dimerization interface formed between residues in the α2 helix of the two HYPE monomers (56).
FIGURE 3.

The dimerization interface. A, superposition of CdFic (monomers are depicted in blue and green) with BtFic (B. thetaiotaomicron; Protein Data Bank code 3CUC) (light brown). The structures are shown in two orientations related by a 90° rotation about the horizontal axis. The left inset shows a close-up view of the CdFic interface with the surface envelope of one monomer shown in green color. The right inset shows the superposition of the CdFic and BtFic interfaces with BtFic shown in light brown color. Only hydrogen bonds from BtFic are shown for clarity using black dashed lines. Both interfaces are symmetric; however, in both insets not all symmetry-related hydrogen bonds are shown for clarity. B, superposition of CdFic dimer (blue and green) against HYPE dimer (H. sapiens; Protein Data Bank code 4U0S) (dark brown). The related but considerably twisted interface of HYPE results in a large overall rotation between its monomers compared with those of CdFic as calculated by DynDom (57).
To corroborate our previous SEC results, we probed the oligomeric state of CdFic in solution by SAXS on purified CdFicSE/AA (Fig. 4A). The pairwise distance distribution function P(r) strongly suggests that CdFic is a dimer in solution as the longest particle dimension (Dmax) is 106.4 Å and the longest dimension of the monomer in the crystals is ∼50 Å. The CdFicSE/AA scattering profile was fitted and compared with the theoretical scattering profile of either the CdFicSE/AA dimer or monomer using CRYSOL (42) (Fig. 4A). The visual inspection shows considerably better fit to the dimer than the monomer, which is also supported by the calculated χ values of χ2 = 3.20 and χ2 = 13.07, respectively. However, the fit includes significant deviations that are likely explained by the presence of the 24-residue flap in each of the monomer units. These two loops of the dimer contribute to scattering of x-rays in solution but are flexible and not visible in the CdFic/CdFicSE/AA crystal structures without ATP (Fig. 1, A and D). These deviations were not resolved by fitting the CdFicSE/AA-ATP dimer containing a well defined and closed flap (χ2 = 3.92), suggesting that the flap does not adopt a closed conformation in solution (Fig. 4B). To determine the solution conformation of the flexible flap, we made use of recent developments that allow for rigid body modeling of known crystal structures to be combined with shape reconstruction of missing loops (58). A loop model was initially computed using Coral (58), resulting in an improved fit (χ2 = 1.78) but still containing significant deviations (data not shown). Subsequently, we used EOM, which enables analysis of flexible protein structures (46, 47). A pool of 10,000 independent models based on the flap sequence was initially generated, and a genetic algorithm was performed comparing the averaged theoretical scattering intensity from an ensemble of conformations with the experimental data. Two models were obtained by the ensemble, and CRYSOL (42) was used to compute a fit to the experimental SAXS data. Visual inspection reveals that the inclusion of the disordered flap modeled by EOM considerably improves the fit over the entire scattering angle range below 0.3 Å−1 (χ2 = 1.13) (Fig. 4B).
FIGURE 4.

SAXS analysis. A, CdFicSE/AA SAXS scattering curve is shown in gray as relative log(intensity) versus inverse scattering angle. The Guinier plot (log I(q) versus q2) and a table with general SAXS parameters are both shown as insets. MW (monomer) is the mass calculated from the sequence, whereas MW (SAXS) is the mass estimated using the Porod volume. CRYSOL (42) fits between experimental SAXS data and the theoretical/calculated scattering profile are shown as colored curves. The fit to the CdFicSE/AA monomer is shown as a blue curve, whereas the fit to the CdFicSE/AA dimer is shown as a green curve. The resulting χ values (χ2) are shown next to the schematic models. B, the same experimental CdFicSE/AA SAXS scattering curve as in A is shown in gray color. The CRYSOL fit to the theoretical/calculated scattering profile of the CdFicSE/AA-ATP dimer structure including an ordered/closed flap is shown as a black curve. The CRYSOL fit to the CdFic dimer structure including the EOM (46, 47)-generated model of the flap is shown as a red curve. C, the three-dimensional shape reconstruction of the ab initio envelope calculated by DAMMIF using P2 symmetry is shown in transparent gray color. The CdFicSE/AA dimer structure with the two monomers shown in green and blue, respectively, is docked in the SAXS envelope. The EOM-generated reconstructions of the flexible flap are colored in red. D, the CdFicSE/AA-ATP crystal structure representing the closed/ordered conformation (red solid line) is aligned against the EOM-computed flap representing the open/flexible conformation (red spheres) with only one of the two EOM models shown for clarity. Superpositions of the BtFic (B. thetaiotaomicron) and the HYPE (H. sapiens) flaps are shown in cyan and yellow, respectively.
Next, we performed three-dimensional ab initio shape reconstructions of the CdFicSE/AA dimer using DAMMIF (43). As models generated with P1 symmetry strongly suggest the presence of an extended two-domain particle, a P2 symmetry constraint was introduced. The mean normalized spatial discrepancy was 0.861, and the model with the lowest discrepancy (normalized spatial discrepancy, 0.734) was used as a representative (χ2 = 1.008). Docking of the CdFicSE/AA crystal structure lacking the flap into the low resolution ab initio envelope shows that the envelope contains a larger volume and that this additional volume is well occupied by the EOM flap reconstructions representing the open flap conformation (Fig. 4C). Superposition of the solution conformation of the flap as determined by SAXS EOM analysis and the ATP-bound CdFicSE/AA crystal structure reveals that the flap adopts an open/flexible conformation in the free state and undergoes considerable transition of ∼27 Å into a closed/ordered conformation upon ATP binding (Fig. 4D).
Cofactor Binding
To characterize ligand binding and specificity of CdFic and CdFicSE/AA, we used DSF (49, 59). By incubating 2 μm CdFic with ATP concentrations ranging between 0 and 17 mm, the melting temperature (Tm) showed a maximum increase of stability (ΔTm) of 3.9 °C (Fig. 5A). The Tm values determined at each ATP concentration were plotted versus the ATP concentration, resulting in a sigmoidal curve with a midpoint value around 2 mm and reaching a plateau at ∼10 mm ATP (Fig. 5B). We also performed identical assays using CdFic with increasing concentrations of ADP, PPi, GTP, CTP, or UTP (Fig. 5, B and C). The largest increase in stability is obtained with ATP, ADP, and PPi followed by GTP, which triggers an intermediate stability shift, whereas CTP/UTP pyrimidines do not significantly induce CdFic stability. Hence, the PPi moiety, which alone is able to induce stability significantly, is not sufficient for pyrimidine nucleotides to stabilize CdFic. Finally, to assess the importance of the inhibitory motif residues Ser-31 and Gluinh-35 on ligand binding, we performed identical assays using the CdFicSE/AA mutant (Fig. 5, D and E). Interestingly, the CdFicSE/AA nucleotide binding profile is almost identical to that of CdFic (Fig. 5F).
FIGURE 5.

Cofactor binding. A, CdFic DSF melting curves showing fraction of unfolded protein versus temperature, averaged from triplicate measurements. Each curve represents a measurement at various ATP concentrations ranging from 0 to 17 mm. For clarity, four different melting curves are highlighted: control (CdFic alone) in gray; 2 mm in cyan; 4 mm in purple, and 17 mm in red. Melting curves for the other ligand concentrations are shown in black. The black dashed line at 0.5 unfolded fraction highlights the points where Tm is extrapolated. The gray and red dashed lines represent the Tm determination for the CdFic control lacking the ligand and the highest ATP concentration (17 mm), respectively. B, the sigmoidal curves represent the plot of the CdFic Tm values as a function of indicated ligand concentration. The vertical lines represent the standard deviation from triplicate measurements. C, DSF melting curve of CdFic averaged from triplicate measurements at 17 mm concentration of ATP, ADP, PPi, GTP, CTP, and UTP. D and E, identical assay and color scheme as for B and C but using the CdFicSE/AA mutant. F, ΔTm values for both CdFic and CdFicSE/AA plotted as bars with ligands colored according to B–E. Error bars represent S.E.
CdFic Autoadenylylation
In the absence of a known physiological protein target, autoadenylylation is a useful read-out to assess adenylylation activity of Fic domain proteins (28). To determine whether CdFic is able to not only bind ATP but also catalyze AMP transfer, we incubated the purified protein with radioactively labeled [α-32P]ATP before visualizing labeled gel bands by phosphorimaging. We found that, despite the presence of an intact proposed inhibitory motif, CdFic is able to catalyze autoadenylylation in vitro (Fig. 6A). We also tested for self-labeling using the CdFicSE/AA inhibitory mutant and found that the activity is essentially identical to that of the wild type protein and therefore independent of the inhibitory motif in accordance with the DSF assay and the CdFicSE/AA-ATP crystal structure. As a negative control, replacement of the highly conserved catalytic Fic motif His-163 with an alanine shows no sign of autoadenylylation activity, confirming that activity is driven by the Fic motif. Also, to determine whether dimerization is important for the activity, we replaced His-57 involved in stabilizing the dimerization interface (Fig. 3A) with alanine, expecting that this would disrupt dimerization. However, SEC revealed that H57A elutes as a single peak corresponding to a dimer as described previously for CdFic and CdFicSE/AA (29). Surprisingly, although dimerization of this mutant is not disrupted, we found that the H57A point mutation enhances self-labeling dramatically (Fig. 6A). Conversely, four different point mutations in the dimerization interface of HYPE, of which only one resulted in a monomeric form, all had a negative impact on autoadenylylation activity (56). We also tested for modification by CMP of CdFic, CdFicSE/AA, or CdFicH57A using [α-32P]CTP, but in accordance with the thermal shift assay we found no detectable self-labeling higher than that of the background even after longer exposure, again using the H163A mutant as a negative control (Fig. 6B). Hence, in agreement with the thermal shift assay as well as the crystal structures, ATP is the preferred substrate over CTP, and CdFic inhibitory motif residue Gluinh does not obstruct either nucleotide binding or autoadenylylation activity. We also assayed the importance of the Arg-200 residue for activity as this arginine forms a salt bridge to the α-phosphate of ATP (Fig. 2E) and is found only in CdFic among the Fic domains structurally determined to date (Fig. 2, D and F). Indeed, we found that the R200A point mutation results in a more than 6-fold reduction in autoadenylylation activity, highlighting its importance for the reaction mechanism of CdFic (Fig. 6C). This result further validates the non-obstructed ATP conformation seen in the CdFicSE/AA-ATP crystal structure. However, although autoadenylylation activity of CdFic is independent of the αinh helix, the activity is still less than that for both NmFic and the dramatically more active αinh mutant NmFicE186G when assayed under identical conditions (Fig. 6D).
To identify the autoadenylylation acceptor site, we performed mass spectrometry analysis of CdFicSE/AA incubated with ATP. The location of the acceptor site was traced to a single tryptic peptide (supplemental Fig. 1, A–C). The MS2 spectra narrowed down the site to either Ser-37 or Thr-38, both residues located in the αinh-α′1 loop. As a control experiment, the same analysis was conducted without ATP incubation, which resulted in the identification of the same, but unmodified, peptide. An additional control experiment was performed by incubation of an isotope-labeled ATP, also identifying Ser-37 or Thr-38 as the acceptor site based on the MS2 spectra (supplemental Fig. 1, D–F). Unfortunately, in both cases, most of the obtained MS2 spectra lacked information about the y13-y14 and b4-b5 ions, which made it difficult to determine the exact location of the AMP group. To determine the exact acceptor site, we substituted either residue Ser-37 or Thr-38 with alanine before assaying the mutants for autoadenylylation activity (Fig. 6E). Although CdFicS37A catalyzes self-labeling comparable with wild type protein, CdFicT38A is inactive, thus identifying Thr-38 as the AMP acceptor site. Two acceptor sites were previously mapped for NmFic with the primary acceptor site being Tyr-183 inside the inhibitory motif,- and an additional acceptor site, Tyr-188, located to the αinh-α′1 loop (21). Superposition of NmFic and CdFic shows that residue Asn-32 in CdFic is situated in a position structurally equivalent to the Tyr-183 primary acceptor site in NmFic (Fig. 6F). The lack of a hydroxyl acceptor at this position may thus explain why this is not a CdFic acceptor site. In contrast, the hydroxyl group of the Tyr-188 secondary acceptor residue in NmFic is structurally in the same position as the hydroxyl group of the Thr-38 acceptor residue in CdFic (Fig. 6, F and G). As the Thr-38 side chain hydroxyl group of CdFic is positioned 15.5 Å away from the ATP α-phosphate and is solvent-exposed, autoadenylylation likely occurs intermolecularly and only after partial rearrangement of the αinh-α′1 loop (Fig. 6H). Interestingly, the Thr-38 side chain forms hydrogen bonds to the side chains of Asp-61 and the dimerization loop residues Arg-56 and His-57. Hence, the disruption of the hydrogen bond between Thr-38 and His-57 in the H57A mutant could result in Thr-38 becoming more solvent-accessible, thereby explaining why this mutant shows a dramatic increase in autoadenylylation activity (Fig. 6A). Although placed near the dimerization interface, the T38A mutant, like H57A, does not disrupt dimer formation.
Finally, Thr-38 is conserved in both HYPE and BtFic (Fig. 6E), which suggests that these two Fic proteins could have autoadenylylation targets similar to CdFic. However, it was recently shown that autoadenylylation of the human Fic protein HYPE occurs outside of the Fic core domain (16). Hence, CdFic is able to catalyze AMP transfer in vitro, and the autoadenylylation activity, in agreement with the CdFicSE/AA-ATP structure, is independent of the inhibitory motif.
Discussion
The results presented here characterize a novel adenylylating class II Fic domain protein from the important human pathogen C. difficile. This is the first Fic domain protein identified with autoadenylylation activity incompatible with the proposed model of inhibition for class I–III Fic domain proteins (21). Although structurally similar to other Fic proteins and exhibiting the presence of αinh, we show that wild type CdFic harbors in vitro autoadenylylation activity, which is not enhanced by introduction of point mutations in the inhibitory motif (Fig. 6A). This is supported by the combined crystal structures of CdFic and CdFicSE/AA-ATP, which reveal that ATP is bound in a conformation that is not obstructed by the inhibitory helix (Fig. 1). The structures of CdFic also provide evidence for a dimer formation and for the movements of the CdFic flap upon ATP binding. We have calculated the low resolution SAXS envelope of the dimer as well as shape reconstructions of the flap, representing the open/disordered conformation of the flap in solution. Superposition against the closed/ordered flap in the crystal structure of CdFicSE/AA-ATP complex highlights the large active site closure that occurs upon ATP binding (Fig. 4D). Moreover, after prolonged incubation with ATP, SEC provides no indication of monomer formation of CdFic, suggesting that autoadenylylation does not disrupt the dimer (results not shown). In the active site, the base and the ribose adopt a canonical position, whereas the phosphates adopt a different path where γ-phosphate coordination is not overlapping with the side chain of Gluinh in the inhibitory motif (Fig. 2, C and G). Notably, the α- and β-phosphates of the ATP are situated on the opposite side of the active site cleft compared with those seen in other Fic domains. This placement of the ATP phosphates is likely explained by the salt bridge formed between the α-phosphate and Arg-200 (Fig. 2E). No other structurally characterized Fic domains are shown to have a similarly positioned arginine in the Fic core on the opposite side of the active site relative to the conserved Fic motif (Fig. 2F). In other structures published to date, the α-phosphate instead forms a salt bridge to the conserved asparagine in the GNG anion hole (Fig. 2D).
Also, the Mg2+ ion in other Fic-ATP-Mg2+ structures is coordinated between the α- and the β-phosphates. However, in the CdFicSE/AA-ATP structure, the Mg2+ ion is instead coordinated by the β- and the γ-phosphates (Fig. 2C). This type of Mg2+ coordination seen in CdFic is in line with the suggested adenylylation reaction mechanism based on the crystal structure of the BepA-Mg2+-PPi complex where Mg2+-PPi appears to be a part of the product (23, 60). Conversely, this particular Mg2+ coordination is also seen in other ATP-binding enzymes where Mg2+ chelates the γ- and β-phosphate groups to lower the threshold either for γ-phosphoryl transfer to a nucleophilic (-OH) group or for hydrolyzing ATP to ADP to release energy to drive a subsequent reaction (61).
We also attempted to soak wild type CdFic crystals with ATP, but because of limited diffraction quality of the crystals we did not succeed to obtain a crystal structure of this complex. However, the biological significance of the CdFicSE/AA-ATP structure is supported by the following observations. (i) A Mg2+ ion is tightly coordinated in the active site. (ii) Replacement of Arg-200 with an alanine significantly reduces autoadenylylation activity, demonstrating the catalytic importance of this residue. (iii) The ATP molecule is not engaged in any crystal packing interactions. (iv) The same ATP conformation was seen in a 3.1-Å structure of the CdFicSE/AA-ATP complex (results not shown), which was solved independently of the presented 2.5-Å complex structure.
In further support of the inhibitory motif-independent mechanism of CdFic, we observe no differences in DSF binding assays of ATP to wild type CdFic and the CdFicSE/AA mutant (Fig. 5). Recently, DSF was demonstrated as an important method for studying inhibitory motif dependence where the human class II Fic domain protein HYPE and its Gluinh mutant exhibited significant differences in thermal shifts (56). HYPE is known to autoadenylylate as well as adenylylate the chaperone BiP, and both activities were shown to be highly dependent on the inhibitory motif (16). Our CdFic DSF results are considerably different from those of HYPE because only ADP, but none of the triphosphate nucleotides or PPi, increases the stability of HYPE. In contrast, the Gluinh mutant of HYPE behaves similarly to both CdFic and CdFicSE/AA as ATP, ADP, and PPi produce the largest thermal shifts, whereas the pyrimidine nucleotides produce the smallest. We also tested other nucleotides in the same assay and found that ATP and ADP induce the highest thermal shifts, suggesting binding specificity for the adenosine base (Fig. 5). In further support of this, we conducted autoadenylylation assays using radioactively labeled CTP and observed no self-labeling (Fig. 6B). These results combined suggest that CdFic is an adenylylation factor in vivo with a nucleotide binding profile of CdFic independent of the inhibitory motif. The structural basis for the non-obstructed autoadenylylation reaction mechanism of CdFic is summarized schematically in Fig. 7.
FIGURE 7.

Non-obstructed ligand binding. Schematic models for inhibitory motif obstructed (left) and non-obstructed (right) autoadenylylation mechanism are shown. In previously described Fic proteins, the inhibitory helix αinh and/or the associated inhibitory residue Gluinh must move away from the obstructive position to accommodate a salt bridge between the terminal phosphate of ATP and the C-terminal arginine of the Fic motif. However, in the non-obstructed autoadenylylation mechanism for CdFic, the ATP is pulled toward the opposite side of the active site cleft. Consequently, the position of Gluinh is not obstructing proper positioning of the ATP phosphates, and the autoadenylylation activity is not inhibited. The rearrangement of the ATP path alters the position of the phosphates relative to the Mg2+ ion. The Mg2+ is shown in red, ATP is in black, the anion hole is in blue, and the inhibitory helix is in dark yellow. Highly conserved residues involved in ATP binding are indicated. For CdFic, Arg-200 that specifically interacts with the α-phosphate is also shown.
It is currently unknown what mechanisms and factors relieve the autoinhibition of class I–III Fic proteins. In addition, the autoinhibition observed in most studies was incomplete as Fic domains in general harbor activity in vitro although typically much less compared with the point mutants of the inhibitory motif (Fig. 6D) (16, 28). Although the autoadenylylation activity for both CdFic and CdFicSE/AA is low compared with NmFicE186G (Fig. 6D), we observed a dramatic increase in activity upon the H57A substitution positioned in the dimerization interface (Fig. 6A). This could be due to a disruption of the hydrogen bond between the side chains of residues His-57 and Thr-38, thereby making the acceptor site more easily accessible for autoadenylylation (Fig. 6G). Alternatively, although SEC reveals that H57A still forms a stable dimer in solution, the substitution may affect the conformation of the dimer. The importance of dimerization for autoadenylylation activity was recently demonstrated for human HYPE, which showed a dramatic decrease in activity when residues inside or near the dimerization loop were substituted (56). Nevertheless, the increase in activity caused by the H57A mutant suggests that an activation mechanism that needs to take place before CdFic can carry out its function might exist and that this activation mechanism is different from the autoinhibition by αinh.
The physiological protein target of CdFic remains unknown. Attempts to identify the target by incubating purified recombinant CdFic, CdFicSE/AA, as well as the more active CdFicH57A together with [α-32P]ATP and lysates of either human Caco-2 or C. difficile cells or with a broad selection of purified Rho-GTPases have so far been unsuccessful. Possibly, the protein target has to be found elsewhere, or an alternative activation mechanism that needs to take place before CdFic can carry out its function might exist. Despite showing that ATP binding and autoadenylylation by CdFic are independent of the inhibitory motif, we cannot rule out the possibility that the αinh of CdFic might still have a function in inhibiting the adenylylation activity against the currently unknown target. However, this would be unusual because the αinh is known to regulate both autoadenylylation and adenylylation in other Fic proteins with known protein targets, such as HYPE (16) and NmFic (62).
In addition, we are unable to completely rule out that CdFic is a kinase or modifies a target protein with an ADP moiety. However, we have looked for possible kinase activity of CdFic in lysed Caco-2 cells with no positive outcome. Furthermore, using mass spectrometry, we detected an AMP moiety attached to Thr-38 of CdFic upon incubation with ATP (Fig. 6E and supplemental Fig. 1), providing important evidence that the scissile bond is situated between the α- and β-phosphates. The AMP acceptor site structurally aligns to one of the two AMP acceptor sites of the inhibitory motif-dependent NmFic (Fig. 6, F and G). This strongly suggests that CdFic catalyzes adenylylation or AMPylation and that the unusual behavior of CdFic in relation to the inhibitory motif is not due to differences in the position of the acceptor site.
Currently, fewer than 10 other unique Fic domain structures with ATP in the binding site are deposited in the Protein Data Bank. Despite this very limited structural knowledge and the high conservation of the Fic motif, Fic domains have been shown to have high diversity. For instance, the Legionella effector AnkX (25) utilizes CDP-choline to catalyze phosphocholine modifications, whereas IbpA has affinity for both ATP and GTP (24). These studies show that the Fic core, which contains weak sequence conservation, is able to confer additional specificity to a structurally conserved Fic motif evolved to coordinate the ribose and the phosphates of nucleotides. In line with this and because the conserved Fic motif in CdFic is structurally similar to other known Fic enzymes, the Fic core of CdFic likely alters the ATP binding of the Fic motif to overcome the obstruction by the inhibitory motif. These new insights into the first characterized Fic domain protein from a Gram-positive bacterium clearly illustrate the diversity of this very large family despite having the same structural fold. They also provide important steps toward revealing the mechanistic function, the protein target, and hence the physiological role of CdFic as well as the potential framework to identify possible other inhibitory motif-independent Fic domain proteins.
Author Contributions
E. D., H. C. v. L., and R. J. conceived and designed the experiments. E. D. performed the SAXS experiments. H. A. designed and performed the DSF experiments. E. D., D. H. W., and R. J. performed the crystallization experiments, x-ray data collection, and structure solution. O. Ø. performed the mass spectrometry analyses. H. C. v. L., J. C., O. I. K., and P. J. H. performed the cloning and site-directed mutagenesis as well as the in vitro adenylylation assays. E. D., H. C. v. L., H. A., and R. J. analyzed the data. E. D. and R. J. wrote the paper. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Wiep Klaas Smits and Sjaak van Voorden (Leiden University Medical Center, Netherlands) for help with cloning. We thank the group of Tilman Schirmer at Biozentrum, University of Basel, for sending us the NmFic and NmFicSE/AA clones. We also acknowledge Giancarlo Tria (Nanyang Technological University, Singapore) for advice regarding EOM SAXS analysis, Seema Mattoo for helpful discussions and carefully reading the manuscript, Tomás Plivelic at SAXS beamline I911-4 MAX-II Laboratory for help with SAXS data collection, the MX beamline staff at I911-2 at MAX-II Laboratory, and the MX beamline staff at ID23-2 and ID29 at European Synchrotron Radiation Facility (Grenoble, France) for help with x-ray diffraction data collection.
This work was supported by Det Frie Forskningsråd Sapere Aude Starting Grant 11-104831/Medical Sciences from the Danish Council for Independent Research. Access to synchrotron beam time was made possible by support from DANSCATT. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1.
The atomic coordinates and structure factors (codes 4X2C, 4X2D, and 4X2E) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Fic
- filamentation induced by cAMP
- αinh
- inhibitory α-helix
- HYPE
- huntingtin yeast interacting protein E
- AU
- asymmetric unit
- SEC
- size exclusion chromatography
- DSF
- differential scanning fluorimetry
- EOM
- ensemble optimization method
- SAXS
- small angle x-ray scattering
- SE/AA
- S31A/Einh35A
- Cd
- C. difficile
- Nm
- N. meningitides
- Bt
- B. thetaiotaomicron
- Hp
- H. pylori.
References
- 1. Poutanen S. M., and Simor A. E. (2004) Clostridium difficile-associated diarrhea in adults. CMAJ 171, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elliott B., Chang B. J., Golledge C. L., and Riley T. V. (2007) Clostridium difficile-associated diarrhoea. Int. Med. J. 37, 561–568 [DOI] [PubMed] [Google Scholar]
- 3. McFarland L. V., Beneda H. W., Clarridge J. E., and Raugi G. J. (2007) Implications of the changing face of Clostridium difficile disease for health care practitioners. Am. J. Infect. Control 35, 237–253 [DOI] [PubMed] [Google Scholar]
- 4. Just I., Selzer J., von Eichel-Streiber C., and Aktories K. (1995) The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J. Clin. Investig. 95, 1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Just I., Wilm M., Selzer J., Rex G., von Eichel-Streiber C., Mann M., and Aktories K. (1995) The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 270, 13932–13936 [DOI] [PubMed] [Google Scholar]
- 6. Lyras D., O'Connor J. R., Howarth P. M., Sambol S. P., Carter G. P., Phumoonna T., Poon R., Adams V., Vedantam G., Johnson S., Gerding D. N., and Rood J. I. (2009) Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuehne S. A., Cartman S. T., Heap J. T., Kelly M. L., Cockayne A., and Minton N. P. (2010) The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713 [DOI] [PubMed] [Google Scholar]
- 8. Lyerly D. M., Saum K. E., MacDonald D. K., and Wilkins T. D. (1985) Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 47, 349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quesada-Gómez C., López-Ureña D., Acuña-Amador L., Villalobos-Zúñiga M., Du T., Freire R., Guzmán-Verri C., del Mar Gamboa-Coronado M., Lawley T. D., Moreno E., Mulvey M. R., de Castro Brito G. A., Rodríguez-Cavallini E., Rodríguez C., and Chaves-Olarte E. (2015) Emergence of an outbreak-associated Clostridium difficile variant with increased virulence. J. Clin. Microbiol. 53, 1216–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yarbrough M. L., Li Y., Kinch L. N., Grishin N. V., Ball H. L., and Orth K. (2009) AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272 [DOI] [PubMed] [Google Scholar]
- 11. Worby C. A., Mattoo S., Kruger R. P., Corbeil L. B., Koller A., Mendez J. C., Zekarias B., Lazar C., and Dixon J. E. (2009) The fic domain: regulation of cell signaling by adenylylation. Mol. Cell 34, 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garcia-Pino A., Zenkin N., and Loris R. (2014) The many faces of Fic: structural and functional aspects of Fic enzymes. Trends Biochem. Sci. 39, 121–129 [DOI] [PubMed] [Google Scholar]
- 13. Müller M. P., Peters H., Blümer J., Blankenfeldt W., Goody R. S., and Itzen A. (2010) The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949 [DOI] [PubMed] [Google Scholar]
- 14. Rahman M., Ham H., Liu X., Sugiura Y., Orth K., and Krämer H. (2012) Visual neurotransmission in Drosophila requires expression of Fic in glial capitate projections. Nat. Neurosci. 15, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ham H., Woolery A. R., Tracy C., Stenesen D., Krämer H., and Orth K. (2014) Unfolded protein response-regulated dFic reversibly AMPylates BiP during endoplasmic reticulum homeostasis. J. Biol. Chem. 289, 36059–36069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanyal A., Chen A. J., Nakayasu E. S., Lazar C. S., Zbornik E. A., Worby C. A., Koller A., and Mattoo S. (2015) A novel link between Fic (filamentation induced by cAMP)-mediated adenylylation/AMPylation and the unfolded protein response. J. Biol. Chem. 290, 8482–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harms A., Stanger F. V., Scheu P. D., de Jong I. G., Goepfert A., Glatter T., Gerdes K., Schirmer T., and Dehio C. (2015) Adenylylation of gyrase and topo IV by FicT toxins disrupts bacterial DNA topology. Cell Rep. 12, 1497–1507 [DOI] [PubMed] [Google Scholar]
- 18. Kinch L. N., Yarbrough M. L., Orth K., and Grishin N. V. (2009) Fido, a novel AMPylation domain common to fic, doc, and AvrB. PLoS One 4, e5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao J., Worby C. A., Mattoo S., Sankaran B., and Dixon J. E. (2010) Structural basis of Fic-mediated adenylylation. Nat. Struct. Mol. Biol. 17, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng F., Yang F., Rong W., Wu X., Zhang J., Chen S., He C., and Zhou J. M. (2012) A xanthomonas uridine 5′-monophosphate transferase inhibits plant immune kinases. Nature 485, 114–118 [DOI] [PubMed] [Google Scholar]
- 21. Engel P., Goepfert A., Stanger F. V., Harms A., Schmidt A., Schirmer T., and Dehio C. (2012) Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110 [DOI] [PubMed] [Google Scholar]
- 22. Goody P. R., Heller K., Oesterlin L. K., Müller M. P., Itzen A., and Goody R. S. (2012) Reversible phosphocholination of Rab proteins by Legionella pneumophila effector proteins. EMBO J. 31, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luong P., Kinch L. N., Brautigam C. A., Grishin N. V., Tomchick D. R., and Orth K. (2010) Kinetic and structural insights into the mechanism of AMPylation by VopS Fic domain. J. Biol. Chem. 285, 20155–20163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mattoo S., Durrant E., Chen M. J., Xiao J., Lazar C. S., Manning G., Dixon J. E., and Worby C. A. (2011) Comparative analysis of Histophilus somni immunoglobulin-binding protein A (IbpA) with other fic domain-containing enzymes reveals differences in substrate and nucleotide specificities. J. Biol. Chem. 286, 32834–32842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mukherjee S., Liu X., Arasaki K., McDonough J., Galán J. E., and Roy C. R. (2011) Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castro-Roa D., Garcia-Pino A., De Gieter S., van Nuland N. A., Loris R., and Zenkin N. (2013) The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat. Chem. Biol. 9, 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Campanacci V., Mukherjee S., Roy C. R., and Cherfils J. (2013) Structure of the Legionella effector AnkX reveals the mechanism of phosphocholine transfer by the FIC domain. EMBO J. 32, 1469–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goepfert A., Stanger F. V., Dehio C., and Schirmer T. (2013) Conserved inhibitory mechanism and competent ATP binding mode for adenylyltransferases with Fic fold. PLoS One 8, e64901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Welner D., Dedic E., van Leeuwen H. C., Kuijper E., Bjerrum M. J., Østergaard O., and Jørgensen R. (2014) Protein expression, characterization, crystallization and preliminary x-ray crystallographic analysis of a Fic protein from Clostridium difficile. Acta Crystallogr. F Struct. Biol. Commun. 70, 827–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York [Google Scholar]
- 37. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 38. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 39. Konarev P. V., Petoukhov M. V., Volkov V. V., and Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 41. Petoukhov M. V., Konarev P. V., Kikhney A. G., and Svergun D. I. (2007) ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, S223–S228 [Google Scholar]
- 42. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 43. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wriggers W. (2010) Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 47. Tria G., Mertens H. D., Kachala M., and Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using x-ray solution scattering. IUCrJ 2, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 49. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 50. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 51. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 52. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 53. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 54. Li Y., Al-Eryani R., Yarbrough M. L., Orth K., and Ball H. L. (2011) Characterization of AMPylation on threonine, serine, and tyrosine using mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 752–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morris A. L., MacArthur M. W., Hutchinson E. G., and Thornton J. M. (1992) Stereochemical quality of protein structure coordinates. Proteins 12, 345–364 [DOI] [PubMed] [Google Scholar]
- 56. Bunney T. D., Cole A. R., Broncel M., Esposito D., Tate E. W., and Katan M. (2014) Crystal structure of the human, FIC-domain containing protein HYPE and implications for its functions. Structure 22, 1831–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Poornam G. P., Matsumoto A., Ishida H., and Hayward S. (2009) A method for the analysis of domain movements in large biomolecular complexes. Proteins 76, 201–212 [DOI] [PubMed] [Google Scholar]
- 58. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boivin S., Kozak S., and Meijers R. (2013) Optimization of protein purification and characterization using Thermofluor screens. Protein Expr. Purif. 91, 192–206 [DOI] [PubMed] [Google Scholar]
- 60. Palanivelu D. V., Goepfert A., Meury M., Guye P., Dehio C., and Schirmer T. (2011) Fic domain-catalyzed adenylylation: insight provided by the structural analysis of the type IV secretion system effector BepA. Protein Sci. 20, 492–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matte A., Tari L. W., and Delbaere L. T. (1998) How do kinases transfer phosphoryl groups? Structure 6, 413–419 [DOI] [PubMed] [Google Scholar]
- 62. Stanger F. V., Burmann B. M., Harms A., Aragão H., Mazur A., Sharpe T., Dehio C., Hiller S., and Schirmer T. (2016) Intrinsic regulation of FIC-domain AMP-transferases by oligomerization and automodification. Proc. Natl. Acad. Sci. U.S.A. 113, E529–E537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wallace A. C., Laskowski R. A., and Thornton J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 64. Gille C., and Frömmel C. (2001) STRAP: editor for STRuctural Alignments of Proteins. Bioinformatics 17, 377–378 [DOI] [PubMed] [Google Scholar]
- 65. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Diederichs K., and Karplus P. A. (1997) Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.