

Abstract
Proline hydroxylation is the most prevalent post-translational modification in collagen. The resulting product trans-4-hydroxyproline (Hyp) is of critical importance for the stability and thus function of collagen, with defects leading to several diseases. Prolyl 4-hydroxylases (P4Hs) are mononuclear non-heme iron α-ketoglutarate (αKG)-dependent dioxygenases that catalyze Hyp formation. Although animal and plant P4Hs target peptidyl proline, prokaryotes have been known to use free l-proline as a precursor to form Hyp. The P4H from Bacillus anthracis (BaP4H) has been postulated to act on peptidyl proline in collagen peptides, making it unusual within the bacterial clade, but its true physiological substrate remains enigmatic. Here we use mass spectrometry, fluorescence binding, x-ray crystallography, and docking experiments to confirm that BaP4H recognizes and acts on peptidyl substrates but not free l-proline, using elements characteristic of an Fe(II)/αKG-dependent dioxygenases. We further show that BaP4H can hydroxylate unique peptidyl proline sites in collagen-derived peptides with asymmetric hydroxylation patterns. The cofactor-bound crystal structures of BaP4H reveal active site conformational changes that define open and closed forms and mimic “ready” and “product-released” states of the enzyme in the catalytic cycle. These results help to clarify the role of BaP4H as well as provide broader insights into human collagen P4H and proteins with poly-l-proline type II helices.
Keywords: Bacillus, collagen, crystal structure, hydroxyproline, mass spectrometry (MS), substrate specificity, Fe(II)/αKG-dependent dioxygenase, prolyl 4-hydroxylase, facial triad
Introduction
Prolyl 4-hydroxylases (P4Hs)2 are ubiquitous enzymes that catalyze the post-translational hydroxylation of proline residues forming 4-hydroxyproline (Hyp) (1). The P4Hs belong to the mononuclear non-heme iron α-ketoglutarate (α-KG) family of oxygenases requiring Fe(II), αKG, and O2 for catalysis (2, 3). These enzymes are characterized by 2His-1-carboxylate (Asp/Glu) facial triad residues that coordinate to the active site iron center (4–7). The common co-substrate αKG binds to iron via its oxo and carboxylate groups and provides reducing equivalents for these enzymes to reduce O2 to a hydroxyl group (2). The oxidation of various substrates containing prolyl residues is accomplished via the activation of a C-H bond of an unactivated alkyl group (8–10). Although one atom from molecular oxygen is incorporated into the substrate, the second atom is utilized in decarboxylation of αKG to form the by-product succinate.
The P4Hs are remarkable in their ability to recognize varied substrates with diverse sequences motifs and are central to the growth, assembly, and function of the host organism (1, 11–13). Although these enzymes exhibit substrate diversity, they share a general reaction mechanism and a conserved fold at their core. A double-stranded β-helix (DSBH) comprising of eight anti-parallel β-strands contains the conserved active site residues required for iron and αKG binding (Fig. 1) (14, 15). Although the DSBH motif is highly conserved in all P4Hs and oxygenases in general, the overall sequence identity is low (11) and the area surrounding the DSBH is the region that gives specificity to each enzyme and substrate (14).
FIGURE 1.

Sequence alignment of BaP4H with vCPH, human C-P4H, and CrP4H and secondary structures defined above the sequence. α Helices are indicated by helical lines with α labels, β strands are indicated by arrows with β labels. Active site residues are highlighted in magenta. The missing loop in BaP4H comprises of residues 70–83. The 310 helices have not been labeled. The sequences were aligned by their secondary structures using ESPript 3.
In humans, there are two types of P4Hs, a collagen P4H (C-P4H) and hypoxia inducible factor-1α (HIF1α)-prolyl hydroxylase domain proteins (PHDs) (13, 16–18). The HIF1α-PHDs are involved in the post-translational hydroxylation of a LXXLAP motif in HIF1α and play a key role in cellular response to hypoxia (19–21). Whereas, C-P4H catalyzes the hydroxylation of proline residues at the Y position of (X-Y-G)n repeats (where, n denotes the number of repeated sequences and X is frequently proline) in collagen and other collagenous domain containing proteins (17, 22). The (P-P-G)n repeats found in collagen adopt a poly-l-proline type II (PPII) conformation in each strand of the collagen triple helix. The PPII helical residues are known to interact with proline recognition domains (PRDs) and are involved in a variety of processes including bacterial and viral pathogenesis, elasticity, transcription, cell motility, and self-assembly (23, 24). The hydroxylation of Y proline residues in collagen is of utmost importance in providing stability to the collagen helical structure. Defects in collagen stability due to insufficient hydroxylation or excessive C-P4H activity can cause diseases like scurvy and fibrosis, respectively (1). Because C-P4H is the rate-limiting enzyme in collagen biosynthesis, therefore, it is an attractive target for developing corresponding mechanism-based activators and inhibitors for scurvy and fibrotic disorders.
Three C-P4H isoenzymes exist in vertebrates and these are all α2β2 tetramers. The β-subunit is identical to protein-disulfide isomerase and is necessary for preventing the aggregation of α-subunit and to retain it in the endoplasmic reticulum (22, 25). The catalytic α-subunit consists of 3 domains: an N-domain responsible for tetramer assembly, a peptide-substrate binding (PSB) domain, and a C-terminal catalytic domain containing the active site (22, 26). The crystal structures of the N and PSB domains have been determined in presence of a C-P4H inhibitor (poly-l-proline) and (P-P-G)3 substrate (26, 27). The two peptides adopt a PPII helical conformation and bind to the PSB domain in a groove lined by conserved tyrosines. The PSB domain of C-P4H consists of α-helices that forms tetratricopeptide repeat motifs. The tetratricopeptide repeat motifs are promiscuous in mediating protein-protein interactions and recognize many ligands including proline-rich peptides (28). Because C-P4H acts on X-P-G repeats in collagen and other proteins with collagen-like sequences, the substrate specificity of this enzyme must arise from residues beyond the conserved region of tetratricopeptide repeat. An open question, which is mainly due to the lack of a structure of C-P4H with an intact catalytic domain, is how the PSB and catalytic domains communicate to position a substrate around the active site for site-specific prolyl hydroxylation. The poor solubility of the α-subunit containing the catalytic site has proved challenging for structural studies and there has been an ongoing effort in pursuit of an improved model for C-P4H.
Previous structures of P4Hs have provided initial insights into enzyme structure and function. A viral collagen P4H (vCPH) has been shown to hydroxylate poly-l-proline and prolines in proteins containing (P-A-P-K)n proline-rich repeats (29, 30). The crystal structures of vCPH with manganese-bound and a zinc-αKG complex show residues involved in metal and αKG binding and related conformational changes when αKG binds (30). Docking simulations using a PKPAPK peptide showed possible interactions with vCPH and the peptide model forms a PPII helix in the PSB groove of this enzyme (30). Plants and green algae have collagenous domain containing glycoproteins with proline-rich motifs where Hyp is important for cell-wall assembly (31, 32). A structure of an algal P4H from Chlamydomonas reinhardtii (CrP4H) complexed with a (S-P)5 peptide is one of the few P4H crystal structures solved to date with a substrate bound in the active site (33). The structure illustrates various enzyme-substrate interactions in the binding groove and in the substrate binding loop (SBL) region adjacent to the active site, with the peptide adopting the PPII helix conformation (34, 35). These highly mobile SBL regions are conserved through P4Hs and in the absence of a substrate one of the loops is usually disordered (30, 36, 37).
Similar to other organisms bacteria contain Hyp, but a distinctive characteristic of bacterial P4Hs is to produce Hyp from free l-proline instead of peptidyl proline (38). On the other hand, Bacillus anthracis P4H (BaP4H) is unique among bacterial P4Hs in that it is reported to bind the human collagen peptide (P-P-G)10 with an affinity similar to that of human C-P4H (1, 36, 39). A sequence alignment of BaP4H with human C-P4H shows ∼30% sequence identity. Structural alignment of BaP4H with CrP4H and vCPH structures shows conserved secondary structure similarities between various P4Hs (Fig. 1). Examining the proteome of B. anthracis reveals collagen-like proteins that contain stretches of (G-X-Y) repeats. A genome-based study of collagen structural motifs in bacterial and viral proteins showed that bacterial collagen structural motifs favor a (G-P-T) triplet unit rather than the second most prevalent (G-P-Q) repeat unit (40). Because the proteome of B. anthracis contains (G-P-T) repeats in some proteins, it has been proposed that a possible physiological substrate for BaP4H could be a protein consisting of (G-P-T)n repeats (36, 41). However, the substrate for BaP4H is not yet fully established.
The previously reported crystal structure of BaP4H is an apo-form of the enzyme lacking necessary cofactors (36, 41). To gain insight into the site-specific prolyl hydroxylation activity of BaP4H, it is important to understand how the active site residues are organized to assemble the cofactors, iron and αKG, for substrate recognition.
In the current study we examine cofactor and substrate binding, including site-specific hydroxylation activity of BaP4H by a combination of biochemical, mass spectrometry, and x-ray crystallographic methods. Attempts toward determining the crystal structure of a substrate-bound form of BaP4H has not been fully successful, therefore molecular docking was used to simulate the binding mode of a collagen-like proline-rich (P-P-G)5 peptide. Here we describe the first biochemical and structural analysis of the cofactor-bound forms of BaP4H, and identified collagen-derived (P-P-G)5,10 peptides as substrates with hydroxylation of proline residues occurring in both X and Y positions. Furthermore, the crystal structures reveal conformational changes upon cofactor binding, and represent “ready” and “product-released” states of the enzyme in the catalytic mechanism (42).
Experimental Procedures
Materials
All buffers, media ingredients, and other reagents were acquired from Sigma and, unless otherwise stated, were of the highest purity available. Solutions were prepared using Nanopure deionized water. N2 (99.98%), NO (>99%), and liquid helium (>99%) were obtained from Praxair (Cedar Rapids, IA).
Protein Expression and Purification
The bap4h gene was synthesized by Genscript (Piscataway, NJ), and then subcloned into the pET28a expression vector using restriction sites NcoI and NdeI. The BaP4H plasmid was transformed into Escherichia coli BL21 star DE3 pLysS cells (Life Technologies). Apo-BaP4H was expressed and purified with a slight modification as reported previously (41).
The BaP4H-(P-P-G)3 construct was made by PCR using appropriate primers to extend the C terminus end to contain a (P-P-G)3 peptide. The PCR product was cut with NcoI and BamHI and ligated back into pET28a. This new plasmid was transformed, expressed, and purified as above.
UV-visible Spectroscopy Measurements
To prevent any turnover, all UV-visible measurements were performed in an anaerobic chamber (Vacuum Atmospheres, Hawthorne, CA) using an Ocean Optics DH-2000-BAL light source (Ocean Optics, Dunedin, FL). Anaerobic solutions were made from dry stocks in a glovebox. Spectra were measured between 250 and 800 nm in a buffer containing 50 mm Tris, pH 7.4, 30 mm NaCl in a 100-μl quartz cuvette. The spectrum of Fe(II)-BaP4H was measured after adding equimolar concentrations of Fe(NH4)2(SO4)2 and 776 μm apo-BaP4H. To detect any spectral changes associated with αKG binding, stoichiometric aliquots of Fe(II) and αKG were added to a 776 μm solution of apo-BaP4H and spectra were recorded after each addition. The difference spectrum of Fe(II)-BaP4H-αKG ternary complex (solid line, Fig. 2A), was obtained by subtracting the Fe(II)-BaP4H spectrum (dashed line, Fig. 2A) from the original ternary complex spectrum to highlight the absorption maximum at 520 nm characteristic of αKG-dependent non-heme iron dioxygenases (43, 44).
FIGURE 2.

Spectral properties and substrate/cofactor binding affinities (Kd) of Fe(II)-BaP4H. A, UV-visible spectra of the reaction of 776 μm apo-BaP4H in the presence of Fe(II) and αKG. Apo-BaP4H, dotted line; Fe(II)-BaP4H, dashed line; Fe(II)-BaP4H-αKG ternary complex, solid line. B, EPR spectra of BaP4H in the presence of NO: top, 100 μm apo-BaP4H with equimolar concentration of Fe(NH4)2(SO4)2; bottom, NO-Fe(II)-BaP4H with 100 μm αKG. C, the intrinsic tryptophan fluorescence was monitored as corresponding peptides were titrated into an anaerobic solution containing 1 μm apo-BaP4H, 3 μm Fe(NH4)2(SO4)2, and 100 μm αKG. The binding affinities for the (P-P-G)10 and (P-P-G)5 peptides were determined to be 0.74 ± 0.11 μm (circles and solid line) and 0.3 ± 0.02 μm (triangles and dashed line), respectively. D, the Kd for l-proline was determined to be 7.9 ± 2.5 μm and was titrated into an anaerobic solution containing 1 μm BaP4H, 3 μm Fe(NH4)2(SO4)2, and 100 μm αKG. E, the Kd (4.2 ± 0.5 μm) for malonate was obtained by titrating it into an anaerobic solution containing 1 μm BaP4H and 3 μm Fe(NH4)2(SO4)2. F, the Kd for Co(II) was determined to be 0.81 ± 0.09 μm. CoCl2 was titrated into an aerobic solution containing 1 μm BaP4H.
To determine the metal stoichiometry, a 100-μl solution containing equimolar concentration of BaP4H and αKG (800 μm) was titrated with a 2 mm stock solution of Fe(NH4)2(SO4)2 anaerobically in a glovebox. Aliquots of Fe(II) were added to this solution of BaP4H/αKG until the absorption at 520 nm had saturated.
Electron Paramagnetic Resonance Spectroscopy (EPR)
X-band EPR spectra were recorded on a Bruker EMX spectrometer (Bruker Biospin Corp., Billerica, MA), equipped with an Oxford ITC4 temperature controller, a Hewlett-Packard model 5340 automatic frequency counter, and Bruker gauss meter. The EPR samples were prepared anaerobically with an enzyme concentration of 100 μm in 50 mm Tris, pH 7.4, 150 mm KCl, 5 mm β-mercaptoethanol. Reduced Fe(II)-BaP4H samples were prepared by adding sodium dithionite to a final concentration of 1 mm. NO gas was bubbled into the protein solutions with cofactors in an anaerobic sealed vial and the solution transferred to an EPR tube via a Hamilton syringe. All spectra were collected at microwave frequency, 9.43 GHz; receiver gain, 2 × 104; modulation frequency, 100 kHz; temperature, 4 K; microwave power, 10 milliwatts; modulation amplitude, 10 G; sweep time, 83.89 s; and 16 scans. The Fe(II)-BaP4H spectrum (data not shown) was recorded using 1 scan keeping all other parameters the same.
Fluorescence Binding Titrations
Fluorescence spectra were recorded at room temperature using a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, Santa Clara, CA). Fluorescence titration was used to determine the binding affinity of substrates and cofactors to BaP4H. The decrease in intensity of intrinsic tryptophan fluorescence at 295 nm served as an indicator of cofactor binding. Titration experiments were performed using an enzyme concentration of 1 μm in 50 mm Tris, pH 7.4, 30 mm NaCl. All titrations were performed anaerobically using solutions that were prepared in a glove box. In a typical anaerobic titration, a degassed protein solution (3 ml) was sealed in a screw top quartz cuvette with a septum to prevent diffusion of O2 into the anaerobic solution. Anaerobic cofactor solutions were added in aliquots via syringe to the quartz cuvette. The wavelength used for excitation was 295 nm and the emission was monitored from 310–380 nm using excitation and emission slit widths of 5 nm. The signal intensity was recorded at the maxima, 329 nm.
Hydroxylation Product Identification by Liquid Chromatography Tandem Mass Spectrometry
All mass spectra LC-MS/MS data were collected on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC 1000 for sample handling and peptide separations. Approximately 200 fmol of peptide resuspended in 5% formic acid + 5% acetonitrile was loaded onto a 125-μm inner diameter fused-silica microcapillary with a needle tip pulled to an internal diameter less than 5 μm. The column was packed in-house to a length of 15 cm with a C18 reverse phase resin (120 Å pore size, 5.0 μm particle size, GP-C18, SePax Technologies). The peptides were separated using a 20-min linear gradient from 3 to 35% buffer B (100% ACN + 0.125% formic acid) equilibrated with buffer A (3% ACN + 0.125% formic acid) at a flow rate of 600 nl/min across the column. The Orbitrap Fusion mass spectrometer was operated in the data-dependent positive ion mode using the top speed strategy. In brief, the scan sequence for the Fusion Orbitrap began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 800–1000 m/z scan range for (P-P-G)10, 600–800 m/z scan range for (P-P-G)5), AGC target 5 × 105, maximum injection time 100 ms, dynamic exclusion of 5 s). “Top speed” (2 s) was selected for MS2 analysis, which consisted of HCD (quadrupole isolation set at 2.0 Da and ion trap analysis, AGC 1 × 105, collision energy 25%, maximum injection time 250 ms). MS2 fragment ions were analyzed in the Orbitrap, resolution 15,000. A suite of in-house software tools were used for .RAW file processing and controlling peptide false discovery rates (45). MS/MS spectra were searched against a custom database containing the (P-P-G)10 sequence and 100 common contaminants with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 1 Da, fragment ion mass tolerance of 0.1 Da, and variable hydroxylation of proline (15.99491 Da). Peptides were filtered to a 1% false discovery rate using linear discriminate analysis (45). We used a modified version of the Ascore algorithm to quantify the confidence with which each hydroxylation site could be assigned to a particular residue. Hydroxylation sites with Ascore values >13 (p ≤ 0.05) were considered confidently localized to a particular residue (45). The enzymatic assays to verify peptide hydroxylation were performed aerobically in 100 mm Tris, pH 7.4, using (10 or 50 μm) apo-BaP4H preincubated anaerobically for 30 min with (10 or 50 μm) Fe(II). A reaction mixture containing 500 μm αKG, ascorbate (10 or 50 μm), and 100 μm of either (P-P-G)5 or (P-P-G)10 were prepared at 20 °C. The reactions were initiated by the addition of Fe(II)-BaP4H solution and incubated for 45 min at 20 °C. The reactions were quenched in 0.5% TFA and vacuum concentrated (SpeedVac concentrator, Savant, SPD131DDA). The dried powder containing the product was collected and stored at 4 °C until it was analyzed by mass spectrometry.
Data Analysis and Equations
Data in Fig. 2 were fit with Sigma Plot 12.0 (Systat Software Inc., Point Richmond, CA) using a ligand-binding model (Equation 1) as described previously (46, 47),
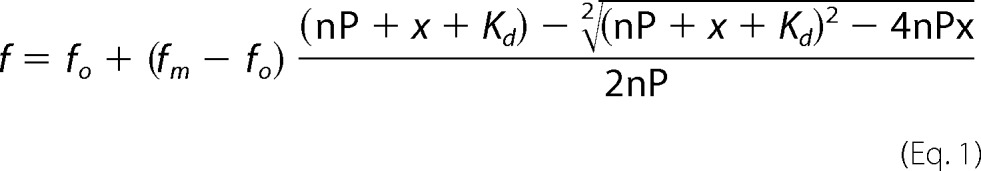 |
where f is the fluorescence signal resulting from binding of the metal cofactor, fo is the signal from a protein solution in the absence of any cofactors, fm corresponds to the maximal quenched fluorescence intensity when a cofactor was bound, Kd is the dissociation constant, P and x are total protein and added metal ion/ligand concentrations, respectively, n is the number of binding sites. A better approximation of the binding affinity can be obtained by using Equation 1 in cases where the protein concentration is higher than the estimated Kd (46, 47).
Crystallization of Apo-BaP4H
Initial crystals of apo-BaP4H were obtained using the hanging-drop vapor-diffusion method with optimization around a previously reported condition (41) by varying pH and PEG concentrations. Crystal drops were set up at 20 °C with 1 μl of 24 mg/ml of apo-BaP4H mixed with 1 μl of precipitant solution containing 0.04 m potassium phosphate monobasic, pH 4.0–7.0, 13–18% PEG 8000, and 20% glycerol. Crystals would appear in 1–2 days and the best crystal used for data collection was obtained from 0.04 m potassium phosphate monobasic, pH 6.0, 14% PEG 8000, and 20% glycerol. Crystals were taken from the precipitant solution and directly frozen in liquid nitrogen.
Crystallization of Co(II)-BaP4H-MLI
A solution of 9 mg/ml of apo-BaP4H, 1 mm CoCl2, and 1 mm l-Pro was incubated on ice for 30 min before setting crystal drops. Crystals were obtained using hanging-drop vapor-diffusion at 20 °C from the PACT screen (Molecular Dimensions) by mixing 1 μl of the above protein solution with 1 μl of reservoir solution. Initial crystals appeared in 0.1 m malonate/imidazole/boric acid, pH 6.0, 25% PEG 1500 and were optimized by varying pH and precipitant concentration. Diffraction quality crystals were obtained from 0.1 m malonate/imidazole/boric acid, pH 6.5, 18% PEG 1500. The crystals of Co(II)-BaP4H-MLI were plate shaped and grew in about 2 days. The crystals were soaked in a cryoprotectant solution of 0.1 m malonate/imidazole/boric acid, pH 6.5, 18% PEG 1500, and 20% glycerol for 5 min and submerged in liquid nitrogen.
Crystallization of Co(II)-BaP4H-PPG
Crystals of BaP4H fused with (P-P-G)3 peptide to the C terminus were obtained from the JCSG screen (Qiagen) using sitting-drop vapor-diffusion at 20 °C. A solution of 12 mg/ml of BaP4H-(PPG)3, 1 mm CoCl2, and 1 mm αKG was incubated on ice for 30 min. Crystal drops were set up by mixing equal volumes of above protein solution with reservoir solution. Crystals were obtained in 0.15 m KBr and 30% PEG 2000 MME. Plate-shaped crystals would appear in about 2 days with dimensions of ∼70 × 70 × 20 μm. The crystals were soaked in cryoprotectant solution containing 0.15 m KBr, 30% PEG 2000 MME, and 20% glycerol for 5 min, prior to freezing in liquid nitrogen.
Data Collection and Analysis
Data sets for apo-BaP4H and Co(II)-BaP4H-PPG were collected at 100 K at the Advanced Light Source, beamline 4.2.2. Data collection parameters were: apo-BaP4H, 0.5° oscillation, 4-s exposure time, 120-mm detector distance; and Co(II)-BaP4H-PPG, 0.1° oscillation, 0.4-s exposure time, 250 mm detector distance. The data set for Co(II)-BaP4H-MLI was collected at 100 K at the Advanced Photon Source, beamline 19-ID-D using 0.5° oscillation, 0.6-s exposure time, and 182-mm detector distance. The data were indexed and integrated using the XDS package (48) and merged and scaled using SCALA (49) (CCP4 suite) (50). Fluorescence scans were taken for cobalt containing crystals and a peak for cobalt was observed at 6.92 KeV. Data collection and refinement statistics are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics
| Apo-BaP4H | Co(II)-BaP4H-MLI | Co(II)-BaP4H-PPG/αKG | |
|---|---|---|---|
| Data Collection | |||
| Space group | P 21 21 21 | P 1 21 1 | P 21 21 21 |
| Unit cell dimensions | |||
| a b c (Å) | 51.6 80.1 103.8 | 42.8 41.5 111 | 50.8 75.6 105.8 |
| α β γ (deg) | 90 90 90 | 90 97 90 | 90 90 90 |
| Beam line | ALS 4.2.2 | APS 19-ID-D | ALS 4.2.2 |
| Wavelength (Å) | 1.00 | 0.979 | 1.00 |
| Resolution range (Å)a | 33.3–1.67 (1.76–1.67) | 55.2–1.70 (1.79–1.70) | 61.5–2.10 (2.21–2.10) |
| Observationsa | 347,255 (32,419) | 155,721 (23,012) | 111,271 (15,669) |
| Unique reflectionsa | 50,418 (6,990) | 42,773 (6,257) | 24,447 (3,527) |
| Redundancya | 6.9 (4.6) | 3.6 (3.7) | 4.6 (4.4) |
| Completeness (%)a | 99.5 (96.5) | 99.6 (100) | 99.8 (100) |
| I/σ (I)a | 27.1 (3.2) | 25.5 (6.6) | 13.1 (3.0) |
| Rmerge (%)a,b | 4.2 (46.0) | 2.6 (16.3) | 6.8 (49.6) |
| Rmeas (%)a,c | 5.0 (65.6) | 3.7 (22.2) | 8.8 (60.7) |
| CC1/2 | 1.000 (0.661) | 0.999 (0.967) | 0.998 (0.911) |
| Refinement | |||
| Rwork, Rfree (%) | 17.5 (21.1) | 21.2 (22.9) | 19.7 (23.4) |
| Resolution range (Å) | 33.3–1.67 | 29.7–1.70 | 61.5–2.10 |
| No. of reflections | 50,232 | 42,742 | 24,291 |
| Number of atoms | |||
| Protein | 3,165 | 3,004 | 3,063 |
| Co(II) | 2 | 2 | |
| αKG | 20 | ||
| PPG | 36 | ||
| Malonate | 14 | ||
| Water | 396 | 254 | 154 |
| Average B-factor (Å2) | |||
| Protein | 16.8 | 20.0 | 29.0 |
| Co(II) | 25.4 | 19.4 | |
| αKG | 24.0 | ||
| PPG | 46.2 | ||
| Malonate | 26.3 | ||
| Water | 33.3 | 33.0 | 34.8 |
| R.m.s. deviation bond lengths (Å) | 0.010 | 0.020 | 0.011 |
| R.m.s. deviation bond angles (degree) | 1.2 | 1.1 | 1.1 |
| Ramachandran favored (%) | 97.5 | 97.7 | 97.6 |
| Ramachandran allowed (%) | 2.5 | 2.3 | 2.4 |
| Clash score | 4.5 | 3.4 | 3.2 |
a Values in parentheses are the highest-resolution shell.
b Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), where Ii(hkl) is the ith measured diffraction intensity and 〈I(hkl)〉 is the mean intensity for the Miller index (hkl).
c Rmeas = Σhkl((n(hkl)/n(hkl)−1))½ Σi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
Structure Determination
Molecular replacement for apo-BaP4H was done with PHASER (51) using PDB code 3ITQ as a starting model. Phases for Co(II)-BaP4H-MLI and Co(II)-BaP4H-PPG were obtained by molecular replacement with our apo-BaP4H structure. Matthews coefficients and estimated solvent contents for apo-BaP4H, Co(II)-BaP4H-MLI, and Co(II)-BaP4H-PPG were 2.15 Å3/Da, 43%; 1.96 Å3/Da, 37%; and 2.03 Å3/Da, 39%, respectively. Refinement was carried out using phenix.refine from the PHENIX software package (52). Structures containing metals had bond length and angle parameters generated by phenix.metal_coordination. Following iterative rounds of refinement using simulated annealing, energy minimization, real space refinement, and B-factor refinement, model building was done in COOT (53). Addition of ligands into distinct positive electron density was based on a simulated annealing omit map generated by omitting the ligand. Water molecules were added into clear densities in later rounds of refinement and composite omit maps were generated to verify the structures. All structures contain a disordered N terminus and flexible loop region that varies slightly for each model. The disordered residues that were not modeled for each structure and chain are as follows: apo-BaP4H chain A 1–10, chain B 1–11 and 73–82; Co(II)-BaP4H-MLI chain A: 1–11 and 72–80, chain B: 1–11 and 71–81; Co(II)-BaP4H-PPG chain A: 1–11, 70–73, and 79–83, chain B: 1–11 and 70–83. The 2nd and 3rd repeat of (P-P-G)3 in the Co(II)-BaP4H-PPG structure were also disordered and not included in the final model. In Co(II)-BaP4H-MLI, no density for proline appeared in the structure and instead density for malonate (from crystallization solution) was observed in the final model. The Ramachandran geometries of each structure were analyzed by PROCHECK (54, 55) (Table 2). All structural figures were made using PyMOL.
Computational Ligand Docking
The Co(II)-BaP4H-PPG structure (excluding the modeled P-P-G repeat from the C terminus end) was used for docking calculations with a peptide from PDB entry 1NAY truncated to (P-P-G)5. The Co(II)-BaP4H-PPG structure and the (P-P-G)5 peptide were submitted to the ZDOCK server (56). ZDOCK was used for initial rigid body and low resolution docking. Most of the outputs from ZDOCK docked the peptide in the peptide binding groove region. The most reasonable conformation (based on CrP4H peptide bound structure, PDB code 3GZE) of peptide docking from ZDOCK was used for flexible docking with the FlexPepDock server (57, 58). The FlexPepDock server performs a rotamer search on protein and treats the peptide as a completely flexible entity. It performs a rigid body optimization and a peptide backbone optimization, thereby optimizing the peptide conformation.
Results
Analysis of Cofactor and Substrate Binding to BaP4H
To probe the nature of spectral changes associated with cofactor and substrate binding to BaP4H, we employed UV-visible and EPR in conjunction with fluorescence saturation studies. The UV-visible spectrum of as-isolated BaP4H exhibited no absorption features above 280 nm. Anaerobic addition of Fe(II) had no effect on the absorption spectrum. However, when αKG was added to complex of Fe(II)-BaP4H the absorbance in the 300–500 nm region developed a broad shoulder with the absorption maximum at 520 nm (Fig. 2A). This feature is due to a charge transfer transition of the αKG-Fe(II)-Ba4H ternary complex and is characteristic of αKG-dependent non-heme iron dioxygenases (43, 44, 59). The absorbance at 520 nm was monitored to determine the stoichiometry of Fe(II) binding to the enzyme. Titrating Fe(II) into an anaerobic solution of BaP4H in the presence of αKG resulted in an increase in absorption at 520 nm and showed a clear inflection point at 1:1 ratio of Fe(II) to BaP4H monomer (data not shown).
To investigate the interaction of ferrous iron with dioxygen in BaP4H we used nitric oxide (NO), which serves as a surrogate of dioxygen and assessed the NO-Fe(II)-BaP4H complex by EPR. BaP4H with reduced Fe(II) exists in an EPR-silent (S = 2) spin state, similar to other non-heme iron proteins (data not shown). When NO interacts with the Fe(II) center, the spin state is changed from S = 2 into an EPR-active S = 3/2 species. EPR signals associated with this species are g = 3.94, 1.97 (Fig. 2B, top) (60–63). Anaerobic addition of NO to the reduced αKG-Fe(II)-BaP4H complex diminished the intensity of the signal at g = 3.91 and the nitrosyl signal at g = 1.98 predominates (Fig. 2B, bottom). In addition, a signal due to oxidized ferric iron is detectable in both NO spectra at g = 4.31. These results suggest that αKG binds to the Fe(II) center and alters the electronic environment without displacing bound NO, as indicated by the small changes in the observed g values.
To determine the binding affinities of various cofactor and potential substrates with BaP4H, intrinsic tryptophan fluorescence served as a probe for ligand binding. Because (P-P-G)10 was suggested to bind to BaP4H with an affinity similar to that of C-P4H (36) we sought to determine the binding affinities of (P-P-G)10 and (P-P-G)5 peptides in addition to l-proline. By measuring the decreasing tryptophan fluorescence intensity at 329 nm, whereas titrating in the appropriate ligand to αKG-Fe(II)-BaP4H ternary complex, the binding affinities were determined. The Kd of (P-P-G)10 and (P-P-G)5 peptides were calculated to be 0.74 ± 0.11 and 0.3 ± 0.02 μm, respectively (Fig. 2C). Although free l-proline binds to Fe(II)-BaP4H (Kd of 7.9 ± 2.5 μm), the affinity is an order of magnitude lower than the peptides (Fig. 2, C and D). The binding affinity of a known P4H inhibitor malonate with the Fe(II)-BaP4H complex was determined to be 4.2 ± 0.5 μm and Co(II) binds to apo-BaP4H with a Kd of 0.81 ± 0.09 μm (Fig. 2, E and F).
Hydroxylation of Human Collagen-like (P-P-G)5,10 Peptides by BaP4H
It has been proposed that BaP4H hydroxylates peptidyl proline but not free l-proline, a common substrate for bacterial P4Hs. To establish the specific targets recognized by BaP4H, we conducted enzymatic assays using (P-P-G)5, (P-P-G)10, and free l-proline. Although BaP4H catalyzes the hydroxylation of both (P-P-G)5 and (P-P-G)10, it is unreactive toward free l-proline.
To identify the hydroxylated peptides from BaP4H-catalyzed reactions, LC-MS/MS was performed on products isolated from reactions carried out under steady state and single turnover conditions using either (P-P-G)10 or (P-P-G)5 peptides. The extracted ion chromatogram (XIC) of the (P-P-G)5 peptide eluted as a sharp peak around 13.3 min (Fig. 3A, black trace) and the hydroxylated-(P-P-G)5 ((P-P-G)5-OH) eluted as a major peak at 12.3 min and a shoulder at 11.8 min (Fig. 3A, green trace). The major peak consists primarily of (P-P-G)5-OH, with prolines hydroxylated in the Y position of the second and third repeats (positions 8 and 5, Table 1). The shoulder consists of (P-P-G)5-OH, with hydroxylated prolines in the fourth repeat (positions 11 and 10, Table 1). The XIC of the (P-P-G)10 peptide eluted as a sharp peak around 16.3 min and the hydroxylated-(P-P-G)10 ((P-P-G)10-OH) eluted as a broad peak from 15.5 to 17.3 min with multiple shoulders (Fig. 3B). Unlike (P-P-G)5-OH we are uncertain of the exact hydroxylated species of (P-P-G)10-OH that corresponds to each of the shoulders in Fig. 3B. This is due to the fact that a small modification such as hydroxylation relative to a large molecule like (P-P-G)10 molecule has little effect on the retention time of the modified peptide. Therefore, it made it difficult to resolve the different hydroxylated forms under our experimental conditions. Although hydroxylated (P-P-G)5 appears at m/z 645.8298 that corresponds to a doubly charged species, the spectrum of (P-P-G)10 contained a peak with m/z 849.4352, corresponding to a triply charged single hydroxylation of (P-P-G)10 (Fig. 3, C and D).
FIGURE 3.

Hydroxylated peptide product identification by LC-MS. A, XIC peak profiles of [(P-P-G)5 +OH]+2 (green) and (P-P-G)5 (black) as separated on a C18 column. B, XIC peak profiles for [(P-P-G)10 + OH]+3 (green) and (P-P-G)10 (black) separated on a C18 column. C, MS analysis of (P-P-G)5 before and after hydroxylation. The top panel shows [(P-P-G)5 + OH]+2, the isotopic mass envelope starts at m/z 645.820. The bottom panel shows the substrate peptide ((P-P-G)5), with an isotopic mass envelope starting at m/z 637.8345. Hydroxylation of (P-P-G)5 results in a m/z change of 7.9945 for the double charged, this is equivalent to a 15.989 Da increase to the peptide mass, which corresponds to incorporation of a single oxygen atom. D, MS analysis of (P-P-G)10 before and after hydroxylation. The top panel shows [(P-P-G)10 + OH]+3, the isotopic mass envelope starts at m/z 849.4371. The bottom panel shows the substrate peptide [(P-P-G)10], with an isotopic mass envelope starting at m/z 844.1041. Hydroxylation of (P-P-G)10 results in a m/z change of 5.3323 for the triply charged ion, this is equivalent to a 15.989 Da increase to the peptide mass, which corresponds to incorporation of a single oxygen atom.
TABLE 1.
Amino acid sequence of (P-P-G)5 and (P-P-G)10 with the unique hydroxylation sites shown
Xcorr values were calculated by the SEQUEST algorithm. The hydroxylation sites and localization scores were determined by a modified version of the Ascore algorithm. The table shows only peptides that have one or more hydroxylation site(s) localized to >99% confidence. The first three hydroxylated peptides for (P-P-G)5 were observed under both steady state and single-turn over conditions and the last two peptides were observed only in the single turnover condition. All peptides listed for (P-P-G)10 were discovered under single turnover conditions. RT, retention time.
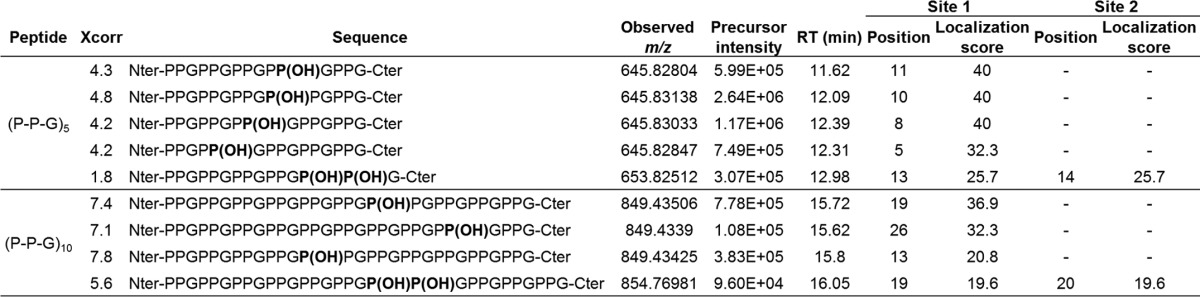
Under steady state conditions hydroxylation sites were localized to both X and Y prolines on the third and fourth repeats of the (P-P-G)5 peptide (Table 1). Whereas, with (P-P-G)10, although hydroxylated peptides were identified, due to low localization scores we were unable to assign specific hydroxylation sites.
The single turnover conditions uncovered additional hydroxylation sites on both the (P-P-G)5 and (P-P-G)10 peptides. The MS2 data of (P-P-G)5 identified two additional peptides: one with a hydroxyl contained on the Y proline of the second repeat; and a second peptide that is doubly hydroxylated with the hydroxyl groups located on adjacent prolines in the fifth repeat (Table 1). The MS2 fragment ion pattern for the Y proline on the fourth repeat is shown in Fig. 4A. Interestingly, hydroxylation localized to the X proline of the fourth repeat was also observed (Table 1). With (P-P-G)10, hydroxylated peptides were indeed detected with acceptable localization site scores (>15), which irrevocably allowed assignment of hydroxylation sites. Preferential hydroxylation of proline residues toward the C terminus end of the (P-P-G)10 peptide were observed, with peptides containing a hydroxyl group on the X proline of the fifth and seventh repeats, and the Y proline of the ninth repeat (Table 1). The MS2 fragment ion pattern for the X proline on the fifth repeat is shown in Fig. 4B. In addition, a doubly hydroxylated peptide containing hydroxyl groups on adjacent prolines of the seventh repeat was observed (Table 1). Although small hydroxylation of the proline residues in the X positions of (P-P-G)10 were observed previously with Arabidopsis thaliana P4H (At-P4H-1), there was greater uncertainty associated with the true degrees of hydroxylation (31). The MS results presented here irrefutably revealed the peptides recognized by BaP4H, including specific prolyl hydroxylation sites.
FIGURE 4.
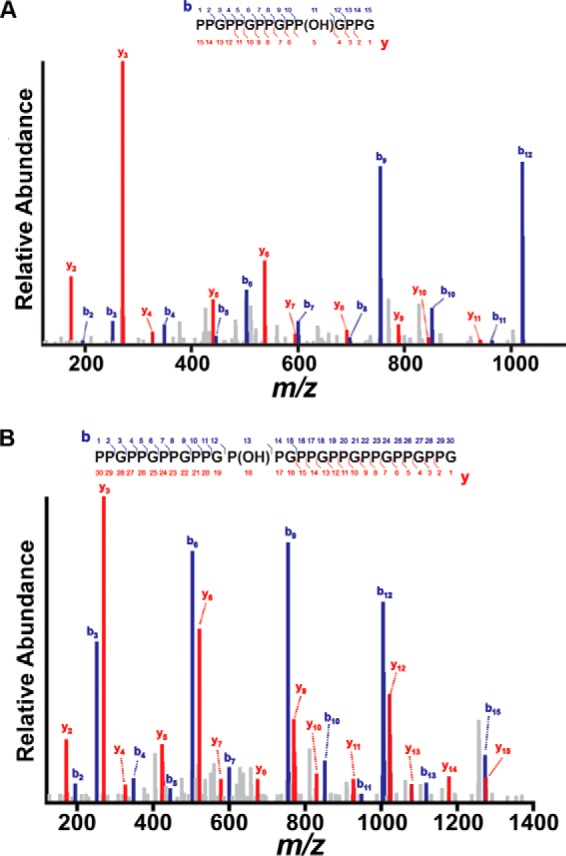
MS2 fragment ion pattern of hydroxylated (P-P-G)5 and (P-P-G)10 peptides. Assignment of b (blue) and y (red) fragment ion series in the MS2 scans from [(P-P-G)5 +OH]+2 (A) and [(P-P-G)10 + OH]+3 (B) precursor ions. Using a variation of the Ascore algorithm, the hydroxylation was localized with >99% confidence to the Y position in the 4th repeat for (P-P-G)5 and to the X position in the 5th repeat for (P-P-G)10. The precursor peptides were selected from the single-turnover experiments.
BaP4H Structural Overview
To establish how cofactors and substrate peptides are assembled, two structures of BaP4H in the presence of Co(II) were determined: one in complex with αKG and a (P-P-G) peptide; and the other structure in complex with a P4H inhibitor, malonate (Figs. 5 and 6, and Table 2). In addition, a structure of apo-BaP4H was obtained (Fig. 7, Table 2) near physiological pH and in a space group different from the previously published apo-BaP4H structure (36). To obtain a peptide-bound structure of BaP4H, a shorter (P-P-G)3 peptide was incorporated into the C terminus end of the protein close to the predicted substrate binding groove. The resulting structure had clear density only for one repeat that is located close to the active site of the symmetry related molecule (Fig. 5). Attempts to determine the Fe(II)-BaP4H structure have failed because of the poor diffraction quality of the resultant crystals.
FIGURE 5.

Structure of the Co(II)-BaP4H-PPG monomer (green) showing the C terminus of a symmetry related molecule (cyan) interacting with the active site of chain A (green). The β-strands of the DSBH-fold (light blue) are labeled βI-βVIII. Other β strands (light orange) are labeled β0, β1, β2, β6, β7, and β8. Missing residues in the substrate binding loop region are indicated by a dashed line. The metal binding residues, His127, Asp129, and His193, αKG (yellow), and the fused (P-P-G) peptide are shown as sticks (oxygen, red; nitrogen, blue). Cobalt is shown as a magenta sphere.
FIGURE 6.

Active site of BaP4H with bound cofactors. A, bidentate binding mode for αKG in Co(II)-BaP4H-PPG structure. B, active site residues of Co(II)-BaP4H-PPG involved in hydrogen bonding interactions between protein, αKG, and water are indicated by black dashes with corresponding distances. The distance between Tyr124 and W1 is 2.8 Å and was excluded from the picture for clarity. C, the PPG peptide (green) at the C terminus interacts with the active site of a symmetry related molecule (cyan). The Y proline (P2) is oriented with the C4 carbon 5.2 Å away from the cobalt metal center. The 2nd and 3rd repeats of (P-P-G)3 were disordered. D, monodentate binding mode for malonate in Co(II)-BaP4H-MLI structure. E, active site residues of Co(II)-BaP4H-MLI showing hydrogen bonding interactions between protein, malonate, and water with labeled distances. αKG, malonate, and protein residues are shown as sticks: carbon (protein backbone color), oxygen (red), nitrogen (blue), water molecules as spheres (cyan), cobalt as a sphere (magenta). The 2Fo − Fc composite omit maps (blue mesh) contoured at 1.0 σ are shown for protein residues in A, C, and D. The Fo − Fc omit maps (also shown as blue mesh) for αKG and MLI are contoured at 3.0 σ in A and D.
FIGURE 7.

A, active site of apo-BaP4H structure with no cofactors bound. B, hydrogen bonding interactions of active site residues of apo-BaP4H between protein and water are indicated by black dashes with corresponding distances. The metal binding residues (His127, Asp129, and His193) and residues involved in hydrogen bonds are shown as sticks: carbon (protein backbone color), oxygen (red), nitrogen (blue). The 2Fo − Fc composite omit maps (blue mesh) are contoured at 1.0 σ around the active site residues.
All three structures solved contain two molecules in the asymmetric unit, therefore, for simplicity all discussion hereafter will be based on chain A from each structure. The overall structures are similar, with root mean square deviations (r.m.s. deviations) of 0.21–0.25 Å over almost 155 Cα atoms. With the exception of chain A in apo-BaP4H, all other chains in the BaP4H structures have about 10–15 disordered residues encompassing the region between residues 70–83, which we refer to as the substrate binding loop (SBL). Although there is electron density around the SBL region in both Co(II)-bound BaP4H structures indicating the presence of flexible amino acids, the density quality is too poor to model and the structures re-converge at residue 84.
Structural alignment between BaP4H and an algal P4H (CrP4H), with an r.m.s. deviation of 0.60 Å over 117 Cα atoms, shows that two short β-strands (β4 and β5) should be located in the disordered SBL region and are flanked between two α-helices (α1 and α2) and the β3 and β6 strands (Fig. 1). Although the apo-BaP4H structure is the only one with a complete loop, subtle differences were seen in the two cobalt-bound structures with partially complete SBLs. The SBL in the apo-form is positioned in an open conformation perpendicular and away from the active site. Although Co(II)-BaP4H-MLI and Co(II)-BaP4H-PPG structures do not have a completely ordered SBL, however, it appears to be adopting a slightly more closed conformation with the SBL oriented toward the protein surface, which could be due to crystallographic contacts with residues that are ordered in the loop region. A truly closed conformation of the SBL would not be expected until a peptide that is not crystallographically constrained is bound.
Active Site Features of Co(II)-BaP4H-PPG and Co(II)-BaP4H-MLI Complexes
Structural comparison of Co(II)-bound forms of BaP4H with our apo-BaP4H shows that regions and residues around the active site within the core β-barrel motif become more organized upon binding of cofactors and/or peptide (Figs. 1 and 5–8). Strands βII and βVII bring triad residues His127 (2.1 Å), Asp129 (2.1 Å), and His193 (2.0 Å) into proximity of coordination to the cobalt atom. The remaining cobalt coordination sites are occupied by either αKG or malonate along with water molecules, resulting in a six-coordinate complex (Fig. 6).
FIGURE 8.
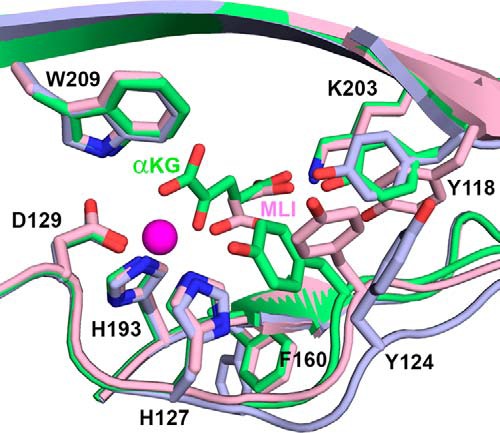
BaP4H structures highlighting conformational changes involving residues Tyr118, Tyr124, and Phe160 upon metal and αKG or malonate binding. Active site comparison between the Co(II)-BaP4H-PPG “closed” active site (green), Co(II)-BaP4H-MLI “partly open” active site (pink), and apo-BaP4H “fully open” active site (light blue). Cobalt is shown as a magenta sphere.
In Co(II)-BaP4H-PPG, αKG coordinates cobalt in a planar fashion via its C1 carboxylate (2.0 Å) and the C2 ketone (2.1 Å) opposite of Asp129 (Fig. 6, A and B). The C5 carboxylate of αKG is anchored in the active site by hydrogen bonding interactions with Tyr118 (2.6 Å), Lys203 (2.9 Å), and Thr159 (2.6 Å). One of the C1 carboxylates is hydrogen bonded to Thr207 (2.6 Å) (Fig. 6B). The bidentate binding of αKG to the metal center and stabilization by lysine, arginine, and threonine interactions is conserved in the Fe(II)/αKG-dependent enzymes, although the positioning of αKG differs among various enzymes (2). Surprisingly, in the Co(II)-BaP4H-PPG structure, the peptide was located in a symmetry related molecule adjacent to the active site (Fig. 5). Although the (P-P-G)3 peptide was disordered with density existing for only one repeat, however, the structure displays that the central Y proline of the peptide points to the cobalt center and is at a distance of 5.2 Å (Fig. 6C), which is slightly longer than that observed for CrP4H (30, 34). In Co(II)-BaP4H-MLI, malonate occupies the αKG binding space in the active site, with its C1 carboxylate located in the same position as the C2 ketone of αKG (Figs. 6 and 8). Malonate coordinates cobalt in a monodentate mode with the C1 carboxylate trans to Asp129 at a distance of 2.6 Å from the cobalt center. The C3 carboxylate forms a salt bridge with Lys203 and is hydrogen bonded to Thr159 (Fig. 6, D and E).
In both Co(II)-bound structures, in addition to the catalytic triad residues and αKG or, malonate that coordinate cobalt, the remaining coordinate sites are occupied by water molecules thus forming a six-coordinate cobalt complex. In the αKG-bound Co(II)-BaP4H-PPG, a water molecule is located 4 Å away from cobalt nearly trans to His193 in both chains in the structure, which is likely the site where dioxygen would bind with iron present (Fig. 6B). In Co(II)-BaP4H-MLI, two water molecules are coordinating cobalt at distances of ∼2.5 Å (Fig. 6, D and E). The apo-BaP4H structure has multiple water molecules spread through the active site. One of the waters (W1) occupies the space where metal would bind and is hydrogen bonded to Asp129 and His193 (Fig. 7A). Two of the other water molecules (W2 and W3) are hydrogen bonding with Thr159 and Lys203, whereas a fourth water (W4) is hydrogen bonding with W2 and W3 (Fig. 7B).
Structures of BaP4H Reveal Open and Closed Active Site Conformations
In the Co(II)-BaP4H-PPG structure, binding of αKG and peptide induces a conformational change in the β-domain involving strands βI, βII, and βIV. These strands are relatively disordered in the apo- and malonate-bound structures, but become more ordered in the Co(II)-BaP4H-PPG structure. Furthermore, the backbone between residues 122–125 in the apo-BaP4H structure is shifted out away from the active site by about 2 Å compared with both cobalt-bound structures (Fig. 8), which indicates that regions of the active site must have greater conformational flexibility.
In the BaP4H structures, we speculate that the residues important for tuning the active site include Tyr118, Tyr124, and Phe160. These residues are located on the “tunable” face of the main β-barrel motif and are around 5–10 Å away from the metal center (Figs. 6, B and E, and 8). Tyr124 exhibits different conformations in the active site. In the apo-form, Tyr124 is 10 Å (measured from the OH to water located at the metal site) away from the metal site and flipped upwards about 90° from the carbon backbone (Figs. 7 and 8). In Co(II)-BaP4H-PPG, this tyrosine is closer to the active site at ∼5.3 Å from cobalt and is oriented such that the plane of aromatic ring is parallel to the metal-binding site (Figs. 6B and 8). In Co(II)-BaP4H-MLI, malonate binding induces a conformational change causing movement within key active site residues (Tyr118, Tyr124, and Phe160). The aromatic ring plane of Tyr124 is oriented perpendicular to the metal binding site and the hydroxyl group is about 7 Å away from cobalt (Figs. 6E and 8). The different open and closed conformations of Tyr124 suggest that it is presumably a catalytically important residue.
Similarly, Tyr118 has multiple conformations and is important in αKG and malonate binding. The apo-form has Tyr118 oriented perpendicular to the αKG site. In Co(II)-BaP4H-PPG, it has a similar orientation but is shifted down by 1.6 Å to position itself about 2.6 Å from the C5 carboxylate of αKG. However, in Co(II)-BaP4H-MLI, Tyr118 is swung outwards away from the active site, at distances of 4.9 and 6.5 Å from malonate C3 and C2 carboxylate ends, respectively. Another residue that is more open in the apo structure is Phe160, located in strand βIV. It protrudes out in apo-BaP4H but is oriented more toward the active site when cobalt is bound (Fig. 8).
Docking of (P-P-G)5 Peptide with Co(II)-BaP4H-PPG Structure
In the absence of a true peptide bound crystal structure of BaP4H, (P-P-G)5 was modeled into the active site of the αKG-bound Co(II)-BaP4H complex. The model shows that the (P-P-G)5 peptide forms an elongated PPII helical conformation similar to that observed in CrP4H (34, 35) (Fig. 9A). The peptide binds BaP4H between β4, β5 (missing loops), and β6 loops and the region between α1, βI, and βII. It interacts with the protein through hydrophobic residues Phe85 from β6 and Phe131 (Fig. 9B). It forms hydrogen bonds with protein residues Glu111, Arg142, Lys163, and Tyr215 through backbone carbonyl atoms and side chains. A proline from the second repeat (P3) is involved in hydrogen bonding with Arg142. The peptide is positioned within the active site of BaP4H with the C4 carbon atom of the Y proline of the second repeat (P4) directed toward the metal center at a distance of 4.6 Å (Fig. 9B), thus indicating that it is a probable hydroxylation site. This model is supported by our mass spectrometry data showing that the Y proline residue in the second repeat of (P-P-G)5 peptide (P4) is hydroxylated by BaP4H (Table 1).
FIGURE 9.

Model of (P-P-G)5 peptide (cyan) into the αKG-bound structure of Co(II)-BaP4H-PPG (green). A, surface representation of BaP4H monomer showing the peptide binding groove residues colored in gray and the overall position of (P-P-G)5. The peptide is shown as cyan sticks with oxygen (red) and nitrogen (blue). The active site region is colored orange. The residues representing the ends of the missing loop are colored violet. Cobalt and αKG are shown as a magenta sphere and yellow sticks, respectively. B, location of the (P-P-G)5 peptide with potential interacting BaP4H residues (green sticks). The C4 position of the Y proline in the second repeat of (P-P-G)5 is located 4.6 Å away from the metal center.
Discussion
The high-resolution MS results demonstrate for the first time that BaP4H can hydroxylate proline residues in both X and Y positions of collagen-like (P-P-G)5 and (P-P-G)10 peptides. Animal C-P4H is known to solely hydroxylate proline in the Y position (64–67). Earlier studies have demonstrated that Hyp in the Y position of collagen repeats contributes greater stability to the collagen triple helix through stereoelectronic effects of the inserted oxygen atom (1). Although preferential hydroxylation of a proline in the X position of (P-P-G)10 has been observed with CrP4H-1A (32), At-P4H-1 acts preferentially but not exclusively on proline in the Y position (31). Interestingly, the MS data revealed asymmetric hydroxylation of (P-P-G)5,10 peptides by BaP4H (Table 1). Both C-P4H (65, 67) and At-P4H-1 (31) displayed an asymmetrical hydroxylation preference in (P-P-G)5,10 peptides, whereas, CrP4H-1A (32) evenly hydroxylated the Pro residues in (P-P-G)10. Furthermore, BaP4H hydroxylates the Y proline in the ninth triplet of (P-P-G)10 in addition to other sites (Table 1), which is consistent with the hydroxylation pattern observed previously using 14C-labeled (P-P-G)10 peptide and either C-P4H (67) or At-P4H-1 (31). With (P-P-G)5, BaP4H catalyzes the hydroxylation of at least one proline residue in the triplets, with the exception of the first repeat (Table 1). Hydroxylation of both X and Y proline residues were identified, which is quite distinct from the hydroxylation pattern of (P-P-G)5 by C-P4H (65). Using 14C-labeled (P-P-G)5, previous studies have shown that C-P4H preferentially hydroxylates the Y proline in the fourth triplet of (P-P-G)5 (65).
Docking studies with (P-P-G)5 illustrates that the peptide is located over the active site with the Y proline residue of the second repeat (P4) in proximity of the metal (Fig. 9). The C-4 carbon atom of this proline is positioned optimally to the metal center for C-H bond activation and subsequent hydroxylation, via a reactive Fe(IV)-oxo intermediate. Although modeling displayed P4 in the proper orientation for hydroxylation, alternatives with prolines from other repeat units of the peptide in correct conformation for hydroxylation are feasible. The MS data revealed that proline residues of the second, fourth, and fifth repeats of the (P-P-G)5 peptide are indeed hydroxylated by BaP4H (Table 1).
Based on the crystal structures of BaP4H, the surface contains a groove across the active site comprised of aromatic and other conserved residues found in Src homology 3 domain proteins known to recognize proline-rich motifs (68). The Src homology 3 domain-like proteins have been reported to occur in prokaryotes (23). Based on structural studies with many protein families, the mechanism of proline-rich peptide recognition is converging, which suggests that a groove composed of aromatic residues is responsible for proline recognition (68). However, the specificity toward a target is mostly dependent on variable loops and neighboring domains (68). In BaP4H, the peptide binding groove along with the SBL most likely contribute to the observed tighter binding affinities for both (P-P-G)10 (Kd, 0.7 μm) and (P-P-G)5 (Kd, 0.3 μm) peptides. These Kd values are comparable with the reported Km for human C-P4H (20 and 170 μm for (P-P-G)10 and (P-P-G)5, respectively (22)), but significantly lower than the Km determined for algal CrP4H (>1.5 mm for (P-P-G)10 (32)) and vCPH (2.9 mm for (P-P-G)10 (29, 30)). Although the binding affinities of BaP4H for both (P-P-G)5,10 peptides are similar, it can catalyze the hydroxylation of (P-P-G)5 with relatively greater ease based on hydroxylated peptides observed in the MS results. It is possible that the longer (P-P-G)10 is more flexible and may not be positioned in the active site in a favorable conformation for hydroxylation. We have also established that BaP4H binds free l-proline but does not catalyze the formation of Hyp. Although there are distinct bacterial P4Hs known to hydroxylate free l-proline (1, 69), it is unclear why BaP4H does not recognize it for hydroxylation. In the absence of a proline- or, a true peptide-bound crystal structure, no additional predictions can be made.
Currently, the structures of only five P4H enzymes have been solved. These are HIFα-PHD2 (70), Pseudomonas aeruginosa PHD (PPHD) (37), CrP4H (34, 35), vCPH (30), and apo-BaP4H (36). The crystal structures of BaP4H with bound cofactors display structural features characteristic of Fe(II)/αKG-dependent enzymes consisting of the 2His-1carboxylate iron-binding motif. Based on sequence and secondary structural similarities with CrP4H, we expect that three β-strands (β3, β4, and β5), including the disordered residues in BaP4H (Fig. 1), are likely involved in substrate recognition and we hypothesize that these residues will become ordered upon peptide binding as observed in CrP4H (35).
The structures of BaP4H provide insight into active site reorganization upon binding of cofactors and the inhibitor, malonate. The binding of cobalt and αKG induces a conformational change bringing the active site from an open state (apo-BaP4H) to a more closed form required for substrate and dioxygen binding prior to catalysis (Fig. 8). It is evident that due to the movement of various important residues, BaP4H can adopt alternate conformations when bound to different ligands. The residue that undergoes the largest movement is Tyr124. Although Tyr124 does not directly interact with αKG, it serves as a lid to keep the active site closed when αKG is present. This tyrosine is conserved across P4Hs and similar movement has been observed in both vCPH and CrP4H (30, 34, 35). It was proposed that this tyrosine could serve as a “conformational switch” to modulate loop movement and substrate binding or release, which is further supported in the BaP4H structures (30, 34, 35). Thus the Co(II)-BaP4H-PPG structure with αKG bound represents the ready or pre-catalytic state of the enzyme.
In BaP4H αKG coordinates cobalt in the “in line” binding mode observed in many Fe(II)/αKG-dependent dioxygenases (2), with the C1 carboxylate opposite of His127 and carbonyl oxygen opposite of Asp129, indicating that the substrate will be positioned near the open site trans to His193 for productive catalysis (Figs. 6, 8, and 9). The αKG binding site in BaP4H uses similar residues that are conserved in the Fe(II)/αKG-dependent enzymes, including other P4Hs. Although BaP4H has two less residues involved in αKG binding compared with vCPH, it is unclear as to why it has a much higher binding affinity for αKG (Kd ∼1 μm) (71) compared with vCPH (Kd ∼700 μm) (30). PHD2 on the other hand has an affinity for αKG (Kd < 2 μm) similar to that determined for BaP4H (72). It was proposed that the weaker binding of αKG in vCPH compared with PHD2 could be due to a difference in the basic residue utilized in binding to the C5 carboxylate of αKG (Lys231 in vCPH versus Arg383 in PHD2 (30). However, BaP4H has Lys203 that interacts with the C5 carboxylate, yet has a high binding affinity toward αKG.
Although malonate is a known P4H inhibitor (73), to the best of our knowledge, this is the first structure of a αKG-dependent dioxygenase with malonate bound. The open and solvent accessible active site of Co(II)-bound BaP4H can accommodate malonate from the crystallization conditions. Malonate occupies the αKG binding space in the active site and mimics binding of the αKG decarboxylation product succinate. The malonate bound form of BaP4H appears to be more open than the αKG-BaP4H-PPG bound form because it simulates a product state of the enzyme post-hydroxylation, where an open form is necessary for product release. The C1 carboxylate of malonate is coordinated to the metal trans to Asp129 (Figs. 6 and 8). The binding mode is similar for this family of enzymes in that the keto group of αKG always binds trans to the carboxylate residue in the catalytic triad (2). Even with two fewer residues responsible for malonate binding to BaP4H compared with αKG, malonate still binds BaP4H with high affinity (Kd ∼4 μm) (Fig. 2E). The slightly lower binding affinity of malonate with BaP4H compared with αKG (Kd ∼1 μm) is somewhat surprising. However, this could lend some evidence that similar metabolites, such as succinate, could provide a feedback type of inhibition to BaP4H. It has been suggested previously that concentrations of αKG could serve as an indicator of the metabolic status of the cell and that the Fe(II)/αKG dependent family of enzymes could possess a wider role in regulation of cellular processes (74, 75). This leads to the conclusion that there does not appear to be an obvious trend in analyzing binding affinities of αKG for different enzymes. A more complex combination of local cellular environment and structural dynamics is most likely involved.
In conclusion, the results described here uncovered that BaP4H can target proline-rich human collagen-like peptides for site-specific hydroxylation. The BaP4H structure-function studies have broader implications in deciphering molecular mechanisms that regulate the PSB domains in P4Hs and PPII substrate binding enzymes, including substrate specificity and the overall reaction mechanisms. Further studies to determine the x-ray crystal structure of active Fe(II)-BaP4H with a peptide substrate bound are underway, in addition to site-directed mutagenesis to confirm the catalytic residues, which altogether would allow the identification of the molecular determinants of substrate binding and site-specific catalysis.
Author Contributions
N. J. S. and M. D. designed the experiments, performed the experiments and data analysis, and wrote the manuscript.
Acknowledgment
We are grateful to Dr. Ryan Kunz at the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School for helping us with the mass spectrometry data collection and analysis. X-ray data were collected at the Advanced Light Source (ALS) and the Advanced Photon Source (APS). Support for the synchrotron sources is provided by the United States Department of Energy.
This work was supported by the University of Iowa College of Liberal Arts and Sciences. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5IAT, 5IAV, and 5IAX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- P4H
- prolyl 4-hydroxylase
- Hyp
- hydroxyproline
- αKG
- α-ketoglutarate
- MLI
- malonate
- PPII
- poly-l-proline type II
- DSBH
- double-stranded β-helix
- PRD
- proline recognition domain
- PSB
- peptide-substrate binding
- SBL
- substrate binding loop
- vCPH
- viral collagen P4H
- XIC
- extracted ion chromatogram
- SBL
- substrate binding loop
- HIF-1α
- hypoxia inducible factor-1α
- PDB
- Protein Data Bank
- PHD
- prolyl hydroxylase domain protein.
References
- 1. Gorres K. L., and Raines R. T. (2010) Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 45, 106–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hausinger R. P. (2004) FeII/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68 [DOI] [PubMed] [Google Scholar]
- 3. Koehntop K. D., Emerson J. P., and Que L. Jr. (2005) The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J. Biol. Inorg. Chem. 10, 87–93 [DOI] [PubMed] [Google Scholar]
- 4. Bollinger J. M. Jr., and Krebs C. (2006) Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 100, 586–605 [DOI] [PubMed] [Google Scholar]
- 5. Bruijnincx P. C., van Koten G., and Klein Gebbink R. J. (2008) Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: recent developments in enzymology and modeling studies. Chem. Soc. Rev. 37, 2716–2744 [DOI] [PubMed] [Google Scholar]
- 6. Costas M., Mehn M. P., Jensen M. P., and Que L. (2004) Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986 [DOI] [PubMed] [Google Scholar]
- 7. Lange S. J., and Que L. Jr. (1998) Oxygen activating nonheme iron enzymes. Curr. Opin. Chem. Biol. 2, 159–172 [DOI] [PubMed] [Google Scholar]
- 8. Bollinger J. M. Jr., and Broderick J. B. (2009) Frontiers in enzymatic C-H-bond activation. Curr. Opin. Chem. Biol. 13, 51–57 [DOI] [PubMed] [Google Scholar]
- 9. Price J. C., Barr E. W., Tirupati B., Bollinger J. M. Jr., and Krebs C. (2003) The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508 [DOI] [PubMed] [Google Scholar]
- 10. Hoffart L. M., Barr E. W., Guyer R. B., Bollinger J. M. Jr., and Krebs C. (2006) Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 103, 14738–14743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loenarz C., and Schofield C. J. (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 4, 152–156 [DOI] [PubMed] [Google Scholar]
- 12. Loenarz C., and Schofield C. J. (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 36, 7–18 [DOI] [PubMed] [Google Scholar]
- 13. Myllyharju J. (2008) Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 40, 402–417 [DOI] [PubMed] [Google Scholar]
- 14. Clifton I. J., McDonough M. A., Ehrismann D., Kershaw N. J., Granatino N., and Schofield C. J. (2006) Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 100, 644–669 [DOI] [PubMed] [Google Scholar]
- 15. Aik W., McDonough M. A., Thalhammer A., Chowdhury R., and Schofield C. J. (2012) Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr. Opin. Struct. Biol. 22, 691–700 [DOI] [PubMed] [Google Scholar]
- 16. Myllyharju J. (2013) Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. (Oxf.) 208, 148–165 [DOI] [PubMed] [Google Scholar]
- 17. Myllyharju J., and Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 18. Schofield C. J., and Ratcliffe P. J. (2004) Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 [DOI] [PubMed] [Google Scholar]
- 19. Aprelikova O., Chandramouli G. V., Wood M., Vasselli J. R., Riss J., Maranchie J. K., Linehan W. M., and Barrett J. C. (2004) Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. J. Cell Biochem. 92, 491–501 [DOI] [PubMed] [Google Scholar]
- 20. Kaelin W. G. Jr., and Ratcliffe P. J. (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 21. Pappalardi M. B., McNulty D. E., Martin J. D., Fisher K. E., Jiang Y., Burns M. C., Zhao H., Ho T., Sweitzer S., Schwartz B., Annan R. S., Copeland R. A., Tummino P. J., and Luo L. (2011) Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites. Biochem. J. 436, 363–369 [DOI] [PubMed] [Google Scholar]
- 22. Hieta R., Kukkola L., Permi P., Pirilä P., Kivirikko K. I., Kilpeläinen I., and Myllyharju J. (2003) The peptide-substrate-binding domain of human collagen prolyl 4-hydroxylases: backbone assignments, secondary structure, and binding of proline-rich peptides. J. Biol. Chem. 278, 34966–34974 [DOI] [PubMed] [Google Scholar]
- 23. Adzhubei A. A., Sternberg M. J., and Makarov A. A. (2013) Polyproline-II helix in proteins: structure and function. J. Mol. Biol. 425, 2100–2132 [DOI] [PubMed] [Google Scholar]
- 24. Berisio R., and Vitagliano L. (2012) Polyproline and triple helix motifs in host-pathogen recognition. Curr. Protein Pept. Sci. 13, 855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Myllyharju J., and Kivirikko K. I. (1999) Identification of a novel proline-rich peptide-binding domain in prolyl 4-hydroxylase. EMBO J. 18, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pekkala M., Hieta R., Bergmann U., Kivirikko K. I., Wierenga R. K., and Myllyharju J. (2004) The peptide-substrate-binding domain of collagen prolyl 4-hydroxylases is a tetratricopeptide repeat domain with functional aromatic residues. J. Biol. Chem. 279, 52255–52261 [DOI] [PubMed] [Google Scholar]
- 27. Anantharajan J., Koski M. K., Kursula P., Hieta R., Bergmann U., Myllyharju J., and Wierenga R. K. (2013) The structural motifs for substrate binding and dimerization of the alpha subunit of collagen prolyl 4-hydroxylase. Structure 21, 2107–2118 [DOI] [PubMed] [Google Scholar]
- 28. Cerveny L., Straskova A., Dankova V., Hartlova A., Ceckova M., Staud F., and Stulik J. (2013) Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect. Immun. 81, 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eriksson M., Myllyharju J., Tu H., Hellman M., and Kivirikko K. I. (1999) Evidence for 4-hydroxyproline in viral proteins: characterization of a viral prolyl 4-hydroxylase and its peptide substrates. J. Biol. Chem. 274, 22131–22134 [DOI] [PubMed] [Google Scholar]
- 30. Longbotham J. E., Levy C., Johannissen L. O., Tarhonskaya H., Jiang S., Loenarz C., Flashman E., Hay S., Schofield C. J., and Scrutton N. S. (2015) Structure and mechanism of a viral collagen prolyl hydroxylase. Biochemistry 54, 6093–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hieta R., Myllyharju J. (2002) Cloning and characterization of a low-molecular weight prolyl 4-hydroxylase from Arabidopsis thaliana: effective hydroxylation of proline-rich, collagen-like, and hypoxia-inducible transcription factor α-like peptides. J. Biol. Chem. 277, 23965–23971 [DOI] [PubMed] [Google Scholar]
- 32. Keskiaho K., Hieta R., Sormunen R., and Myllyharju J. (2007) Chlamydomonas reinhardtii has multiple prolyl 4-hydroxylases, one of which is essential for proper cell wall assembly. Plant Cell 19, 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horita S., Scotti J. S., Thinnes C., Mottaghi-Taromsari Y. S., Thalhammer A., Ge W., Aik W., Loenarz C., Schofield C. J., and McDonough M. A. (2015) Structure of the ribosomal oxygenase OGFOD1 provides insights into the regio- and stereoselectivity of prolyl hydroxylases. Structure 23, 639–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koski M. K., Hieta R., Böllner C., Kivirikko K. I., Myllyharju J., and Wierenga R. K. (2007) The active site of an algal prolyl 4-hydroxylase has a large structural plasticity. J. Biol. Chem. 282, 37112–37123 [DOI] [PubMed] [Google Scholar]
- 35. Koski M. K., Hieta R., Hirsilä M., Rönkä A., Myllyharju J., and Wierenga R. K. (2009) The crystal structure of an algal prolyl 4-hydroxylase complexed with a proline-rich peptide reveals a novel buried tripeptide binding motif. J. Biol. Chem. 284, 25290–25301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Culpepper M. A., Scott E. E., and Limburg J. (2010) Crystal structure of prolyl 4-hydroxylase from Bacillus anthracis. Biochemistry 49, 124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scotti J. S., Leung I. K., Ge W., Bentley M. A., Paps J., Kramer H. B., Lee J., Aik W., Choi H., Paulsen S. M., Bowman L. A., Loik N. D., Horita S., Ho C. H., Kershaw N. J., Tang C. M., Claridge T. D., Preston G. M., McDonough M. A., and Schofield C. J. (2014) Human oxygen sensing may have origins in prokaryotic elongation factor Tu prolyl-hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 111, 13331–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adefarati A. A., Hensens O. D., Jones E. T., and Tkacz J. S. (1992) Pneumocandins from Zalerion arboricola: V. glutamic acid- and leucine-derived amino acids in pneumocandin A0 (L-671,329) and distinct origins of the substituted proline residues in pneumocandins A0 and B0. J. Antibiot. 45, 1953–1957 [DOI] [PubMed] [Google Scholar]
- 39. Kukkola L., Koivunen P., Pakkanen O., Page A. P., and Myllyharju J. (2004) Collagen prolyl 4-hydroxylase tetramers and dimers show identical decreases in Km values for peptide substrates with increasing chain length: mutation of one of the two catalytic sites in the tetramer inactivates the enzyme by more than half. J. Biol. Chem. 279, 18656–18661 [DOI] [PubMed] [Google Scholar]
- 40. Rasmussen M., Jacobsson M., and Björck L. (2003) Genome-based identification and analysis of collagen-related structural motifs in bacterial and viral proteins. J. Biol. Chem. 278, 32313–32316 [DOI] [PubMed] [Google Scholar]
- 41. Miller M. A., Scott E. E., and Limburg J. (2008) Expression, purification, crystallization and preliminary x-ray studies of a prolyl-4-hydroxylase protein from Bacillus anthracis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 788–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ryle M. J., Liu A., Muthukumaran R. B., Ho R. Y., Koehntop K. D., McCracken J., Que L. Jr., and Hausinger R. P. (2003) O2- and α-ketoglutarate-dependent tyrosyl radical formation in TauD, an R-keto acid-dependent non-heme iron dioxygenase. Biochemistry 42, 1854–1862 [DOI] [PubMed] [Google Scholar]
- 44. Ryle M. J., Koehntop K. D., Liu A., Que L. Jr., and Hausinger R. P. (2003) Interconversion of two oxidized forms of taurine/α-ketoglutarate dioxygenase, a non-heme iron hydroxylase: evidence for bicarbonate binding. Proc. Natl. Acad. Sci. U.S.A. 100, 3790–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brummett A. E., Schnicker N. J., Crider A., Todd J. D., and Dey M. (2015) Biochemical, kinetic, and spectroscopic characterization of Ruegeria pomeroyi DddW-A mononuclear iron-dependent DMSP lyase. PloS One 10, e0127288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knauer S. H., Hartl-Spiegelhauer O., Schwarzinger S., Hänzelmann P., and Dobbek H. (2012) The Fe(II)/alpha-ketoglutarate-dependent taurine dioxygenases from Pseudomonas putida and Escherichia coli are tetramers. FEBS J. 279, 816–831 [DOI] [PubMed] [Google Scholar]
- 48. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., and Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 54. Laskowski R. A., Macarthur M. W., Moss D. S., and Thornton J. M. (1993) Procheck: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 55. Ramachandran G. N., Ramakrishnan C., and Sasisekharan V. (1963) Stereochemistry of Polypeptide Chain Configurations. J. Mol. Biol. 7, 95–99 [DOI] [PubMed] [Google Scholar]
- 56. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. London N., Raveh B., Cohen E., Fathi G., and Schueler-Furman O. (2011) Rosetta FlexPepDock web server-high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 39, W249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raveh B., London N., and Schueler-Furman O. (2010) Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 78, 2029–2040 [DOI] [PubMed] [Google Scholar]
- 59. Grzyska P. K., Ryle M. J., Monterosso G. R., Liu J., Ballou D. P., and Hausinger R. P. (2005) Steady-state and transient kinetic analyses of taurine/αketoglutarate dioxygenase: effects of oxygen concentration, alternative sulfonates, and active-site variants on the FeIV-oxo intermediate. Biochemistry 44, 3845–3855 [DOI] [PubMed] [Google Scholar]
- 60. Coufal D. E., Tavares P., Pereira A. S., Hyunh B. H., and Lippard S. J. (1999) Reactions of nitric oxide with the reduced non-heme diiron center of the soluble methane monooxygenase hydroxylase. Biochemistry 38, 4504–4513 [DOI] [PubMed] [Google Scholar]
- 61. Gaffney B. J. (2009) EPR of mononuclear non-heme iron proteins. Biol. Magn. Reson. 28, 233–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lancaster J. R. Jr., Langrehr J. M., Bergonia H. A., Murase N., Simmons R. L., and Hoffman R. A. (1992) EPR detection of heme and nonheme iron-containing protein nitrosylation by nitric oxide during rejection of rat heart allograft. J. Biol. Chem. 267, 10994–10998 [PubMed] [Google Scholar]
- 63. Orville A. M., and Lipscomb J. D. (1993) Simultaneous binding of nitric oxide and isotopically labeled substrates or inhibitors 3,4-dioxygenase by reduced protocatechuate. J. Biol. Chem. 268, 8596–8607 [PubMed] [Google Scholar]
- 64. Hutton J. J. Jr., Kaplan A., and Udenfriend S. (1967) Conversion of the amino acid sequence Gly-Pro-Pro in protein to Gly-Pro-Hyp by collagen proline hydroxylase. Arch. Biochem. Biophys. 121, 384–391 [DOI] [PubMed] [Google Scholar]
- 65. Kivirikko K. I., Suga K., Kishida Y., Sakakibara S., and Prockop D. J. (1971) Asymmetry in the hydroxylation of (Pro-Pro-Gly) 5 by protocollagen proline hydroxylase. Biochem. Biophys. Res. Commun. 45, 1591–1596 [DOI] [PubMed] [Google Scholar]
- 66. Berg R. A., and Prockop D. J. (1973) Purification of 14C protocollagen and its hydroxylation by prolyl-hydroxylase. Biochemistry 12, 3395–3401 [DOI] [PubMed] [Google Scholar]
- 67. Berg R. A., Kishida Y., Sakakibara S., and Prockop D. J. (1977) Hydroxylation of (Pro-Pro-Gly)5 and (Pro-Pro-Gly)10 by prolyl hydroxylase: evidence for an asymmetric active site in the enzyme. Biochemistry 16, 1615–1621 [DOI] [PubMed] [Google Scholar]
- 68. Zarrinpar A., and Lim W. A. (2000) Converging on proline: the mechanism of WW domain peptide recognition. Nat. Struct. Biol. 7, 611–613 [DOI] [PubMed] [Google Scholar]
- 69. Adefarati A. A., Giacobbe R., Hensens O. D., and Tkacz J. S. (1991) Biosynthesis of L-671,329, an echinocandin-type antibiotic produced by Zalerion arboricola: origins of some of the unusual amino acids and the dimethylmyristic acid side chain. J. Am. Chem. Soc. 113, 3542–3545 [Google Scholar]
- 70. McDonough M. A., Li V., Flashman E., Chowdhury R., Mohr C., Liénard B. M., Zondlo J., Oldham N. J., Clifton I. J., Lewis J., McNeill L. A., Kurzeja R. J., Hewitson K. S., Yang E., Jordan S., Syed R. S., and Schofield C. J. (2006) Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U.S.A. 103, 9814–9819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schnicker N. J., and Dey M. (2016) Structural analysis of cofactor binding for a prolyl 4-hydroxylase from the pathogenic bacterium Bacillus anthracis. Acta Cryst. D72, 675–681 [DOI] [PubMed] [Google Scholar]
- 72. McNeill L. A., Flashman E., Buck M. R., Hewitson K. S., Clifton I. J., Jeschke G., Claridge T. D., Ehrismann D., Oldham N. J., and Schofield C. J. (2005) Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol. Biosyst. 1, 321–324 [DOI] [PubMed] [Google Scholar]
- 73. Majamaa K., Hanauske-Abel H. M., Günzler V., and Kivirikko K. I. (1984) The 2-oxoglutarate binding site of prolyl4-hydroxylase Identification of distinct subsites and evidence for 2-oxoglutarate decarboxylation in a ligand reaction at the enzyme-bound ferrous ion. Eur. J. Biochem. 138, 239–245 [DOI] [PubMed] [Google Scholar]
- 74. Yuan H. X., Xiong Y., and Guan K. L. (2013) Nutrient sensing, metabolism, and cell growth control. Mol. Cell 49, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ploumakis A., and Coleman M. L. (2015) OH, the places you'll go! hydroxylation, gene expression, and cancer. Mol. Cell 58, 729–741 [DOI] [PubMed] [Google Scholar]