

Abstract
The majority of excitatory synapses in the brain exist on dendritic spines. Accordingly, the regulation of dendritic spine density in the hippocampus is thought to play a central role in learning and memory. The development of novel methods to control spine density could, therefore, have important implications for treatment of a host of neurodegenerative and developmental cognitive disorders. Herein, we report the design and evaluation of a new class of benzothiazole amphiphiles that exhibit a dose-dependent response leading to an increase in dendritic spine density in primary hippocampal neurons. Cell exposure studies reveal that the increase in spine density can persist for days in the presence of these compounds, but returns to normal spine density levels within 24 h when the compounds are removed, demonstrating the capability to reversibly control spinogenic activity. Time-lapse imaging of dissociated hippocampal neuronal cultures shows that these compounds promote a net increase in spine density through the formation of new spines. Biochemical studies support that promotion of spine formation by these compounds is accompanied by Ras activation. These spinogenic molecules were also capable of inhibiting a suspected mechanism for dendritic spine loss induced by Alzheimer-related aggregated amyloid-β peptides in primary neurons. Evaluation of this new group of spinogenic agents reveals that they also exhibit relatively low toxicity at concentrations displaying activity. Collectively, these results suggest that small molecules that promote spine formation could be potentially useful for ameliorating cognitive deficiencies associated with spine loss in neurodegenerative diseases such as Alzheimer disease, and may also find use as general cognitive enhancers.
Keywords: Alzheimer disease, dendritic spine, drug design, neurodegeneration, neuron, Benzothiazoles, RasGRF1, Spinogenic
Introduction
Dendritic spines are specialized protrusions responsible for receiving excitatory synaptic inputs, providing an important function in communication between neurons (1–3). The morphology of dendritic spines and their overall density correlates with synaptic function and is strongly implicated in memory and learning (1, 4, 5). Consequently, alteration or misregulation of dendritic spines can influence synaptic function and plays a major role in various neurological and psychiatric disorders such as autism, fragile X syndrome, Parkinson disease (PD), and Alzheimer disease (AD)2 (4, 6–12). For example, in AD there is mounting evidence suggesting deficits begin with alterations of hippocampal synaptic function correlated with accumulation of aggregated amyloid-β (Aβ) peptide prior to neuronal loss (13–16). Therefore, treatment strategies that target the initial synaptic loss, rather than late stage disease intervention, may provide a better prognosis for the treatment of AD. Furthermore, since most cognitive disorders elicit abnormalities in the form and function of dendritic spines, it would be desirable to target them directly using a small molecule to alter or alleviate these spine changes.
We previously reported the design, synthesis, and evaluation of two oligo(ethylene glycol) derivatives of benzothiazole aniline (BTA), BTA-EG6 and BTA-EG4, which exhibited a variety of advantageous properties for the potential treatment of neurodegenerative diseases such as AD (17–19). Interestingly, BTA-EG4 showed the capability to improve memory and learning in cognitive performance tests in both wild-type mice and in a mouse model for AD (18, 19). This in vivo activity of BTA-EG4 was also accompanied by a phenotypic increase in dendritic spine density (18, 19). Because of the scarcity of small molecules known to increase dendritic spine density, this rare feature of benzothiazole amphiphiles is of particular interest and could be utilized as a tool to help study the relationship between spines and cognitive function.
The in vivo results of BTA-EG4 suggest that it may provide broad therapeutic benefits for improving cognitive function in AD as well as in other dendritic spine-related diseases (20–22). However, we also observed toxicity of this compound in SH-SY5Y neuroblastoma cells that correlated with its ability to partition in membranes and induce membrane lysis (23). Toxicity is one of the biggest issues at every stage of drug development (24) and the toxicity of BTA-EG4 precluded our capability to adequately evaluate the extent of its spinogenic biological activity.
To further evaluate the ability of the benzothiazole agents to promote spinogenesis, herein we designed and characterized three novel structural variants of the BTA compounds. These new benzothiazole amphiphiles (BAMs) (1–3, Fig. 1) exhibit substantially less toxicity compared with the parent BTA compound. We show that these new BAM agents are capable of promoting an increase in dendritic spine density and can serve as either a pre- or co-treatment to negate the overall net spine loss induced by the presence of aggregated Aβ peptides. Additionally, these compounds are capable of directly inhibiting aggregated Aβ from inducing spine loss mediated by a Cofilin-dependent pathway. This spinogenic activity was dose-dependent in primary neurons, and, using BAM1-EG6 (1) as a representative example, we demonstrate that the increase in spine density is reversible by removal of the compound from the cellular environment. Time-dependent imaging studies of primary neurons treated with BAM1-EG6 reveal that these benzothiazoles can increase spine density through promoting the formation of new spines. Signal transduction studies support that these molecules promote spine formation by involving the activation of the Ras and do not directly affect overall F/G actin ratios or ARP2 expression levels in primary neurons. Taken together, these results demonstrate that these BAM agents represent new potential tools to study the relationship between dendritic spines and cognitive behavior and may open up a new avenue to explore the use of spinogenic agents for the treatment of neurodegenerative and other spine-related cognitive disorders.
FIGURE 1.

Design and synthesis of benzothiazole amphiphiles (BAMs) (1–3). A, BAMs (1–3) were designed to exhibit decreased hydrophobicity and hydrogen-bonding capabilities compared with the parent compound, BTA-EG6. B, synthetic scheme for the preparation of BAM 1–3. Abbreviations for all synthetic steps are as follows: potassium carbonate (K2CO3), potassium iodide (KI), potassium hydroxide (KOH), ionic liquid or 1-pentyl-3-methylimidazolium bromide ([pmIm]Br, microwave (MW), tetrahydrofuran (THF), meta-chloroperoxybenzoic acid (mCPBA), dichloromethane (DCM), trifluoroacetic anhydride (TFAA), sodium hydroxide (NaOH), methanol (MeOH), sodium hydride (NaH), dimethylformamide (DMF), 17-iodo-3,6,9,12,15-pentaoxaheptadecan-1-ol (EG6-I).
Experimental Procedures
Materials
3-(4,5-Dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT) cell proliferation assay (Product No: 30-1010K) was purchased from American Type Culture Collection (ATCC) (Manassas, VA). PierceTM BCA Assay kit (23225), Active Ras Pull-down and Detection Kit (16117), and PierceTM phosphatase inhibitor tablets (88667) were purchased from Thermo Scientific. G-actin/F-actin In Vivo Assay Kit (BK037) was purchased from Cytoskeleton, Inc. Protease inhibitor tablets (05892791001) were purchased from Roche. Primary antibodies used were: rabbit anti-Synapsin (EMD Millipore AB1543), mouse anti-PSD95 (EMD Millipore CP35), mouse anti-RasGRF1 (BD 610149), mouse anti-Ras (Thermo 1862335), mouse anti-GAPDH (Sigma G8795), rabbit anti-actin (Cytoskeleton AAN01), rabbit anti-Arp2 (Santa Cruz sc-15389), mouse anti-Aβ (6E10) (Biolegend® 803001), rabbit anti-p-Cofilin (Santa Cruz Biotechnology sc-12912-R) and rabbit anti-Cofilin (Santa Cruz Biotechnology sc-33779). Secondary antibodies used were as follows: goat anti-mouse AlexaFluor 568 (Invitrogen A110040), goat anti-rabbit AlexaFluor 647 (Invitrogen A21244), ECLTM horseradish peroxidase (HRP)-linked anti-mouse (GE NA931), and anti-rabbit (GE NA934). Amersham BiosciencesTM ECLTM Prime Western blotting Detection Reagent (RPN2232) was purchased from GE Healthcare. Diphytanoylphosphatidyl-choline (DiPhyPC) was purchased from Avanti Polar Lipids, Inc. Synthetic Aβ-(1–42) peptide was purchased from PL Lab. All chemical reagents were purchased and used as is from Sigma Aldrich or Fisher unless otherwise stated.
Compounds
BTA-EG6 was synthesized as previously reported (25). The general synthetic procedures we used to prepare benzothiazole amphiphiles (BAMs 1–3) are outlined in Fig. 1B. For the synthesis of the benzothiazole core for BAM2, commercially available 4-hydroxy benzaldehyde (4) was alkylated with 2-chloro-N-methylacetamide (5) via an in situ Finklestein reaction (26). The aryl ether (6) underwent a rearrangement under basic conditions to yield 4-N-(methylamino) benzaldehyde (7) (27). Microwave irradiation in ionic liquid ([pmim]Br) (28) of 2-aminothiophenol (8) with benzaldehyde (7) afforded benzothiazole (9). An analogous microwave-assisted reaction (29) between 8 and 12 gave 2-(4-(methylthio)phenyl)benzo[d]thiazole (13) in good yield. The methylthiol group on 13 was then oxidized to the sulfoxide via mCPBA oxidation to yield 2-(4-(methylsulfinyl)phenyl)-benzo[d]thiazole (14). Pummerer rearrangement (30, 31) of compound 14 gave the α-acyloxy-thioether (15), which was converted to the thiol (16). Compounds 10 (commercially available), 9, and 16 were then reacted with EG6-Iodide (11) (23) under standard nucleophilic substitution conditions to yield BAM1-EG6 (1), BAM2-EG6 (2), and BAM3-EG6 (3), respectively. Extended detailed descriptions of syntheses and characterization of compounds can be found in the supplemental information.
Measurement of Fluorescence Emission Spectra
The emission spectra of benzothiazoles in different environments was evaluated as previously described (23). Briefly, BAMs 1–3 and BTA-EG6 were diluted to a final concentration of 50 μm in deionized H2O, pure octanol, and a liposome suspension. The liposomes were prepared from a total lipid concentration of 10 mm diphytanoylphosphatidylcholine (DiPhyPC) in water by the gentle dehydration/rehydration method followed by tip sonication (32). 200 μl of each sample was transferred to a cuvette (Helma Analytics, Quartz SUPRASIL (QS), 10 mm), and the fluorescence emission spectrum was measured in a PTI spectrofluorometer (0.5 nm step size) in water, octanol and an aqueous liposome suspension for BAMs 1–3 and BTA-EG6. Maximal excitation and emission values (λmax) for all compounds were as follows: BTA-EG6 (Ex/Em 355/420 nm), BAM1-EG6 (Ex/Em 355/420 nm), BAM2-EG6, (Ex/Em 365/428 nm), and BAM3-EG6 (Ex/Em 335/398 nm). Each experiment was repeated at least three separate times and error bars denote standard deviation from the mean. Data were processed using Origin 7.0 (MicroCal Software, Inc., Northampton, MA).
Estimation of Log P and Solvent Accessible Surface Area (SASA)
Log P values were calculated using molinspiration cheminformatics software and solvent accessible surface area (SASA) values were calculated with PyMOL.
Neuronal Cultures
Rat dissociated hippocampal neurons from postnatal day 1 Sprague-Dawley® outbred rats of both sexes were plated at a density of 45,000 cells/cm2 onto poly-d-lysine-coated coverslips or 96-well plates. Neurons were maintained in B27 supplemented Neurobasal medium (Invitrogen) until days in vitro (DIV) 18–23, as previously described (33, 34). Overall neuronal health was monitored visually weekly and throughout the experimental process.
Measurement of Cell Viability in the Presence of BAMs 1–3 and BTA-EG6
An MTT cell viability assay was performed. Briefly, 21 DIV rat dissociated hippocampal neurons in 96-well plates were dosed with 100 μl of various samples solutions of either BTA-EG6 or BAMs 1–3 with final concentrations of 0 to 250 μm. Cells were exposed to these solutions for 24 h at 37 °C, 5% CO2. An MTT cell viability kit (ATCC, Product No: 30–1010K) was then used to determine cell viability. Briefly, 20 μl of the provided MTT reagent was added per well and cells were placed in the incubator for 3 h. The insoluble intracellular purple formazan was then dissolved by the addition of 100 μl of detergent reagent provided and let solubilize overnight at room temperature. The cell viability was determined by measuring the absorbance at 570 nm using a Spectramax 190 microplate reader (Molecular Devices). All results are presented as percent reduction of MTT relative to untreated cells (100% viability), and all wells were blanked with absorbance values from the wells containing medium, MTT reagent and detergent only.
Western Blot Analysis
Rat dissociated neurons were lysed with RIPA buffer containing both protease and phosphatase inhibitors. Protein concentration was determined by BCA assay and proteins were separated by SDS-PAGE followed by transfer onto nitrocellulose membranes. Membranes were blocked with either 5% BSA or milk in Tris-buffered saline with Tween 20 (TBST), followed by incubation with primary antibodies overnight at 4 °C with shaking. Proteins were visualized using the appropriate HRP-labeled secondary by ECL and detection was carried out on film (Freedom Imaging, SRX-101A). The intensity of each band was then quantified using ImageJ software.
Measurement of Cellular Active Ras Levels
To analyze the levels of active Ras in the rat primary neuronal lysates treated with BTA-EG6, BAMs 1–3 or control, we used an Active-Ras Pulldown and Detection Kit (Thermo) according to the manufacturer's guidelines.
Measurement of Cellular F-Actin/G-Actin Ratio
To analyze the levels of filamentous (F-actin) and globular (G-actin) in rat primary neuronal lysates treated with BTA-EG6, BAMs 1–3 or control, we used a G-actin/F-actin in Vivo Assay Kit (Cytoskeleton) according to the manufacturer's guidelines.
Neuronal Treatments
For all neuronal treatments, the following general protocol was followed: Briefly, 18–23 DIV rat dissociated hippocampal neurons were dosed with various concentrations (0–50 μm final concentration) of BAMs 1–3 or BTA-EG6 (with 0.1% final DMSO concentration) for various incubation times (24–72 h depending on the experiment). After dosing and at the desired time point, the medium was removed, and cells were rinsed with PBS-MC (phosphate-buffered saline, 1 mm MgCl2 and 0.1 mm CaCl2). Following rinsing, cells were fixed with 4% paraformaldehyde (PFA)/sucrose in PBS for 10 min at room temperature. After fixation, coverslips were carefully rinsed (3× PBS-MC) and then mounted onto slides (Polysciences Inc., 18606) for imaging.
Colocalization of PSD95 and Synapsin
After following the general procedure for neuronal treatment, fixed cells were then permeabilized, blocked in 5% BSA/PBS-MC, and incubated overnight at 4 °C with anti-Synapsin (1:1000) and anti-PSD95 (1:1000). After incubation with Alexafluor conjugated secondary antibodies, the coverslips were mounted for imaging.
Sindbis Production
PalGFP SinRep5 DNA was obtained as a generous gift from Takahiro Furuta (Kyoto University, Kyoto, Japan) (35). Recombinant Sindbis virion production was accomplished through RNA transcription using the SP6 mMessage mMachine kit (Ambion, Austin, TX). Electroporation of RNA into baby hamster kidney cells (BHK) was completed using a BTX ECM 600 electroporator (BTX, Holliston, MA) at 220V, 129Ω, and 1050μF. After 24 h, virion was collected and concentrated by centrifugation at 20,000 rpm for 90 min using a Beckman Coulter Optima MAX Ultracentrifuge (Beckman Coulter, Indianapolis, IN). The treated neurons were infected with palGFP expressing sindbis 18 h prior to fixation. The palGFP (membrane targeting) signal was imaged directly in fixed neurons.
Confocal Microscopy and Dendritic Spine Analysis
For all imaging of neurons, we used a Leica DMI6000 inverted microscope outfitted with a Yokogawa Spinning disk confocal head, an Orca ER High Resolution CCD camera (6.45 μm/pixel at 1×) (Hamamatsu), Plan Apochromat 63×/1.4 na objective, and PerkinElmer solid-state laser with 488 nm excitation. Confocal z-stacks were acquired in all experiments, and all imaging was acquired in the dynamic range of 8-bit acquisition (0–255 pixel intensity units, respectively) with Volocity (PerkinElmer) imaging software. Imaged dendrites from one secondary dendrite per cell (after 1 branch) at a distance of 40–80 μm from the soma were straightened using ImageJ. We estimated spine density as the number of manually counted spines divided by dendrite segment length. The analyzer was blind to treatment and statistical significance was determined between experimental conditions by either unpaired t tests (two groups) or by ANOVA and indicated post hoc multiple-comparison test (>2 experimental conditions).
Real Time Imaging of Spine Changes in Rat Primary Hippocampal Neurons
For this study, 21 DIV neurons plated in 35 mm dishes (MatTek) were rinsed 3x with an excess of HBSS and then left in HBSS for the duration of imaging. For live imaging we kept cells at 37 °C and used a Leica DMI6000 inverted microscope outfitted with a Yokogawa Spinning disk confocal head, an Orca ER High Resolution CCD camera (6.45 μm/pixel at 1×) (Hamamatsu), Plan Apochromat 63×/1.4 na objective, and PerkinElmer solid-state laser with 488 nm excitation. The spine changes on the same neuron were monitored 1 h before dosing (−60 min) and up to 3 h after dosing (+180 min). Dosing occurred at t = 0 and consisted of either 0 (for control) or 5 μm BAM1-EG6. For each condition, neurons from three different neuronal preparations (prep) were used and two neurons per prep were monitored. Confocal z-stacks were acquired in all experiments, and all imaging was acquired in the dynamic range of 8-bit acquisition (0–255 pixel intensity units, respectively) with Volocity (PerkinElmer) imaging software. For analysis, imaged dendrites were straightened using ImageJ and the same dendrite length was analyzed for each condition. Spines gained were counted as any new spines found at each respective time point and spines lost were counted as spines that disappeared from the analyzed segment. The analyzer was blind to treatment.
Preparation and Characterization of Aggregated Aβ-(1–42)
Aggregated Aβ-(1–42) was prepared as previously described (36). Briefly, Aβ-(1–42) was initially solubilized in 100% 1,1,1,3,3,3,-hexafluoro-2-propanal (HFIP) to 1 mm concentration at RT for 21 h with shaking. The solution was sonicated and vortexed before it was diluted in cold nanopure water (2:1 H2O:HFIP). Aliquoted fractions were lyophilized for 2 days, followed by storage at −80 °C until use. Solutions of Aβ were obtained by dissolving Aβ in sterile PBS to a concentration of 100 μm and incubated at 37 °C for 3 days before use. Western blot analysis of the 3-day incubated Aβ was carried out to determine composition. We estimated the relative abundance of monomers, oligomers, and protofibrils of Aβ using ImageJ and percentage of each composition was calculated by dividing the intensity of each aggregation state over the total intensity for Aβ in the lane. This preparation of Aβ lead to a composition of ∼12% monomers, ∼16% low MW oligomers, and ∼72% mixture of soluble protofibrils/fibrils (data not shown). Aggregated Aβ was also characterized by EM, MALDI-TOF, and binding by Thioflavin T (data not shown).
Rescue of Net Aβ-induced Spine Loss in Rat Primary Hippocampal Neurons
We followed the same general dosing procedure as described for neuronal treatment, except with the following noted changes. For Aβ co-treatment, 18 DIV rat dissociated hippocampal neurons were dosed with a final concentration of 1 μm aggregated Aβ-(1–42) with or without the presence of 1 or 5 μm of BAMs 1–3 or BTA-EG6 for 3 days. Control cells were treated with vehicle control only (0.1% DMSO) for the three-day period. For Aβ pre-treatment, rat dissociated hippocampal neurons were dosed with 5 μm of BAMs 1–3 or BTA-EG6 for 24 h. After a wash out of compounds, neurons were dosed with a final concentration of 1 μm of aggregated Aβ-(1–42) for 3 days. For both treatments, after dosing medium was removed, cells were rinsed with PBS-MC, fixed with 4% paraformaldehyde (PFA)/sucrose in PBS for 10 min at room temperature, and mounted onto slides (Polysciences Inc., 18606) for imaging. All analysis was done blinded, and each experiment was repeated at least three separate times using neurons from three different preparations.
Results
Design and Evaluation of Benzothiazole Amphiphiles (BAMs) 1–3 with Decreased Partitioning in Membranes Compared with BTA-EG6
The toxicity of BTA-EG4,6 agents were previously reported to correlate with their capability to partition in membranes (23). We hypothesized that altering the hydrophobic core of these molecules would decrease their energetic driving force to partition into membranes, thereby reducing their concomitant toxicity. To test this hypothesis, we used BTA-EG6 as a lead compound for the design of three new compounds 1–3 (Fig. 1A). We designed these new benzothiazoles to test whether moving the 6-methyl group in BTA-EG6 to the aniline nitrogen or complete removal of the 6-methyl group would reduce the hydrophobicity of the compounds enough to significantly reduce membrane partitioning and toxicity to cells. We also tested the importance of retaining the aniline nitrogen in 1 by modifying it to a sulfur group as in BAM3-EG6 (3).
Calculations (Fig. 2C) suggested that small modifications to BTA-EG6 could affect its hydrophobic character as determined by their octanol-water partitioning coefficient (log P) and their solvent accessible surface area (SASA) without the need to modify the core benzothiazole structure, which is presumably required to impart spinogenic properties. We, therefore, examined the hydrophobic character of the new BAMs 1–3 relative to the parent compound, BTA-EG6, by taking advantage of the solvatochromic nature of these compounds. In this assay, the fluorescence emission spectra were measured for each compound in octanol, water and an aqueous suspension of liposomes to mimic cell membranes. All compounds exhibited a shift of maximum fluorescence emission to shorter wavelengths in a more hydrophobic environment (i.e. in octanol compared with water) (Fig. 2, A and C). The emission maximum in an aqueous suspension of liposomes was measured for all compounds and it was found that compounds 1–3 exhibited λmax that reflected a more polar, water-like environment compared with BTA-EG6, with changes of emission max of 29–34% from water (relative to λmax in pure octanol) compared with a 50% change in λmax for BTA-EG6 (Fig. 2, A and C). These results demonstrate that the novel structural modifications in BAMs 1–3 decreased their membrane partitioning compared with BTA-EG6.
FIGURE 2.

Physical and toxic properties of BAMs 1–3 and BTA-EG6. A, fluorescence emission properties of BAMs 1–3 and BTA-EG6 in water, octanol, or an aqueous solution of liposomes. B, viability of primary neurons as a function of increasing concentration of BTA-EG6 or BAMs 1–3. C, table of calculated hydrophobic parameters, measured membrane partitioning properties, and IC50 values of toxicity in rat dissociated primary neurons of BAMs 1–3 and BTA-EG6. a, log P values were calculated from molinspirations cheminformatics software. b, SASA values were calculated with PyMOL.
BAMs 1–3 Exhibit a Decreased Toxicity Compared to BTA-EG6
An MTT cell proliferation assay was performed to compare the toxicity of compounds 1–3 to the parent compound, BTA-EG6. In this assay, BTA-EG6 exhibited moderate toxicity to rat primary neurons with an IC50 of 9.8 μm after 24-h exposure (Fig. 2, B and C). Satisfyingly, we found that all BAMs 1–3 were significantly less toxic than BTA-EG6, with IC50 values ranging from 44–67 μm (Fig. 2, B and C).
Effects of BAMs 1–3 on Dendritic Spine Density
BTA-EG6 was first used to assess increases in spine density in primary hippocampal neurons as a control due to its previously published ability to increase spine density (37). To visualize all spines, we used a virally transfected membrane-targeting palGFP that is known to reliably fluorescently label dendritic spines (38–40). Moreover, expression was limited to less than 18 h to minimize any artifacts from viral transduction. After confirming an observed increase in dendritic spine density in BTA-EG6-treated neurons over treatment with vehicle control (Fig. 3), neurons were next treated with 1 or 5 μm concentrations of benzothiazoles 1–3. All new compounds 1–3 showed a dose-dependent increase in spine density after a 24 h exposure (Fig. 3). In addition, compounds 1–3 were able to produce a statistically significant increase in net spine density at a lower concentration compared with BTA-EG6, suggesting the structural differences (and possibly the decreased hydrophobic character) of 1–3 compared with BTA-EG6 results in overall increased spinogenic activity. There was no observed change in spine density when the cells were treated with the vehicle control (0.1% DMSO).3
FIGURE 3.
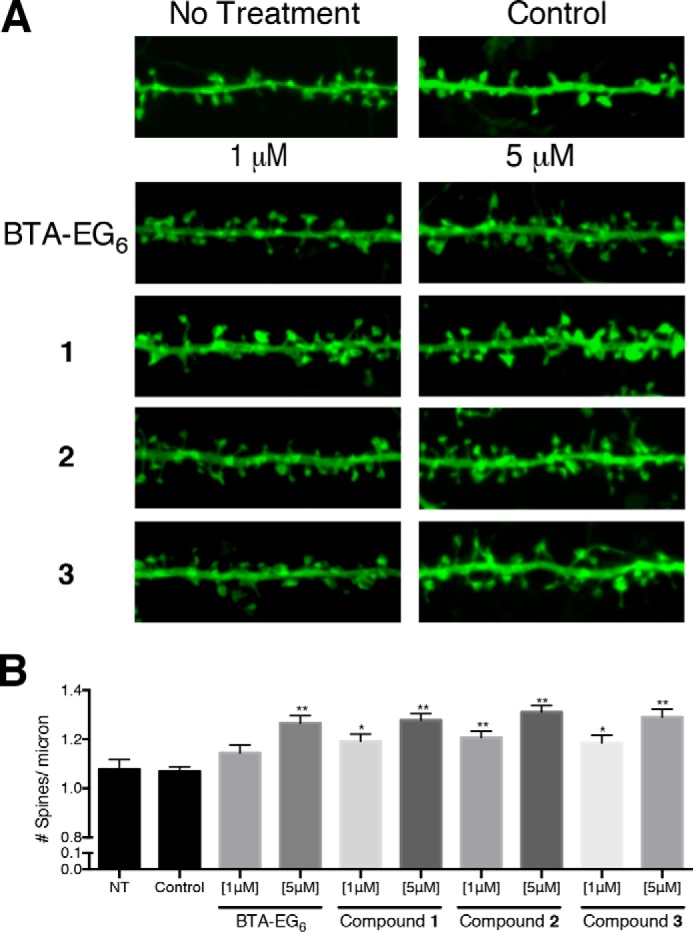
Spinogenic properties of BTA-EG6 and BAMs 1–3 observed in rat primary hippocampal neurons. A, representative spine segments (23 microns) for BTA-EG6 and BAMs 1–3 compared with control (0.1% DMSO). B, quantitative representation of spine number per micron for all compounds compared with control. Data are expressed as mean values ± S.E., n ≥ 54, *, p < 0.001; **, p <0.0001 as determined by unpaired t test compared with control.
Using BAM1-EG6 as a representative compound for this class of benzothiazoles, we next examined the effects of BAM agents on the density of neuronal puncta containing pre- and post-synaptic markers. We dosed neurons for 24 h with and without BAM1-EG6 (5 μm) and then examined the colocalization of PSD95 (post-synaptic marker) and Synapsin (pre-synaptic marker). In addition to the increase in dendritic spine density (Fig. 3), we also observed an increase in the density of colocalized PSD95-Synapsin puncta in neurons dosed with BAM1-EG6 over the control (Fig. 4).
FIGURE 4.

Analysis of the localization of synaptic proteins of neurons exposed to BAM1-EG6. A, representative images of dendrites (pal-GFP) PSD95 (post-synaptic marker) and Synapsin (pre-synaptic marker) from neurons dosed with control (0.1% DMSO) or BAM1-EG6. Scale bar, 20 microns B, representative images of spine segments (20 microns) for cells treated with BAM1-EG6 compared with control (0.1% DMSO) labeled with pal-GFP (green), PSD95 (red), and Synapsin (blue). C, quantitative representation of spine number per micron for BAM1-EG6 compared with control. D, quantitative representation of amount of colocalized PSD95 and Synapsin puncta per micron for neurons treated with BAM1-EG6 compared with control. Data are expressed as mean values ± S.E., n ≥ 48; **, p <0.0001 as determined by unpaired t test compared with control.
The cumulative distribution of spine length and width was also measured for neurons dosed with BAM1-EG6. No observable difference in average spine length or width was found compared with cells treated with vehicle control (Fig. 5, A and B).
FIGURE 5.

Examination of the spinogenic properties of BAM1-EG6 observed in rat primary hippocampal neurons. Cumulative distribution of spine length (A) or width (B) of control cells versus cells treated with compound BAM1-EG6 (1 μm). C, concentration-dependent effects of neurons dosed for 24 h with 1–25 μm BAM1-EG6 on spine density. D, kinetics of spine density increase in cells exposed to BAM1-EG6 compared with vehicle control (0.1% DMSO). Neurons were dosed and then fixed at 2, 4, 12, 24, 48, and 72 h. E, effects of removal of BAM1-EG6 on dendritic spine number after treatment of cells for 24 h. After 24 h, BAM1-EG6 was washed out (w.o.) and spine changes were monitored for an additional 24 and 48 h (48 and 72 h total time). The dendritic spine density 24 h after removal of BAM1-EG6 is indistinguishable from control cells. F, effect on spine density increases of adding additional doses of BAM1-EG6 every 24 h over a total incubation time of 72 h. Neurons were dosed at 24 h (1×), 48 h (2×), and 72 h (3×) with no observable additional increase of dendritic spine density compared with the 1× dose. Data are expressed as mean values ± S.E., n ≥ 54. **, p ≤ 0.0001; n.s., not significant, as determined by unpaired t test compared with control. Arrows denote time points when aliquots of cells were fixed and analyzed.
To evaluate the maximum effect of the BAM agents on spine density increase, we dosed primary neurons for 24 h in the presence of 1–25 μm BAM1-EG6. The maximum observed increase in spine density was ∼20%, occurring with a dose of 5 μm with no further increase at higher concentrations (Fig. 5C).
A time course of spinogenic activity was also examined in three separate experiments: In the first experiment, BAM1-EG6 was exposed to primary neurons at a constant concentration (5 μm) in the culture medium for up to 72 h. At various time points, we fixed cells and measured spine density (as estimated by spine number per μm). This experiment revealed a trend of increasing average spine density between 2 and 24 h, with statistically significant spine density increase reached after a 12 h exposure of the neurons to BAM1-EG6 (Fig. 5D). The spinogenic activity of the benzothiazoles reached equilibrium within 24 h, and this maximal increase of ∼20% in spine density levels (compared with treatment with vehicle) persisted for up to 3 days upon exposure to a constant concentration of the BAM1-EG6.
In a second experiment, we evaluated whether the spine density increases induced by the benzothiazoles persisted after the compounds have been removed from the culture medium. Primary hippocampal neurons were dosed for 24 h with 5 μm BAM1-EG6, resulting in the expected ∼20% increase in spine density levels compared with treatment with vehicle alone (0.1% DMSO). The cells were then rinsed and the culture medium was replaced with compound-free medium, and we monitored the average spine density on the cells over an additional 48 h. The initial spine increase after 24 h exposure to BAM1-EG6 did not persist once we removed the compound, with the density of spines returning back to normal levels (i.e. to the spine density observed in control cells) within 24 h of removal of BAM1-EG6 (Fig. 5E).
In a third experiment, we monitored the effect on spine density in primary neurons by adding fresh aliquots of BAM1-EG6 every 24 h to the culture medium over a 72 h period. We incubated primary neurons initially in culture medium containing a final concentration of 5 μm BAM1-EG6 (1× dose). At 24 h (2× dose) and 48 h (3× dose) of incubation, an additional 2 μl of a 5 mm BAM1-EG6 DMSO stock (final concentration 5 μm, 0.1% DMSO) was added to the culture medium. We found that further addition of BAM1-EG6 every 24 h (which putatively increased the final concentration of compound after every addition) did not result in further increases in dendritic spine density above the original observed increase of ∼20% after 24 h exposure of 5 μm BAM1-EG6 (Fig. 5F).
BAMs 1–3 Promote the Formation of New Dendritic Spines
An observed increase in dendritic spine density by benzothiazoles 1–3 could arise either by promoting the formation of new spines or by increasing the stability of previously formed dendritic spines. To help elucidate which mechanistic pathway BAMs promotes dendritic spine density alterations, we monitored the changes in spine number in real time by periodically capturing live confocal images of primary neurons over a 4-h time period. To account for baseline changes in spine dynamics, neurons were monitored 1 h prior to dosing. Live imaging then continued up to 3 h after dosing with either compound 1 (5 μm) or the vehicle control to gain insight into the spine changes induced by compound 1. We observed that dosing with compound 1 led to a statistical increase in new spines compared with the control 60 min after dosing, while no significant change in spine loss was observed over the same time period (Fig. 6).
FIGURE 6.

Live cell imaging showing the increase in formation of new spines upon dosing with BAM1-EG6 (compound 1). A, representative segments (20 microns) of live cells before (−time) and after dosing (+time) with BAM1-EG6 (5 μm) or vehicle control (0. 1% DMSO). * denotes new spines. B, quantitative representation of the total dendritic spines gained or lost per 20 micron segments for either BAM1-EG6 or vehicle control. n = 6; *, p < 0.01 compared with control at same time point by unpaired t test.
BAMs 1–3 Promote Ras Signaling
Ras and RasGRF1, a guanine nucleotide exchange factor involved in Ras signaling, are important intermediates in the regulation of spine density (42). Previous work has reported that BTA-EG4 and BTA-EG6 could promote spine density increases in vitro in murine primary hippocampal neurons (37) and BTA-EG4 could promote spine density increases in vivo in the hippocampus of wt mice and a 3× tg mouse model for AD (18, 19). The increase in spine density in neurons by BTA-EG4,6 correlated with an increase in expression of RasGRF1 compared with control cells (37). In order to test whether the spinogenic activity induced by compounds 1–3 operated along a similar mechanistic path as BTA-EG4,6, we analyzed the effects of these compounds on the expression level of both RasGRF1 and active Ras in rat-dissociated primary neurons. When we exposed primary neurons to 5 μm concentrations of compounds 1–3 or BTA-EG6, we observed about a 2-fold increase in RasGRF1 expression levels (Fig. 7A). Additionally, an ∼1.5-fold increase in active Ras/total Ras was also observed for compounds 1–3 or BTA-EG6 compared with control cells (Fig. 7B).
FIGURE 7.
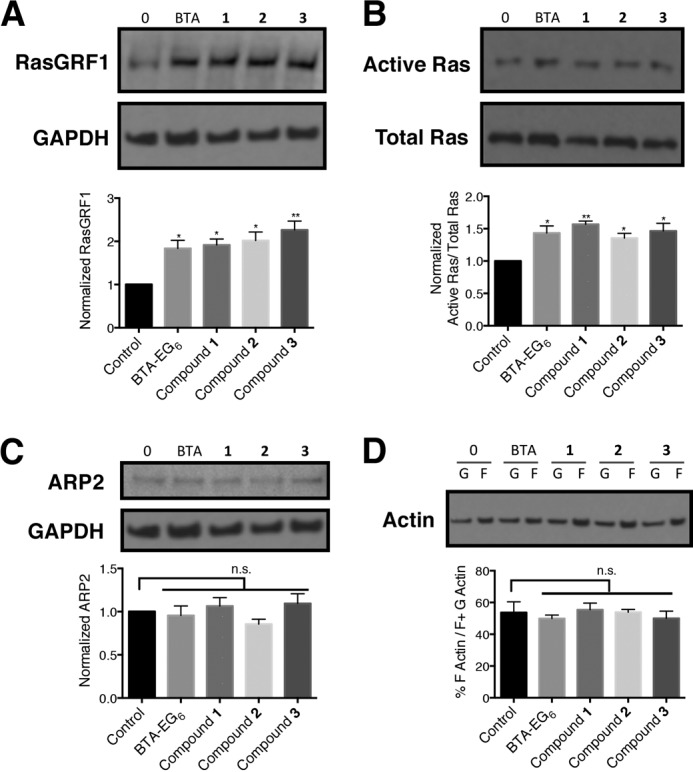
Analysis of protein expression, activation, and dynamics in primary neurons exposed to benzothiazole agents. Relative expression levels of A) RasGRF1, B) active Ras, and C) ARP2 in primary neurons upon dosing with 5 μm BTA-EG6 and BAMs 1–3. D, ratio of F-actin (filamentous actin) over total actin (F- plus G-actin (globular actin)) in primary neurons treated with BTA-EG6 and BAMs 1–3 (5 μm) or vehicle control. Data are expressed as mean values ± S.E., n = 3 or more for each concentration. n.s., not significant compared with control *, p < 0.05 compared with control; **, p < 0.01 compared with control as determined by unpaired t test.
BAMs 1–3 Do Not Directly Affect Overall Cellular Cytoskeletal Actin Dynamics or Actin-related Protein-2 (ARP2)
Due to the rapid changes in dendritic spine dynamics as seen in both live and fixed cell imaging, we next sought to examine the effects of BAMs on cytoskeletal rearrangement. The actin-related protein -2/3 (ARP2/3) complex plays a central role in the regulation of actin organization (43). Therefore, we first inspected whether the BAM agents affected ARP2 expression levels in primary neurons. We did not find a statistical difference in ARP2 expression between the control cells and cells treated with 5 μm BAMs 1–3 or BTA-EG6 (Fig. 7C).
We next examined whether the BAM agents had an effect on overall actin polymerization within neurons. We utilized a G-actin/F-actin assay kit, which enabled us to probe both F- and G-actin levels from primary lysates that had been dosed with 5 μm BAMs 1–3 and BTA-EG6 for 24 h. We did not observe a significant difference in the ratio of F-actin over total actin levels in neurons treated with any of the compounds compared with control neurons (Fig. 7D).
BAMs 1–3 Inhibit Aβ-induced Cofilin Activation and Negate Aβ-induced Net Spine Loss
Since BAMs 1–3 were able to increase dendritic spine density in primary hippocampal neurons (Fig. 3), we examined whether co- or pre-treatment of neurons with these compounds could negate any observed net spine loss in neurons exposed to aggregated amyloid-β (Aβ-(1–42)), the toxic peptide cleavage product of the amyloid precursor protein (APP) associated with AD.
For co-treatment, we treated primary neurons for 3 days with medium containing aggregated Aβ-(1–42) with or without the presence of BAMs 1–3 or BTA-EG6. We observed around a 20% decrease in spine density in primary neurons that were incubated in the presence of 1 μm aggregated Aβ-(1–42) alone for 3 days (Fig. 8). In contrast, when we treated primary neurons with 1 μm Aβ-(1–42) and 1 or 5 μm concentrations of BAMs 1–3 or BTA-EG6, we observed a net increase in dendritic spine density by ∼20% compared with control (Fig. 8A). Furthermore, the observed net increase in spine density in cells treated simultaneously with Aβ-(1–42) and BAMs 1–3 or BTA-EG6 were ∼40% higher than in cells treated with Aβ-(1–42) alone.
FIGURE 8.

Compounds counteract Aβ associated net spine loss in rat primary hippocampal neurons. A, representative spine segments (23 microns) of primary neurons dosed with Aβ, or Aβ plus BTA-EG6, or BAMs 1–3 compared with control (0.1% DMSO) and quantitative representation of spine number per micron for all dosing experiments compared with control. B, representative spine segments (23 microns) of primary neurons pre-treated with 5 μm BTA-EG6 or BAMs 1–3 for 24 h followed by addition of aggregated Aβ. C, relative expression levels of p-Cofilin/total Cofilin in primary neurons treated with Aβ alone or Aβ plus BTA-EG6 or BAMs 1–3 compared with control expression levels. Data are expressed as mean values ± S.E., n = 42. ##, p ≤ 0.01, as determined by unpaired t test compared with control. *, p ≤ 0.001; *, p ≤ 0.0001 as determined by unpaired t test compared with cells treated with Aβ alone.
For the pre-treatment regiment, we pre-treated primary neurons for 1 day in medium containing BAMs 1–3 or BTA-EG6 (or vehicle control). This pre-treatment presumably increased the dendritic spine density in cells exposed to compounds (Fig. 3). Next, we replaced the medium with medium containing 1 μm Aβ-(1–42) and incubated for an additional 3 days. When we pre-treated primary neurons with 5 μm concentrations of BAMs 1–3 or BTA-EG6, we observed no statistical difference in overall spine number compared with neurons that were not exposed to compounds or Aβ peptide (Fig. 8B). These results demonstrate that pre-treatment with BTA-EG6 and benzothiazoles 1–3 effectively protects cells from an overall net decrease in dendritic spine density induced by aggregated Aβ-(1–42) peptides.
We next examined whether the BAM agents could specifically inhibit a mechanistic pathway for Aβ-induced spine loss. Cofilin promotes actin depolymerization and its activation is promoted through dephosphorylation of p-Cofilin (44). Shatz and coworkers recently reported that Aβ aggregates induce spine loss through a pathway that involves activation of Cofilin-dependent depolymerization of actin upon binding of Aβ aggregates to the PirB receptor (45). Here, we observed a statistical decrease in p-Cofilin/total Cofilin levels in primary neurons that had been dosed with aggregated Aβ alone (Fig. 8C), which agrees well with the previous report by Shatz. However, when neurons had been dosed with BAMs 1–3 or BTA-EG6 together with aggregated Aβ, the ratio of p-Cofilin/total Cofilin was the same as cells that were not exposed to aggregated Aβ peptides (Fig. 8C). This results supports that the BAM agents are able to block the Aβ-induced activation of Cofilin, thereby blocking a specific mechanism for Aβ-induced spine loss.
Discussion
Many cognitive disorders are accompanied with loss of dendritic spines, yet there are few examples of molecules that promote the formation of new dendritic spines. The capability to promote increases in spine density through external administration of a drug could lead to a better understanding of the underlying circuitry affecting cognitive behavior, and, ultimately, to novel approaches for treatment of cognitive disorders.
We previously reported that oligo(ethylene glycol) derivatives of benzothiazole could insert into planar lipid bilayers and induce membrane lysis (23). The concentration required to observe lysis in membranes was roughly the same as the concentration required to observe cytotoxicity of the compounds in human SH-SY5Y neuroblastoma cells (IC50 ∼60 μM), suggesting lysis of cells as the significant factor for the apparent toxicity (46) of the BTA-EG4,6 compounds at high micromolar concentrations (23). Hence, we hypothesized that altering the hydrophobic core of these molecules would decrease their energetic driving force to partition into membranes, thereby reducing their toxicity.
With a goal of generating structural analogs of BTA-EG6 with reduced hydrophobic character, we designed and synthesized benzothiazole analogs 1–3 (Fig. 1). This new set of benzothiazoles exhibited reduced partitioning in membranes and lower overall toxicity in primary neurons (Fig. 2) compared to parent BTA-EG6. Interestingly, we did not find a correlation between the calculated log P values (a typical measure of hydrophobicity) and toxicity. Instead, we found a similar trend between solvent-accessible surface area (SASA) and toxicity in BTA-EG6 and all of the BAM derivatives. The SASA could, therefore, represent a more useful and quantifiable alternative parameter to Log P for guiding the development of additional members of this class of spinogenic compounds with low toxicity.
While reducing toxicity of benzothiazole agents is an important step toward improving their biocompatibility, it is also important to assess whether the new BAMs 1–3 retain the potential beneficial biological activity of the parent compound. The capability of BTA-EG4,6 to promote an increase in dendritic spine density is a distinctive and extremely rare property for any small molecule reported to date. The results demonstrate that the new benzothiazoles 1–3 are indeed capable of promoting dose-dependent increases in dendritic spine density in primary hippocampal neurons (Fig. 3), with maximal spine density increases of ∼20% after 24 h of exposure to cells.
The activity of BAM1-EG6 was further examined as a representative for this class of compounds to further evaluate the spinogenic effects of this class of benzothiazoles. Analysis of the colocalization of both pre- and post-synaptic markers, Synapsin and PSD95, in neurons exposed to BAM1-EG6 showed both an increase in the spines density and an increase in the density of colocalized PSD95/Synapsin puncta (Fig. 4). This result supports that the promotion of new dendritic spines may correlate with an increase in the number of synapses formed in neurons exposed to the BAM agents. Furthermore, the analysis of the cumulative distribution of spine width and length of cells exposed to BAM1-EG6 showed no difference when compared to control cells (Fig. 5, A and B), demonstrating that the increase in spine density by BAM agents does not affect the overall distribution of spine morphology in the cells (47).
Temporal studies showed that the spine density increased within 12 h in neurons and stably persisted for 72 h in the presence of BAM1-EG6. However, the spine density increase in cells exposed to BAM1-EG6 returned to basal levels (i.e. levels comparable to control neurons) within 24 h of removal of this compound from the medium (Fig. 5, D and E). This capability of BAM agents to reversibly control the magnitude of spine density changes in primary neurons may be very attractive as a tool for further studies on the relationship between dendritic spines and other parameters related to neural circuitry.
Live cell imaging and biochemical studies support that these benzothiazoles promote the formation of new dendritic spines in neurons (Fig. 6) through a mechanism that is accompanied by an increase in both RasGRF1 expression and active Ras levels (Fig. 7, A and B). Previous studies showed that shRNA knockdown of RasGRF1 in primary neurons completely blocked the effect of BTA-EG4 on spine density increases (18), further supporting that Ras signaling is essential for the spinogenic activity of the BAM agents.
The kinetic data gained from the live cell imaging and from imaging of fixed cells after various short exposures to the BAM agents indicate that changes in spine dynamics begin within the first couple hours, with significant net increases in dendritic spine density observed by 12 h (Figs. 6 and 5D). The surprising speed at which we observe changes in spine dynamics suggests that cytoskeletal reorganization (48, 49) plays an important role in the mechanism of action of the BAM agents. While we did not find a statistical difference in either the global F-/G-actin ratios or ARP2 expression levels in neurons exposed to the BAM agents (Fig. 7, C and D), these results do not rule out the possibility that the BAM agents act on more localized cytoskeletal machinery that affects dendritic spine dynamics.
Finally, we showed that co- and pre-treatment of neurons with BAMs 1–3 and BTA-EG6 were able to negate the net dendritic spine loss observed in the presence of aggregated Aβ (Fig. 8, A and B). Importantly, we found that the BAM agents could block Aβ-induced activation of Cofilin (Fig. 8C), which has been implicated as a specific mechanism for Aβ-induced synaptic dysfunction in AD through the binding of aggregated Aβ to the PirB receptor (45). Since BTA-EG6 was previously shown to bind to aggregated Aβ and act as a general inhibitor of protein-amyloid interactions (17, 25), we hypothesize that the BAM agents inhibit Aβ-induced spine loss by binding to Aβ and inhibiting its interaction with the PirB receptor (45) (and, thus, lead to the downstream inhibition of Cofilin activation by aggregated Aβ). Collectively, these results support a dual mode of action of BTA-EG6 and the BAM agents on the spine density of primary neurons exposed to aggregated Aβ: 1) these compounds are capable of directly inhibiting Aβ from inducing spine loss through a Cofilin-dependent pathway, and 2) these compounds can act on an Aβ-independent pathway to promote the formation of dendritic spines. These results, therefore, demonstrate that BAMs 1–3 have potential to counteract one of the earliest observed pathological events associated with AD (16, 41).
In conclusion, we used rational design to develop a novel set of benzothiazole amphiphiles 1–3 with improved biocompatibility compared with the previously reported BTA-EG4,6 compounds (17–19). These new compounds were capable of 1) promoting dose-dependent increases in dendritic spine density, 2) temporally and reversibly controlling elevated spine levels, and 3) protecting against Aβ-induced dendritic spine loss. Current efforts are focused on identifying the cellular target for the BAM agents and elucidating additional mechanistic details leading to the spinogenic activity of these compounds. These novel benzothiazoles represent a significant step toward the development of new tools to study and treat spine related disorders, and may also lead to a new class of general cognitive enhancers.
Author Contributions
J. L. C., G. N. P., and J. Y. designed the research. J. L. C. and T. S. C. synthesized all compounds. J. L. C. evaluated and analyzed compound physical and toxic properties. L. D. and G. N. P. prepared and maintained neuronal cultures; J. L. C. and L. D. performed and analyzed all dendritic spine experiments. J. L. C. and J. Y. wrote the paper. All authors reviewed and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Yongxuan Su for use of the molecular mass spectrometry facility (MMSF) at UCSD.
This work was supported in part by the UCSD Alzheimer's Disease Research Center (National Institutes of Health Grant 3P50 AG005131). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental information.
DMSO was used for all compounds due to its necessity to solubilize BTA-EG4, which was used as an internal standard (data not shown).
- AD
- Alzheimer disease
- BAM
- benzothiazole amphiphile
- DIV
- days in vitro
- BTA
- benzothiazole aniline
- EG
- ethylene glycol
- Aβ
- amyloid-β
- SASA
- solvent-accessible surface area.
References
- 1. Nimchinsky E. A., Sabatini B. L., and Svoboda K. (2002) Structure and function of dendritic spines. Annu. Rev. Physiol. 64, 313–353 [DOI] [PubMed] [Google Scholar]
- 2. Matsuzaki M., Honkura N., Ellis-Davies G. C. R., and Kasai H. (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kasai H., Matsuzaki M., Noguchi J., Yasumatsu N., and Nakahara H. (2003) Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368 [DOI] [PubMed] [Google Scholar]
- 4. Bourne J. N., and Harris K. M. (2008) Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moser M. B., Trommald M., and Andersen P. (1994) An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc. Natl. Acad. Sci. U.S.A. 91, 12673–12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Penzes P., Cahill M. E., Jones K. A., VanLeeuwen J.-E., and Woolfrey K. M. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai K.-O., and Ip N. Y. (2013) Structural plasticity of dendritic spines: the underlying mechanisms and its dysregulation in brain disorders. Biochim. Biophys. Acta 1832, 2257–2263 [DOI] [PubMed] [Google Scholar]
- 8. Fiala J. C., Spacek J., and Harris K. M. (2002) Dendritic Spine Pathology: Cause or Consequence of Neurological Disorders? Brain Res. Rev. 39, 29–54 [DOI] [PubMed] [Google Scholar]
- 9. Schulz-Schaeffer W. J. (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 120, 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Spronsen M., and Hoogenraad C. C. (2010) Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kolomeets N. S., Orlovskaya D. D., Rachmanova V. I., and Uranova N. A. (2005) Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse 57, 47–55 [DOI] [PubMed] [Google Scholar]
- 12. Glantz L. A., and Lewis D. A. (2000) Decreased Dendritic Spine Density on Prefrontal Cortical Pyramidal Neurons in Schizophrenia. Arch. Gen. Psychiatry 57, 65–73 [DOI] [PubMed] [Google Scholar]
- 13. Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., and Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 [DOI] [PubMed] [Google Scholar]
- 14. Selkoe D. J. (2002) Alzheimer's disease is a synaptic failure. Science 298, 789–791 [DOI] [PubMed] [Google Scholar]
- 15. Walsh D. M., and Selkoe D. J. (2004) Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44, 181–193 [DOI] [PubMed] [Google Scholar]
- 16. Jacobsen J. S., Wu C.-C., Redwine J. M., Comery T. A., Arias R., Bowlby M., Martone R., Morrison J. H., Pangalos M. N., Reinhart P. H., and Bloom F. E. (2006) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Habib L. K., Lee M. T. C., and Yang J. (2010) Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 285, 38933–38943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Megill A., Lee T., DiBattista A. M., Song J. M., Spitzer M. H., Rubinshtein M., Habib L. K., Capule C. C., Mayer M., Turner R. S., Kirkwood A., Yang J., Pak D. T. S., Lee H.-K., and Hoe H.-S. (2013) A Tetra(Ethylene Glycol) Derivative of Benzothiazole Aniline Enhances Ras-Mediated Spinogenesis. J. Neurosci. 33, 9306–9318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song J. M., DiBattista A. M., Sung Y. M., Ahn J. M., Turner R. S., Yang J., Pak D. T. S., Lee H.-K., and Hoe H.-S. (2014) A tetra(ethylene glycol) derivative of benzothiazole aniline ameliorates dendritic spine density and cognitive function in a mouse model of Alzheimer's disease. Exp. Neurol. 252, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Penzes P., Cahill M. E., Jones K. A., VanLeeuwen J.-E., and Woolfrey K. M. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith D. L., Pozueta J., Gong B., Arancio O., and Shelanski M. (2009) Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. U.S.A. 106, 16877–16882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Selkoe D. J. (2013) The therapeutics of alzheimer's disease where we stand and where we are heading. Ann. Neurol. 74, 328–336 [DOI] [PubMed] [Google Scholar]
- 23. Prangkio P., Rao D. K., Lance K. D., Rubinshtein M., Yang J., and Mayer M. (2011) Self-assembled, cation-selective ion channels from an oligo(ethylene glycol) derivative of benzothiazole aniline. Biochim. Biophys. Acta 1808, 2877–2885 [DOI] [PubMed] [Google Scholar]
- 24. Kramer J. A., Sagartz J. E., and Morris D. L. (2007) The application of discovery toxicology and pathology towards the design of safer pharmaceutical lead candidates. Nat. Rev. Drug Discov. 6, 636–649 [DOI] [PubMed] [Google Scholar]
- 25. Inbar P., Li C. Q., Takayama S. A., Bautista M. R., and Yang J. (2006) Oligo(ethylene glycol) derivatives of thioflavin T as inhibitors of protein-amyloid interactions. Chembiochem 7, 1563–1566 [DOI] [PubMed] [Google Scholar]
- 26. Finkelstein H. (1910) Darstellung organischer Jodide aus den entsprechenden Bromiden und Chloriden. Berichte der Dtsch. Chem. Gesellschaft 43, 1528–1532 [Google Scholar]
- 27. Chittiboyina A. G., Venkatraman M. S., Mizuno C. S., Desai P. V., Patny A., Benson S. C., Ho C. I., Kurtz T. W., Pershadsingh H. A., and Avery M. A. (2006) Design and synthesis of the first generation of dithiolane thiazolidinedione- and phenylacetic acid-based PPARγ agonists. J. Med. Chem. 49, 4072–4084 [DOI] [PubMed] [Google Scholar]
- 28. Namboodiri V. V., and Varma R. S. (2002) Solvent-free sonochemical preparation of ionic liquids. Org. Lett. 4, 3161–3163 [DOI] [PubMed] [Google Scholar]
- 29. Ranu B. C., and Jana R. (2006) Ionic Liquid as Catalyst and Reaction Medium – A simple, efficient and green procedure for Knoevenagel Condensation of aliphatic and aromatic carbonyl compounds using a task-specific basic ionic liquid. Eur. J. Org. Chem. 2006, 3767–3770 [Google Scholar]
- 30. Schaumann E. (ed.) (2007) Sulfur-Mediated Rearrangements I, Springer Berlin Heidelberg, Berlin, Heidelberg [Google Scholar]
- 31. Yang J., Gabriele B., Belvedere S., Huang Y., and Breslow R. (2002) Catalytic oxidations of steroid substrates by artificial cytochrome P-450 enzymes. J. Org. Chem. 67, 5057–5067 [DOI] [PubMed] [Google Scholar]
- 32. Huang C.-H. (1969) Phosphatidylcholine vesicles. Formation and physical characteristics. Biochemistry 8, 344–352 [DOI] [PubMed] [Google Scholar]
- 33. Djakovic S. N., Schwarz L. A., Barylko B., DeMartino G. N., and Patrick G. N. (2009) Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 26655–26665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cartier A. E., Djakovic S. N., Salehi A., Wilson S. M., Masliah E., and Patrick G. N. (2009) Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 29, 7857–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furuta T., Tomioka R., Taki K., Nakamura K., Tamamaki N., and Kaneko T. (2001) In vivo transduction of central neurons using recombinant Sindbis virus: Golgi-like labeling of dendrites and axons with membrane-targeted fluorescent proteins. J. Histochem. Cytochem. 49, 1497–1508 [DOI] [PubMed] [Google Scholar]
- 36. Zhao X., and Yang J. (2010) Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem. Neurosci. 1, 655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee N. J., Song J. M., Cho H.-J., Sung Y. M., Lee T., Chung A., Hong S.-H., Cifelli J. L., Rubinshtein M., Habib L. K., Capule C. C., Turner R. S., Pak D. T. S., Yang J., and Hoe H.-S. (2016) Hexa (ethylene glycol) derivative of benzothiazole aniline promotes dendritic spine formation through the RasGRF1-Ras dependent pathway. Biochim. Biophys. Acta 1862, 284–295 [DOI] [PubMed] [Google Scholar]
- 38. Hsieh H., Boehm J., Sato C., Iwatsubo T., Tomita T., Sisodia S., and Malinow R. (2006) AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodrigues E. M., Scudder S. L., Goo M. S., and Patrick G. N. (2016) Aβ-induced synaptic alterations require the E3 ubiquitin ligase Nedd4–1. J. Neurosci. 36, 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scudder S. L., Goo M. S., Cartier A. E., Molteni A., Schwarz L. A., Wright R., and Patrick G. N. (2014) Synaptic strength is bidirectionally controlled by opposing activity-dependent regulation of Nedd4–1 and USP8. J. Neurosci. 34, 16637–16649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serrano-Pozo A., Frosch M. P., Masliah E., and Hyman B. T. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krapivinsky G., Krapivinsky L., Manasian Y., Ivanov A., Tyzio R., Pellegrino C., Ben-Ari Y., Clapham D. E., and Medina I. (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40, 775–784 [DOI] [PubMed] [Google Scholar]
- 43. Goley E. D., and Welch M. D. (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 7, 713–726 [DOI] [PubMed] [Google Scholar]
- 44. Sumi T., Matsumoto K., Takai Y., and Nakamura T. (1999) Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147, 1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim T., Vidal G. S., Djurisic M., William C. M., Birnbaum M. E., Garcia K. C., Hyman B. T., and Shatz C. J. (2013) Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science 341, 1399–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prangkio P., Yusko E. C., Sept D., Yang J., and Mayer M. (2012) Multivariate analyses of amyloid-β oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS ONE 7, e47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peters A., and Kaiserman-Abramof I. R. (1970) The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am. J. Anat. 127, 321–355 [DOI] [PubMed] [Google Scholar]
- 48. Matus A., Brinkhaus H., and Wagner U. (2000) Actin dynamics in dendritic spines: a form of regulated plasticity at excitatory synapses. Hippocampus 10, 555–560 [DOI] [PubMed] [Google Scholar]
- 49. Halpain S. (2000) Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 23, 141–146 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.