

Abstract
KYE28 (KYEITTIHNLFRKLTHRLFRRNFGYT-LR), the representative sequence of helix D of heparin co-factor II, was demonstrated to be potent against agronomically important Gram-negative plant pathogens Xanthomonas vesicatoria and Xanthomonas oryzae, capable of inhibiting disease symptoms in detached tomato leaves. NMR studies in the presence of lipopolysaccharide provided structural insights into the mechanisms underlying this, notably in relationship to outer membrane permeabilization. The three-dimensional solution structure of KYE28 in LPS is characterized by an N-terminal helical segment, an intermediate loop followed by another short helical stretch, and an extended C terminus. The two termini are in close proximity to each other via aromatic packing interactions, whereas the positively charged residues form an exterior polar shell. To further demonstrate the importance of the aromatic residues for this, a mutant peptide KYE28A, with Ala substitutions at Phe11, Phe19, Phe23, and Tyr25 was designed, which showed attenuated antimicrobial activity at high salt concentrations, as well as lower membrane disruption and LPS binding abilities compared with KYE28. In contrast to KYE28, KYE28A adopted an extended helical structure in LPS with extended N and C termini. Aromatic packing interactions were completely lost, although hydrophobic interaction between the side chains of hydrophobic residues were still partly retained, imparting an amphipathic character and explaining its residual antimicrobial activity and LPS binding as observed from ellipsometry and isothermal titration calorimetry. We thus present key structural aspects of KYE28, constituting an aromatic zipper, of potential importance for the development of novel plant protection agents and therapeutic agents.
Keywords: antimicrobial peptide (AMP), lipopolysaccharide (LPS), membrane biophysics, nuclear magnetic resonance (NMR), structure-function
Introduction
Recent years have seen a profound increase in the occurrence of multidrug resistant Gram-negative bacterial strains (1, 2). Consequently, infections due to these strains pose a considerable, and increasing, threat to human health and economy. This is the case also within agriculture, where large amount of crops are lost every year due to bacterial infections, putting global food security at stake (3). For example, Xanthomonas species are associated with bacterial spot disease in a wide variety of agriculturally important crops, particularly tomato and pepper. They affect leaves, stem, and fruits (4), and are found to be most destructive to crops that are grown in warm and humid weather conditions, leading to a substantial crop loss on a global scale (5). Xanthomonas has been found to display streptomycin resistance since the 1960s (4), thus directing control and prevention toward large scale use of copper-based compounds, mancozeb and/or maneb (6). The latter compounds are carcinogenic as well as toxic to the environment (7). In addition, extensive application of copper compounds has, in itself, led to the development of bacterial resistance in many strains of Xanthomonas (8). Counteracting this, genetic breeding techniques have been successfully applied but are time consuming and suffer from drawbacks that include hybrid weakness and pre- and post-zygotic barriers that prevent attainment of fertile hybrids (9, 10). Thus, to reduce both environmental and health hazards, new environmentally friendly compounds are needed. Besides, new approaches of plant disease management are also required to meet the current demand of crops.
Within the broader context of multidrug resistance in bacteria, antimicrobial peptides (AMPs)4 have attracted considerable attention as potential anti-infective drug alternatives (11, 12). AMPs are small peptides, ≤50 amino acids, displaying diverse structures as well as biological functions (2, 13). They are found in a wide variety of organisms, forming a key part of the innate immunity system, in which they provide antimicrobial properties against a broad range of pathogens (12, 14). Most AMPs are cationic and amphipathic in nature, interacting with, and disrupting, negatively charged microbial membranes (2, 15, 16). Apart from their antimicrobial activities, some AMPs have also been found to display other host defense functions, such as modulation of pro- or anti-inflammatory pathways along with activation of dendritic cells, helping in wound healing and angiogenesis (17, 18).
Central for the antimicrobial and anti-inflammatory effects of AMPs against Gram-negative bacteria is their interaction with bacterial lipopolysaccharide (LPS), a key component in the outer leaflet of the outer membrane of Gram-negative bacteria (19–21). Through electrostatic and steric effects, LPS restricts the entry of AMPs to their site-of-action, i.e. the inner membrane (22–24). Furthermore, LPS triggers inflammation, causing deregulated expression of proinflammatory cytokines (25). In contrast to AMP interactions with lipid membranes, AMP-LPS interactions remain poorly understood, despite their importance. Further studies on these are therefore needed. In particular, structural elucidations of AMPs in the context of LPS- and membrane-mimicking environments are key for the identification of specific motifs resulting in potent antimicrobial and anti-inflammatory effects against Gram-negative infections.
Earlier studies on heparin co-factor II (HC-II) have demonstrated that helices A and D are responsible for its observed antimicrobial property (26). More recently, the 28-residue peptide KYE28 (KYEITTIHNLFRKLTHRLFRRNFGYTLR) representing the helix D of HC-II (Fig. 1) has been reported to be highly active against the two Gram-negative bacteria Pseudomonas aeruginosa and Escherichia coli (27). In addition, KYE28 displays immunomodulatory effects, mediating a reduction in proinflammatory cytokines following either endotoxin injection or bacterial infection, although the mechanisms underlying this remain unclear (27).
FIGURE 1.
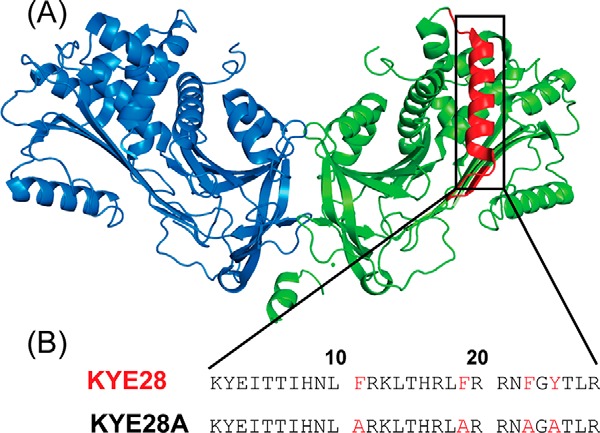
Peptide design. A, crystal structure of dimeric heparin co-factor II (HCII), showing helix D highlighted in red. B, sequence of helix D of HCII corresponding to KYE28, as well as of its alanine substituted mutant KYE28A.
Taken together, the considerations above initiated our interest in studying LPS binding of KYE28, as well as its influence on devastating Gram-negative plant pathogens belonging to the genus Xanthomonas. To place these investigations in a wider context, results obtained on these plant pathogens were compared with those for more widely investigated clinical pathogens, Klebsiella pneumoniae and Salmonella typhi. Importantly, KYE28 was found to be very active against all tested plant pathogens. In addition to elucidating the membrane disruption underlying this antimicrobial effect through an array of biophysical methods, we also performed NMR studies to elucidate the three-dimensional structure of KYE28 in LPS for better understanding the key residues involved in stabilization of its adopted conformation, and to provide an understanding of the structure-activity correlation. Our studies indicated the importance of the aromatic residues of KYE28 for its interaction with LPS and resulting antimicrobial and anti-inflammatory properties. Therefore, we infer that the aromatic zipper, together with background hydrophobic interactions, is crucial for endowing KYE28 with its bioactivity.
Experimental Procedures
Chemicals
Both peptides (KYE28 and KYE28A, the latter with Ala substitutions at Phe11, Phe19, Phe23, and Tyr25) were synthesized by Biopeptide Co., San Diego, CA, and were of >95% purity, as evidenced by mass spectral analysis (MALDI-TOF Voyager). 1 mm stock solutions of peptides were prepared by weighing the peptide in autoclaved distilled water or 10 mm phosphate buffer, pH 7.4, the concentration confirmed by measuring UV spectroscopy with respect to molar absorptive coefficient (ϵ). These stocks were used for all experiments if not otherwise specified. MTT (3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide), E. coli LPS (0111:B4), P. aeruginosa serotype 10 LPS, and E. coli lipid A F583 (Rd mutant) were obtained from Sigma, whereas DOPG (1,2-dioleoyl-sn-glycero-3-phosphoglycerol, monosodium salt), DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), E. coli total lipid extract, and 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO) were from Avanti Polar Lipids (Alabaster, CA) and of >99% purity. All other chemicals used were of analytical grade.
Microbial Strains
Fungal strains Candida albicans SC5314 and Cryptococcus neoformans var. grubii H99 were a gift from Prof. Kaustuv Sanyal, JNCASR, India. The agronomical bacterial strains Xanthomonas vesicatoria and Xanthomonas oryzae were provided by Dr. Christian Lindermayer, Helmholtz-Münich, Germany, and Prof. Sampa Das, Bose Institute, India, respectively. The other bacterial strains, K. pneumoniae and S. typhi, were obtained from ATCC.
Media Composition
YPD broth (1% yeast extract, 1% peptone, 2% dextrose) was used to grow C. albicans and Cryptococcus neoformans var. grubii, whereas X. vesicatoria, K. pneumoniae, and S. typhi were grown in Nutrient broth and Xanthomonas oryzae were grown in PS broth (1% peptone, 1% sucrose) K. pneumoniae and S. typhi were grown at 37 °C. All other strains were incubated at 28 °C with 150 rpm for 24–48 h.
Antimicrobial Activity
To determine the minimal inhibitory concentration (MIC), a microtiter broth dilution method was utilized, as previously described (28). In brief, overnight cultures of the indicated microbes were washed twice and re-suspended in either 10 mm phosphate buffer, pH 7.4, alone or in the presence of 150 mm NaCl, and further diluted to 105 cfu/ml. The peptide was dissolved in the same buffer to a stock concentration of 1 mm. 50 μl of microbial suspension was added to the corresponding well of a 96-well microtiter plate (polypropylene) together with 50 μl of peptide solution to obtain a final peptide concentration ranging from 1 to 100 μm. The plate was subsequently incubated at 37 °C with rotation for 3 h, after which 200 μl of media was added and incubated at the temperature indicated above for 24 to 48 h. The MIC was obtained as the lowest concentration of the peptide where no visual microbial growth was detected. 10 μm Polymyxin B was used as a positive control. Microbial suspension in the absence of peptide was used as the negative control.
NF-κB Activation Assay
THP1-XblueTM-CD14 cells (1 × 106/ml) were stimulated with 100 ng/ml of E. coli LPS (InvivoGen), with or without the immediate addition of KYE28 or KYE28A. NF-κB activation was determined 20 h later by using the Quanti-BlueTM assay (InvivoGen). In brief, through activation of the cells by various antagonists, NF-κB/AP-1 activation is induced, leading to the secretion of embryonic alkaline phosphatase into the cell supernatant. For detection, the cell supernatant and a secretion of embryonic alkaline phosphatase detection reagent (QUANTI-BlueTM) were mixed and absorbance was measured at 600 nm. Results given represent mean values from quadruple measurements.
MTT Assay
Sterile filtered MTT solution (5 mg/ml in PBS) was stored protected from light at −20 °C until use. THP-1 cells from the above experiment were washed, and 20 μl of the MTT solution was added to each well and the plates were incubated for 1 h in CO2 at 37 °C. The MTT-containing medium was then removed by aspiration. The blue formazan product generated was dissolved by the addition of 100 μl of 100% DMSO per well, and the plates gently swirled for 10 min at room temperature to dissolve the precipitate. The absorbance was monitored at 550 nm, and results given represent the mean values from quandruple measurements.
Circular Dichroidism (CD) Spectroscopy
A Jasco 815 spectrometer was used to study the secondary structures of the peptides in buffer solution as well as in the presence of LPS. Both peptide and LPS stocks were prepared either in 10 mm phosphate buffer, pH 7.4, or in the same buffer with pH 4.5. 25 μm peptide was titrated with increasing concentrations of LPS, ranging between 25 and 75 μm, and the spectra were recorded at room temperature (25 °C) with accumulations of three scans and at a speed of 100 nm/min. Spectra were scanned from 190 to 260 nm, with a data interval of 1 nm. A cuvette having a 0.1-cm path length was used for all measurements. The spectra obtained were corrected using a baseline spectrum in each case. The ellipticity in millidegrees was plotted against wavelength in nm.
NMR Studies
1 mm KYE28/KYE28A solution in 10 mm phosphate buffer, pH 4.5, and 10% D2O were used to obtain one-dimensional proton NMR spectra at 25 °C, using Bruker Avance III 700 MHz NMR spectrometer equipped with a 5-mm cryoprobe. 4,4-Dimethyl-4-silapentane-5-sulfonate sodium salt (DSS) was used as an internal standard (0.0 ppm) in all NMR experiments. A series of one-dimensional titrations were performed upon addition of increasing concentrations of LPS ranging from 3 to 24 μm (E. coli LPS or P. aeruginosa LPS), until line broadening was observed in the proton spectra. Upon significant line broadening (occurring at a LPS:peptide molar ratio of ∼1:25), two-dimensional homonuclear total correlation spectroscopy (TOCSY) (with mixing time of 80 ms), and transferred nuclear Overhauser effect spectroscopy (trNOESY) experiments with four different mixing times, viz. 75, 100, 150, and 200 ms, were carried out to rule out the possibility of spin diffusion. In addition, trNOESY experiments were carried out for the same sample of the KYE28-LPS complex in 100% D2O for unambiguous assignments of aromatic/aliphatic or aromatic/aromatic NOEs. 24 and 16 scans were performed either for NOESY (peptide in aqueous solution) or trNOESY (LPS-peptide complex with a molar ratio of 1:25) and TOCSY, respectively, per t1 increment. 16 Dummy scans were performed in each case and a spectral width of 12 ppm was maintained for both experiments along both dimensions. 456 increments in t1 and 2048 data points in t2 dimension along with States TPPI (29) for quadrature detection in t1 dimension and WATERGATE for water suppression were employed (30). TOCSY and NOESY or trNOESY spectra were processed using 4K (t2) × 1K (t1) data matrices after zero filling. The experiment was processed with Bruker TOPSPIN software and was analyzed using Sparky.
Saturation transfer difference (STD) NMR experiments were conducted with 0.5 mm KYE28/KYE28A dissolved in 100% D2O, pH 4.5. LPS stock was also prepared in D2O and pH was adjusted to 4.5. A standard STD pulse program (31, 32) using a saturation frequency of −1 ppm and an off-resonance frequency of 40 ppm yielded the spectra. The experiments were conducted at 25 °C. The saturation frequency selectively saturated LPS in solution (on-resonance), whereas the off resonance was such a frequency where neither LPS nor the peptide saw any resonances. An STD experiment was carried out for peptides in the absence and presence of LPS. Total 40 Gaussian-shaped pulses (49 ms, 1 ms delay between pulses) with a saturation time of 2 s was employed (33). Subtraction of off-resonance from the on-resonance spectra yielded the saturation transfer difference spectrum (34). 1024 and 512 scans were performed for both STD and reference spectra, respectively, maintaining a spectral width of 12 ppm in each case. A relaxation delay of 2 s was used and 16 dummy scans were carried out for both STD and reference spectra. Before conducting Fourier transformation, an exponential line broadening function of 3 Hz was multiplied with STD one-dimensional spectra.
Structure Calculation
The two-dimensional trNOESY spectra of KYE28 or KYE28A in LPS, recorded at 150 ms mixing time, were used to obtain the upper bound distance constraints from the NOE build up rate, as it contained no spin diffusion. The upper bound distance constraints were calculated with respect to the NOE intensities between the resolved ring protons of either Phe-11 (viz. Phe11-2H and Phe11-3H for KYE28 and ring protons) or Tyr2 (viz. Tyr2-2H and Tyr2-3H for KYE28A). The 2.0-Å distance was fixed for lower distance constraints. The calculated distance was then categorized as strong (2.5 Å), medium (2.6–3.5 Å), and weak (3.6–5.0 Å) for further structure calculation using CYANA. It is worth mentioning that the folding of the structure depends mainly only on medium- and long-range NOEs. The backbone dihedral angles Φ and ψ were varied from −30 to −120 and −120 to +120, respectively, for all nonglycine residues to reduce conformational search. The anti-parallel dimeric structure was also calculated as previously described (35) by connecting two monomer units with a glycine linker consisting of 10 glycines. Sequential, intra-residual, and medium-range NOEs were used to stabilize the monomeric units, whereas long-range NOEs were used as inter-monomeric NOEs. Successive refinements were performed as previously described (36). The 20 lowest energy structures were chosen from the 100 calculated structures to represent the ensemble structure. Procheck (37), PyMOL, and MOLMOL software was used for quality check and visualization of the PDB structures, respectively.
DPH Fluorescence Assay
Fluorescence spectroscopy was used to determine peptide interaction with E. coli LPS. A fluorescence emission spectrum of DPH was obtained in the presence of LPS bicelles, as well as in the presence of increasing peptide concentrations. For the preparation of bicelles, 2 mg of LPS from E. coli 0111:B4 was dissolved in buffer (10 mm phosphate buffer, pH 7.4, 150 mm NaCl) along with 1.51 mg of CHAPSO, keeping q ratio = 0.25. The sample was next subjected to five freeze-thaw cycles and used for further assay.
Bicelle disruption experiments were performed on a Varian Cary Eclipse fluorimeter, using excitation at 358 nm, emission wavelengths of 400 to 600 nm, excitation and emission slit widths of 5 nm, and a photomultiplier detector voltage of 700 V. Measurements were carried out in a 600-μl sample volume containing DPH in bicellar buffer (10 mm phosphate buffer, pH 7.4, 150 mm NaCl), which did not show any fluorescence readings. 5 μm LPS bicelle was added, which caused a slight fluorescence increase (F0). To induce the bicelle-disrupting action of the peptide, exposing the hydrophobic acyl group chains of the LPS moieties, the peptide was added in increasing concentrations from 5 μm to a final concentration of 35 μm. After peptide addition, the increase in fluorescence (F) was monitored after 5 min. The difference, δ, was calculated as the total fluorescence induced by disruption of the bicelle at a particular concentration of the peptide.
NPN Dye Uptake Assay
Overnight cultures of X. vesicatoria were washed in 10 mm Tris buffer, pH 7.2, containing 150 mm NaCl and re-suspended to a final concentration of 0.5 optical density. 0.5 mm of the fluorescent dye, 1-N-phenylnapthylamine (NPN) was used as the working solution. NPN, from the above working solution, was added to the reaction volume, consisting of 0.5 optical density in 600 μl of buffer to a final concentration of 10 μm, and allowed to stabilize. Subsequently, increasing concentrations of the peptide were added to the reaction volume, from 5 to 30 μm. An increase in fluorescence intensity, on account of the outer membrane permeabilization, was measured using a Varian Cary Eclipse fluorimeter with excitation and emission wavelengths of 350 and 420 nm, respectively. The excitation and emission slit width was 5 nm, whereas the photomultiplier detector voltage used was 700 V. 1% Triton X-100 was used as a positive control, relative to which % of NPN dye uptake was calculated.
Liposome Preparation and Leakage Assay
Model liposomes investigated were anionic (DOPE/DOPG, 75/25, mol/mol), extensively used as a model system in the AMP literature, and previously demonstrated to give similar results regarding peptide interactions as E. coli lipid extract membranes (38). In contrast to the latter, DOPE/DOPG offers methodological advantages. Because of the long, symmetric, and unsaturated acyl chains of these phospholipids, membrane cohesion is good, which facilitates stable, unilamellar, and largely defect-free liposomes and well defined supported lipid bilayers, allowing detailed data on leakage and adsorption density to be obtained. The lipid mixture was dissolved in chloroform, after which solvent was removed by evaporation under vacuum overnight. Subsequently, 10 mm Tris buffer, pH7.4, was added together with 0.1 m carboxyfluorescein (CF) (Sigma). After hydration, the lipid mixture was subjected to eight freeze-thaw cycles, consisting of freezing in liquid nitrogen and heating to 60 °C. Unilamellar liposomes of about Ø140 nm were generated by multiple extrusions (30 passages) through polycarbonate filters (pore size 100 nm) mounted in a LipoFast miniextruder (Avestin, Ottawa, Canada) at 22 °C. Untrapped CF was removed by two subsequent gel filtrations (Sephadex G-50, GE Healthcare, Uppsala, Sweden) at 22 °C, with Tris buffer as eluent. CF release from the liposomes was determined by monitoring the emitted fluorescence at 520 nm from a liposome dispersion (10 μm lipid in 10 mm Tris, pH 7.4). An absolute leakage scale was obtained by disrupting the liposomes at the end of each experiment through addition of 0.8 mm Triton X-100 (Sigma). A SPEX-fluorolog 1650 0.22-m double spectrometer (SPEX Industries, Edison, NJ) was used for the liposome leakage assay. Measurements were performed in triplicate at 37 °C.
Ellipsometry
Peptide adsorption to supported lipid bilayers was studied in situ by null ellipsometry, using an Optrel Multiskop (Optrel, Kleinmachnow, Germany) equipped with a 100-milliwatt Nd:YAG laser (JDS Uniphase, Milpitas, CA). All measurements were carried out at 532 nm and an angle of incidence of 67.66° in a 5-ml cuvette under stirring (300 rpm). Both the principles of null ellipsometry and the procedures used have been described in detail before (39). In brief, by monitoring the change in the state of polarization of light reflected at a surface in the absence and presence of an adsorbed layer, the mean refractive index (n) and layer thickness (d) of the adsorbed layer can be obtained. From the thickness and refractive index the adsorbed amount (Γ) was calculated according to,
 |
where n0 is the refractive index of the bulk solution (1.3347), and dn/dc the refractive index increment (0.154 cm3/g). Corrections were routinely done for changes in bulk refractive index caused by changes in temperature and excess electrolyte concentration.
LPS-coated surfaces were prepared by adsorbing E. coli LPS to methylated silica surfaces (surface potential, obtained from electro-osmosis, −40 mV, contact angle 90° (40) from 5 mg/ml of LPS stock solution in water at a concentration of 0.4 mg/ml over a period of 2 h. This results in a hydrophobically driven LPS adsorption of 1.48 ± 0.38 mg/m2, corresponding to a plateau in the LPS adsorption isotherm under these conditions, with an approximate area per acyl group of 200 Å2, assuming an LPS molecular weight of 104, and 6 acyl chains per LPS. Non-adsorbed LPS was removed by rinsing with Tris buffer at 5 ml/min for 30 min, and allowing buffer stabilization for 20 min. The LPS surfaces thus obtained displayed similar peptide adsorption as surfaces for which LPS was incorporated into preformed DOPE/DOPG lipid bilayers (41). Peptide addition was performed at different concentrations of 0.01, 0.1, 0.5, and 1 μm, and the adsorption monitored for 1 h after each addition. All measurements were performed in at least duplicate at 25 °C.
For lipid A deposition, this was solubilized with 0.25 wt % triethylamine under vigorous vortexing and heating to 60 °C for 10 min. Lipid A was adsorbed at methylated silica surfaces for 2 h from 5 mg/ml of lipid A stock solution in 0.25% triethylamine at a concentration of 0.2 mg/ml in 10 mm Tris, pH 7.4, containing 150 mm NaCl. Non-adsorbed lipid A was subsequently removed by rinsing with the same buffer at 5 ml/min for 15 min, followed by buffer stabilization for 20 min. This results in a lipid A adsorption of 0.8 ± 0.2 mg/m2. Peptide addition was subsequently performed to 0.01, 0.1, 0.5, and 1 μm, and adsorption was monitored for 1 h after each addition. (Absence of transient maxima in the time-resolved adsorption curves after peptide addition indicated competitive displacement to be unlikely for the LPS and lipid A substrates.) All measurements were performed in at least duplicate at 25 °C.
Supported lipid bilayers were generated by liposome adsorption. Anionic DOPE/DOPG (75/25 mol/mol) liposomes were prepared as described above, but the dried lipid films were resuspended in Tris buffer only with no CF present. To avoid adsorption of peptide directly at the silica substrate (surface potential −40 mV, contact angle <10°) (39) through any defects of the supported lipid layer, poly-l-lysine (molecular mass = 170 kDa, Sigma) was preadsorbed from water prior to lipid addition to an amount of 0.045 ± 0.01 mg/m2, followed by removal of nonadsorbed poly-l-lysine by rinsing with water at 5 ml/min for 20 min (42). Water in the cuvette was then replaced by buffer also containing 150 mm NaCl, followed by addition of liposomes in buffer at a lipid concentration of 20 μm, and subsequently by rinsing with buffer (5 ml/min for 15 min) when the liposome adsorption had stabilized. The final layer formed had structural characteristics (thickness 4 ± 1 nm, mean refractive index 1.47 ± 0.03), suggesting that a layer fairly close to a complete bilayer is formed. After lipid bilayer formation, the cuvette content was replaced by 10 mm Tris buffer at a rate of 5 ml/min over a period of 30 min. After stabilization for 40 min, peptide was added to a concentration of 0.01 μm, followed by three subsequent peptide additions to 0.1, 0.5, and 1 μm, in all cases monitoring the adsorption for 1 h. All measurements were made in at least duplicate at 25 °C.
Isothermal Titration Calorimetry (ITC)
A VP-ITC microcalorimeter (Micro Cal Inc., Northampton, MA) was used to perform the ITC experiments in triplicates to assess the binding interaction of KYE28/KYE28A with LPS. Peptide and lipid stocks were prepared in 10 mm phosphate buffer, pH 7.4, and degassed. A sample volume of 300 μl of 50 μm LPS was titrated with KYE28/KYE28A at rate of 2 μl/injection at an interval of 180 s, at 308 K, in a set of 20 injections. The raw data were plotted using MicroCal Origin 5 software to obtain thermodynamic parameters, including the association constant (Ka), change in enthalpy (ΔH), free energy of binding (ΔG), and entropy (ΔS). The data were fitted in a single site binding model using Equations 3 and 4.
Detached Leaf Assay
Healthy leaves from 60-day-old tomato plants were excised with a sterile blade and placed on wet tissue in separate sterile Petri plates. The leaves were grouped into three, representing control, infected (bacteria alone), and treated (bacteria + peptide) sets. A log phase X. vesicatoria culture, washed twice and re-suspended in 10 mm phosphate buffer corresponding to 4 optical density was used to set up the reaction. 50 μl of bacterial suspension was incubated with 50 μl of peptide at 50 μm concentration. A control was maintained containing 50 μl of bacterial suspension and 50 μl of buffer. All reactions were incubated at 28 °C for 3 h. 10 μl of the reaction mixture was loaded on the lower surface of each leaf and spread all over the surface. Autoclaved buffer was applied to the control set of leaves. Only bacterial suspension was applied to the infected set. The peptide-treated bacterial suspension was applied to the treated set of leaves. The leaves were kept at 25 °C in a photoperiod of 12 h of light and 12 h of dark. The leaves were observed for symptom development until 10 to 12 days post-infection. The experiment was conducted in triplicates. Three biological replicates were used.
Results and Discussion
Solution Structure of KYE28 in LPS Micelle
Recent investigations on KYE28 had shown potent activity of the peptide against Gram-negative E. coli and P. aeruginosa. It demonstrated a reduction in the levels of LPS-induced pro-inflammatory cytokines both in vitro and in mouse models. In addition, the peptide was able to neutralize LPS itself very effectively. To analyze the structural parameters responsible for these activities, we here carried out a structural characterization of KYE28 in LPS. First, we investigated the secondary structure of the peptide in the presence of LPS using CD spectroscopy. The CD spectrum of KYE28 in aqueous solution showed a strong negative band near 200 nm corresponding to random coil conformation of the peptide in free state at both pH 7.4 (physiological pH) as well as at pH 4.5 (NMR experimental condition). This is in accordance with previous reports that suggest that many AMPs assume random coil conformations in aqueous solution (41). Upon addition of LPS at a concentration greater than its CMC, a drastic change occurred in the CD profile at both pH conditions (pH 4.5 and 7.4), indicating structural changes in KYE28 upon binding to LPS (Figs. 2A, left panel, and supplemental S1, upper panel). A positive maxima at 195 nm and two strong negative peaks, one at 208 nm and a shoulder of lower intensity at 222 nm, were observed, suggesting an α-helical conformation of the peptide in LPS under both pH conditions. Deconvolution of the CD spectrum using CDNN software (43) showed a helical content of ∼20%, turn conformation of 18%, parallel and antiparallel β sheet conformation of 10 and 12%, respectively, and random coil conformation of ∼40% (Fig. 2A, right panel). However, it may be noted that deconvolution with CDNN is merely supportive and only included for comparison purposes, considering that such evaluations may be skewed to some extent by the databases being based on globular proteins rather than linear amphipathic peptides. To gain further insight into the KYE28 structure in LPS at the atomic resolution, small amounts of LPS micelles were added to a sample containing KYE28 and monitored using one-dimensional 1H NMR spectra (44). In doing so, line broadening was observed for peptide signals (without causing any chemical shift perturbation), indicating fast conformational exchange between free and bound forms of the peptide. It is important to note here that LPS forms high molecular weight aggregates/micelles at very low concentrations (14 μg/ml) (45). This motivated us to determine the three-dimensional structure of KYE28 bound to LPS micelles with the well known trNOESY method used to study macromolecule-ligand interaction having Kd values in the millimolar to micromolar range (22, 46). The two-dimensional 1H-1H NOESY spectra of KYE28 in aqueous solution showed only intra-residual and sequential NOEs between the backbone and side chain protons (supplemental Fig. S2), supporting lack of any folded conformation, as obtained from CD spectrum as well. In contrast, the trNOESY spectrum of KYE28 in the presence of LPS was characterized by a large number of medium- and long-range NOE cross-peaks (Fig. 2B and supplemental Fig. S2). Additionally, the trNOESY spectra of KYE28 in both E. coli and P. aeruginosa LPS showed a perfect superimposition, indicating that the peptide adopted a similar structure in both contexts, irrespective of different O-antigen moieties (Fig. 2C). As seen in Fig. 2, B and D, medium-range NOEs αN (i, i+2/i+3/i+4) were seen between residues Tyr2/Thr6, Ile4/Ile7, Ile4/His8, Thr6/His8, His8/Leu10, His16/Phe19, Asn22/Gly24, and Phe23/Tyr25. In addition, there were several side chain NOEs between aliphatic amino acid residues and aromatic ring protons (Fig. 2B). There were also a large number of long-range unambiguously assigned trNOE contacts between Thr6/Tyr25, Leu10/Phe19, Phe11/Phe19, Leu18/Phe23, Phe19/Tyr25, Tyr25/Tyr2, Thr26/Tyr2, and Leu27/Tyr2 (Fig. 2B, supplemental Table S1). Note that the aromatic peaks for Phe residues 11, 19, and 23 of KYE28 were unambiguous in this sample condition. A closer look at the fingerprint regions of the spectra revealed several αN (i,i+2/i+3/i+4) trNOEs for the residues from Lys1 to Phe11 and from His16 to Gly24, signifying α helical pattern in these regions. Also, a plot of ΔHα (deviation in chemical shift values) of the CαH against the amino acid residues showed consecutive upfield shifted values from the chemical shifts of random coil conformation, for residues Lys1 to Phe11 and His16 to Gly24, confirming α helical populations in these segments (supplemental Fig. S3). In addition, NN(i,i+2) trNOEs between Ile7/Asn9, Asn9/Phe11, and Phe19/Arg21 (Fig. 2, B and D) were also observed. Collectively, aromatic amino acid residues (Tyr2, Phe11, Phe19, Phe23, and Tyr25) and hydrophobic amino acid residues (Ile7, Leu10, and Leu27) of KYE28 showed major hydrophobic interactions (Fig. 2E).
FIGURE 2.

Structural characteristics of KYE28 in LPS. A, CD spectra of KYE28 alone and in the presence of LPS, showing adoption of an α-helical pattern of KYE28 upon addition of LPS (left panel). Also shown is a bar plot showing the content of the secondary structure of KYE28 in LPS obtained from deconvolution of the CD spectra using CDNN software (right panel). B, different regions of the trNOESY spectra of KYE28 in LPS showing important medium-, long-range, sequential NH-NH, and CαH NOE contacts. The experiment was performed using Bruker Avance III 700 MHz NMR spectrometer (150 ms NOESY mixing time and 25 °C). C, an overlay of the trNOESY spectra of KYE28 in E. coli 0111:B4 LPS and P. aeruginosa LPS, showing almost identical patterns, indicating similar structural conformation in both types of LPS (irrespective of the O-antigen moieties). D, bar diagram showing sequential and medium-range NOEs of KYE28 in LPS. The bar thickness indicates the peak intensity assigned as strong, medium, and weak. E, histogram depicting the number of trNOEs of KYE28 in LPS with respect to residue numbers. Residues marked in red depict the residues involved in hydrophobic stabilization.
The three-dimensional structure of KYE28 in LPS was calculated using the NOE distance constraints obtained from the trNOESY spectra. Long-range NOE contacts between residues located at the two termini, notably Tyr2/Tyr25, Thr6/Tyr25, Tyr2/Thr26, and Tyr2/Leu27, suggested the possibility of formation of an anti-parallel dimer of KYE28. Therefore, we determined an antiparallel dimer conformation of KYE28 using the above short-, medium-, and long-range NOEs. However, the backbone and side chain root mean square deviation values of the calculated anti-parallel dimer conformation clearly indicated the lack of a well defined structure, as also evident from the poor superimposition of the backbone and side chain of 20 ensemble structures (supplemental Fig. S4, A and B). Only some of the long-range NOE contacts, such as Leu10/Phe19 and Phe11/Phe19, were satisfied in the antiparallel dimer conformation when used as inter-monomeric contacts (supplemental Fig. S4 C, marked in blue). In contrary, the majority of the prominent as well as unambiguously assigned long-range NOEs such as Tyr2/Tyr25, Thr6/Tyr25, Leu18/Phe23, Phe11/Tyr25, and Phe19/Tyr25 (supplemental Fig. S4C, marked in red) were incompatible and were not satisfied in the anti-parallel dimer conformation (supplemental Fig. S4B) due to the large violations in distance constraints (>1 Å) upon structure calculation. Consequently, no NOE cross-peaks were observed, corresponding to the short inter-proton distances between two monomeric units of the anti-parallel dimer conformation, viz. CαH/CαH or NH/NH NOE contacts (Fig. 2B). A parallel dimeric structure was similarly not feasible using the set of observed NOEs in the spectra.
Considering the incompatibility of the experimental results with an anti-parallel dimeric structure, we calculated a monomeric structure using the obtained NOE constraints. Interestingly, it gave rise to a structure closely resembling a “helix-loop-helix” motif, stabilized by an aromatic zipper (Fig. 3B) that showed a good convergence of backbone atoms (Cα, N, and C′) of all the 20 lowest energy structures (Fig. 3A), with an average backbone and heavy atom root mean square deviation values of 0.66 and 1.46 Å, respectively (Table 1). The LPS-bound folded structure of KYE28 was characterized by an N-terminal helical region Lys1-Phe11, followed by a loop, a short helical stretch, and an extended C terminus (Fig. 3B). This is in agreement with the deconvoluted CD data denoting the percentage secondary structure of KYE28 in LPS. The structure was stabilized by aromatic packing interactions majorly between Tyr2/Tyr25, Phe11/Phe19, Phe19/Phe23, Phe19/Tyr25, as well as by hydrophobic interactions between Tyr2/Leu27, Thr6/Tyr25, Ile7/Phe11, Leu10/Phe19, Leu18/Phe23, Phe23/Phe19, and Leu27/Tyr25 (Fig. 3B). This also correlates well with recent findings of a partial activity loss for KYE21 and NLF20, fragments obtained from KYE28 lacking the C-terminal and N-terminal ends, respectively (41). It is interesting to note that the positively charged Arg and Lys residues remain flexible and oriented at one particular face (Fig. 3B, highlighted), forming a polar exterior shell that may interact with the negatively charged phosphate and carboxyl groups of LPS. All aliphatic and aromatic residues, in turn, were packed together forming a core that may help the peptide to penetrate into the lipid bilayer. The electrostatic potential map also supports this observation (Fig. 3C).
FIGURE 3.

Bioactive conformation of KYE28 in LPS. A, an ensemble of KYE28 in LPS showing the superposition of the backbone atoms (N, Cα, and C′) of the 20 lowest energy structures calculated using CYANA 2.1 software. The PDB acquisition code is 2NAT. B, schematic representation of a representative structure of the LPS-bound form of KYE28, showing the orientation of the positive charges forming a cationic face highlighted in violet (left panel) and the hydrophobic residues forming an inner core through interaction between its side chains shown in stick representation thus adopting an amphipathic orientation (middle panel). Shown also is a schematic representation of KYE28 showing the aromatic zipper composed of Tyr2, Phe11, Phe19, Phe23, and Tyr25, highlighted in spheres, which drives the stabilization of the folded conformation of KYE28 in LPS (right panel). C, electrostatic surface potential of KYE28 in LPS micelles in two different orientations at an angle of 180° to each other showing the surface distribution of charges.
TABLE 1.
Summary of structural statistics for the 20 lowest energy ensemble structures of KYE28 and KYE28A in LPS
| Distance restrains | KYE28 | KYE28A |
|---|---|---|
| Intra-residue (i–j = 0) | 74 | 60 |
| Sequential (|i–j| = 1) | 112 | 111 |
| Medium-range (2≤ |–j|≤4) | 26 | 18 |
| Long-range (|i–j| ≥5) | 18 | 0 |
| Total | 230 | 189 |
| Angular restraints | 40 | 40 |
| Φ | 27 | 27 |
| Ψ | 27 | 27 |
| Distance restraints from violations (≥ 0.4 Å) | 0 | 0 |
| Deviation from mean structure (Å) | ||
| Average back bone to mean structure | 0.66 ± 0.24 | 2.55 ± 0.68 |
| Average heavy atom to mean structure | 1.46 ± 0.33 | 3.44 ± 0.62 |
| Ramachandran plot for mean structure | ||
| % Residues in the most favourable and additionally allowed regions | 100 | 100 |
| % Residues in the generously allowed Region | 0 | 0 |
| % Residues in the disallowed region | 0 | 0 |
The aromatic-aromatic or aromatic-aliphatic packing, which resulted in bringing the two terminal ends in close proximity to each other, as determined using trNOESY methods in the presence of LPS, is common for many AMPs. One such example is the recently reported 16-residue AMP, designed from the dengue viral fusion peptide, which also had a single N-terminal helical turn and a looped out structure, with N and C termini brought close to each other via packing interactions between the centrally located Trp5 and Phe12 (28). Similarly, MSI-594 (36), consisting of 24 amino acid residues (PDB acquisition code 2K98), or paradaxin (22), consisting of 33 amino acid residues (PDB acquisition code 2KNS), in LPS also adopt helical hairpin conformations to stabilize the peptide through Phe and other aliphatic amino residues.
Therefore, AMPs with helical hairpin or helix-loop-helix motifs, stabilized via aromatic-aliphatic residue interactions seem to be favorable for permeabilization of the outer membrane of Gram-negative bacteria. Furthermore, helical conformations have been previously suggested to stabilize folded structures of AMPs on the bacterial membrane surface, helping in anchoring, which subsequently leads to their translocation and membrane disruption (47, 48). Based on this, we propose that the N-terminal helical portion of KYE28 helps in anchoring the lipid membrane, translocating the peptide into the outer leaflet and stabilizing it through hydrophobic interactions between the acyl chains of LPS and the peptide hydrophobic core.
Design and Antimicrobial Activities of the Mutant Peptide KYE28A
To further probe the importance of aromatic packing interactions involved in the antimicrobial activity of KYE28, we carried out Ala substitutions at the critical aromatic residues Phe11, Phe19, Phe23, and Tyr25, which were primarily seen to be involved in stabilizing the folded conformation of KYE28 bound to LPS. This resulted in the analogue KYE28A (Fig. 1). Next, to monitor the consequences of these substitutions, antimicrobial assay of both the parent peptide KYE28 and the mutant KYE28A were performed against Gram-negative plant pathogens X. oryzae and X. vesicatoria in addition to S. typhi, K. pneumoniae, and the fungal pathogens C. albicans and C. neoformans. It should here be noted that X. oryzae and X. vesicatoria are devastating plant pathogens causing wilts and bacterial spots in their host plants leading to considerable food-crop losses worldwide (3, 4). The MIC values demonstrated that KYE28 efficiently inhibits all the tested strains at micromolar concentrations, both in the absence and presence of high salt concentrations (150 mm) (Table 2). Notably, KYE28 was most active against X. vesicatoria and X. oryzae, having a MIC of 1 and 5 μm, respectively, although it displayed 2–5-fold higher MIC values for the other tested pathogens in the presence of salt. In contrast, KYE28A required 3- and 25-fold higher concentrations to completely inhibit X. oryzae and X. vesicatoria in the absence of salt. Similarly, it required more than 2–5-fold higher concentrations to inhibit C. neoformans, K. pneumonia, and S. typhi, respectively, in the absence of salt (Table 2). In contrast to KYE28, KYE28A could not inhibit any of the pathogens in the presence of salt up to a concentration of 50 μm, except for X. oryzae (Table 2).
TABLE 2.
MICs of the peptides KYE28 and KYE28A (assay conducted with 105 cells/ml)
| Strains | KYE28 |
KYE28A |
||
|---|---|---|---|---|
| In presence of 150 mm NaCl | 10 mm Phosphate buffer | In presence of 150 mm NaCl | 10 mm Phosphate buffer | |
| C. neoformans | 50 μm | 25 μm | >50 μm | 50 μm |
| C. albicans | >50 μm | 25 μm | >50 μm | >50 μm |
| S. typhi | 50 μm | 10 μm | >50 μm | >50 μm |
| K. pneumoniae | 25 μm | 10 μm | >50 μm | >50 μm |
| X. vesicatoria | 1 μm | 1 μm | >50 μm | 25 μm |
| X. oryzae | 5 μm | 5 μm | 50 μm | 15 μm |
KYE28 Exerts Antimicrobial Activity through Membrane Disruption
Further biophysical studies were performed with both the native and mutant peptide to assess the membrane destabilizing behavior of the peptides. An increased DPH fluorescence in the presence of LPS bicelles, or in the presence of E. coli total lipid extract bicelles, upon peptide addition was taken as a measure of bicelle disruption mediated by peptide anchoring and translocation. In aqueous solution, DPH showed only weak fluorescence, which, however, is enhanced by peptide-induced disruption of bicelles and exposure of the hydrophobic acyl chains of the lipids to DPH. The native peptide KYE28 was found to cause disruption of both E. coli total lipid extract bicelles (Fig. 4A, left panel) and LPS bicelles (Fig. 4A, right panel) in a concentration-dependent manner, evident from a steady increase in the DPH fluorescence intensity. In contrast, KYE28A caused disruption of the bicelles to a much lower extent (Fig. 4A), showing an ∼6-fold lower increase in DPH intensity in the case of E. coli total lipid extract bicelle and ∼12-fold lower increase in the case of LPS bicelle. It may be noted that KYE28 showed a greater increase in DPH intensity in LPS bicelles, starting from 20 μm and reaching 2-fold higher values at 35 μm peptide concentration when compared with E. coli total lipid extract bicelles. This may be attributed to either a preferential binding of KYE28 to LPS bicelles causing increased disruption, or effects of the difference in the hydrophobic environments displayed by LPS and the E. coli total lipid extract component lipids with which DPH interacts upon bicelle disruption. KYE28A, however, showed similar patterns of fluorescence increase in both cases. Next, outer membrane permeabilization of X. vesicatoria was determined using NPN dye uptake assay. NPN, being a neutral hydrophobic fluorescent probe, generally remains excluded by the outer membrane of Gram-negative bacteria. However, the dye exhibits an increase in fluorescence intensity on interaction with, and disruption of, the parted outer membrane. A steady increase in NPN fluorescence was observed in a concentration-dependent manner, showing a maximum of around 80% increase in NPN uptake upon addition of 30 μm KYE28 peptide (Fig. 4B), whereas a 66% increase in NPN uptake was observed for as low as 1 μm KYE28 (i.e. its MIC) (Fig. 4B). In contrast, KYE28A showed only 40% increase in NPN uptake at a peptide concentration of 30 μm (Fig. 4B). Taken together, these data further support the importance of the aromatic residues in KYE28 that upon Ala substitution resulted in attenuated activity. In addition, carboxyfluorescein leakage from DOPE/DOPG (75/25 mol/mol) vesicles as a consequence of peptide binding and vesicle disruption was studied. KYE28 and KYE28A both displayed concentration-dependent permeabilization of DOPE/DOPG liposomes (Fig. 4C). Quantitatively, KYE28 was found to be only slightly more potent than KYE28A in causing membrane rupture, demonstrating that under such low ionic strength conditions, electrostatics dominate as the driving force for lipid membrane de-stabilization as both KYE28 and KYE28A have the same number of positively charged Arg and Lys residues in place. This is in agreement with the antimicrobial activity assay data, where KYE28A displayed some antimicrobial effect in the absence of salt, but failed to do so at the same concentrations in the presence of salt.
FIGURE 4.

Membrane permeabilization effects of KYE28 and KYE28A. A, bar plots showing the increase in DPH fluorescence as a function of peptide concentration upon addition of peptide to E. coli total lipid extract bicelle (left) and LPS bicelle (right) as a measure of bicelle disruption. KYE28 is seen to cause a pronounced concentration-dependent increase in DPH fluorescence in both E. coli total lipid extract bicelles and LPS bicelles, much larger than those caused by KYE28A. B, plot showing percentage of NPN dye uptake by X. vesicatoria cells upon addition of KYE28 and KYE28A. More than 2-fold higher uptake is seen for KYE28 as compared with KYE28A. C, percentage of dye leakage from DOPE/DOPG (75/25 mol/mol) vesicles as a function of peptide concentration. Both KYE28 and KYE28A induced comparable dye leakage from vesicles, KYE28 producing slightly greater effects.
Binding Interactions of KYE28 and KYE28A to Lipid Bilayers
To elucidate binding interactions between KYE28/KYE28A and LPS or DOPE/DOPG bilayers, ellipsometry measurements were carried out. In analogy to anionic DOPE/DOPG membranes, both KYE28 and KYE28A display extensive binding to E. coli LPS, as well as to its lipid A moiety (Fig. 5, A and B). This is reasonable, because both peptides are strongly positively charged, and KYE28 also quite hydrophobic, whereas LPS carries a negative charge due to its phosphate and carboxylate groups (49) and its lipid A moiety is obviously also hydrophobic. In analogy to other hydrophobically modified biopolymers, e.g. peptidoglycans (50), LPS is likely to interact with the hydrophobic (θ ≈ 90°) and negatively charged (z ≈−40 mV) methylated silica surface through its lipid A moiety. At the relatively high LPS densities reached in these experiments (1.4 mg/m2), the lipid A moiety will therefore be partly screened by the LPS carbohydrate chains, hence not fully accessible to peptide binding. Because the carbohydrate moieties are much longer than lipid A, they are expected to have a larger peptide binding capacity than lipid A (51). Despite this, KYE28 binding is quite comparable for LPS and lipid A, suggesting a higher binding affinity of this peptide to lipid A. KYE28A, on the other hand, displays a considerably lower binding affinity for lipid A, an obvious consequence of the deletion of aromatic amino acids in this peptide. As can be seen in Fig. 5C, both KYE28 and KYE28A display concentration-dependent binding to DOPE/DOPG bilayers, reaching adsorption densities at 1 μm of 330 and 276 nmol/m2, corresponding to about 10 and 12 lipid molecules per peptide, respectively. The lower membrane binding displayed by KYE28A is due to the absence of the aromatic residues, in line with previous findings of the membrane-binding driving force provided by such aromatic amino acid residues (52). ITC experiments corroborated this. Downward profiles of ITC suggest an exothermic or enthalpy driven reaction in both the cases of KYE28 (Fig. 5D) and KYE28A (Fig. 5E) binding to LPS. Quantitatively, however, the binding interaction of KYE28 to LPS was found to be substantially stronger, having a dissociation constant of 35 μm, a ∼5 fold lower value compared with that of KYE28A. Again, this signifies a stronger binding of KYE28 to LPS because of the aromatic residues Tyr2, Phe11, Phe19, Phe23, and Tyr25 that were crucial in mediating hydrophobic interactions with the acyl chains of LPS. All the thermodynamic parameters of KYE28/KYE28A binding to LPS are summarized in Fig. 5, D and E.
FIGURE 5.

Binding of KYE28 and KYE28A to LPS, lipid A, and DOPE/DOPG. Ellipsometry results on the binding of LPS (A), lipid A (B), and DOPE/DOPG bilayers (C). Also shown are plots of the heat of reaction, obtained from ITC experiments, for the binding between KYE28 (D) or KYE28A (E) and LPS, as a function of peptide/LPS molar ratio. The data were fitted into single site binding equation using MicroCal Origin 7 software after performing corrections for heat of dilution of peptide. Average values of triplicates have been reported with error.
Structural Aspects of KYE28A in LPS
To elucidate the consequences of alanine mutations on the structural aspects of KYE28A, CD experiments were performed in the presence of LPS. The CD spectrum of KYE28A alone in aqueous solution was characteristic of a random coil nature. However, upon addition of LPS, a positive maxima at 195 nm and two negative maxima at 208 and 222 nm appeared, a signature of α helix conformation. Surprisingly, the molar ellipticity peak at 222 nm was lower in intensity and broader in nature, indicating a large extent of dynamicity in the α-helical content (Fig. 6A). CD data deconvolution reflected an α-helical content, amounting to 28% of the total. In addition, 37.5% random coil conformation, 15.5% turn conformation, and nearly 10 and 9% in parallel and antiparallel β sheet conformation, respectively, were observed for KYE28A in LPS (Fig. 6A). To correlate the structural attributes of KYE28A with its observed attenuation in antimicrobial activity in the presence of salt and retention of only partial activity in the absence of salt, we elucidated its three-dimensional structure, using a two-dimensional 1H-1H trNOESY experiment in the presence of LPS. The trNOESY spectra revealed only sequential αN/NN(i,i+1) and medium-range αN(i,i+2/i+3) NOE contacts (Fig. 6, B and D). No long-range NOEs were observed (Fig. 6E). The superposition of aromatic regions of the trNOESY spectra of KYE28 (marked in red) and KYE28A (marked in blue) in the presence of LPS (Fig. 6C) thus indicated that there were no long-range NOE contacts of KYE28A in the presence of LPS. αN(i,i+3/i+4) NOE contacts were seen between residues Ile4/Ile7, Thr6/Asn9, Lys13/His16, Lys13/Arg17, and between Leu14/Arg17 or Leu14/Leu18 (Fig. 6B), which is a signature of α-helix formation. An αN(i,i+2) NOE contact was also seen between Ile4 and Thr6 (Fig. 6B). The ΔHα values of CαH were furthermore, seen to be shifted upfield in LPS for residues Tyr2 to His8 and Leu14 to Phe19 (supplemental Fig. S3), signifying that these regions were α-helical in nature. However, smaller deviations in the chemical shift values for KYE28A compared with KYE28 indicated that the KYE28A structure was a loosely bound one. This data agrees well with the CD results (Fig. 6A). As a consequence, KYE28A showed fewer NOE cross-peaks in LPS (Fig. 6E) compared with KYE28 in LPS (Fig. 2E). This indicated the absence of a well defined structure of KYE28A in the presence of LPS, as also evident from an absence of convergence of the 20 lowest energy structures (Fig. 6F), as well as high root mean square deviation values for both backbone and heavy side chain atoms (Table 1). With these reservations, the three-dimensional structure of KYE28A (Fig. 6G) showed an N-terminal helical segment from residues Tyr2 to Leu18 and an extended C terminus, where the α-helix discontinuation at Ala19 could be due to the presence of the two consecutive positively charged Arg residues of Arg20 and Arg21. This is supported by reports on a centrosymmetric designed α-helical peptide motif of 7 residues according to which a hydrophobic residue at the 12th position and a positively charged residue at the 9th and 13th position would favor the formation of a continuous helix (53). The presence of an Asn residue, Asn22, may also contribute to the break as Asn is usually found in turns or in turns of β sheets due to the fact that its side chain can form hydrogen bond with the peptide backbone, thereby disrupting the α-helical structure (54). The peptide structure revealed that the positively charged residues Arg12, Lys13, Arg17, and Arg20 were present at the central region of KYE28A and oriented toward one face of the helix, maintaining a specific pattern dictated by a motif 12RXXXH16XXXR20 and 13KXXXR17XXXR21 (Fig. 6G). This motif is thought to be primarily responsible for governing electrostatic interactions with negatively charged phosphate head groups of LPS (23). It is furthermore, interesting to note the orientation of His16, which allowed it to be placed in the polar face of the molecule, aiding it to interact with the negatively charged phosphate head group of LPS. Overall, stabilization of the KYE28A structure in the presence of LPS is predominantly electrostatically driven, due to the presence of 8 positively charged amino acid residues in its sequence. In contrast, the absence of aromatic residues in KYE28A led to a destabilization of its hydrophobic core and formation of a longer helical segment, mediated by the substituted alanine. It is to be noted that alanine is well known to have a high α-helical propensity among all amino acids (53, 55). Apart from the strong electrostatic interaction between positively charged amino acid residues of KYE28A and phosphate head groups of LPS, the peptide was also stabilized by hydrophobic interactions such as 4IXXI7XXXA11XXL14XXXL18in the hydrophobic face of the structure. This observation of binding of either KYE28 or KYE28A to LPS is well supported by ellipsometry measurements, showing similar affinities of KYE28 and KYE28A to LPS only at low ionic strength conditions, as well as substantially lower affinity for KYE28A to lipid A.
FIGURE 6.

Structural characterization of KYE28A in LPS. A, CD spectra of KYE28A alone and in LPS, showing adoption of an α-helical pattern of KYE28A upon addition of LPS (left panel). A bar plot showing the content of secondary structure of KYE28A in LPS obtained from deconvolution of the CD spectra using CDNN software is also included (right panel). B, different regions of the trNOESY spectra of KYE28A in LPS showing important medium- and long-range NOE contacts. The experiments were performed using Bruker Avance III 700 MHz NMR spectrometer (150 ms NOESY mixing time and at 25 °C). C, an overlay of the aromatic region of the trNOESY spectra of KYE28 (red) and KYE28A (blue) in E. coli 0111:B4 LPS showing a complete loss of NOEs in case of KYE28A. D, bar diagram showing sequential and medium-range NOEs of KYE28A in LPS. The bar thickness indicates the peak intensity assigned as strong, medium, and weak. E, histogram depicting the number of trNOEs of KYE28A in LPS with respect to residue numbers. F, an ensemble of KYE28A in LPS showing the superposition of the backbone atoms (N, Cα, and C′) of the 20 lowest energy structures calculated using CYANA (PDB acquisition code 2NAU). G, representative schematic structure of KYE28A in LPS showing the orientation of the positive charges (sphere representation) over one side of the helical segment (left panel) maintaining a specific pattern dictated by 12RXXXH16XXXR20 or 13KXXXR17XXXR21 motif, signifying interaction with negatively charged monophosphate and diphosphate groups of LPS through either electrostatic interaction or hydrogen bonding. The hydrophobic residues remain oriented toward the other end maintaining an amphipathic orientation (left panel). H, electrostatic surface potential of KYE28A in LPS micelles in two different orientations at an angle of 180° to each other showing the surface distribution of charges.
Overall, this study provides molecular insights into the mechanism of action of KYE28. An aromatic zipper was found to be principally responsible for its biological activities, whose substitution with alanine in KYE28A led to an amphipathic but bent helical structure (Fig. 7A) with much reduced activity both in terms of antimicrobial effects in presence of high salt concentrations and LPS binding and neutralization. Again, this is analogous to findings on a mutant of a designed peptide, MSI-594, for which a Phe5 was substituted with an Ala, leading to disruption of the helix-loop-helix motif and formation of a curved helix (Fig. 7B) (36). Thus, it was found that the Phe5 residue in MSI-594 plays a crucial role in inter-helical packing interactions, removal of which completely abolished its broad spectrum antimicrobial activity against Gram-negative bacteria and led to reduced binding affinity to LPS. Similarly, in Pardaxin, a naturally occurring antimicrobial peptide found in sole fishes, Phe15 was reported to play a major role in mediating hydrophobic contacts between the nonpolar faces of the two helical segments (24) (Fig. 7C).
FIGURE 7.

Comparison of structural motifs of KYE28/KYE28A with MSI-594/MSI-594F5A and Pardaxin to elucidate importance of hydrophobic cluster. A, NMR derived three-dimensional structure of KYE28 and KYE28A showing the importance of the aromatic zipper (sphere representation) in stabilization of the helix-loop-helix motif; B, NMR derived three-dimensional structure of MSI-594 (PDB accession code 2K98) showing a similar helix-loop-helix motif stabilized by F5 (44). The removal of F5 (PDB accession code 2L36) (36) leads to an opened out curved helix yet again indicating the importance of the hydrophobic interactions mediated by the aromatic residue in forming the helix loop helix motif. C, NMR derived helical hairpin structure of Pardaxin in LPS (PDB accession code 2KNS). Phe15 plays an important role to stabilize the structure by connecting the two helical segments (22).
Residue-specific Interactions of KYE28/KYE28A with LPS
Residue-specific interactions of KYE28 and KYE28A with LPS were probed using STD NMR spectroscopy. This method is well known to recognize residues or groups in a ligand molecule that are involved in direct interaction with their high molecular weight binding partners. Fig. 8 shows the reference one-dimensional proton spectrum of KYE28 (Fig. 8A) and KYE28A (Fig. 8B) and their corresponding STD spectra. In case of KYE28, large STD effects were observed for the aromatic proton resonances resonating between 6.7 to 7.5 ppm due to the ring protons of Tyr2/His8/Phe11/His16/Phe19/Phe23/Tyr25. Among these STD effects, unambiguous assignments were possible for Phe (Phe11, Phe19, and Phe23) aromatic protons of KYE28. This signifies the close proximity of these residues to LPS micelles. In addition, strong STD effects were observed between 0.8 and 1.3 ppm, corresponding to the alkyl side chain groups of Ile/Leu residues. Moderate STD effects were also observed between 1.4 and 1.9 ppm, which could originate from the alkyl side chain groups of the positively charged Lys/Arg residues. Moderate to low STD effects were also observed for the β protons of the aromatic residues resonating between 2.8 and 3.3 ppm. This provides additional evidence for the involvement of the aromatic residues in stabilization of the bound conformation of KYE28 in LPS micelles through hydrophobic interactions. In contrast, the STD spectrum of KYE28A quite obviously lacked most of the STD effects from the aromatic resonances. Instead, weak STD effects were observed for the ring proton and β protons of Tyr2. Strong STD effects were observed between 0.8 and 1.3 ppm, corresponding to the alkyl side chain groups of Ile/Leu and the substituted Ala, showing their involvement in the interaction and stabilization of KYE28A in LPS micelles. Moderate STD effects, corresponding to the side chain groups of the positively charged residues, similar to those of KYE28, were also observed between 1.4 and 1.9 ppm. Due to severe signal overlap in certain regions of the one-dimensional STD spectra in both cases, however, residue-specific information could not be obtained for all participating residues of KYE28/KYE28A. Nevertheless, total STD effects from the methyl protons of Leu/Ile (resonating between 0.8 and 1 ppm) and β/γ protons of Leu/Ile/Lys/Arg (resonating between 1 and 1.8 ppm) in the case of KYE28 were found to be 2-fold higher than for KYE28A, signifying that these residues in KYE28 were more closely placed in the LPS micellar environment. Taken together, these studies reveal the importance of the aromatic, hydrophobic, and positively charged residues in stabilization of the bound form of KYE28 in LPS.
FIGURE 8.
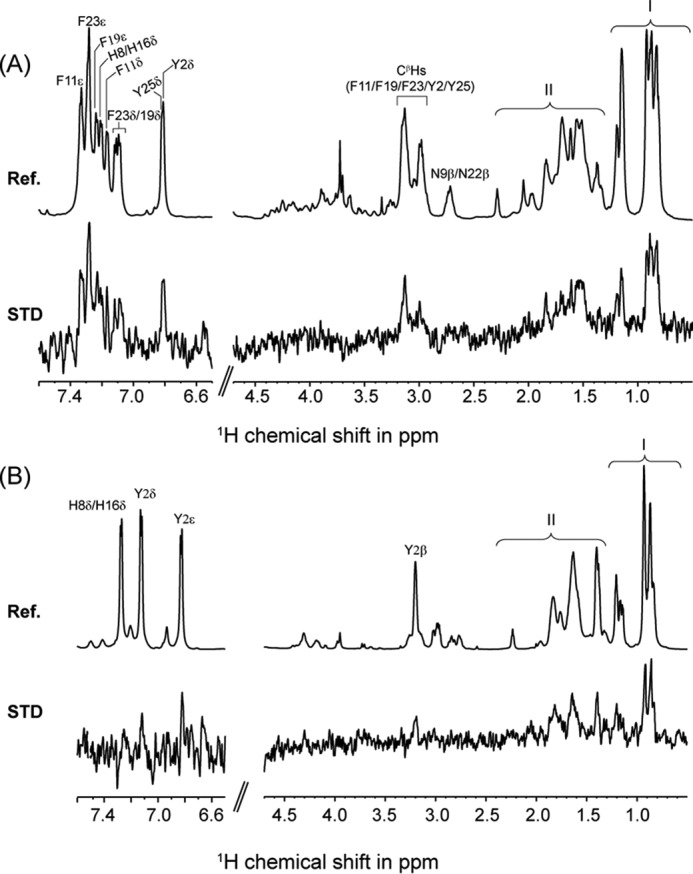
STD NMR and docking studies of interaction between KYE28/KYE28A and LPS. A, reference 1H NMR spectra and STD NMR spectra of KYE28 in LPS. The spectra show side chain resonances of aromatic amino acids Tyr2, His8, Phe11, His16, Phe19, Phe23,and Tyr25. Spectral regions indicated by I and II represent overlap in the signal intensities of the side chain resonances of Ile4, Ile7, Leu10, Leu14, Leu18, and Leu27, and positively charged residues Lys1, Arg12, Lys13, Arg20, Arg21, and Arg28, respectively. B, reference 1H NMR spectra and STD NMR spectra of KYE28A in LPS. The spectra shows side chain resonances of aromatic amino acid residue Tyr2, His8, and His16. Spectral regions indicated by I and II represent overlap in the signal intensities of the side chain resonances of Ile4, Ile7, Leu10, Ala11, Leu14, Leu18, Ala19, Ala23, Ala25, and Leu27, and positively charged residues Lys1, Arg12, Lys13, Arg20, Arg21, and Arg28, respectively.
Anti-inflammatory Effects of KYE28 and KYE28A
To assess to what extent the differences in LPS binding between KYE28 and KYE28A influence the anti-inflammatory effects of these peptides, the ability of the peptides to block LPS-triggered NF-κB activation in macrophages was investigated. As shown in Fig. 9, KYE28 suppressed NF-κB activation in a dose-dependent manner, with sizeable suppression already at 0.5–1 μm, i.e. around the MIC of this peptide. In contrast, KYE28A displayed essentially no blocking of NF-κB up to a peptide concentration of 50 μm. As shown by the corresponding MTT assay results, these effects are indeed due to differences in peptide blocking of LPS activation, and not due to peptide toxicity effects. Thus, the differences observed regarding peptide binding amount/affinity from ellipsometry and ITC, and regarding conformational aspects observed by the different NMR methodologies, do indeed translate to the anti-inflammatory function of these peptides. Conversely, this correlation between biological function and peptide structure on LPS binding suggests that the structural zipper motif identified may be a promising feature to aim for in the development of novel antimicrobial and anti-inflammatory peptide therapeutics and plant protection peptides.
FIGURE 9.

Suppression of LPS-triggered NF-κB activation by KYE28/KYE28A. A, effects of the indicated peptides on macrophages. RAW264.7 macrophages were incubated with LPS from E. coli in the presence of peptides at the indicated concentrations, followed by monitoring of NF-κB activation. B, MTT assay results on peptide toxicity, demonstrating that macrophages were not damaged by the peptides at the concentrations investigated.
Ex Vivo Inhibition of X. vesicatoria Infection by KYE28
The low MIC of KYE28 against the plant pathogen X. vesicatoria in the presence of high salt concentrations motivated us to study its effects in inhibiting disease in plants to obtain initial information on the feasibility of its use in treating plant diseases. Inhibition of X. vesicatoria disease symptoms in detached tomato leaves, as obtained from the detached leaf assay, revealed that upon treatment with KYE28 at a concentration of 50 μm, the disease symptoms were not seen in the treated set of leaves. This is in stark contrast to the infected leaves that had visible black spots on the leaf surface, a characteristic of X. vesicatoria infection (Fig. 10). Potent activities of the peptide against X. vesicatoria even in the presence of high salt concentrations may have important biological implications in application of the peptides to alleviate symptoms in diseased plants. Therefore, inhibition of X. vesicatoria disease in detached tomato leaves is encouraging enough to study the effects of the peptide in treating plant disease through external application or genetic engineering. Thus, there is further scope in analyzing the efficacy of the peptide in treating plant disease in vivo (development of transgenic line over-expressing the KYE28 peptide).
FIGURE 10.

KYE28 inhibition of X. vesicatoria disease symptoms in detached tomato leaves. Detached tomato leaves applied with autoclaved water (control), 2 optical density X. vesicatoria cell suspension in 10 mm phosphate buffer treated with 50 μm KYE28 (treated), and 2 optical density cell suspension alone (infected), 12 days post-infection showing black spots on the infected set of leaves.
In conclusion, rationally designed peptides provide a promising approach toward development of novel therapeutics endowed with improved antimicrobial and anti-inflammatory properties (56–58). In this broader context, the structural insights provided by KYE28 in LPS, identifying an aromatic zipper motif playing a crucial role in endowing KYE28 with potent antimicrobial activity, may help in the development of potent new antimicrobials with enhanced biological properties.
Author Contributions
A. B. and M. M. designed the research; A. D., D. B., S. S., A. G., A. S., M. M., and A. B. performed the experiments and analyzed the results; A. D., D. B., A. B., and M. M. wrote the manuscript. All authors reviewed the manuscript; M. M. and A. B. arranged funding for this work.
Supplementary Material
Acknowledgments
We acknowledge DBT, Government of India, for infrastructure development funding (BT/PR3106/INF/22/138/2011) to Bose Institute for purchasing 700 MHz NMR spectrometer with cryoprobe. Ann-Charlotte Strömdahl and Lise-Britt Walberg are gratefully acknowledged for skillful technical assistance. We thank Prof. Pallob Kundu, Bose Institute, India, for stimulating discussions on plant experiments. We are grateful to Dr. Christian Lindermayer (Helmholtz-Münich, Germany) and Prof. Sampa Das (Bose Institute, India) for providing Xanthomonas bacterial strains. Dr. Kaustuv Sanyal (JNCASR, India) is thanked for providing Candida and Cryptococcus strains. The Central Instrument Facility (CIF) as well as the Madhyamgram Experimental Farm (MEF) of the Bose Institute are greatly acknowledged.
This work was supported in part by the Swedish Research Council Projects 2012–1842 (to M. M.) and 2012–1883 (to A. S.), Bose Institute Plan Project-II (to A. B.), India, and the Knut and Alice Wallenberg Foundation (to A. S.). M. M. and A. S. are founders of Ximmune AB, developing anti-inflammatory peptide therapeutics.

This article contains supplemental Table S1 and Figs. S1–S4.
The atomic coordinates and structure factors (codes 2NAT and 2NAU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AMP
- antimicrobial peptide
- HC-II
- heparin co-factor II
- trNOESY
- transferred nuclear Overhauser effect spectroscopy
- STD
- saturation transfer difference
- DOPG,1,2-dioleoyl-sn-glycero-3-phosphoglycerol
- monosodium salt
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- TEA
- triethyl amine
- CF
- carboxyfluorescein
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate
- DPH
- 1,6-diphenyl-1,3,5-hexatriene
- MTT
- 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide
- MIC
- minimal inhibitory concentration
- NPN
- 1-N-phenylnapthylamine
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank.
References
- 1. Levy S. B., and Marshall B. (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10, S122–129 [DOI] [PubMed] [Google Scholar]
- 2. Hancock R. E., and Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 3. Strange R. N., and Scott P. R. (2005) Plant disease: a threat to global food security. Annu. Rev. Phytopathol. 43, 83–116 [DOI] [PubMed] [Google Scholar]
- 4. Potnis N., Timilsina S., Strayer A., Shantharaj D., Barak J. D., Paret M. L., Vallad G. E., and Jones J. B. (2015) Bacterial spot of tomato and pepper: diverse Xanthomonas species with a wide variety of virulence factors posing a worldwide challenge. Mol. Plant Pathol. 16, 907–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Timilsina S., Jibrin M. O., Potnis N., Minsavage G. V., Kebede M., Schwartz A., Bart R., Staskawicz B., Boyer C., Vallad G. E., Pruvost O., Jones J. B., and Goss E. M. (2015) Multilocus sequence analysis of xanthomonads causing bacterial spot of tomato and pepper plants reveals strains generated by recombination among species and recent global spread of Xanthomonas gardneri. Appl. Environ. Microbiol. 81, 1520–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marco G., and Stall R. (1983) Control of bacterial spot of pepper initiated by strains of Xanthomonas campestris pv. vesicatoria that differ in sensitivity to copper. Plant Dis. 67, 779–781 [Google Scholar]
- 7. Makovitzki A., Viterbo A., Brotman Y., Chet I., and Shai Y. (2007) Inhibition of fungal and bacterial plant pathogens in vitro and in planta with ultrashort cationic lipopeptides. Appl. Environ. Microbiol. 73, 6629–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abbasi P. A., Khabbaz S. E., Weselowski B., and Zhang L. (2015) Occurrence of copper-resistant strains and a shift in Xanthomonas spp. causing tomato bacterial spot in Ontario. Can. J. Microbiol. 61, 753–761 [DOI] [PubMed] [Google Scholar]
- 9. Manzur J. P., Fita A., Prohens J., and Rodríguez-Burruezo A. (2015) Successful wide hybridization and introgression breeding in a diverse set of common peppers (Capsicum annuum) using different cultivated Ají (C. baccatum) accessions as donor parents. PLoS ONE 10, e0144142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuligowska K., Lütken H., Christensen B., Skovgaard I., Linde M., Winkelmann T., and Müller R. (2015) Evaluation of reproductive barriers contributes to the development of novel interspecific hybrids in the Kalanchoë genus. BMC Plant Biol. 15, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hancock R. E. (1997) Peptide antibiotics. Lancet 349, 418–422 [DOI] [PubMed] [Google Scholar]
- 12. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 13. Mangoni M. L., Epand R. F., Rosenfeld Y., Peleg A., Barra D., Epand R. M., and Shai Y. (2008) Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J. Biol. Chem. 283, 22907–22917 [DOI] [PubMed] [Google Scholar]
- 14. Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 15. Salnikov E. S., Aisenbrey C., Balandin S. V., Zhmak M. N., Ovchinnikova T. V., and Bechinger B. (2011) Structure and alignment of the membrane-associated antimicrobial peptide arenicin by oriented solid-state NMR spectroscopy. Biochemistry 50, 3784–3795 [DOI] [PubMed] [Google Scholar]
- 16. Lee D. K., Bhunia A., Kotler S. A., and Ramamoorthy A. (2015) Detergent-type membrane fragmentation by MSI-78, MSI-367, MSI-594, and MSI-843 antimicrobial peptides and inhibition by cholesterol: a solid-state nuclear magnetic resonance study. Biochemistry 54, 1897–1907 [DOI] [PubMed] [Google Scholar]
- 17. Epand R. M., and Vogel H. J. (1999) Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1462, 11–28 [DOI] [PubMed] [Google Scholar]
- 18. Hancock R. E., and Scott M. G. (2000) The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. U.S.A. 97, 8856–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hancock R. E. (1984) Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38, 237–264 [DOI] [PubMed] [Google Scholar]
- 20. Shai Y. (2002) Mode of action of membrane active antimicrobial peptides. Biopolymers 66, 236–248 [DOI] [PubMed] [Google Scholar]
- 21. Matsuzaki K. (1998) Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1376, 391–400 [DOI] [PubMed] [Google Scholar]
- 22. Bhunia A., Domadia P. N., Torres J., Hallock K. J., Ramamoorthy A., and Bhattacharjya S. (2010) NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: mechanism of outer membrane permeabilization. J. Biol. Chem. 285, 3883–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhunia A., Mohanram H., Domadia P. N., Torres J., and Bhattacharjya S. (2009) Designed β-boomerang antiendotoxic and antimicrobial peptides: structures and activities in lipopolysaccharide. J. Biol. Chem. 284, 21991–22004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papo N., and Shai Y. (2005) A molecular mechanism for lipopolysaccharide protection of Gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 280, 10378–10387 [DOI] [PubMed] [Google Scholar]
- 25. Cohen J. (2002) The immunopathogenesis of sepsis. Nature 420, 885–891 [DOI] [PubMed] [Google Scholar]
- 26. Kalle M., Papareddy P., Kasetty G., Tollefsen D. M., Malmsten M., Mörgelin M., and Schmidtchen A. (2013) Proteolytic activation transforms heparin cofactor II into a host defense molecule. J. Immunol. 190, 6303–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalle M., Papareddy P., Kasetty G., van der Plas M. J., Mörgelin M., Malmsten M., and Schmidtchen A. (2014) A peptide of heparin cofactor II inhibits endotoxin-mediated shock and invasive Pseudomonas aeruginosa infection. PLoS ONE 9, e102577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Datta A., Ghosh A., Airoldi C., Sperandeo P., Mroue K. H., Jiménez-Barbero J., Kundu P., Ramamoorthy A., and Bhunia A. (2015) Antimicrobial peptides: insights into membrane permeabilization, lipopolysaccharide fragmentation and application in plant disease control. Sci. Rep. 5, 11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marion D., Ikura M., Tschudin R., and Bax A. (1989) Rapid recording of 2D NMR spectra without phase cycling: application to the study of hydrogen exchange in proteins. J. Magn. Reson. 85, 393–399 [Google Scholar]
- 30. Sklenar V., Piotto M., Leppik R., and Saudek V. (1993) Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. Ser. A 102, 241–245 [Google Scholar]
- 31. Bhunia A., Bhattacharjya S., and Chatterjee S. (2012) Applications of saturation transfer difference NMR in biological systems. Drug Discov. Today 17, 505–513 [DOI] [PubMed] [Google Scholar]
- 32. Mayer M., and Meyer B. (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117 [DOI] [PubMed] [Google Scholar]
- 33. Bhunia A., and Bhattacharjya S. (2011) Mapping residue-specific contacts of polymyxin B with lipopolysaccharide by saturation transfer difference NMR: insights into outer-membrane disruption and endotoxin neutralization. Biopolymers 96, 273–287 [DOI] [PubMed] [Google Scholar]
- 34. Mayer M., and Meyer B. (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 35. Saravanan R., and Bhattacharjya S. (2011) Oligomeric structure of a cathelicidin antimicrobial peptide in dodecylphosphocholine micelle determined by NMR spectroscopy. Biochim. Biophys. Acta 1808, 369–381 [DOI] [PubMed] [Google Scholar]
- 36. Domadia P. N., Bhunia A., Ramamoorthy A., and Bhattacharjya S. (2010) Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: role of the helical hairpin conformation in outer-membrane permeabilization. J. Am. Chem. Soc. 132, 18417–18428 [DOI] [PubMed] [Google Scholar]
- 37. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 38. Schmidtchen A., Ringstad L., Kasetty G., Mizuno H., Rutland M. W., and Malmsten M. (2011) Membrane selectivity by W-tagging of antimicrobial peptides. Biochim. Biophys. Acta 1808, 1081–1091 [DOI] [PubMed] [Google Scholar]
- 39. Malmsten M. (1994) Ellipsometry studies of protein layers adsorbed at hydrophobic surfaces. J. Colloid Interface Sci. 166, 333–342 [Google Scholar]
- 40. Malmsten M., Burns N., and Veide A. (1998) Electrostatic and hydrophobic effects of oligopeptide insertions on protein adsorption. J. Colloid Interface Sci. 204, 104–111 [DOI] [PubMed] [Google Scholar]
- 41. Singh S., Papareddy P., Kalle M., Schmidtchen A., and Malmsten M. (2013) Importance of lipopolysaccharide aggregate disruption for the anti-endotoxic effects of heparin cofactor II peptides. Biochim. Biophys. Acta 1828, 2709–2719 [DOI] [PubMed] [Google Scholar]
- 42. Ringstad L., Schmidtchen A., and Malmsten M. (2006) Effect of peptide length on the interaction between consensus peptides and DOPC/DOPA bilayers. Langmuir 22, 5042–5050 [DOI] [PubMed] [Google Scholar]
- 43. Bera S., Ghosh A., Sharma S., Debnath T., Giri B., and Bhunia A. (2015) Probing the role of Proline in the antimicrobial activity and lipopolysaccharide binding of indolicidin. J. Colloid Interface Sci. 452, 148–159 [DOI] [PubMed] [Google Scholar]
- 44. Bhunia A., Ramamoorthy A., and Bhattacharjya S. (2009) Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chemistry 15, 2036–2040 [DOI] [PubMed] [Google Scholar]
- 45. Yu L., Tan M., Ho B., Ding J. L., and Wohland T. (2006) Determination of critical micelle concentrations and aggregation numbers by fluorescence correlation spectroscopy: aggregation of a lipopolysaccharide. Anal. Chim. Acta 556, 216–225 [DOI] [PubMed] [Google Scholar]
- 46. Post C. B. (2003) Exchange-transferred NOE spectroscopy and bound ligand structure determination. Curr. Opin. Struct. Biol. 13, 581–588 [DOI] [PubMed] [Google Scholar]
- 47. Porcelli F., Buck-Koehntop B. A., Thennarasu S., Ramamoorthy A., and Veglia G. (2006) Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 45, 5793–5799 [DOI] [PubMed] [Google Scholar]
- 48. Ramamoorthy A., Thennarasu S., Lee D. K., Tan A., and Maloy L. (2006) Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 91, 206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Snyder D. S., and McIntosh T. J. (2000) The lipopolysaccharide barrier: correlation of antibiotic susceptibility with antibiotic permeability and fluorescent probe binding kinetics. Biochemistry 39, 11777–11787 [DOI] [PubMed] [Google Scholar]
- 50. Malmsten M., Claesson P., and Siegel G. (1994) Forces between proteoheparan sulfate layers adsorbed at hydrophobic surface. Langmuir 10, 1274–1280 [Google Scholar]
- 51. Junkes C., Harvey R. D., Bruce K. D., Dölling R., Bagheri M., and Dathe M. (2011) Cyclic antimicrobial R-, W-rich peptides: the role of peptide structure and E. coli outer and inner membranes in activity and the mode of action. Eur. Biophys. J. 40, 515–528 [DOI] [PubMed] [Google Scholar]
- 52. Schmidtchen A., Pasupuleti M., Mörgelin M., Davoudi M., Alenfall J., Chalupka A., and Malmsten M. (2009) Boosting antimicrobial peptides by hydrophobic oligopeptide end tags. J. Biol. Chem. 284, 17584–17594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J., Chou S., Xu L., Zhu X., Dong N., Shan A., and Chen Z. (2015) High specific selectivity and membrane-active mechanism of the synthetic centrosymmetric α-helical peptides with Gly-Gly pairs. Sci. Rep. 5, 15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jeremy B. M., John T. L., and Lubert S. (2002) Biochemistry, 5 Ed., W. H. Freeman, New York [Google Scholar]
- 55. Wang G., Li X., and Wang Z. (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37, D933–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zweytick D., Deutsch G., Andrä J., Blondelle S. E., Vollmer E., Jerala R., and Lohner K. (2011) Studies on lactoferricin-derived Escherichia coli membrane-active peptides reveal differences in the mechanism of N-acylated versus nonacylated peptides. J. Biol. Chem. 286, 21266–21276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scorciapino M. A., Pirri G., Vargiu A. V., Ruggerone P., Giuliani A., Casu M., Buerck J., Wadhwani P., Ulrich A. S., and Rinaldi A. C. (2012) A novel dendrimeric peptide with antimicrobial properties: structure-function analysis of SB056. Biophys. J. 102, 1039–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scorciapino M. A., and Rinaldi A. C. (2012) Antimicrobial peptidomimetics: reinterpreting nature to deliver innovative therapeutics. Front. Immunol. 3, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.