

Abstract
The receptor for advanced glycation end products (RAGE) is a multiligand transmembrane receptor that can undergo proteolysis at the cell surface to release a soluble ectodomain. Here we observed that ectodomain shedding of RAGE is critical for its role in regulating signaling and cellular function. Ectodomain shedding of both human and mouse RAGE was dependent on ADAM10 activity and induced with chemical activators of shedding (ionomycin, phorbol 12-myristate 13-acetate, and 4-aminophenylmercuric acetate) and endogenous stimuli (serum and RAGE ligands). Ectopic expression of the splice variant of RAGE (RAGE splice variant 4), which is resistant to ectodomain shedding, inhibited RAGE ligand dependent cell signaling, actin cytoskeleton reorganization, cell spreading, and cell migration. We found that blockade of RAGE ligand signaling with soluble RAGE or inhibitors of MAPK or PI3K blocked RAGE-dependent cell migration but did not affect RAGE splice variant 4 cell migration. We finally demonstrated that RAGE function is dependent on secretase activity as ADAM10 and γ-secretase inhibitors blocked RAGE ligand-mediated cell migration. Together, our data suggest that proteolysis of RAGE is critical to mediate signaling and cell function and may therefore emerge as a novel therapeutic target for RAGE-dependent disease states.
Keywords: ADAM, cell migration, cell signaling, matrix metalloproteinase (MMP), receptor for advanced glycation end products (RAGE), S100 proteins, ADAM, ectodomain shedding
Introduction
Receptor for advanced glycation end products (RAGE)3 is a transmembrane, multiligand receptor expressed by most cells but is characterized by its up-regulation in a range of inflammatory disease states including diabetes, various cancers, and cardiovascular disease (1). RAGE activation through ligand binding transduces intracellular signaling and in turn induces cell migration, invasion, and adhesion (1). Work from multiple groups has demonstrated using rodent models that blocking RAGE signaling impairs the development of numerous pathologic states and therefore highlights RAGE as an attractive therapeutic target (2–5). The most widely used means for blocking RAGE signaling is with soluble RAGE (sRAGE), the recombinantly produced extracellular domain of RAGE. Soluble RAGE acts as a decoy receptor and therefore neutralizes RAGE ligands (2, 6). Endogenous sRAGE isoforms, which have been identified in human and mouse sera, are generated through two distinct biological mechanisms: the alternative splicing of the transmembrane region of RAGE leading to a secreted isoform (esRAGE/RAGEv1) and the cleavage of extracellular domain (ectodomain or ECD) at the cell surface by ectodomain shedding (1, 7–10). Ectodomain shedding is a tightly regulated process and is mediated by various metalloproteinases (11, 12). Shedding is essential for normal physiological function but can be deregulated in various pathological disorders where altered levels of metalloproteinases are found (11, 12). In fact, many transmembrane proteins are known to undergo ectodomain shedding including cell adhesion molecules, ligands for growth factor receptors, immunoglobulins, and various enzymes (10–13). Ectodomain shedding of cell surface receptors can affect a number of processes: the loss of cell-cell or cell-matrix interactions, the production of a soluble ectodomain that acts as a agonist/antagonist for the cell surface protein, and the release of the intracellular domain (ICD) of the receptor to induce cell signaling (10–13). For RAGE, it has been proposed that shedding acts to release the soluble ECD, which in turn acts as an inhibitor of RAGE signaling through sequestering RAGE ligand from binding cell surface intact RAGE. RAGE shedding is dependent on ADAM10 activity and can be induced with phorbol 12-myristate 13-acetate (PMA) and calcium ionophores in a PKCα/β1-dependent manner (8–10). The importance of RAGE shedding is underscored by clinical cohort studies that show serum sRAGE levels are correlated with clinical disease states including cardiovascular disease, diabetes, cancer, and various inflammatory disease states (14–19). Although a few studies have explored the regulation of RAGE ectodomain shedding (8–10), no studies to date have investigated the biological function of RAGE ectodomain shedding.
In the current study, we investigated in depth the regulation of RAGE ectodomain shedding and for the first time investigated the impact RAGE ectodomain shedding has on cellular signaling and function. Our data identify RAGE ectodomain shedding as an integral part of its function and a target for designing novel therapeutics for use in RAGE-associated pathologic states.
Experimental Procedures
Generation of Stable Cell Lines
HEK-293 and C6 cells were obtained from American Type Culture Collection (ATCC) and used to generate cells stably expressing human RAGE (GenBankTM accession number AY755619), mouse RAGE (GenBank accession number EU520325), mouse RAGEv4 (GenBank accession number EU570243), and empty vector (mock) by transfection with FuGENE 6 (Promega) and antibiotic selection with G418 or Zeocin as described previously for RAGE and its splice variants (7, 20). Stable expression was confirmed by Western blotting using anti-RAGE polyclonal antibodies as described below. An N-terminal His-tagged RAGE construct was provided by Dr. Nam-Ho Huh (21) that was previously engineered to express a His6-2HA tag in between the N-terminal signal sequence and the rest of the RAGE cDNA. Cells were maintained in DMEM (Gibco) supplemented with 10% FBS (Atlanta Biologicals) along with penicillin/streptomycin (Gibco) and MycoZap (Lonza) antibiotics. All cell counting was performed using a Countess® automated cell counter (Invitrogen) according to the manufacturer's instructions.
Ectodomain Shedding Assays
Stably transfected HEK-293 cells (293-RAGE, 293-mRAGE, 293-mRAGE splice variant 4 (mRAGEv4), and 293-mock empty vector-transfected) were seeded at 2.5 × 105 cells/well on poly-d-lysine-coated 6-well plates and grown for 2 days until 70–80% confluence was reached. The cells were then serum-starved overnight (0.5% FBS in DMEM) and the following day incubated for 1 h with fresh serum-free DMEM prior to experiments. All medium changes were performed with a sterile 1× PBS rinse. Shedding assays were performed in 2 ml of serum-free DMEM with or without shedding compounds for a period of 1 h. In experiments using inhibitors of proteinases or signaling pathways, cells were preincubated with compound for 1 h prior to shedding assays to allow uptake into cells. The cell medium was then removed, and fresh medium containing inhibitor and shedding stimulator was added as above for 1 h.
After shedding assays were completed, conditioned medium was removed from cells and centrifuged for 5 min at 1000 rpm to remove cell debris. Cell lysate was processed as described below for Western blotting. Shedding inducers used included PMA, 4-aminophenylmercuric acetate (APMA), ionomycin, cantharidin, calyculin A, and sodium pervanadate (all obtained from Sigma). Pervanadate was freshly prepared for each experiment by mixing 100 mm hydrogen peroxide and 100 mm sodium orthovanadate, pH 10, at 1:1 as described previously (22). RAGE ligands used for shedding assays included CML-human serum albumin (prepared as we have described previously (3)) and s100B (Calbiochem). Inhibitors used included BB94 (Santa Cruz Biotechnology), Gö6976 (Millipore), GFX109203X (Millipore), SB203580 (Sigma), SP600125 (Sigma), U0126 (Cell Signaling Technology), LY-294002 (Sigma), and GI254023X (Tocris). EGTA was obtained from Sigma and prepared in water.
Western Blotting Analysis
Cell lysate was collected immediately after experiments by rinsing in ice-cold PBS followed by lysis with Mammalian Protein Extraction Reagent (MPER) (Thermo Scientific) containing protease (Sigma) and phosphatase inhibitors (Sigma) according to the manufacturers' instructions. Conditioned medium for Western blotting was concentrated by cold acetone precipitation at −20 °C for 2 h followed by centrifugation at 16,000 × g for 10 min and resuspension of protein pellet in Western blotting sample buffer (Invitrogen). Protein concentration in cell lysate was determined by the Bradford assay (Pierce), and lysates were run on SDS-polyacrylamide gels as described previously (23). Antibodies used were as follows: human RAGE monoclonal antibody (Millipore; MAB5328), RAGE polyclonal (Santa Cruz Biotechnology; H300), β-actin (Millipore; MAB1501), and anti-HisG (Invitrogen; R940-25).
sRAGE ELISA
Soluble RAGE levels were measured in whole conditioned medium by ELISAs using the respective human and mouse RAGE DuoSet kits (R&D Systems) according to the manufacturer's instructions. ELISA microplates (R&D Systems; DY990) were coated overnight at room temperature with capture antibody in PBS. Plates were blocked for 2 h at room temperature with Reagent Diluent 2 (R&D Systems) before incubation of samples for 2 h at room temperature. RAGE detection was performed using a RAGE streptavidin-labeled antibody diluted and incubated for 2 h at room temperature followed by streptavidin-HRP (R&D Systems) binding. ELISA plates were extensively washed between all incubations with a PBS, 0.05% Tween 20 solution. For detection, 1-Step Ultra TMB-ELISA Substrate Solution (Thermo Scientific) was added before quenching with 2 n H2SO4 (Sigma). ELISA plates were measured using a Bio-Rad iMark 1.04.02 at 450 nm subtracting from 595 nm background. To quantitate RAGE levels in conditioned media, each ELISA experiment contained a human or mouse RAGE standard as provided with the kit. Data were analyzed using Microplate Manager Version 6.1.
Cell Migration Assays
For functional cell assays, the C6 cell line, which is an established model for RAGE signaling and cell function (3, 4, 24), was used. Cell migration assays were performed using C6-mRAGE-, C6-mRAGEv4-, and C6-mock-transfected cells with transwell migration chambers as described previously (4). 5 × 103 cells were seeded in the upper chamber of 8-μm porous transwell inserts (ThinCerts, Greiner) in serum-free DMEM and incubated in 24-well plates with 5 μg/ml S100B or 1% FBS used as a chemoattractant for 24 h. For collagen I, transwell inserts were coated with 10 μg/ml for 1 h at 37 °C before use in migration assays. For experiments involving inhibitors, these were added to the upper and lower chambers of transwell experiments (U0126, 10 μm; LY-294002, 10 μm; GI254023X, 5 μm; DAPT, 10 μm; BB94, 10 μm; sRAGE (R&D Systems), 5 μg/ml). Following incubation, cells were fixed with methanol for 10 min and stained with 2% crystal violet in 2% ethanol solution. Non-migrated cells were removed from transwell chambers with a cotton swab. To quantify the cells, the cell stain was extracted with 10% acetic acid, transferred to a 96-well plate, and measured at 595 nm using an iMark microplate reader.
Cell Adhesion/Spreading Assay
Cell spreading assays were performed by seeding cells (C6-mRAGE, C6-mRAGEv4, and C6-mock) in serum-free medium on culture slides coated with either collagen I or PBS. Culture slides were coated with either 5 μg/ml collagen I or PBS control for 1 h at 37 °C followed by two washes in PBS. C6-mRAGE, -mRAGEv4, or -mock cells were then seeded in wells for 2 h at 37 °C. Unbound cells were washed from the plates with PBS, and attached cells were fixed with 4% paraformaldehyde for 15 min at room temperature. The surface area of cells was determined in at least 50 cells per view in triplicate manually outlined using ImageJ software as described previously (25–27). For immunofluorescence assays, fixed cells were stained for actin and vinculin using the Actin Cytoskeleton/Focal Adhesion Staining kit (Millipore; FAK100).
Cell Signaling Assays
For cell signaling assays, C6-mRAGE, -mRAGEv4, or -mock cells were harvested and seeded onto 6-well plates previously coated with 5 μg/ml collagen I as above for 2 h. Cells lysis was performed as described above, and Western blotting was performed using antibodies against phospho-/total proteins including Src (p416 (6943P), p527 (2105P), and total (2123P)), Akt (phospho (4060P)/total (4691P)), ERK1/2 (phospho (4370P)/total (9102P)), and p38 (phospho (4511P)/total (8690P)) (all from Cell Signaling Technology). Densitometry was used to quantify phospho- and total protein levels using ImageJ, and relative activity was assessed by calculating the ratio of phospho- to total protein. Western blot images shown are representative of three independent experiments.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism version 6.00 for Windows (GraphPad Software, San Diego, CA). Student's t test was used for comparison between experiments involving two groups, and one-way ANOVA was used for analysis of experiments involving three or more groups. A probability value of p < 0.05 was considered to be of significance. For all experiments where statistical analysis was performed, three independent experiments were analyzed.
Results
Human and Mouse RAGE Undergo Constitutive Shedding in a Similar Manner
Previous studies have demonstrated that human RAGE undergoes ectodomain shedding in both a constitutive (non-stimulated) and inducible (stimulated) manner (8–10); however, this has not been established for mouse RAGE. Although multiple sized products of RAGE have been detected in murine tissue, whether these are a result of ectodomain shedding has not been established experimentally (9, 28).
We therefore first tested whether human (herein hRAGE) and mouse RAGE (herein mRAGE) undergo constitutive shedding in a similar manner (Fig. 1A). HEK-293 cells stably expressing hRAGE displayed a doublet band of ∼52 kDa (Fig. 1B), whereas mRAGE doublet bands were only detectable at lower exposures of Western blots (data not shown). Prior studies have confirmed these doublet bands to be the non- and N-glycosylated forms of RAGE (9, 10). In a similar manner, conditioned medium displayed doublet bands for hRAGE (∼48 kDa) and mRAGE (∼45 kDa) (Fig. 1B). ELISA of conditioned medium revealed hRAGE and mRAGE levels to be ∼5000 and ∼1500 pg/ml, respectively, which is within the physiological range seen in human serum (14). Our data confirm in the same cellular context that both hRAGE and mRAGE undergo constitutive shedding.
FIGURE 1.

RAGE undergoes constitutive and inducible ectodomain shedding in HEK-293 cells. A, schematic of RAGE showing the major protein domains (ECD, transmembrane region (TM), and ICD). Protein sizes for full-length and cleaved ECD human or mouse RAGE are shown in kDa. B, stably transfected HEK-293 cells expressing empty vector control, hRAGE, or mRAGE were incubated for 24 h in serum-free medium to allow constitutive shedding to occur. Blotting was performed on concentrated conditioned medium and total cell lysate with anti-RAGE antibodies. Cell lysates were probed with anti-β actin as a loading control. Images are representative of three independent experiments. C and D, stably transfected HEK-293 cells expressing empty vector, hRAGE, or mRAGE were serum-starved overnight, and fresh medium was added for experiments. RAGE shedding was induced with PMA (200 nm), ionomycin (Iono) (1 μm), or APMA (25 μm) with non-stimulated (NS) control treated with vehicle (DMSO) for 1 h. Medium and lysate were collected as above, and Western blotting (WB) was performed for RAGE. Images are representative of three independent experiments. E and F, human and mouse RAGE shedding was measured by ELISA using conditioned medium collected as above. In addition to PMA, ionomycin, and APMA stimulation, stimulation of shedding by 1% FBS was measured by ELISA. Data are means ± S.E. from three independent experiments. Statistical difference between groups was assessed by one-way ANOVA compared with non-stimulated where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. G, RAGE shedding was measured by ELISA after incubation of HEK-293 hRAGE-expressing cells with RAGE ligands (S100B and CML-human serum albumin; 1 μg/ml) or RAGE inhibitor (FPS-ZM1; 1 μm). Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA compared with non-stimulated where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. H, shedding of RAGE was induced with the phosphatase inhibitors cantharidin, calyculin A, and sodium pervanadate. Stably transfected HEK-293 cells expressing hRAGE were serum-starved overnight, and fresh medium was added for experiments. Shedding was induced for 1 h with cantharidin (500 μm), calyculin A (50 nm), and sodium pervanadate (10 μm) with non-stimulated (NS) control treated with vehicle (DMSO). Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA compared with non-stimulated where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E.
Human and Mouse RAGE Similarly Undergo Inducible Shedding in Response to Diverse Stimuli
Ectodomain shedding is a tightly regulated process that can be induced by a wide range of stimuli. For the majority of receptors tested to date, factors that induce ectodomain shedding include phorbol esters, calcium ionophores, serum factors, the organomercurial compound APMA that activates latent metalloproteinases, and various phosphatase inhibitors including cantharidin, calyculin A, and sodium pervanadate (12, 13, 29, 30). Recently it was shown that shedding of hRAGE could be induced by calcium ionophores (calcimycin and ionomycin), but contrasting data were seen whether hRAGE shedding could be activated by PMA (8–10). To test the effects of these compounds on hRAGE and mRAGE shedding, cells were incubated for 1 h with PMA (200 nm), ionomycin (1 μm), APMA (25 μm), or FBS (1%). Western blotting analysis of conditioned medium revealed a similar pattern of induction for both hRAGE and mRAGE; PMA displayed a lower induction of shedding, whereas ionomycin and APMA strongly induced shedding of RAGE (Fig. 1, C and D). ELISA of neat conditioned medium revealed a similar pattern for both hRAGE and mRAGE as seen by Western blotting with a strong induction seen with of ionomycin/APMA and less intense induction with PMA (Fig. 1, E and F). Incubation with serum revealed a similar induction of shedding for both hRAGE and mRAGE (Fig. 1, E and F). As conflicting data exist for the effect of RAGE ligands on RAGE ECD shedding (8, 9), we tested the effect of CML-advanced glycation end product and s100B on RAGE shedding. Both CML-advanced glycation end product and s100B induced RAGE shedding but to a lesser degree than ionomycin/APMA (Fig. 1G). To test whether inhibitors of RAGE ligand binding impacted shedding of RAGE, we tested the effect of the RAGE inhibitor FPS-ZM1 on shedding. FPS-ZM1 was developed to interact with the ligand binding domain of the receptor to block RAGE signaling (31). FPS-ZM1 treatment for 1 h led to a striking decrease in RAGE ECD shedding compared with control (Fig. 1G). These data together demonstrate human and mouse RAGE to be proteolytically processed to a similar level by diverse stimuli including RAGE ligands.
RAGE Shedding Is Mediated by Metalloproteinases and Is Induced by Distinct Signaling Pathways
RAGE ectodomain shedding has been shown to be a metalloproteinase-dependent mechanism; however, the regulation of the upstream signaling mechanism has not been fully explored (8–10). Various studies have demonstrated that protein phosphatase inhibitors are strong inducers of protein ectodomain shedding of other receptors due to their activation of numerous protein kinases (12, 13, 29, 30). As the role of serine/threonine and tyrosine phosphatases in regulating RAGE shedding has not been studied before, we tested whether phosphatase inhibition could activate RAGE shedding. Cantharidin and calyculin A are natural compounds derived from the blister beetle and marine sponge Discodermia calyx, respectively, known to inhibit the PP1/PP2A family of serine/threonine phosphatases (32, 33), whereas pervanadate is a potent inhibitor of tyrosine protein phosphatases (34). As similar data were seen between hRAGE and mRAGE with all other inducers of shedding, we tested the effect of phosphatase inhibitors on hRAGE shedding in HEK-293 cells. Stimulation of cells with cantharidin, calyculin A, and pervanadate resulted in the strong induction of RAGE ectodomain shedding as assessed by ELISA (Fig. 1H). Together, these results demonstrate that the inhibition of protein phosphatases promotes strong shedding of RAGE and suggest a role of inside-out signaling in the regulation of RAGE ectodomain shedding.
Zhang et al. (10) previously demonstrated a role for PKC signaling (PKCα and PKCβI) in PMA-induced shedding but found a lesser inhibition of ionophore-induced RAGE ectodomain shedding by PKC inhibition. We observed that cells treated with the PKC inhibitor Gö6976 displayed markedly reduced PMA-induced shedding of RAGE (Fig. 2A). Furthermore, to control for off-target effects of PKC inhibitors, we confirmed our data with the PKCα and PKCβI inhibitor GFX109203. Treatment of cells with GFX109203 also inhibited PMA-induced shedding of RAGE (Fig. 2A). In contrast, ionomycin-induced ectodomain shedding of RAGE was not affected by either PKC inhibitor (Fig. 2B). To demonstrate that ionomycin-induced shedding could be inhibited, cells were treated with the metalloproteinase inhibitor BB94 (batimastat), which completely inhibited RAGE shedding for both PMA and ionomycin (Fig. 2, A and B). We next tested the effect of calcium chelation with EGTA on PMA- and ionomycin-induced shedding. Treatment of cells with EGTA did not impair PMA-induced shedding (Fig. 2A) but completely impaired ionomycin-induced shedding (Fig. 2B). These data therefore demonstrate PMA stimulation to be independent of calcium influx and that different signaling pathways exist to activate RAGE shedding.
FIGURE 2.

Regulation of RAGE ectodomain shedding in HEK-293 cells. A and B, stably transfected HEK-293 cells expressing hRAGE were serum-starved overnight and treated for 1 h with the PKC inhibitors Gö6976 (2 μm) and GFX109203X (2.5 μm) or metalloproteinase inhibitor BB94 (10 μm). Fresh medium was added, and shedding was induced for 1 h with either PMA (200 nm) (A) or ionomycin (1 μm) (B) with inhibitors re-added at the time of the experiment. The calcium chelator EGTA (10 mm) was added at the time of stimulation for 1 h. RAGE shedding was quantified in conditioned medium by human ELISA. Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. C and D, stably transfected HEK-293 cells expressing hRAGE were serum-starved overnight and treated for 1 h with either the p38 inhibitor SB203580 (10 μm), SAPK/JNK inhibitor SP600125 (100 μm), MEK inhibitor U0126 (10 μm), or phosphoinositide 3-kinase inhibitor LY-294002 (50 μm). Fresh medium was then added, and shedding was induced for 1 h with either PMA (200 nm) or ionomycin (1 μm) with inhibitors re-added at the time of the experiment. RAGE shedding was quantified in conditioned medium by human ELISA. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. E and F, stably transfected HEK-293 cells expressing hRAGE were serum-starved overnight and treated for 1 h with either the ADAM10 inhibitor GI254023X (5–20 μm) or metalloproteinase inhibitor BB94 (10 μm). Fresh medium was then added, and shedding was induced for 1 h with either PMA (200 nm) or ionomycin (1 μm) with inhibitors re-added at the time of the experiment. RAGE shedding was quantified in conditioned medium by human ELISA. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. NS, not-stimulated; Iono, ionomycin.
Because it appears that PMA and Ca2+ influx (by ionomycin) stimulate RAGE shedding by distinct signaling pathways, we probed the role of other signaling pathways in this process. The mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways have been shown to regulate shedding of other proteins (30, 34, 35). Treatment of cells with inhibitors for components of the MAPK pathway including p38 (SB203580), MEK (U0126), and SAPK/JNK (SP600125) did not significantly affect PMA- or ionomycin-induced RAGE shedding (Fig. 2, C and D). In contrast, treatment of cells with a phosphoinositide 3-kinase inhibitor (LY-294002) led to reduced RAGE ectodomain shedding after stimulation with ionomycin but not PMA (Fig. 2, C and D).
Prior studies have shown through gene knockdown and chemical inhibitors that the major metalloproteinase mechanism responsible for ectodomain shedding of RAGE involves ADAM10 (8–10). We next tested whether ADAM10 is the major sheddase activated by these different signaling pathways. Inhibition of ADAM10 with GI254023X repressed RAGE ECD shedding for both PMA and Ca2+ influx (by ionomycin) (Fig. 2, E and F). These data together indicate that distinct signaling pathways involving PKCα/PKCβI or phosphoinositide 3-kinase regulate activation of RAGE ectodomain shedding and that this is an ADAM10 metalloproteinase-dependent mechanism.
Identification of the RAGE Cleavage Site
Typically, ectodomain shedding of cell surface receptors occurs at the extracellular juxtamembrane (JM) region. For RAGE, because the full-length RAGE protein is around 50–55 kDa and the cleaved form of RAGE is about 48 kDa, this has led to the assumption that the cleavage site must be within the extracellular JM region. However, it is not known whether RAGE can be cleaved at multiple sites throughout the ECD, producing smaller ectodomain products. To first test whether ectodomain shedding occurs at a single cleavage site/region, we tested for RAGE cleavage products using an N-terminally epitope-tagged construct as described previously (21) (Fig. 3A). This cDNA construct of RAGE was engineered to express a His6-2HA tag in between the N-terminal signal sequence and the rest of the RAGE cDNA (Fig. 3A) (21). This allows epitope tag detection of all ECD cleavage products independently of RAGE antibody epitope recognition issues. Stimulation of HEK-293 cells expressing His6-2HA-tagged RAGE was performed with PMA, ionomycin, and APMA. Western blotting using anti-His epitope antibodies revealed a single cleaved product under all conditions at ∼52 kDa (Fig. 3B). These data provide strong evidence to suggest that there is only a single cleavage site of RAGE and that this occurs at/around the JM region.
FIGURE 3.

RAGE ectodomain shedding occurs within a single region of the extracellular domain. A, schematic of the His6-2HA N-terminally tagged human RAGE construct and isoform used in this study. The RAGE ECD, transmembrane region (TM), and ICD are shown. The predicted cleavage site at the JM domain should result in a shed ECD RAGE product of ∼50 kDa as indicated by the arrow. B, HEK-293 cells were transfected with a His6-2HA N-terminally tagged human RAGE construct, and 48 h later shedding was induced with PMA (200 nm), ionomycin (1 μm), or APMA (25 μm) with non-stimulated (NS) control treated with vehicle (DMSO) for 1 h. Blots were performed on conditioned medium and total cell lysate with anti-His and anti-RAGE antibodies. Cell lysates were probed with anti-β actin as a loading control. Images are representative of three independent experiments. WB, Western blotting; Iono, ionomycin.
Prior work has suggested that RAGE is proteolytically processed between amino acids Gly331 and Ser332 in the JM region of the extracellular domain as protein sequencing of soluble RAGE isolated from mouse lung was found to have the Gly331 at the C terminus (10, 28). To further explore this potential site of RAGE ectodomain shedding, we analyzed protein sequences of RAGE from numerous species to investigate conservation of the potential cleavage region. As RAGE is restricted to mammals (36), we aligned the RAGE protein sequence of mouse, pig, horse, and various primates (rhesus macaque and chimpanzee) with human. As shown in Fig. 4A, this region of RAGE is highly conserved with the Gly331-Ser332 proposed cleavage site being conserved across all species analyzed.
FIGURE 4.

Identification of a protease-resistant RAGE splice variant. A, RAGE protein sequences around the proposed cleavage site were obtained from UniProt (72) and aligned using Jalview (61). These included mouse (Q62151_MOUSE), pig (A5A8Y1_PIG), horse (F6W335_HORSE), rhesus monkey (F1ABQ1_MACMU), human (Q15109_HUMAN), and chimpanzee (H2R7G0_PANTR) obtained from UniProt (72). Conserved amino acids are shown below the alignments; a “+” is shown if two or more residues are equally conserved. Amino acid residues are colored according to the ClustalX color scheme where a color is only applied if the amino acid meets the criteria specific for the residue type at the alignment position (64). The proposed cleavage site is shown above, and the exon coding for each region is shown below. B, schematic of the RAGE ectodomain shedding site. The RAGE ECD, transmembrane region (TM), and ICD are shown as a schematic with the sequence alignment of the JM region and transmembrane region shown below. Protein alignment of mRAGE with mRAGEv4 shows the 9 amino acids missing in mRAGEv4 as a result of alternative splicing of exon 9 of RAGE with the proposed cleavage site indicate by an arrow. C, stably transfected HEK-293 cells expressing mRAGE or mRAGEv4 were assessed for constitutive shedding. RAGE was detected by Western blotting using polyclonal antibodies raised against the total ECD of RAGE. Cells were incubated for 24 h in serum-free medium to allow constitutive shedding to occur, and blotting was performed on conditioned medium and total cell lysate with anti-RAGE antibodies. Cell lysate were probed with anti-β actin as a loading control. Images are representative of three independent experiments. D, cell surface RAGE expression of HEK-293 cells (vector control, mRAGE, or mRAGEv4) was assessed by flow cytometry. Cells were incubated sequentially with anti-RAGE and Alexa Fluor 647-labeled secondary antibodies, and cell surface RAGE expression was detected using a FACSCalibur flow cytometer. Images are representative of three independent experiments. E, stably transfected HEK-293 cells expressing mRAGE or mRAGEv4 were serum-starved overnight, and fresh medium was added for experiments. RAGE shedding was induced with PMA (200 nm), ionomycin (1 μm), or APMA (25 μm) with non-stimulated (NS) control treated with vehicle (DMSO) for 1 h. Medium and lysate were collected as above, and Western blotting was performed for RAGE. Images are representative of three independent experiments. F, RAGE shedding of HEK-293 cells (vector control, mRAGE, or mRAGEv4) was quantified by ELISA using total unconcentrated conditioned medium collected in D. In addition to PMA, ionomycin (Iono), and APMA stimulation, 1% FBS-stimulated shedding was measured by ELISA. Data are means ± S.E. from three independent experiments (n = 3). Error bars represent S.E.
As point mutations of ADAM cleavage sites in target proteins have shown minimal effects on ectodomain shedding in vitro (37–40) and cleavage site sequence specificity of ADAM proteins has not provided a clear consensus sequence (37–40), we investigated other approaches to functionally characterize the proposed cleavage site of RAGE. Previous studies from our group have revealed that RAGE alternative splicing affects various regions of the ECD (7, 20). Interestingly, in lung, kidney, and heart, one of these splice variants lacks exon 9 (mRAGEv4) and is the most prevalent murine isoform after the canonical full-length isoform of RAGE (20). Protein alignment of mRAGE and mRAGEv4 revealed that mRAGEv4 appears to lack 9 amino acids that overlap with the proposed cleavage site (Fig. 4B).
We therefore tested whether mRAGEv4 displayed impaired ectodomain shedding compared with canonical RAGE. We generated HEK-293 cells stably expressing mRAGEv4 (Fig. 4C) and validated by flow cytometry that mRAGEv4 is indeed expressed on the cell surface (Fig. 4D). Under constitutive conditions, we were unable to detect any shed ectodomain product for mRAGEv4 in conditioned medium by either Western blotting or ELISA in contrast to mRAGE (Fig. 4C). We next tested whether shedding of mRAGEv4 could be induced with PMA, ionomycin, APMA, or serum. As seen with constitutive shedding, induced mRAGE shedding could readily be detected by both Western blotting and ELISA (Fig. 4, E and F). In contrast, for mRAGEv4, we could not detect any shed ectodomain product in conditioned medium by either Western blotting or ELISA (Fig. 4, E and F). Together, these data demonstrate that mRAGEv4 is an endogenous ectodomain cleavage-resistant isoform of RAGE. This therefore presented us with an endogenous control to explore the cellular function of RAGE ectodomain shedding.
Inhibition of RAGE Ectodomain Shedding Impairs Cell Migration
RAGE has been shown to affect various cell properties, most notably cell migration and adhesion (1). To test whether ectodomain shedding of RAGE is important for cell migration, we investigated its effect using C6 glioma cells. The C6 cell line has been demonstrated to be an excellent model of RAGE signaling and cell function in numerous studies (3, 4, 24). C6 cells that stably expressed mRAGE, mRAGEv4, or empty vector control (mock-transfected) were generated. Western blotting of cell lysate confirmed that both mRAGE and mRAGEv4 were expressed in total cell lysates in C6 cells for multiple stably transfected cell clones (Fig. 5 A). Importantly, we did not detect any cleaved product from mRAGEv4 C6 cells in conditioned medium (Fig. 5A). We tested the effects of RAGE ectodomain shedding in transwell migration assays using various RAGE ligands. We first tested the effects of collagen I on RAGE-mediated cell migration. It has been shown that not only does RAGE bind directly and specifically to various collagens but RAGE/collagen binding enhances cell migration and adhesion (36, 41, 42). C6 cells expressing mRAGE displayed increased collagen-stimulated cell migration compared with mock-transfected cells (Fig. 5B). In contrast, cells expressing mRAGEv4 displayed migration comparable with mock cells (Fig. 5B).
FIGURE 5.

RAGE ectodomain shedding affects cell migration of C6 glioma cells. A, stably transfected C6 cells (vector control, mRAGE, or mRAGEv4) were generated and assessed for RAGE expression and shedding. Shedding of mRAGE from independently generated stable transfectants (denoted as mRAGE-A or -B and mRAGEv4-A or -B) was assessed by Western blotting (WB) using polyclonal antibodies raised against the total ECD of RAGE. Cells were incubated for 24 h in serum-free medium to allow constitutive shedding to occur. Blotting was performed on conditioned medium and total cell lysate with anti-RAGE antibodies. Cell lysates were probed with anti-β actin as a loading control. Images are representative of three independent experiments. B, C6 glioma cells expressing mRAGE, mRAGEv4, or empty vector (mock) were assessed for ligand-induced migration toward collagen I in transwell migration assays. Independently generated stable transfectants (denoted as mRAGE-A or -B and mRAGEv4-A or -B) were tested. Following 24h of migration, cells were fixed in crystal violet solution, and dye was extracted and quantified using spectrophotometry. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. C and D, C6 glioma cells stably expressing mRAGE, mRAGEv4, or empty vector (mock) were assessed for ligand-induced migration in transwell migration assays with S100B (C) or FBS (D). Cells were seeded into the upper chamber of a Boyden transwell filter and allowed to migrate toward 5 μg/ml S100B or 1% FBS stimulant for 24 h. Cells were fixed in crystal violet solution, and dye was extracted and quantified using spectrophotometry. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E.
To ensure these cleavage resistance effects were not ligand-specific, we next tested the effects of other RAGE ligands including the well characterized RAGE ligand s100B and 1% FBS. Recent studies have shown that serum is a rich source of numerous RAGE ligands (43–45); furthermore, various studies have shown serum to induce RAGE-dependent cell migration and signaling effects (23, 46–51). For both s100B and 1% FBS, C6 glioma cells expressing cleavage-resistant mRAGEv4 displayed lower levels of cell migration compared with both mock and mRAGE-expressing cells (Fig. 5, C and D). These data demonstrate RAGE ectodomain shedding to be important to facilitate RAGE ligand-mediated cell migration.
Inhibition of RAGE Ectodomain Shedding Impairs Cell Adhesion, Spreading, and Actin Cytoskeletal Reorganization
Integral to the process of cell migration is the ability of cells to adhere and spread on extracellular matrix components. Recent studies have revealed that RAGE promotes cell adhesion and spreading through direct binding to various extracellular matrix proteins including collagens I and IV (36, 41, 42). As ectodomain shedding of proteins has been shown to be critical in this cellular process, we tested the effect of RAGE cleavage on cell spreading and actin cytoskeletal reorganization. Cell spreading of C6-mRAGE-expressing cells displayed increased spreading on collagen versus non-coated plates (Fig. 6, A and B). C6-mRAGE cells also had higher levels of cell spreading on both uncoated plastic and collagen I-coated dishes compared with C6-mock cells (Fig. 6, A and B). In contrast, mRAGEv4-expressing C6 cells displayed less spread area compared with mRAGE cells on both uncoated and collagen I-coated dishes (Fig. 6, A and B).
FIGURE 6.

RAGE ectodomain shedding affects cell spreading and actin cytoskeleton rearrangement. A and B, cell adhesion/spreading assays were performed by seeding C6 glioma cells expressing mRAGE, mRAGEv4, or empty vector (mock) onto cell culture dishes coated with either PBS (control) or collagen I for 2 h. Independently generated stable transfectants (denoted as mRAGE-A or -B and mRAGEv4-A or -B) were tested. Adhered cells were fixed and imaged using Olympus CK2 (400× magnification shown in A) with surface area estimated using ImageJ. Surface area was calculated as a percentage of control PBS cells. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E. C, C6 glioma cells expressing mRAGE, mRAGEv4, or empty vector (mock) were seeded on collagen-coated cell culture dishes for 2 h, fixed, and stained with rhodamine-conjugated phalloidin (actin)/anti-vinculin with GFP secondary antibody/DAPI. Cells were examined and photographed using an Olympus FV1000 microscope. Cells were examined and photographed using fluorescence microscopy. Images are representative of three independent experiments.
To further establish the mechanisms underlying these differences in cell migration and adhesion, we analyzed the actin cytoskeleton organization by rhodamine-conjugated phalloidin and vinculin staining in cells by immunofluorescence. Compared with mock cells, mRAGE-overexpressing cells displayed a well spread and polarized cell morphology, demonstrated by the presence of prominent actin stress fibers (Fig. 6C). Further broad cellular staining of focal adhesions (vinculin), indicative of the highly migratory phenotype of RAGE-expressing cells, was seen (Fig. 6C). In striking contrast, mRAGEv4-expressing cells displayed condensed actin stress fiber staining mainly around the cell periphery accompanied with a similar and lesser staining pattern of focal adhesions, indicating impaired adhesion, spreading, and migration (Fig. 6C). These data demonstrate RAGE ectodomain shedding to be important for the adhesion of cells to the extracellular matrix and their reorganization of the actin cytoskeleton.
Inhibition of RAGE Ectodomain Shedding Impairs Cell Signaling
Next we examined the effect of RAGE ectodomain shedding on downstream signaling. RAGE has been shown to signal through diverse pathways involved in cell migration including various MAPKs (ERK1/2 and p38), PI3K/Akt, and Src (2, 3, 52–56). We tested the effect of RAGE ectodomain shedding on cell signaling by plating cells on collagen I and assessing ligand activation of downstream signaling pathways. Compared with mock-expressing C6 cells, RAGE-overexpressing cells displayed increased activation of Src (both p416Src and p527Src), Akt, and ERK1/2 pathways (Fig. 7). By contrast, mRAGEv4-expressing cells displayed impaired activation of signaling compared with RAGE-overexpressing cells (Fig. 7). We did not detect significant changes in the p38 pathway for mock, mRAGE, or mRAGEv4 (Fig. 7E). Together, these data demonstrate that RAGE ectodomain shedding is a critical mechanism to induce cell signaling.
FIGURE 7.

RAGE ectodomain shedding affects cell signaling in C6 glioma cells. C6 glioma cells expressing mRAGE, mRAGEv4, or empty vector (mock) were seeded on collagen-coated cell culture dishes for 2 h. Cell lysates were then subject to immunoblotting with antibodies to phospho- (p) and total Src (B), Akt (C), ERK1/2 (D), and p38 (E). Samples were also subjected to Western blotting for RAGE and actin controls (A). Densitometry for three independent experiments was performed using ImageJ. Activation was calculated by normalization to total proteins levels, with percentage of induction calculated relative to control cells. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Images are representative of three independent experiments. Error bars represent S.E.
RAGE-mediated Cell Migration through Ectodomain Shedding Is Dependent on Activation of MAPK and PI3K Signaling, Metalloproteinases, and γ-Secretase
Finally, we tested the mechanisms by which ectodomain shedding of RAGE activates cell migration. As the MAPK and Akt/PI3K pathways are involved in RAGE shedding and down-regulated in mRAGEv4-expressing cells, we first tested how inhibitors of these pathways affect cell migration through RAGE. In mRAGE-expressing cells, both MEK (U0126) and PI3K (LY294002) inhibitors significantly reduced migration of mRAGE cells to basal mock-transfected cell levels (Fig. 8A). By contrast, both MEK (U0126) and PI3K (LY294002) inhibition had no effect on RAGE ligand-mediated cell migration of mRAGEv4- or mock-transfected cells (Fig. 8A).
FIGURE 8.

Cell migration induced by RAGE ectodomain shedding is dependent on signaling through ADAM10 and γ-secretase. C6 glioma cells expressing mRAGE, mRAGEv4, or empty vector (mock) were assessed for collagen I-induced migration in the presence of inhibitors of MEK (10 μm U0126) or PI3K (10 μm LY294002) (A), sRAGE (5 μg/ml) (B), metalloproteinases (10 μm BB94) (C), ADAM10 (5 μm GI254023X) (D), and γ-secretase (10 μm DAPT) (E). Following 24 h of migration, cells were fixed in crystal violet solution, and dye was extracted and quantified using spectrophotometry. Data are means ± S.E. from three independent experiments (n = 3). Statistical difference between groups was assessed by one-way ANOVA where * denotes significant differences (p < 0.05) between groups. Error bars represent S.E.
We next tested whether exogenous treatment of cells with the RAGE ectodomain (sRAGE) could affect ligand-stimulated cell migration in mRAGE and proteolytically resistant mRAGEv4 C6 cells. Treatment of C6-mRAGE with 5 μg/ml recombinant sRAGE significantly impaired RAGE ligand-mediated cell migration compared with vehicle-treated C6-mRAGE cells (Fig. 8B). Treatment of both C6-mock and C6-mRAGEv4 cells with sRAGE did not affect cell migration (Fig. 8B).
To determine whether ectodomain shedding of RAGE regulates cell migration through a metalloproteinase-dependent mechanism, we performed experiments using broad metalloproteinase (BB94)- and ADAM10-specific (GI254023X) inhibitors. In mRAGE-expressing cells, both inhibitors BB94 and GI254023X significantly reduced migration of mRAGE cells to basal mock-transfected cell levels (Fig. 8, C and D). By contrast, neither BB94 nor GI254023X affected cell migration of mRAGEv4- or mock-transfected cells (Fig. 8, C and D).
Finally, we studied the role of γ-secretases in regulating RAGE-mediated cell migration. γ-Secretases regulate cell signaling by cleavage of the residual membrane-bound C-terminal fragments formed by ectodomain shedding (8). Following cleavage of the C-terminal fragments by γ-secretase, this results in the release of the ICD of receptors, which can activate signaling (8). Prior studies have indicated that following ectodomain shedding the remaining membrane-bound C-terminal fragment of RAGE is processed by γ-secretase (8, 10). However, no studies have investigated whether γ-secretase regulates RAGE function. Using the γ-secretase inhibitor DAPT, we demonstrated that DAPT significantly reduced migration of mRAGE-expressing cells (Fig. 8E), whereas DAPT had no effect on the RAGE ligand-induced migration of either mock- or mRAGEv4-expressing cells (Fig. 8E). Together, these mechanistic studies demonstrate that not only is RAGE ectodomain shedding central to the mediating the effects of RAGE but it is dependent on an inside-out signaling mechanism.
Discussion
In the current report, we have studied the mechanisms regulating RAGE ectodomain shedding and importantly its role in cell function. First, human and mouse RAGE proteins undergo shedding in a similar constitutive and inducible manner. Second, RAGE shedding is regulated by distinct signaling pathways dependent on the stimuli. Third, we confirm that the site of cleavage occurs in the protein region close to the membrane, and RAGE ectodomain shedding can be blocked by a splice variant lacking this region of RAGE. Most importantly, we demonstrate ectodomain shedding of RAGE to be a critical mechanism in the biology of RAGE and its ability to induce cell signaling and movement.
Ectodomain shedding is an important post-translational mechanism that increases the range of functions of cell surface molecules (11, 12). For RAGE, various studies have demonstrated that this cell surface receptor undergoes ectodomain shedding in response to various stimuli and is a metalloproteinase-dependent mechanism (8–10). However, no studies to date have addressed whether this process is important for RAGE function. Although numerous studies have shown a role for RAGE and its soluble ECD in pathogenic states, less is known about the normal biology of RAGE. RAGE has been shown to be a pattern recognition receptor and to bind multiple ligands (4, 5, 36, 41, 42, 57–59). Interestingly, RAGE is most closely related to genes coding for immunoglobulin cell adhesion molecules including activated leukocyte cell adhesion molecule and basal cell adhesion molecule (36), molecules with both ligand signaling properties and key functions in cell adhesion and migration. Therefore, we hypothesized RAGE ectodomain shedding to be an important mechanism to mediate the biological activity of RAGE.
To understand further how RAGE ectodomain shedding is regulated, we first tested whether RAGE ectodomain shedding is a conserved mechanism between human and mouse RAGE. Both human and mouse RAGE shedding was found to be induced in a similar magnitude by PMA, ionomycin, and APMA, suggesting common cleavage site mechanisms. As these compounds represent non-physiological stimuli, we demonstrated that serum can induce RAGE ECD shedding. Serum factors that induce ectodomain shedding include proteinaceous factors (growth factors and cytokines) as well as lipid-based factors (lysophosphatidic acid and ceramide) (11, 12). Furthermore, serum is a rich source of RAGE ligands, which may activate ectodomain shedding of RAGE (9, 60). To investigate whether RAGE ligands induce shedding of RAGE, we tested two highly characterized RAGE ligands for their ability to induce shedding of RAGE. Prior studies have again been conflicting in the role of RAGE ligands as inducers of RAGE ectodomain shedding (8, 25, 51). In our studies, both CML and S100B induced shedding of RAGE ectodomain, albeit to a lesser degree than chemical inducers of shedding. To further validate these data with RAGE ligands, we demonstrated that incubation of cells with the inhibitor FPS-ZM1 (31) led to striking impairment of RAGE shedding.
To explore the intracellular signaling mechanisms regulating induction of RAGE ectodomain shedding, we began by investigating the effects of protein phosphatase inhibitors on RAGE shedding. These compounds also mimic the effects of physiological stimuli by inducing signal transduction pathways triggered downstream of sheddase enzymes (34). We found that, in a similar manner to other stimuli of ectodomain shedding used, all protein phosphatase inhibitors induced RAGE ECD shedding (including pervanadate, calyculin A, and cantharidin). Although these inhibitors target different phosphatase types (serine/threonine versus tyrosine), we observed a similar effect, indicating the role of various diverse signaling pathways on activating RAGE shedding. Prior work has shown that PMA-stimulated RAGE ectodomain shedding is inhibited by the PKC inhibitor Gö6976 (10). Furthermore, Zhang et al. (10) observed that PKCα/PKCβI inhibition with Gö6976 led to a decrease in shedding due to calcium influx. We observed that PKCα/PKCβI inhibition with both Gö6976 and GFX109203X inhibited PMA-induced shedding but had no effect on RAGE shedding induced by calcium influx. Furthermore, inhibition of calcium influx by EGTA affected ionomycin- but not PMA-induced RAGE shedding, indicating key differences in the regulation of RAGE shedding. These data are consistent with studies of other receptors that display divergent mechanisms of regulation of ectodomain shedding including L-selectin, CD44, Syndecan-1, KIM-1, HER2, and ERB4 (30, 34, 35, 62). Indeed, for CD44, shedding induced by calcium influx is insensitive to PKCα and PKCβI inhibition, whereas PMA-induced shedding similarly is PKCα/PKCβI-dependent and calcium influx/EGTA-independent (62). Furthermore, we cannot rule out that these effects may be mediated by other PKC isoforms as multiple isoforms beyond PKCα/PKCβI have been implicated in ectodomain shedding (30, 63). To further probe the different signaling pathways leading to RAGE shedding, we studied the role of MAPK and PI3K signaling. For other cell surface proteins regulated by shedding, these pathways have been shown to be a key mechanism leading to the activation of sheddases (30, 34, 35). In our experiments, PMA-induced shedding was not affected by inhibitors against p38, MEK, SAPK/JNK, or PI3K. However, calcium influx-induced shedding was inhibited by PI3K inhibitors.
Prior studies have clearly shown that RAGE ectodomain shedding is mediated by a metalloproteinase-dependent mechanism, most likely ADAM10 (8, 10). Using multiple approaches including siRNA knockdown, ADAM10-specific chemical inhibitors, and cells from ADAM10 knock-out mice, it appears that ADAM10 is the major RAGE sheddase (8–10). Our data support these findings as we observed that ADAM10 inhibition with GI254023X prevented RAGE shedding induced by both PMA and ionomycin. These data together highlight divergent mechanisms of regulating RAGE shedding (Fig. 9A).
FIGURE 9.
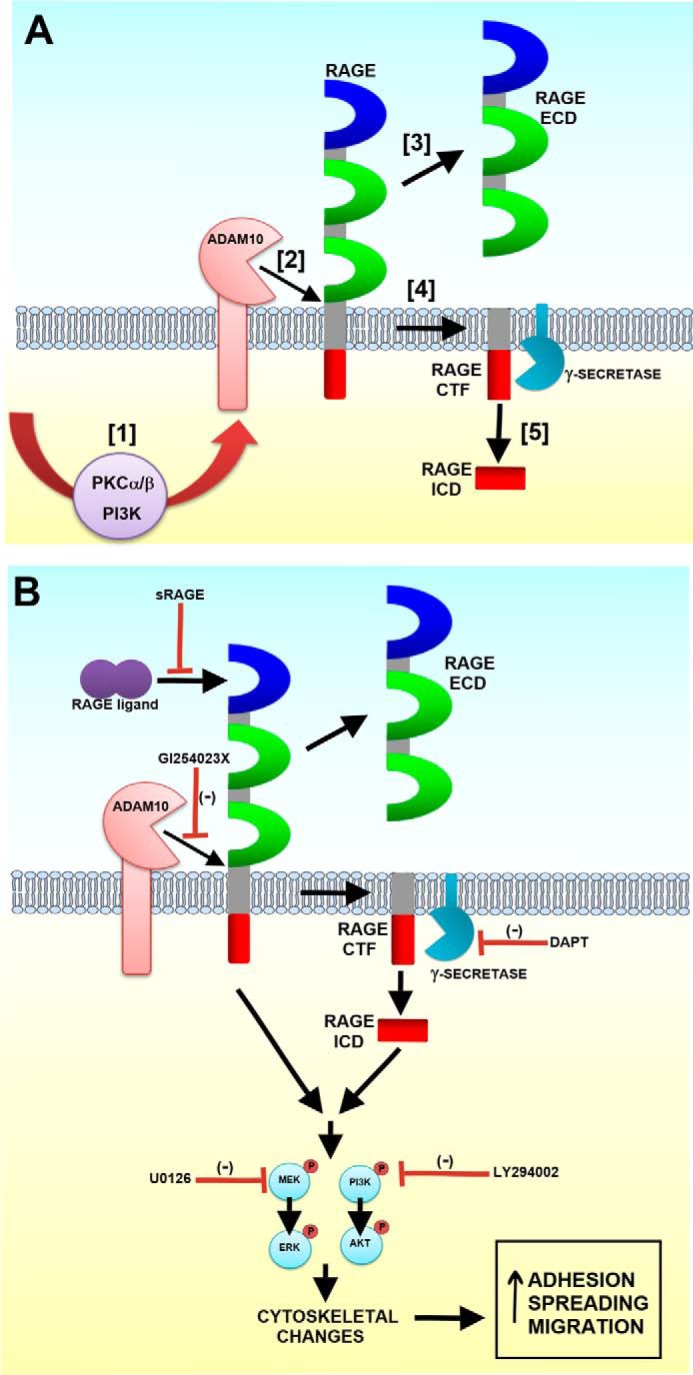
Schematic model of the proposed mechanisms of RAGE ectodomain shedding and its regulation of cell function. A, following cell exposure to stimuli or calcium influx, PKC and PI3K signaling is activated (1). This in turn activates ADAM10 (2), which cleaves RAGE at the juxtamembrane region, releasing the RAGE ECD (3). A membrane-tethered C-terminal fragment (CTF) remains (4), and it can be cleaved by a γ-secretase to release the RAGE ICD (5). B, inhibition of pathways up- and downstream of RAGE ectodomain shedding affect cell migration. These include the inhibition of RAGE ligand binding (sRAGE), activation of proteases including ADAM10 (GI254023X) and γ-secretase (DAPT), and downstream signaling pathways (MEK and PI3K inhibition).
Although RAGE ectodomain shedding has been described previously (8, 10), little is known about the cleavage site of RAGE. To attempt to characterize this site, we performed a number of experiments to identify the site of cleavage. Using an N-terminally epitope-tagged RAGE construct, we observed only one cleavage product even under conditions of various shedding stimulants. This supports previous data suggesting that RAGE ectodomain shedding occurs around the JM domain (10, 28). For most receptors studied to date, the cleavage site for sheddases occurs around 6–15 amino acids upstream of the start of the transmembrane region (11, 12). As point mutations of potential ADAM cleavage sites in target proteins have shown limited effects on shedding in vitro (37–40), we tried alternative approaches to characterizing the site and function of RAGE ectodomain shedding. Sequencing of murine soluble RAGE isolated from lung has suggested a putative cleavage site between Gly331 and Ser332 of RAGE (28). This site occurs in/around the short exon 9 sequence of both human and mouse RAGE. Alignment of the protein sequence of RAGE from multiple species revealed this site and the surrounding amino acid residues to be strongly conserved for RAGE, suggesting that a consensus sequence may exist for shedding. Analysis of previously isolated alternative splice variants of RAGE revealed that a variant exists that lacks exon 9 (mRAGEv4) and importantly is missing the proposed cleavage site (20). The mRAGEv4 splice variant is highly prevalent in lung, kidney, and heart (20); however, we did not previously identify this variant in human tissue (7). BLAST analysis of human RAGE cDNA revealed various cDNAs for human and primate RAGE comparable with mRAGEv4 that lacks exon 9 (data not shown). We found that, under both constitutive and inducible conditions, the mRAGEv4 splice variant proved to be protease-resistant in multiple cell models. Therefore, these data confirm the cleavage site of RAGE to be upstream of ∼12 amino acids of the transmembrane domain. In previous studies, it was difficult to identify the precise site of cleavage (10). It may be for RAGE as for other receptors that, rather than a precise consensus sequence of cleavage, the secondary structure of the receptor proximal to the transmembrane region is more important. As there is no clear consensus in existing ADAM10 targets, recent studies have utilized peptide libraries and proteomics to attempt to identify an ADAM10 and -17 cleavage consensus motif (65, 66). It was found for ADAM10 that this sheddase prefers a Leu or aromatic amino acid (Phe, Tyr, or Trp) in the residue after cleavage (termed the P1′ site) (65, 66). However, this consensus does not cover all ADAM10 targets including RAGE, which contains a Ser in this position. Further analysis of known proteolytic substrate sites using the MEROPS database did not reveal any further protease sites around the JM region of RAGE (67). Analysis of secondary structure of mRAGE versus mRAGEv4 splice variant did not reveal changes in secondary structures such as α-helix or β-sheets. The removal of these 9 amino acids may therefore be critical for solvent accessibility between the JM region and the transmembrane domain. These data would suggest that this, rather than altering a protein cleavage site motif, could affect accessibility of the protease to its substrate site on RAGE. The production of alternative splice isoforms that are resistant to ectodomain shedding is not unique to RAGE. Indeed, it has been shown that alternative splicing of the ERBB4 gene results in a JM region variant that is protease-resistant (68). Moreover, as seen for RAGE in this study, expression of the non-cleavable ERBB4 splice variant altered cell growth and survival (68). This therefore raises the intriguing question whether this splice variant of RAGE that prevents shedding could affect cell function.
To understand the role of ectodomain shedding, we utilized the protease-resistant RAGEv4 splice variant to determine how it affected cell function compared with normal RAGE. One of the major functions of RAGE shown to date is its regulation of cell migration (1). We therefore investigated how ectodomain shedding of RAGE impacts cell migration. We found that expression of non-cleavable mRAGEv4 in C6 cells impaired cell migration in response to various RAGE ligands including S100B, serum, and collagen I. This was verified not to be a clonal cell resulting from stable transfection, as multiple clones of mRAGE and mRAGEv4 gave similar results. Cell migration is a complex, multifaceted process involving changes in actin cytoskeletal remodeling, focal adhesion dynamics, and cell spreading. We tested whether shedding of RAGE alters cell migration by affecting cell adhesion and spreading through altering signaling and rearrangement of the actin cytoskeleton. Expression of ectodomain shedding-resistant RAGE (mRAGEv4) impaired cell spreading and in turn was reflected by a condensed actin stress fiber staining mainly around the cell periphery and lesser staining of focal adhesions. These data are consistent with a lower migratory cell phenotype. To understand the signaling mechanisms affected by RAGE ectodomain shedding, we investigated how RAGE signaling was affected by impairment of cleavage of its ectodomain. Cells expressing ectodomain shedding-resistant RAGE displayed impaired signaling through MAPK and Akt/PI3K pathways.
It is also possible that the results seen here are due to other effects of the mRAGEv4 splice variant. First, it is possible that mRAGEv4 affects RAGE dimerization and subsequent signaling; however, this is unlikely as the region involved in the dimerization process is upstream of this region (69). Second, it is possible that mRAGEv4 affects RAGE ligand binding and signaling through alteration of the secondary structure of the RAGE ECD. This again is unlikely as the majority of ligand binding sites occur in the V-domain of RAGE, and only deletion of this domain drastically affects RAGE function as seen here (3, 31, 45, 70, 71). These data together therefore suggest ectodomain shedding of RAGE to be a mechanism of multiple functions in addition to merely generating the soluble ectodomain.
Our data suggest that shedding of RAGE may be a prerequisite to induce cell signaling and affect cell function (Fig. 9B). To further explore the mechanistic regulation of RAGE ectodomain shedding and how it affects cell migration, we tested the role of various pathways in this process. Exogenous treatment of cells with the sRAGE ectodomain did not affect mRAGEv4 cell migration but, as expected, impaired mRAGE cell migration. Inhibitors of both MAPK and Akt/PI3K impaired RAGE-mediated migration of C6 cells but had no effect on protease-resistant mRAGEv4-expressing cells. Similarly, broad metalloproteinase and ADAM10 inhibitors blocked mRAGE-dependent cell migration without affecting mRAGEv4 cell migration. As the non-cleavable RAGE isoform blocks these cell processes, this therefore suggests production of the soluble ECD to act as a decoy; it is not the mechanism responsible for inhibiting migration and adhesion. Our data would therefore indicate that RAGE ectodomain shedding may exist to allow the release and subsequent processing of its ICD (Fig. 9). This process, known as regulated intramembrane proteolysis, typically follows ectodomain shedding and has been demonstrated to occur and be important for signaling of numerous receptors (11, 12). In fact, for RAGE, it has been shown that following ectodomain shedding the remaining C-terminal fragment is further processed by a γ-secretase (8, 10). Furthermore, ectopic expression of the ICD results in a predominant nuclear expression, suggesting a direct role in gene regulation (8). Cell migration assays demonstrated that that this process is required for RAGE-mediated cell function as γ-secretase impaired mRAGE-expressing cells from migrating but did not alter mRAGEv4-mediated migration. These data show for the first time that processing of RAGE by γ-secretase following ectodomain shedding is a central mechanism by which RAGE mediates signaling.
In conclusion, shedding of RAGE is a precisely regulated process involving multiple signaling pathways and stimuli. Our data suggest that RAGE shedding is a critical mechanism not only to affect cell signaling but also to mediate cell functional changes. Further studies are required to explore how the RAGE ICD formed by γ-secretase is regulated and to determine its function. Collectively, these data suggest that proteolytic regulation of RAGE is a central mechanism in RAGE biology and describe a new function by which RAGE can mediate diverse functional roles in both physiology and disease.
Author Contributions
A. B. conducted most of the experiments, analyzed the results, and wrote the paper. T. K. conducted experiments and analyzed the results. J. J. conducted experiments and analyzed the results. E. H. conducted experiments and analyzed the results. R. L. was involved in the design of experiments and critical review of the manuscript. B. I. H. conceived the idea for the project and wrote the paper with A. B.
Acknowledgment
We thank Dr. Nam-Ho Huh for providing the N-terminal His-tagged RAGE construct.
This work was supported by a career catalyst research grant from Susan G. Komen for the Cure and by American Cancer Society Institutional Research Grant 98-277-10. The authors declare that they have no conflicts of interest with the contents of this article.
- RAGE
- receptor for advanced glycation end products
- ADAM
- a disintegrin and metalloproteinase
- sRAGE
- soluble RAGE
- ECD
- extracellular domain
- ICD
- intracellular domain
- PMA
- phorbol 12-myristate 13-acetate
- APMA
- 4-aminophenylmercuric acetate
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- ANOVA
- analysis of variance
- hRAGE
- human RAGE
- mRAGE
- mouse RAGE
- CML
- carboxymethyllysine
- JM
- juxtamembrane
- mRAGEv4
- mRAGE splice variant 4.
References
- 1. Kalea A. Z., Schmidt A. M., and Hudson B. I. (2009) RAGE: a novel biological and genetic marker for vascular disease. Clin. Sci. 116, 621–637 [DOI] [PubMed] [Google Scholar]
- 2. Taguchi A., Blood D. C., del Toro G., Canet A., Lee D. C., Qu W., Tanji N., Lu Y., Lalla E., Fu C., Hofmann M. A., Kislinger T., Ingram M., Lu A., Tanaka H., et al. (2000) Blockage of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 405, 354–360 [DOI] [PubMed] [Google Scholar]
- 3. Hudson B. I., Kalea A. Z., Del Mar Arriero M., Harja E., Boulanger E., D'Agati V., and Schmidt A. M. (2008) Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 283, 34457–34468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalea A. Z., See F., Harja E., Arriero M., Schmidt A. M., and Hudson B. I. (2010) Alternatively spliced RAGEv1 inhibits tumorigenesis through suppression of JNK signaling. Cancer Res. 70, 5628–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Logsdon C. D., Fuentes M. K., Huang E. H., and Arumugam T. (2007) RAGE and RAGE ligands in cancer. Curr. Mol. Med. 7, 777–789 [DOI] [PubMed] [Google Scholar]
- 6. Park L., Raman K. G., Lee K. J., Lu Y., Ferran L. J. Jr., Chow W. S., Stern D., and Schmidt A. M. (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 4, 1025–1031 [DOI] [PubMed] [Google Scholar]
- 7. Hudson B. I., Carter A. M., Harja E., Kalea A. Z., Arriero M., Yang H., Grant P. J., and Schmidt A. M. (2008) Identification, classification, and expression of RAGE gene splice variants. FASEB J. 22, 1572–1580 [DOI] [PubMed] [Google Scholar]
- 8. Galichet A., Weibel M., and Heizmann C. W. (2008) Calcium-regulated intramembrane proteolysis of the RAGE receptor. Biochem. Biophys. Res. Commun. 370, 1–5 [DOI] [PubMed] [Google Scholar]
- 9. Raucci A., Cugusi S., Antonelli A., Barabino S. M., Monti L., Bierhaus A., Reiss K., Saftig P., and Bianchi M. E. (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 22, 3716–3727 [DOI] [PubMed] [Google Scholar]
- 10. Zhang L., Bukulin M., Kojro E., Roth A., Metz V. V., Fahrenholz F., Nawroth P. P., Bierhaus A., and Postina R. (2008) Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 283, 35507–35516 [DOI] [PubMed] [Google Scholar]
- 11. Garton K. J., Gough P. J., and Raines E. W. (2006) Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 79, 1105–1116 [DOI] [PubMed] [Google Scholar]
- 12. Hayashida K., Bartlett A. H., Chen Y., and Park P. W. (2010) Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. 293, 925–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jia H. P., Look D. C., Tan P., Shi L., Hickey M., Gakhar L., Chappell M. C., Wohlford-Lenane C., and McCray P. B. Jr. (2009) Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L84–L96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hudson B. I., Moon Y. P., Kalea A. Z., Khatri M., Marquez C., Schmidt A. M., Paik M. C., Yoshita M., Sacco R. L., DeCarli C., Wright C. B., and Elkind M. S. (2011) Association of serum soluble receptor for advanced glycation end-products with subclinical cerebrovascular disease: the Northern Manhattan Study (NOMAS). Atherosclerosis 216, 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Falcone C., Emanuele E., D'Angelo A., Buzzi M. P., Belvito C., Cuccia M., and Geroldi D. (2005) Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler. Thromb. Vasc. Biol. 25, 1032–1037 [DOI] [PubMed] [Google Scholar]
- 16. Thomas M. C., Söderlund J., Lehto M., Mäkinen V. P., Moran J. L., Cooper M. E., Forsblom C., Groop P. H., and FinnDiane Study Group (2011) Soluble receptor for AGE (RAGE) is a novel independent predictor of all-cause and cardiovascular mortality in type 1 diabetes. Diabetologia 54, 2669–2677 [DOI] [PubMed] [Google Scholar]
- 17. Montaner J., Perea-Gainza M., Delgado P., Ribó M., Chacón P., Rosell A., Quintana M., Palacios M. E., Molina C. A., and Alvarez-Sabín J. (2008) Etiologic diagnosis of ischemic stroke subtypes with plasma biomarkers. Stroke 39, 2280–2287 [DOI] [PubMed] [Google Scholar]
- 18. Yokota C., Minematsu K., Tomii Y., Naganuma M., Ito A., Nagasawa H., and Yamaguchi T. (2009) Low levels of plasma soluble receptor for advanced glycation end products are associated with severe leukoaraiosis in acute stroke patients. J. Neurol. Sci. 287, 41–44 [DOI] [PubMed] [Google Scholar]
- 19. Park H. Y., Yun K. H., and Park D. S. (2009) Levels of soluble receptor for advanced glycation end products in acute ischemic stroke without a source of cardioembolism. J. Clin. Neurol. 5, 126–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalea A. Z., Reiniger N., Yang H., Arriero M., Schmidt A. M., and Hudson B. I. (2009) Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 23, 1766–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sakaguchi M., Murata H., Yamamoto K., Ono T., Sakaguchi Y., Motoyama A., Hibino T., Kataoka K., and Huh N. H. (2011) TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One 6, e23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Codony-Servat J., Albanell J., Lopez-Talavera J. C., Arribas J., and Baselga J. (1999) Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res. 59, 1196–1201 [PubMed] [Google Scholar]
- 23. Jules J., Maiguel D., and Hudson B. I. (2013) Alternative splicing of the RAGE cytoplasmic domain regulates cell signaling and function. PLoS One 8, e78267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grobben B., De Deyn P. P., and Slegers H. (2002) Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 310, 257–270 [DOI] [PubMed] [Google Scholar]
- 25. Naik M. U., Stalker T. J., Brass L. F., and Naik U. P. (2012) JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood 119, 3352–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collec E., El Nemer W., Gauthier E., Gane P., Lecomte M. C., Dhermy D., Cartron J. P., Colin Y., Le van Kim C., and Rahuel C. (2007) Ubc9 interacts with Lu/BCAM adhesion glycoproteins and regulates their stability at the membrane of polarized MDCK cells. Biochem. J. 402, 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vega F. M., Colomba A., Reymond N., Thomas M., and Ridley A. J. (2012) RhoB regulates cell migration through altered focal adhesion dynamics. Open Biol. 2, 120076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanford L. E., Enghild J. J., Valnickova Z., Petersen S. V., Schaefer L. M., Schaefer T. M., Reinhart T. A., and Oury T. D. (2004) Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J. Biol. Chem. 279, 50019–50024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirata M., Umata T., Takahashi T., Ohnuma M., Miura Y., Iwamoto R., and Mekada E. (2001) Identification of serum factor inducing ectodomain shedding of proHB-EGF and studies of noncleavable mutants of proHB-EGF. Biochem. Biophys. Res. Commun. 283, 915–922 [DOI] [PubMed] [Google Scholar]
- 30. Killock D. J., and Ivetić A. (2010) The cytoplasmic domains of TNFα-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem. J. 428, 293–304 [DOI] [PubMed] [Google Scholar]
- 31. Deane R., Singh I., Sagare A. P., Bell R. D., Ross N. T., LaRue B., Love R., Perry S., Paquette N., Deane R. J., Thiyagarajan M., Zarcone T., Fritz G., Friedman A. E., Miller B. L., and Zlokovic B. V. (2012) A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 122, 1377–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W., Xie L., Chen Z., Zhu Y., Sun Y., Miao Y., Xu Z., and Han X. (2010) Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 101, 1226–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suganuma M., Fujiki H., Furuya-Suguri H., Yoshizawa S., Yasumoto S., Kato Y., Fusetani N., and Sugimura T. (1990) Calyculin A, an inhibitor of protein phosphatases, a potent tumor promoter on CD-1 mouse skin. Cancer Res. 50, 3521–3525 [PubMed] [Google Scholar]
- 34. Zhang Z., Humphreys B. D., and Bonventre J. V. (2007) Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J. Am. Soc. Nephrol. 18, 2704–2714 [DOI] [PubMed] [Google Scholar]
- 35. Fitzgerald M. L., Wang Z., Park P. W., Murphy G., and Bernfield M. (2000) Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 148, 811–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sessa L., Gatti E., Zeni F., Antonelli A., Catucci A., Koch M., Pompilio G., Fritz G., Raucci A., and Bianchi M. E. (2014) The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS One 9, e86903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brakebusch C., Varfolomeev E. E., Batkin M., and Wallach D. (1994) Structural requirements for inducible shedding of the p55 tumor necrosis factor receptor. J. Biol. Chem. 269, 32488–32496 [PubMed] [Google Scholar]
- 38. Müllberg J., Oberthür W., Lottspeich F., Mehl E., Dittrich E., Graeve L., Heinrich P. C., and Rose-John S. (1994) The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J. Immunol. 152, 4958–4968 [PubMed] [Google Scholar]
- 39. Tang P., Hung M.-C., and Klostergaard J. (1996) Length of the linking domain of human pro-tumor necrosis factor determines the cleavage processing. Biochemistry 35, 8226–8233 [DOI] [PubMed] [Google Scholar]
- 40. Chen A., Engel P., and Tedder T. F. (1995) Structural requirements regulate endoproteolytic release of the L-selectin (CD62L) adhesion receptor from the cell surface of leukocytes. J. Exp. Med. 182, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Demling N., Ehrhardt C., Kasper M., Laue M., Knels L., and Rieber E. P. (2006) Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 323, 475–488 [DOI] [PubMed] [Google Scholar]
- 42. Milutinovic P. S., Englert J. M., Crum L. T., Mason N. S., Ramsgaard L., Enghild J. J., Sparvero L. J., Lotze M. T., and Oury T. D. (2014) Clearance kinetics and matrix binding partners of the receptor for advanced glycation end products. PLoS One 9, e88259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamagishi S., Adachi H., Nakamura K., Matsui T., Jinnouchi Y., Takenaka K., Takeuchi M., Enomoto M., Furuki K., Hino A., Shigeto Y., and Imaizumi T. (2006) Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism 55, 1227–1231 [DOI] [PubMed] [Google Scholar]
- 44. Jäckel A., Deichmann M., Waldmann V., Bock M., and Näher H. (1999) S-100β protein in serum, a tumor marker in malignant melanoma—current state of knowledge and clinical experience. Hautarzt 50, 250–256 [DOI] [PubMed] [Google Scholar]
- 45. Pietzsch J., and Hoppmann S. (2009) Human S100A12: a novel key player in inflammation? Amino Acids 36, 381–389 [DOI] [PubMed] [Google Scholar]
- 46. Arumugam T., Ramachandran V., Gomez S. B., Schmidt A. M., and Logsdon C. D. (2012) S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 18, 4356–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang R., Loux T., Tang D., Schapiro N. E., Vernon P., Livesey K. M., Krasinskas A., Lotze M. T., and Zeh H. J. 3rd (2012) The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. U.S.A. 109, 7031–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kang R., Tang D., Schapiro N. E., Livesey K. M., Farkas A., Loughran P., Bierhaus A., Lotze M. T., and Zeh H. J. (2010) The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 17, 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xiong F., Leonov S., Howard A. C., Xiong S., Zhang B., Mei L., McNeil P., Simon S., and Xiong W. C. (2011) Receptor for advanced glycation end products (RAGE) prevents endothelial cell membrane resealing and regulates F-actin remodeling in a beta-catenin-dependent manner. J. Biol. Chem. 286, 35061–35070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Z., Immel D., Xi C. X., Bierhaus A., Feng X., Mei L., Nawroth P., Stern D. M., and Xiong W. C. (2006) Regulation of osteoclast function and bone mass by RAGE. J. Exp. Med. 203, 1067–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zen K., Chen C. X., Chen Y. T., Wilton R., and Liu Y. (2007) Receptor for advanced glycation endproducts mediates neutrophil migration across intestinal epithelium. J. Immunol. 178, 2483–2490 [DOI] [PubMed] [Google Scholar]
- 52. Kislinger T., Fu C., Huber B., Qu W., Taguchi A., Du Yan S., Hofmann M., Yan S. F., Pischetsrieder M., Stern D., and Schmidt A. M. (1999) Nϵ-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274, 31740–31749 [DOI] [PubMed] [Google Scholar]
- 53. Huang J. S., Guh J. Y., Chen H. C., Hung W. C., Lai Y. H., and Chuang L. Y. (2001) Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 81, 102–113 [DOI] [PubMed] [Google Scholar]
- 54. Yeh C. H., Sturgis L., Haidacher J., Zhang X. N., Sherwood S. J., Bjercke R. J., Juhasz O., Crow M. T., Tilton R. G., and Denner L. (2001) Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-κB transcriptional activation and cytokine secretion. Diabetes 50, 1495–1504 [DOI] [PubMed] [Google Scholar]
- 55. Lander H. M., Tauras J. M., Ogiste J. S., Hori O., Moss R. A., and Schmidt A. M. (1997) Activation of the receptor for advanced glycation end products triggers a p21ras-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 272, 17810–17814 [DOI] [PubMed] [Google Scholar]
- 56. Huttunen H. J., Fages C., and Rauvala H. (1999) Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 274, 19919–19924 [DOI] [PubMed] [Google Scholar]
- 57. Ichikawa M., Williams R., Wang L., Vogl T., and Srikrishna G. (2011) S100A8/A9 activate key genes and pathways in colon tumor progression. Mol. Cancer Res. 9, 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Björk P., Källberg E., Wellmar U., Riva M., Olsson A., He Z., Törngren M., Liberg D., Ivars F., and Leanderson T. (2013) Common interactions between S100A4 and S100A9 defined by a novel chemical probe. PLoS One 8, e63012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leclerc E., Fritz G., Weibel M., Heizmann C. W., and Galichet A. (2007) S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J. Biol. Chem. 282, 31317–31331 [DOI] [PubMed] [Google Scholar]
- 60. Lam J. K., Wang Y., Shiu S. W., Wong Y., Betteridge D. J., and Tan K. C. (2013) Effect of insulin on the soluble receptor for advanced glycation end products (RAGE). Diabet. Med. 30, 702–709 [DOI] [PubMed] [Google Scholar]
- 61. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagano O., Murakami D., Hartmann D., De Strooper B., Saftig P., Iwatsubo T., Nakajima M., Shinohara M., and Saya H. (2004) Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J. Cell Biol. 165, 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kveiborg M., Instrell R., Rowlands C., Howell M., and Parker P. J. (2011) PKCα and PKCδ regulate ADAM17-mediated ectodomain shedding of heparin binding-EGF through separate pathways. PLoS One 6, e17168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thompson J. D., Gibson T. J., and Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2, Unit 2.3 [DOI] [PubMed] [Google Scholar]
- 65. Tucher J., Linke D., Koudelka T., Cassidy L., Tredup C., Wichert R., Pietrzik C., Becker-Pauly C., and Tholey A. (2014) LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res. 13, 2205–2214 [DOI] [PubMed] [Google Scholar]
- 66. Caescu C. I., Jeschke G. R., and Turk B. E. (2009) Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rawlings N. D., Barrett A. J., and Finn R. (2016) Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Veikkolainen V., Vaparanta K., Halkilahti K., Iljin K., Sundvall M., and Elenius K. (2011) Function of ERBB4 is determined by alternative splicing. Cell Cycle 10, 2647–2657 [DOI] [PubMed] [Google Scholar]
- 69. Wei W., Lampe L., Park S., Vangara B. S., Waldo G. S., Cabantous S., Subaran S. S., Yang D., Lakatta E. G., and Lin L. (2012) Disulfide bonds within the C2 domain of RAGE play key roles in its dimerization and biogenesis. PLoS One 7, e50736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma W., Lee S. E., Guo J., Qu W., Hudson B. I., Schmidt A. M., and Barile G. R. (2007) RAGE ligand upregulation of VEGF secretion in ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 48, 1355–1361 [DOI] [PubMed] [Google Scholar]
- 71. Ma W., Rai V., Hudson B. I., Song F., Schmidt A. M., and Barile G. R. (2012) RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 274, 72–82 [DOI] [PubMed] [Google Scholar]
- 72. Bairoch A., Apweiler R., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M. J., Natale D. A., O'Donovan C., Redaschi N., and Yeh L. S. (2005) The Universal Protein Resource (UniProt). Nucleic Acids Res. 33, D154–D159 [DOI] [PMC free article] [PubMed] [Google Scholar]