

Abstract
The first exon of the Huntingtin protein (Httex1) is one of the most actively studied Htt fragments because its overexpression in R6/2 transgenic mice has been shown to recapitulate several key features of Huntington disease. However, the majority of biophysical studies of Httex1 are based on assessing the structure and aggregation of fusion constructs where Httex1 is fused to large proteins, such as glutathione S-transferase, maltose-binding protein, or thioredoxin, or released in solution upon in situ cleavage of these proteins. Herein, we report an intein-based strategy that allows, for the first time, the rapid and efficient production of native tag-free Httex1 with polyQ repeats ranging from 7Q to 49Q. Aggregation studies on these proteins enabled us to identify interesting polyQ-length-dependent effects on Httex1 oligomer and fibril formation that were previously not observed using Httex1 fusion proteins or Httex1 proteins produced by in situ cleavage of fusion proteins. Our studies revealed the inability of Httex1–7Q/15Q to undergo amyloid fibril formation and an inverse correlation between fibril length and polyQ repeat length, suggesting possible polyQ length-dependent differences in the structural properties of the Httex1 aggregates. Altogether, our findings underscore the importance of working with tag-free Httex1 proteins and indicate that model systems based on non-native Httex1 sequences may not accurately reproduce the effect of polyQ repeat length and solution conditions on Httex1 aggregation kinetics and structural properties.
Keywords: circular dichroism (CD), electron microscopy (EM), fibril, Huntington disease, protein aggregation, protein purification, protein structure
Introduction
Huntington disease (HD)2 is a fatal neurodegenerative disorder caused by a CAG expansion within the first exon (Exon 1) of the huntingtin gene, IT15 (1). The CAG repeat length ranges between 6 and 35 in healthy subjects, whereas patients with HD exhibit lengths of 36 or greater resulting in the synthesis of a mutant Huntingtin protein (Htt) with an expanded polyglutamine (polyQ) domain (2). The length of the polyQ tract directly correlates with disease severity and is inversely correlated with disease age of onset (3). HD patients suffer from motor impairments, cognitive decline, and depression due to neurodegeneration in the striatum and cortex (4–8).
The accumulation of N-terminal fragments of mutant Htt in the nucleus and cytoplasm of striatal neurons led to the hypothesis that these fragments play causative roles in the neurodegeneration and pathology observed in HD (9–13). This hypothesis is supported by the observation that the overexpression of the expanded Exon 1 in R6/2 transgenic mice recapitulates several key symptoms of HD (14). Subsequent studies demonstrated that aberrant splicing in HttQ150 knock-in mice gave rise to a short mRNA, which translates into the Huntingtin Exon 1 protein (Httex1), thus linking the genetic cause of HD to the generation of a highly toxic N-terminal Htt fragment (15). Together, these findings highlight the importance of this region in HD pathogenesis and underscore the critical importance of investigating the structure, aggregation, and functional properties of Httex1 and its role in health and disease.
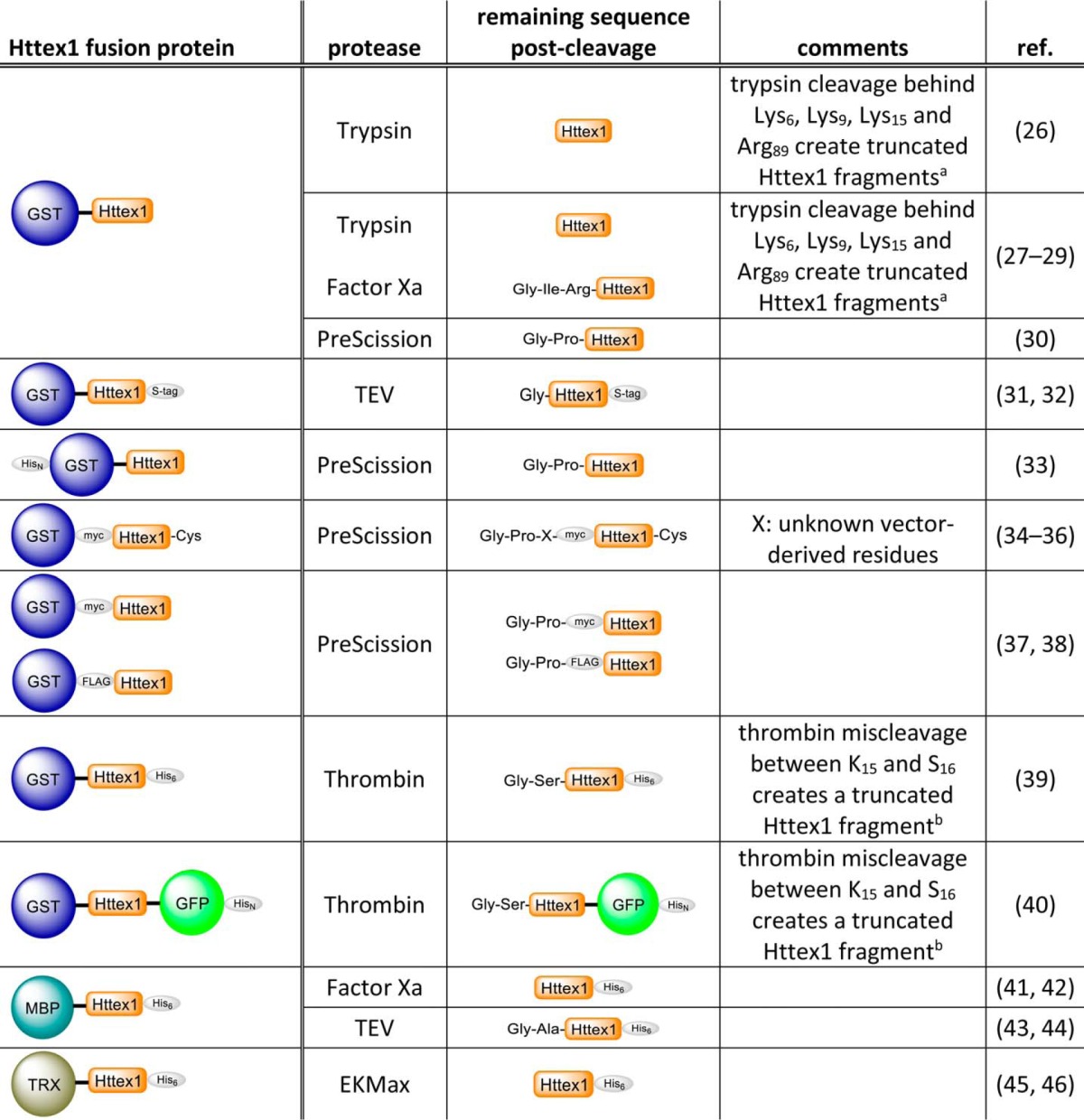
The amino acid sequence of Httex1 is dominated by the polyQ tract that is flanked by the N-terminal domain consisting of the first 17 amino acids (Nt17 domain) and the proline-rich domain (16). With the exception of the Nt17 domain, the remainder of the protein is rich in glutamine and proline residues. The homotypic and uncharged nature of Httex1 renders the protein unstable and prone to aggregation, especially when containing an expanded polyQ domain of >36Q. These properties present significant challenges for producing Httex1 by standard recombinant bacterial and mammalian expression systems. Consequently, the majority of in vitro studies investigating the structure and aggregation properties of Httex1 have been conducted using synthetic polyQ or Httex1-like peptides containing additional solubilizing amino acids such as lysine residues (17–22) or relied upon artificial fusion constructs whereby the polyQ domain (23–25) or Httex1 itself (see Table 1, Refs. 26–46) are fused to large solubilizing protein tags, such as glutathione S-transferase (GST), maltose-binding protein (MBP), or thioredoxin. Typically, the fusion protein is cleaved in situ by the addition of a protease to release and initiate the aggregation of Httex1. Enzyme-mediated cleavage of Httex1 fusion proteins often results in the incorporation of additional amino acids at the N or C terminus of the protein. This could alter the biophysical and biochemical properties of Httex1 because of the important role of the Nt17 domain and proline-rich domain in regulating the conformational and aggregation properties of the protein (18, 20, 22, 32, 47, 49). Moreover, it has been shown that commonly used enzymes such as trypsin and thrombin may lead to cleavages within the Nt17 domain and result in the generation of undesired Httex1 fragments (19, 28, 50). The net effect of incomplete and/or unspecific enzymatic cleavage of Httex1 fusion proteins is the generation of heterogeneous protein mixtures that precludes accurate interpretation and comparison of aggregation and structural data across different laboratories. Therefore, the use of fusion constructs as precursors for the generation of Httex1 requires enzymes that are highly specific and efficient to ensure complete cleavage and to minimize changes to the protein sequence due to the nature of the enzyme used, engineered cleavage site, or the presence of undesired cleavage products.
TABLE 1.
A list of the most common Httex1 fusion constructs and proteases used for in vitro aggregation studies
The cited fusion constructs were chosen from studies that focused on the characterization of the aggregation propensity and aggregate structure of Httex 1 in vitro. HisN, polyhistidine tag; S-tag, S-peptide epitope tag.
To overcome these limitations, our group and others explored different strategies for the generation of tag-free native Httex1 proteins. Singer et al. (51) used total chemical synthesis to produce Httex1, whereas our group employed a semisynthetic strategy to produce unmodified and site-specifically phosphorylated Httex1 (50). However, both approaches demand special technical expertise and equipment that are not commonly accessible in many research groups. Herein, we present the first recombinant approach that allows the efficient production and purification of authentic tag-free Httex1 in milligram quantities from Escherichia coli. This intein-based strategy eliminates the need for proteolytic enzymes and has been successfully used to produce Httex1 proteins with polyQ repeats ranging from 7Q to at least 49Q. Biophysical characterization of these proteins revealed that they exhibit the expected aggregation properties of tag-free Httex1 proteins produced by chemical or semisynthetic strategies (50, 52). The use of tag-free Httex1 proteins enabled us to decipher subtle effects of concentration and polyQ-repeat length on Httex1 oligomerization and fibril formation, which were not observed previously using Httex1 fusion constructs or Httex1-based peptide model systems containing non-native sequences. This includes an inverse correlation between polyQ repeat length and fibril length. Finally, unlike previous reports using polyQ peptides, we did not observe amyloid fibril formation by Httex1 proteins with polyQ repeats of 7 or 15Q, even at high protein concentrations. These findings underscore the importance of using tag-free native Httex1 proteins to investigate the sequence and molecular determinants of Httex1 aggregation and to elucidate the structural basis of Httex1 toxicity.
Experimental Procedures
Materials
pTWIN1 vector (N6951S) and ER2566 E. coli (E6901S) competent cells were purchased from New England BioLabs. His6-Ssp-Httex1-QN cDNA was synthesized by GeneArt®. Isopropyl-β-d-thiogalactopyranoside was ordered from Applichem (A1008,0025). Phenylmethanesulfonyl fluoride (PMSF) was purchased from Axonlab (A0999.0005). CLAP protease inhibitor (1000×) was made of 2.5 mg/ml leupeptin, chymostatin, antipain, and pepstatin A from Applichem (A2183, A2144, A2129, A2205) in DMSO. Amicon Ultra-15 centrifugal filters with a molecular weight cutoff of 3 (UFC900324) and the primary mouse anti-Huntingtin monoclonal antibody (MAB5492) were purchased from Millipore. Secondary goat anti-mouse labeled with Alexa680 was purchased from Invitrogen (A-21057). The PageRuler prestained protein ladder (26617), SeeBlue Plus2 pre-stained protein standard (LC5925), and the SnakeSkin dialysis tubing with a molecular weight cutoff of 3.5 kDa (68035) were purchased from Thermo Scientific, and Dulbecco's buffer substance (PBS) was purchased from Serva (47302.03). Microcon centrifugal filters with a molecular weight cutoff of 100 were obtained from Millipore (MRCF0R100), and PES syringe filter membranes with a pore size of 0.45 μm were from Techno Plastic Products (TPP-99745). Formvar carbon film on 200-mesh copper grids (FCF200-Cu) and uranyl formate (16984-59-1) from Electron Microscopy Sciences were used for sample preparation for negative-stain transmission electron microscopy (TEM).
Cloning and Expression
His6-Ssp-Httex1-QN cDNA was subcloned into the pTWIN1 vector using NdeI/PstI restriction sites by GeneArt®. Chemo-competent E. coli BER2566 cells (eBiolabs) were transformed with resulting vectors pTWIN1-His6-Ssp-Httex1-QN. Isolated single colonies were inoculated in 500 ml of lysogeny broth (100 μg/ml ampicillin) at 37 °C overnight with 180 rpm shaking. For expression of His6-Ssp-Httex1-QN, 8 liters of lysogeny broth (100 μg/ml ampicillin) were mixed with the overnight culture to obtain an A600 nm of 0.05. Cells were grown at 37 °C until an A600 nm ∼0.1 was reached; the temperature of the incubator was then set to 14 °C. At an A600 nm between 0.3 and 0.4 (not higher than 0.5), expression was induced with 0.4 mm isopropyl-β-d-thiogalactopyranoside overnight. Cells were harvested by centrifugation (4 °C, 1935 × g, 15 min) and snap-frozen in liquid nitrogen and stored at −20 °C or lysed immediately for purification.
Purification of Huntingtin Exon 1
After expression, cell pellets were resuspended in 50 ml of buffer A (20 mm HEPES, 0.5 m NaCl, pH 8.5) containing 0.3 mm PMSF and 1× chymostatin/leupeptin/antipain/pepstatin. Cells were lysed on ice by sonication (4 min, pulse on 59 s, pulse off 30 s, 70% amplitude) using a vibra cell VCX130 from Sonics. Cell debris was separated from the crude cell extract by centrifugation (2 × 30 min, 4 °C, 27,216 × g). The cell extract was filtered through 0.45-μm syringe filter membranes and then applied to a 5-ml Histrap column (GE Healthcare, 17-5248-02) at a flow rate of 1 ml/min. Nonspecifically bound proteins were removed by washing the column with 10 column volumes of buffer A at a flow rate of 2 ml/min. Remaining impurities were removed with 3 column volumes of 5% buffer B (20 mm HEPES, 0.5 m NaCl, 0.5 m imidazole, pH 8.5), whereas His6-Ssp-Httex1-QN was eluted using a gradient from 5 to 30% buffer B in 5 column volumes. Elution fractions were analyzed by SDS-PAGE and analytical C8 reversed-phase ultra-high performance liquid chromatography (RP-UHPLC). Fractions containing the fusion protein were pooled, and splicing was induced by adjusting the pH to 7.0 and incubating the protein at room temperature. Splicing was monitored by analytical C8 RP-UHPLC, SDS-PAGE, and Western blot (WB). Httex1 with expanded polyQ lengths were purified after a maximum of 4–6 h of splicing at room temperature, whereas unexpanded Httex1 proteins were purified after 12–16 h. Protein solutions were filtered through 0.45-μm syringe filter membranes and injected into a preparative C4 reversed-phase high performance liquid chromatography (RP-HPLC) column (00G-4168-P0-AX, Jupiter C4, 10 μm, 300 Å, 21.2-mm inner diameter × 250-mm length) pre-equilibrated with 95% buffer C (degassed water, 0.1% trifluoroacetic acid (TFA)) and 5% buffer D (degassed acetonitrile, 0.1% v/v TFA). Spliced Httex1-QN eluted during a gradient of 30–40% buffer D in 40 min at 15 ml/min. Collected fractions were analyzed by liquid chromatography mass spectrometry (LC/MS) using a Thermo Scientific LTQ ion trap mass spectrometer and pooled accordingly for lyophilization. Purity of lyophilized protein was analyzed by LC/MS using a C3 poroshell 300SB 1.0 × 75-mm 5-μm column from Agilent (5–95% acetonitrile in 5 min, flow rate of 0.3 ml/min, injection volume of 10 μl), analytical RP-UHPLC, SDS-PAGE, and WB. All obtained LC/MS spectra were deconvoluted with MagTran software version 1.03b from Amgen.
Native Purification of Huntingtin Exon 1
The elution fractions of the nickel affinity chromatography containing His6-Ssp-Httex1–23Q were pooled, and the pH was set to 7.0 to initiate splicing. The protein was dialyzed against 3 liters of buffer E (20 mm Tris, 0.5 m NaCl, pH 7.0) using SnakeSkin dialysis tubing with a molecular weight cutoff of 3.5 (Thermo Fisher, 68035) for 12–16 h at room temperature to remove the imidazole. After dialysis/splicing the Ssp DnaB intein was removed from the desired splicing product Httex1–23Q by nickel affinity chromatography. Httex1 was collected in the flow-through, concentrated using centrifugal filter units with a molecular weight cutoff of 3 (Millipore, UFC900324), and injected into a Superose 6 10/300 GL column (GE Healthcare, 17-5172-01) equilibrated with buffer F (20 mm Tris, 0.2 m NaCl, pH 7.4) for final purification.
In Vitro Aggregation of Huntingtin Exon 1
Lyophilized Httex1-QN was disaggregated by the addition of TFA following a modified protocol from O'Nuallain et al. (53). After allowing for evaporation for 15 min the remaining acid was removed under a stream of dry nitrogen. Finally, Httex1-QN proteins were dissolved in 10 mm PBS to obtain a final concentration of 3.5, 7, 15, 30, 60, or 120 μm. The pH was adjusted to 7.2–7.4 using 1 m NaOH. Proteins were kept on ice or at 4 °C during further preparation to avoid aggregation. Protein solutions were filtered through 100-kDa centrifugal filter units into 1.5-ml Eppendorf tubes to remove any remaining preformed aggregates. To assess the aggregation of Httex1–43Q in the presence of proteins that are commonly used as fusion tags, equimolar amounts of MBP or GST (PRO-616 and ENZ-393 from PROSPEC) were added to the filtered protein solution, respectively. Before initiating aggregation, aliquots were taken for time point (0 h) and for amino acid analysis to confirm the protein concentration determined by a RP-UHPLC standard curve. Aggregation was induced by transferring the samples to an incubator at 37 °C (without agitation). For each time point the amount of soluble protein and the change in secondary structure were assessed. The secondary structure was determined by circular dichroism (CD) spectroscopy using 100 μl of each protein solution. To analyze the soluble fraction, aliquots of each sample were spun down at 20,817 × g at 4 °C for 20 min, after which 4 μl were injected into a C8 RP-UHPLC column. The formation of oligomers and fibrils during the aggregation process was followed by negative-stain TEM.
SDS-PAGE and WB
Samples for SDS-PAGE were mixed with 4× Laemmli and loaded onto 15% polyacrylamide gels. Electrophoresis was performed at 180 V for 1 h, and semidry transfer was performed using 200 mA at 25 V for 1 h on a nitrocellulose membrane using systems from Bio-Rad. Gels were stained with a Coomassie R-450 solution and destained in water. WB nitrocellulose membranes were incubated in Odyssey blocking solution from Licor (20–30 min at room temperature) and later blotted with primary mouse anti-Htt MAB5492 (1 h at room temperature or 12–16 h at 4 °C) and secondary Alexa680-conjugated goat anti-mouse (45 min at room temperature) for detection by an Odyssey Infrared Imager system from Licor.
Analytical RP-UHPLC
Soluble fractions were injected into a C8 Waters Acquity UPLC BEH300 1.7-μm 300 Å 2.1 × 150-mm column (10–90% acetonitrile in 2.75 min, preheated to 40 °C, flow rate of 0.6 ml/min) connected to an Acquity H-class UPLC system from Waters. Proteins were eluted at 0.6 ml/min with a gradient from 10% to 90% acetonitrile (0.1% v/v TFA) over 2.75 min. Acquired chromatograms at 214 nm were used to determine purity and to identify splicing products. Moreover, the amount of soluble protein was calculated from the peak area using the Empower Software from Waters.
CD Spectroscopy
Samples were analyzed by a J-815 CD spectrometer from Jasco using a 1-mm quartz cuvette. The CD spectra were acquired from 195–250-nm at 25 °C, and data points were acquired continuously every 0.2 nm at a speed of 50 nm/min with a digital integration time of 2 s and a bandwidth of 1.0 nm. 3–5 spectra of each sample were obtained and averaged. A sample containing buffer-only was used for background subtraction. The obtained spectra were further processed by smoothing using a binomial filter with a convolution width of 99 data points. Note that samples with a protein concentration higher than 15 μm were diluted before the CD measurement.
TEM
Samples were deposited on Formvar/carbon-coated 200-mesh copper grids (Electron Microscopy Sciences) for 1 min at room temperature, then the grids were blotted with filter paper, washed twice with ultrapure water, and stained with a 0.75% w/v uranyl formate solution in water (Electron Microscopy Sciences) for 1 × 15 s and 1 × 30 s. The blotted off grids were air-dried and imaged using a Tecnai Spirit BioTWIN microscope at 80 kV (LaB6 gun, 0.34-nm line resolution) equipped with a 4k × 4k Eagle CCD camera with high sensitivity scintillator from FEI. Fibril lengths were quantified using ImageJ software (54).
Amino Acid Analysis
Before aggregation of Httex1-QN, 3–5 μg of each protein were dried in an evacuated centrifuge and sent for amino acid analysis to the Functional Genomic Center Zurich (FGCZ) at the Eidgenössische Technische Hochschule Zürich (ETH Zurich) to determine the exact sample concentration.
Results
An Intein-based Strategy for the Expression and Purification of Tag-free Huntingtin Exon 1
To achieve the production of authentic tag-free Httex1, we fused the Ssp DnaB intein containing a His6 tag for nickel affinity purification to the N terminus of Httex1 (Fig. 1, A and B). The purpose of the Ssp DnaB intein was 2-fold: 1) to improve the stability and solubility of Httex1 during expression in E. coli and 2) to allow for pH controlled splicing of the fusion protein leading to the generation of the native Httex1 sequence (tag-free Httex1). The His6-Ssp-Httex1-QN fusion proteins were expressed at low temperature to prevent protein aggregation and premature protein splicing (Fig. 1C). The fusion proteins were isolated from cell lysates by nickel affinity purification yielding ∼7 mg of protein per liter of bacterial culture, which is 1.4–2.7-fold higher compared with the widely used purification strategy developed by Scherzinger et al. (27). The Ssp DnaB-mediated splicing of His6-Ssp-Httex1-QN was induced at ambient temperature and neutral pH, thus allowing for the production of the aggregation-prone Httex1 under mild conditions. In contrast to most enzymatic cleavages, the splicing process is autocatalytic and traceless, generating the native Httex1 protein sequence without any additional amino acids within several hours. The splicing reaction of His6-Ssp-Httex1-QN was monitored by WB and RP-UHPLC (Fig. 1, D–E). Generally, the incubation time for His6-Ssp-Httex1–43Q was limited to 6 h to prevent aggregation of the splicing product, Httex1–43Q. Splicing products were separated by preparative C4 RP-HPLC, yielding Httex1 with >95% purity based on analysis of purified fractions by RP-UHPLC (Fig. 1F). The purity and integrity of the protein was further assessed by SDS-PAGE, WB, and LC/MS (Fig. 1, G and H). Due to its high glutamine and proline content, Httex1 is predominantly uncharged and exhibits a low SDS binding capacity and, thus, its apparent size on denaturing gels is larger than its calculated molecular weight. The purified proteins were snap-frozen in liquid nitrogen and lyophilized to prevent aggregation and allow for long term storage. Using these expression and purification protocols, we achieved yields of 1.0–1.5 mg of pure Httex1 with polyQ-lengths ranging from 7Q to 49Q per liter of bacterial culture (Fig. 1I).
FIGURE 1.

Steps involved in the intein-based expression and purification strategy for the generation of tag-free Httex1-QN proteins. A, scheme of the intein-based expression and purification strategy. B, the amino acid sequence of the His6-Ssp-Httex1-QN fusion constructs (His6-Ssp in green, Httex1 in orange). The Httex1 proteins are devoid of the first methionine, as it was reported to be cleaved off in vivo (48). C, the expression of His6-Ssp-Httex1–23Q/43Q (arrow) was analyzed by SDS-PAGE. D, the generation of Httex1–23Q/43Q (star) by splicing of the His6-Ssp-Httex1–23Q/43Q fusion proteins (arrow) monitored by WB using monoclonal anti-Htt MAB5492. E, the splicing of His6-Ssp-Httex1–43Q (1) into His6-Ssp (2), and Httex1–23Q/43Q (3) was assessed by analytical RP-UHPLC. AU, absorbance units. F–H, the purity and integrity of Httex1–23Q/43Q was determined using RP-UHPLC (F), SDS-PAGE and WB (G), and LC/MS (H). Expected molecular masses for Httex1–23Q/43Q are 9944 Da and 12506 Da. I, purity analysis of Httex1–7Q/15Q/29Q/37Q/49Q by SDS-PAGE and RP-UHPLC.
Biophysical Characterization of Tag-free Wild Type and Mutant Huntingtin Exon 1
With the purified recombinant tag-free Httex1 proteins in hand, we sought to characterize the structural and polyQ-dependent aggregation properties of the wild type (23Q) and mutant (43Q) proteins. Before assessing the aggregation properties, lyophilized Httex1–23Q/43Q proteins were disaggregated using TFA, resolubilized in PBS buffer, and filtered to remove any remaining preformed aggregates. To ensure that the HPLC purification, lyophilization, and disaggregation procedure did not affect the structure of Httex1, we performed a control experiment where we replaced the RP-HPLC purification step by size-exclusion chromatography to obtain native Httex1–23Q. The CD spectra of size-exclusion chromatography-purified Httex1–23Q and HPLC-purified and disaggregated Httex1–23Q protein were identical and revealed a predominantly disordered structure for both proteins (Fig. 2).
FIGURE 2.
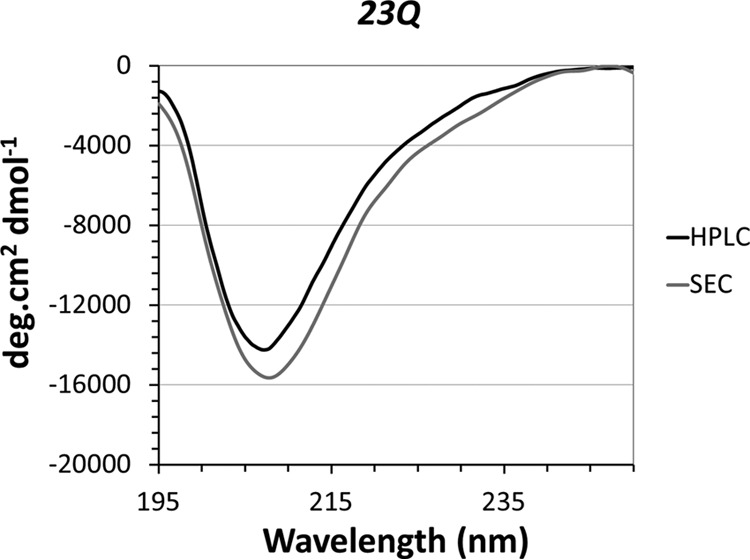
The secondary structure of Httex1–23Q is not altered by the RP-HPLC purification or disaggregation procedure. The secondary structure of Httex1–23Q purified by size-exclusion chromatography (SEC) was assessed by CD (gray line) and compared with the structure of HPLC-purified and disaggregated Httex1–23Q (black line).
The aggregation of Httex1–23Q and Httex1–43Q was monitored in PBS at physiological pH and protein concentrations of 7 or 3.5 μm, respectively. The loss of soluble protein was determined using RP-UHPLC, the secondary structure was analyzed by CD, and the morphological characteristics of the aggregates formed by Httex1–23Q/43Q were assessed by negative-stain TEM. For Httex1–23Q, a loss of 20–30% of soluble protein was observed after 168 h at 37 °C by RP-UHPLC (Fig. 3A). Despite the moderate aggregation of Httex1–23Q, the protein exhibited a broad minimum at 205 nm in the CD spectra throughout the experiment (Fig. 3B), consistent with a predominantly disordered secondary structure. Notably, the minimum of the CD signal for Httex1 was slightly red-shifted compared with what has been reported for polyQ peptides whose minimum is usually observed at ∼200 nm (18, 55). This shift is probably due to the influence of the proline-rich domain (18) and the Nt17 domain, both of which have been reported to adopt partial or transient helical structures (56, 57). TEM imaging revealed the presence of large amounts of oligomeric species ranging from 15 to 25 nm in diameter throughout the aggregation process (Fig. 3C). These oligomeric structures are of similar size as those observed for myc-Httex1–20Q by Legleiter et al. (36) using atomic force microscopy. After 72 h at 37 °C, Httex1–23Q formed proto-fibrillar structures with an average length of 45–55 nm next to the oligomers (Fig. 3C). These structures seemed to elongate and grow into short immature fibrils of 100–200 nm in length after 168 h (Fig. 3C). Nonetheless, the majority of recombinant Httex1–23Q (60%) remained soluble for as long as 2 weeks at 37 °C and existed predominantly as non-amyloidogenic monomers or oligomers as discerned by the absence of a β-sheet signal in the CD spectra (Fig. 3B).
FIGURE 3.

The aggregation properties of recombinant wild type (23Q) and mutant (43Q) tag-free Httex1. A, the kinetics and extent of Httex1 aggregation were monitored by measuring the amounts of soluble Httex1–23Q/43Q over time using analytical RP-UHPLC. All data (23Q, n = 3–4; 43Q, n = 2–3) were normalized to t0h and are represented as the mean ± S.D. The resulting data points were fitted single exponentially. B, the secondary structure of Httex1–23Q/43Q was analyzed by CD during incubation at 37 °C. C, aggregates formed by Httex1–23Q/43Q were imaged using negative-stain TEM.
Next, we characterized the aggregation of the mutant protein, Httex1–43Q. In contrast to Httex1–23Q, >50% of Httex1–43Q aggregated within 12 h at 37 °C (Fig. 3A), and complete aggregation was observed after 36 h; <5% of soluble protein could be detected by RP-UHPLC (Fig. 3A). Consistent with the rapid depletion of soluble protein, recombinant Httex1–43Q readily formed mature amyloid-like fibrils after 24 h at 37 °C with lengths ranging from 200 to 300 nm (Fig. 3C). A shift of the minimum from 205 nm to 215 nm in the CD spectra of Httex1–43Q between 24 and 36 h confirmed the structural transition from a predominantly disordered to a β-sheet-rich structure (Fig. 3B). The structural transition toward a β-sheet-rich conformation is in agreement with previous in vitro aggregation studies using synthetic Httex1, thioredoxin/GST-polyQ, and GST/MBP-Httex1 fusion proteins with expanded polyQ tracts, which readily formed amyloid-like fibrils with a high β-sheet content as determined by CD, nuclear magnetic resonance (NMR), and Fourier transform infrared spectroscopy (23, 24, 33, 41, 52).
Wild Type Httex1 Forms Mature Amyloid Fibrils at High Protein Concentrations
Httex1 proteins and model peptides aggregate and form fibrils in a concentration-dependent manner, with the critical concentration for aggregation decreasing with increasing polyQ repeat length (21, 22, 28). To determine whether our recombinant and tag-free Httex1–23Q can adopt a β-sheet-rich structure and form mature amyloid-like fibrils at higher protein concentrations, we assessed the aggregation of Httex1–23Q at 15, 30, 60, and 120 μm. At the highest concentrations, 120 and 60 μm, complete Httex1–23Q aggregation occurred within 120 and 336 h, respectively, whereas at 15 and 30 μm the protein aggregated much slower with >50% of the protein remaining in solution after 300 h (Fig. 4A). Fig. 4B demonstrates that a concentration of 15 μm was sufficient to induce a complete transition from disordered to β-sheet-rich conformations after several weeks of incubation at 37 °C as discerned by the single minima at 215 nm, which was similar for all 4 protein concentrations. Over this concentration range, Httex1–23Q formed highly ordered fibrils with variable lengths ranging from 200 nm to 1 μm (Fig. 4C).
FIGURE 4.

Concentration dependence of Httex1–23Q aggregation. A, the aggregation propensity of Httex1–23Q at 15, 30, 60, and 120 μm was monitored by measuring the soluble protein fraction over time using analytical RP-UHPLC. All data were normalized to t0h and are represented as the mean ± S.D. (15/30/120 μm, n = 3; 60 μm, n = 3–5). B, the CD spectra of Httex1–23Q at the final aggregation time points. C, TEM images of fibrils formed by Httex1–23Q at the end point of aggregation.
PolyQ Repeat Length Inversely Correlates with Fibril Length
Given the observation that higher concentrations drive the formation of mature Httex1 fibrils, we then sought to determine if Httex1 with polyQ repeats of <23Q (Httex1–15Q and -7Q) have the intrinsic propensity to form mature Httex1 fibrils. The aggregation of Httex1–15Q/7Q was assessed at 60 μm to allow for a reasonable time scale of aggregation. At a concentration of 60 μm, Httex1–15Q/7Q exhibited very slow aggregation propensities compared with Httex1–23Q. 20–30% of Httex1–7Q and 70% of Httex1–15Q aggregated only after 1482 h of incubation at 37 °C, whereas Httex1–23Q exhibited nearly complete aggregation after 336 h under the same conditions (Fig. 5A). The CD spectra of Httex1–7Q/15Q did not show a conformational transition to a β-sheet-rich structure after 1482 h, which was unexpected for Httex1–15Q, as 70% of soluble protein was depleted (Fig. 5B). TEM imaging revealed that Httex1–15Q formed very large oligomers or amorphous aggregates, whereas Httex1–7Q formed small oligomeric structures (Fig. 5C). To compare the fibrillization propensity of wild type Httex1 (23Q) with Httex1 containing intermediate and expanded polyQ domains, we assessed the aggregation of Httex1–29Q, -37Q, and -43Q at 15 μm, due to the increased aggregation propensity of expanded Httex1 proteins. In contrast to Httex1–23Q, the Httex1 proteins with polyQ lengths of 29–43Q readily aggregated at a concentration of 15 μm. Complete aggregation of Httex1–37Q/29Q was reached after 124.5–242 h (Fig. 5D). This corresponds to a 4–9 times faster aggregation compared with Httex1–23Q at 15 μm, which was fully aggregated only after 1176 h at 37 °C. Notably, the mutant Httex1–43Q protein aggregated ∼200 times faster compared with Httex1–23Q at 15 μm (Fig. 5D). Interestingly, although all three Httex1–29Q/37Q/43Q proteins underwent a conformational shift to β-sheet and exhibited a strong fibrillization as discerned by CD and TEM only the CD spectra of Httex1–43Q showed a pronounced maximum at 200 nm, indicating that the aggregates formed by this mutant have a higher β-sheet content (Fig. 5, E and F). Moreover, the fibrils formed by Httex1–29Q/37Q/43Q were shorter compared with those formed by Httex1–23Q at 15 μm (Fig. 5F). To confirm this observation we quantified the length of ∼400 fibrils for each protein (i.e. 23–43Q) (Fig. 5G). The mean fibril length for Httex1–23Q was 522 nm, whereas a fibril length of 284 nm for Httex1–29Q, 268 nm for Httex1–37Q, and 183 nm for Httex1–43Q was observed. The inverse correlation between the polyQ-length and fibril length suggests structural differences between Httex1 proteins with different polyQ repeat lengths. Additional studies are required to assess these structural differences at the single fibril level and their implications for Httex1 aggregation, toxicity, and propagation.
FIGURE 5.

PolyQ-length dependence of Httex1 aggregation. A, the aggregation propensity of Httex1–23Q (black, n = 3–5), Httex1–15Q (blue, n = 3), and Httex1–7Q (green, n = 3) at 60 μm. The amount of soluble protein was assessed by analytical RP-UHPLC, and all data were normalized to t0h and are represented as the mean ± S.D. B, the CD spectra of Httex1–23Q (black), Httex1–15Q (blue), and Httex1–7Q (green) at the initial and final aggregation time points. C, TEM images of the aggregates formed by Httex1–23Q/15Q/7Q at the final aggregation time points. The white arrow indicates one of the oligomeric structures formed by Httex1–7Q. D, the aggregation propensity of Httex1–23Q (black, n = 3), Httex1–29Q (purple, n = 5), Httex1–37Q (green, n = 5), and Httex1–43Q (blue, n = 5) at 15 μm. All data were normalized to t0h and are represented as the mean ± S.D. E, the CD spectra of Httex1–23Q (black), Httex1–29Q (purple), Httex1–37Q (green), and Httex1–43Q (blue) at the initial and final aggregation time points. F, TEM images of fibrils formed by Httex1–23Q/29Q/37Q/43Q at 15 μm at the end point of aggregation. G, quantification of fibril lengths for Httex1–23Q (black, n = 419), Httex1–29Q (purple, n = 437), Httex1–37Q (green, n = 483), and Httex1–43Q (blue, n = 380) at the end point of aggregation. Solid lines represent Gaussian fits of the fibril lengths.
The Presence of MBP or GST Does Not Alter the Aggregation Properties of Httex1–43Q
The in vitro aggregation studies on Httex1 fusion proteins have prompted speculations that the fusion tag might affect the aggregation properties of Httex1 after the enzymatic cleavage in situ. To determine if the presence of the commonly used fusion tags GST and MBP influences the aggregation properties of Httex1 after the enzymatic cleavage in situ, we compared the aggregation propensity and the structural properties of Httex1–43Q aggregates in the absence or presence of equimolar amounts of purified GST or MBP. Under these controlled conditions, which assume quantitative cleavage of the fusion protein, we could not detect a significant effect of MBP or GST on the aggregation profile of Httex1–43Q or its ability to form fibrils (Fig. 6, A and B). These results suggest that GST and MBP do not significantly alter the aggregation properties of Httex1 in a homogenous aggregation mixture.
FIGURE 6.
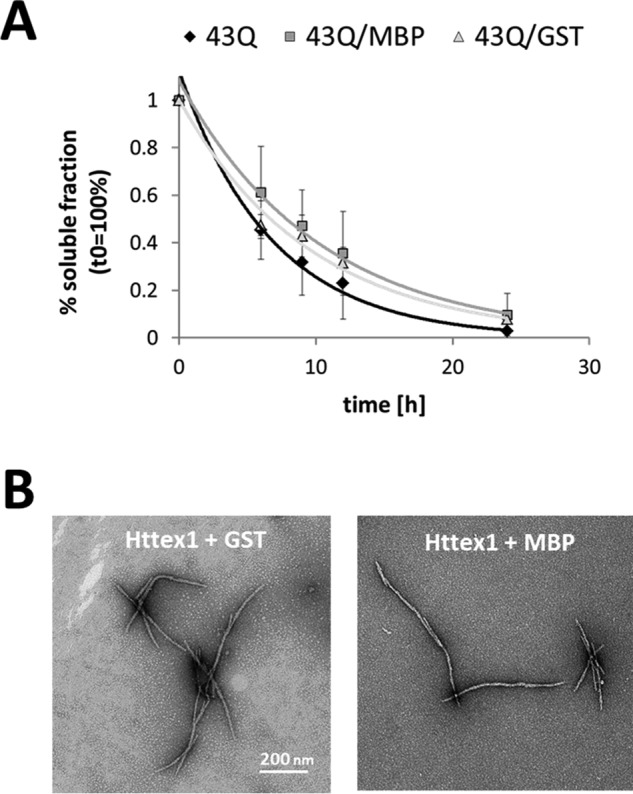
Aggregation of mutant Httex1 in the presence of MBP and GST. A, the soluble fraction of Httex1–43Q incubated at 37 °C (black) and co-incubated with MBP (dark gray) or GST (light gray) was determined by analytical RP-UHPLC. All data (n = 2) were normalized to t0h and are represented as the mean ± S.D. The resulting data points were fitted single exponentially. B, TEM images of Httex1–43Q aggregates formed after co-incubation with MBP or GST for 24h at 37 °C.
Discussion
To date, the majority of in vitro biophysical and biochemical aggregation studies on Httex1 proteins have relied on the use of artificial fusion constructs or incorporation of additional charged amino acids to stabilize and facilitate the purification and handling of Httex1 proteins, especially those containing expanded (>36Q) polyQ repeats (Table 1). There are several disadvantages for using this strategy. First, the presence of highly structured proteins, such as thioredoxin, GST, or MBP, makes it difficult to monitor and quantify subtle changes in secondary structure that occur during the early events of Httex1 oligomerization and fibril formation or in response to sequence modifications (e.g. post-translational modifications) (23, 41, 58). Second, fusing these proteins to mutant Httex1 proteins has been shown to alter their aggregation properties and favors the formation of round particles or amorphous aggregates (27, 39, 41, 59). Third, the use of enzymatic cleavage to remove these proteins or other solubilizing motifs often leaves behind additional amino acids in the protein sequence (Table 1) and/or occasionally results in the generation of undesired cleavage products (19, 28, 50).
The sample heterogeneity, i.e. the presence of the enzyme, uncleaved fusion protein, or miscleavage products, introduced by the use of different fusion proteins and enzymes could account for the differences in the aggregation properties of tag-free Httex1 and Httex1 proteins generated by in situ enzymatic cleavage. Indeed, comparison of the Httex1 aggregation studies reported here and in the literature revealed that the tag-free homogeneous Httex1 proteins exhibit faster aggregation kinetics and a higher propensity to form amyloid-like fibrils compared with several Httex1 fusion proteins or Httex1 proteins produced by in situ cleavage of fusion proteins (28, 39, 41). Interestingly, Httex1 proteins generated by enzymatic cleavage often form heterogeneous fibrils that exhibit the tendency to associate into bundles (26–28, 39, 40, 45), unlike the tag-free Httex1 proteins produced by the intein strategy herein (Figs. 3–6), semisynthesis, or chemical synthesis, which form very uniform and smooth amyloid-like fibrils (50, 52). Moreover, several studies based on in situ cleavage of Httex1 fusion proteins have reported that Httex1 proteins with non-pathogenic polyQ repeats do not form fibrils (26–28, 37, 38), whereas studies using synthetic (52), semisynthetic (50), or recombinant tag-free Httex1 proteins have shown that Httex1 with 23Q at concentrations ranging from 15 to 120 μm can form fibrils in vitro (Fig. 4). These findings suggest that delays in enzymatic cleavage, sequence modifications, and/or sample heterogeneity influence greatly the aggregation kinetics and morphology of Httex1 fibrils. Consistent with these observations, recent studies showed that removal of the cleaved fusion proteins before initiating Httex1 aggregation resulted in an increased aggregation propensity of Httex1 (44). This further underscores the critical importance of using homogeneous preparations of native Httex1 to ensure accurate assessment of changes in the aggregation kinetics and structural properties of Httex1.
To test whether the presence of the commonly used fusion tags (GST and MBP) alters the aggregation properties of Httex1, we compared the aggregation propensity and aggregate structure of Httex1–43Q in the absence and presence of MBP or GST. Fig. 6 shows that co-incubation of tag-free Httex1 with purified GST or MBP did not affect the aggregation properties of Httex1. This finding supports our hypothesis that the sample heterogeneity introduced by the incomplete enzymatic cleavage and/or the presence of enzymes or miscleavage products could be responsible for the observed differences in the aggregation properties of tag-free Httex1 and Httex1 generated by enzymatic cleavage.
To enable the generation of homogeneous preparations of native Httex1, we sought to develop expression systems and cleavage protocols that would enable rapid and complete enzymatic cleavage of the fusion Httex1 proteins while preserving the integrity of the native Httex1 sequence. Our intein-based Httex1 expression strategy meets these criteria and addresses the limitation of previous strategies by employing the Ssp DnaB intein technology, which eliminates the need for using enzymes. The use of the N-terminal His6-Ssp-Httex1-QN fusion proteins allows for the generation of the native sequence of Httex1-QN without any additional amino acids by pH- and temperature-sensitive autocatalytic splicing. The Ssp DnaB intein is easily removed after splicing using RP-HPLC or affinity chromatography, thus enabling the rapid production of highly pure and homogeneous Httex1 proteins for aggregation and structural studies (Fig. 1).
The recombinant tag-free Httex1 proteins produced by the intein strategy exhibited similar aggregation and structural properties compared with tag-free Httex1 produced by semisynthesis (50) or microwave-assisted solid-phase peptide synthesis (51, 52). The Httex1 proteins derived from all three strategies have a predominantly disordered structure before aggregation and form virtually identical amyloid-like fibrils upon incubation at 37 °C. Nevertheless, the aggregation propensity of our recombinant Httex1–43Q was different from that of Httex1–42Q produced by solid-phase peptide synthesis by Sahoo et al. (52) despite using similar protein concentrations (synthetic: 5.5 μm; recombinant: 3.5 μm), disaggregation (TFA), and incubation conditions (PBS buffer, 37 °C). In their study, 25–30% of the synthetic Httex1 remained soluble after 70–75 h (52), whereas complete aggregation and disappearance of soluble protein was observed for the recombinant tag-free Httex1–43Q within 24–36 h (Fig. 3). In an attempt to conduct a direct and thorough comparison of the synthetic and recombinant Httex1–43Q proteins, we sought to produce this protein using the synthetic protocol described by Singer and Sahoo et al. (51, 52). Despite extensive efforts to follow the published procedure and the use of different synthetic strategies and peptide synthesizers, we were not able to achieve the chemical synthesis of Httex1. These difficulties highlight the comparative advantage of our recombinant intein-based approach, which can be adopted by most laboratories with access to basic protein expression and purification setups.
The use of recombinant tag-free Httex1 produced via the intein-based strategy has enabled us to make previously undescribed observations relating to the effect of polyQ repeat length on the structural properties and morphology of Httex1 aggregates. Previous studies using model polyQ peptide systems or partial Httex1 sequences demonstrated that non-pathogenic polyQ repeats even as short as 15Q can aggregate in vitro (22, 55, 60, 61), whereas Httex1 proteins with non-pathogenic polyQ repeats produced from fusion proteins by in situ enzymatic cleavage often failed to form fibrils in vitro (26–28, 37, 38). When Httex1 with wild type polyQ repeats (25Q) is purified after the enzymatic cleavage (44) or is produced as a tag-free protein by the intein strategy (23Q), it aggregates and forms amyloid-like fibrils in a concentration-dependent manner (Fig. 4). Interestingly, including an additional purification step after the enzymatic cleavage also resulted in a fast fibrillization of recombinant Httex1 with a polyQ-length of 17Q at 20 μm generated from a MBP-Httex1–17Q-His6 fusion protein (44). However, in our hands no abundant fibril formation was observed for Httex1–7Q/15Q even after weeks of incubation at 60 μm (Fig. 5). Instead, large non-fibrillar and globular structures exhibiting a predominantly disordered conformation were observed (Fig. 5). This discrepancy in the aggregation behavior might be caused by the additional non-native amino acids at the N (Gly-Ala dipeptide) and C termini (His6 tag) of the Httex1–17Q protein used by Monsellier et al. (44) and the fact that other polyQ model systems lack significant domains of Httex1 and/or incorporate non-native sequences (20–22, 55, 60, 62). These findings indicate that differences in the primary sequence can have a strong effect on the aggregation properties of Httex1, which could make it difficult to compare aggregation data from different laboratories. Finally, careful examination of our TEM images revealed for the first time an inverse correlation between fibril length and polyQ repeat number. Interestingly, Httex1 with polyQ repeats above the pathogenic threshold formed fibrils with a mean length of 183 nm, whereas Httex1 with polyQ repeats of 23Q formed fibrils with a broad length distribution with a mean length of 522 nm. Httex1 with polyQ repeat lengths ranging from 29Q to 37Q formed fibrils with mean average lengths of 268–284 nm. These observations suggest that the structural properties of the final fibril structures are strongly dependent on the polyQ repeat length. The formation of shorter fibrils by mutant Httex1 would result in a larger numbers of fibrils, which could significantly enhance the seeding capacity of mutant Httex1 compared to Httex1 with non-pathogenic repeats. Noteworthy, the fibril length distributions of 23Q at 15 and 60 μm are superimposable at the end point of aggregation, indicating that the observed inverse correlation is independent of the protein concentration but could be caused by a structural-based difference in the nucleation capacity of wild type and mutant Httex1.
In conclusion, we have shown that our intein-based expression strategy enables the rapid generation of milligram quantities of highly pure Httex1 with a wide range of polyQ repeats (7–49Q). Our findings further underscore the importance of working with tag-free Httex1 proteins and indicate that model systems based on non-native Httex1 sequences may not accurately reproduce the effect of polyQ repeat length and solution conditions on Httex1 aggregation kinetics and structural properties. The availability of homogeneous preparations of tag-free Httex1 proteins produced using our intein-based strategy should facilitate future studies aimed at elucidating the secondary and tertiary structural changes that occur during the various stages of Httex1 oligomerization and fibrillogenesis using high resolution techniques, including NMR, small-molecule Förster resonance energy transfer and x-ray crystallography. Moreover, the availability of tag-free Httex1 in milligram quantities should advance the development of assays to identify ligands or proteins that modulate the aggregation and toxic properties of Httex1 and to discover of novel imaging agents for detection and monitoring Huntingtin aggregation in vivo.
Author Contributions
S. V. and H. A. L. designed the experiments and wrote the paper. H. A. L. conceived and coordinated the study. S. V. conducted and analyzed the experiments. J. B. W. performed the quantification of fibril lengths. A. A. and Z.-M. W. helped to design the gene constructs and together with J. B. W. provided technical and intellectual guidance. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Stefan Kochanek from the Department of Gene Therapy at the Universitätsklinikum Ulm for scientific discussions. We thank Sean M. Deguire, Bruno Fauvet, Mohammed-Bilal Fares, and Elizabeth Doherty for critical review of the manuscript.
This work was supported primarily by grants from the CHDI foundation (A_7627) and the Swiss National Science Foundation (31003A-146680). The authors declare that they have no conflicts of interest with the contents of this article.
- HD
- Huntington disease
- Exon 1
- first exon
- polyQ
- polyglutamine
- Htt
- Huntingtin protein
- Httex1
- Huntingtin Exon1 protein
- Nt17 domain
- N-terminal domain consisting of the first 17 amino acids of the Htt protein
- MBP
- maltose-binding protein
- WB
- Western blot
- RP-UHPLC
- reversed-phase ultra-high performance liquid chromatography
- TEM
- transmission electron microscopy.
References
- 1. Macdonald M. E., Ambrose C. M., Duyao M. P., Myers R. H., Lin C., Srinidhi L., Barnes G., Taylor S. A., James M., Groat N., Macfarlane H., Jenkins B., Anderson M. A., Wexler N. S., Gusella J. F., et al. (1993) A novel gene containing a trinucleotide that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Rubinsztein D. C., Leggo J., Coles R., Almqvist E., Biancalana V., Cassiman J. J., Chotai K., Connarty M., Crauford D., Curtis A., Curtis D., Davidson M. J., Differ A. M., Dode C., Dodge A., et al. (1996) Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Genet. 59, 16–22 [PMC free article] [PubMed] [Google Scholar]
- 3. Lee J.-M., Ramos E. M., Lee J.-H., Gillis T., Mysore J. S., Hayden M. R., Warby S. C., Morrison P., Nance M., Ross C. A., Margolis R. L., Squitieri F., Orobello S., Di Donato S., Gomez-Tortosa E., et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78, 690–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacDonald M. E., Gines S., Gusella J. F., and Wheeler V. C. (2003) Huntington's Disease. Neuromolecular Med. 4, 7–20 [DOI] [PubMed] [Google Scholar]
- 5. Vonsattel J. P., and DiFiglia M. (1998) Huntington Disease. J Neuropathol. Exp. Neurol. 57, 369–384 [DOI] [PubMed] [Google Scholar]
- 6. Halliday G. M., McRitchie D. A., Macdonald V., Double K. L., Trent R. J., and McCusker E. (1998) Regional specificity of brain atrophy in Huntington's disease. Exp. Neurol. 154, 663–672 [DOI] [PubMed] [Google Scholar]
- 7. Huntington G. (2003) On chorea. George Huntington, M.D. J. Neuropsychiatry Clin. Neurosci. 15, 109–112 [DOI] [PubMed] [Google Scholar]
- 8. Vonsattel J. P., Myers R. H., Stevens T. J., Ferrante R. J., Bird E. D., and Richardson E. P. Jr. (1985) Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44, 559–577 [DOI] [PubMed] [Google Scholar]
- 9. DiFiglia M. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 10. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., and Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 11. Lunkes A., Lindenberg K. S., Ben-Haïem L., Weber C., Devys D., Landwehrmeyer G. B., Mandel J.-L., and Trottier Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269 [DOI] [PubMed] [Google Scholar]
- 12. Kegel K. B., Meloni A. R., Yi Y., Kim Y. J., Doyle E., Cuiffo B. G., Sapp E., Wang Y., Qin Z.-H., Chen J. D., Nevins J. R., Aronin N., and DiFiglia M. (2002) Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 277, 7466–7476 [DOI] [PubMed] [Google Scholar]
- 13. Miller J. P., Holcomb J., Al-Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S., Botas J., Hughes R. E., and Ellerby L. M. (2010) Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington's disease. Neuron 67, 199–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., and Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 [DOI] [PubMed] [Google Scholar]
- 15. Sathasivam K., Neueder A., Gipson T. A., Landles C., Benjamin A. C., Bondulich M. K., Smith D. L., Faull R. L., Roos R. A., Howland D., Detloff P. J., Housman D. E., and Bates G. P. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tartari M., Gissi C., Lo Sardo V., Zuccato C., Picardi E., Pesole G., and Cattaneo E. (2008) Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive polyQ in sea urchin. Mol. Biol. Evol. 25, 330–338 [DOI] [PubMed] [Google Scholar]
- 17. Chen S., and Wetzel R. (2001) Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 10, 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhattacharyya A., Thakur A. K., Chellgren V. M., Thiagarajan G., Williams A. D., Chellgren B. W., Creamer T. P., and Wetzel R. (2006) Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 355, 524–535 [DOI] [PubMed] [Google Scholar]
- 19. Mishra R., Hoop C. L., Kodali R., Sahoo B., van der Wel P. C., and Wetzel R. (2012) Serine phosphorylation suppresses huntingtin amyloid accumulation by altering protein aggregation properties. J. Mol. Biol. 424, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thakur A. K., Jayaraman M., Mishra R., Thakur M., Chellgren V. M., Byeon I.-J., Anjum D. H., Kodali R., Creamer T. P., Conway J. F., Gronenborn A. M., and Wetzel R. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kar K., Jayaraman M., Sahoo B., Kodali R., and Wetzel R. (2011) Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 18, 328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crick S. L., Ruff K. M., Garai K., Frieden C., and Pappu R. V (2013) Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 20075–20080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bulone D., Masino L., Thomas D. J., San Biagio P. L., and Pastore A. (2006) The interplay between PolyQ and protein context delays aggregation by forming a reservoir of protofibrils. PLoS ONE 1, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagai Y., Inui T., Popiel H. A., Fujikake N., Hasegawa K., Urade Y., Goto Y., Naiki H., and Toda T. (2007) A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 14, 332–340 [DOI] [PubMed] [Google Scholar]
- 25. Masino L., Kelly G., Leonard K., Trottier Y., and Pastore A. (2002) Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 513, 267–272 [DOI] [PubMed] [Google Scholar]
- 26. Georgalis Y., Starikov E. B., Hollenbach B., Lurz R., Scherzinger E., Saenger W., Lehrach H., and Wanker E. E. (1998) Huntingtin aggregation monitored by dynamic light scattering. Proc. Natl. Acad. Sci. U.S.A. 95, 6118–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G. P., Davies S. W., Lehrach H., and Wanker E. E. (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 90, 549–558 [DOI] [PubMed] [Google Scholar]
- 28. Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G. P., Lehrach H., and Wanker E. E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington ' s. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muchowski P. J., Schaffar G., Sittler A., Wanker E. E., Hayer-Hartl M. K., and Hartl F. U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heiser V., Scherzinger E., Boeddrich A., Nordhoff E., Lurz R., Schugardt N., Lehrach H., and Wanker E. E. (2000) Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 6739–6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bennett E. J., Bence N. F., Jayakumar R., and Kopito R. R. (2005) Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365 [DOI] [PubMed] [Google Scholar]
- 32. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., and Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., and Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wacker J. L., Zareie M. H., Fong H., Sarikaya M., and Muchowski P. J. (2004) Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 11, 1215–1222 [DOI] [PubMed] [Google Scholar]
- 35. Legleiter J., Lotz G. P., Miller J., Ko J., Ng C., Williams G. L., Finkbeiner S., Patterson P. H., and Muchowski P. J. (2009) Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 284, 21647–21658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Legleiter J., Mitchell E., Lotz G. P., Sapp E., Ng C., DiFiglia M., Thompson L. M., and Muchowski P. J. (2010) Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Busch A., Engemann S., Lurz R., Okazawa H., Lehrach H., and Wanker E. E. (2003) Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: a potential mechanism for loss of huntingtin function in Huntington's disease. J. Biol. Chem. 278, 41452–41461 [DOI] [PubMed] [Google Scholar]
- 38. Bounab Y. (2010) CRMP1 Protein Complexes Modulate PolyQ-Mediated Htt Aggregation and Toxicity in Neurons. Ph.D. thesis, Humboldt-Universität zu Berlin [Google Scholar]
- 39. Nucifora L. G., Burke K. A., Feng X., Arbez N., Zhu S., Miller J., Yang G., Ratovitski T., Delannoy M., Muchowski P. J., Finkbeiner S., Legleiter J., Ross C. A., and Poirier M. A. (2012) Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J. Biol. Chem. 287, 16017–16028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dahlgren P. R., Karymov M. A., Bankston J., Holden T., Thumfort P., Ingram V. M., and Lyubchenko Y. L. (2005) Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomedicine 1, 52–57 [DOI] [PubMed] [Google Scholar]
- 41. Poirier M. A., Li H., Macosko J., Cai S., Amzel M., and Ross C. A. (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277, 41032–41037 [DOI] [PubMed] [Google Scholar]
- 42. Duim W. C., Chen B., Frydman J., and Moerner W. E. (2011) Sub-diffraction imaging of huntingtin protein aggregates by fluorescence blink-microscopy and atomic force microscopy. Chemphyschem 12, 2387–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pieri L., Madiona K., Bousset L., and Melki R. (2012) Fibrillar α-synuclein and Huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monsellier E., Redeker V., Ruiz-Arlandis G., Bousset L., and Melki R. (2015) Molecular interaction between the chaperone Hsc70 and the N-terminal flank of Huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bugg C. W., Isas J. M., Fischer T., Patterson P. H., and Langen R. (2012) Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 287, 31739–31746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Isas J. M., Langen R., and Siemer A. B. (2015) Solid-state nuclear magnetic resonance on the static and dynamic domains of Huntingtin exon-1 fibrils. Biochemistry 54, 3942–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Atwal R. S., Xia J., Pinchev D., Taylor J., Epand R. M., and Truant R. (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry, and toxicity. Hum. Mol. Genet. 16, 2600–2615 [DOI] [PubMed] [Google Scholar]
- 48. Arnesen T., Starheim K. K., Van Damme P., Evjenth R., Dinh H., Betts M. J., Ryningen A., Vandekerckhove J., Gevaert K., and Anderson D. (2010) The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol. Cell. Biol. 30, 1898–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dehay B., and Bertolotti A. (2006) Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 281, 35608–35615 [DOI] [PubMed] [Google Scholar]
- 50. Ansaloni A., Wang Z.-M., Jeong J. S., Ruggeri F. S., Dietler G., and Lashuel H. A. (2014) One-pot semisynthesis of exon 1 of the Huntingtin protein: new tools for elucidating the role of posttranslational modifications in the pathogenesis of Huntington's disease. Angew. Chem. Int. Ed. Engl. 53, 1928–1933 [DOI] [PubMed] [Google Scholar]
- 51. Singer D., Zauner T., Genz M., Hoffmann R., and Zuchner T. (2010) Synthesis of pathological and nonpathological human exon 1 huntingtin. J. Pept. Sci. 16, 358–363 [DOI] [PubMed] [Google Scholar]
- 52. Sahoo B., Singer D., Kodali R., Zuchner T., and Wetzel R. (2014) Aggregation behavior of chemically synthesized, full-length huntingtin exon1. Biochemistry 53, 3897–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O'Nuallain B., Thakur A. K., Williams A. D., Bhattacharyya A. M., Chen S., Thiagarajan G., and Wetzel R. (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 413, 34–74 [DOI] [PubMed] [Google Scholar]
- 54. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen S., Berthelier V., Yang W., and Wetzel R. (2001) Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 311, 173–182 [DOI] [PubMed] [Google Scholar]
- 56. Kim M. W., Chelliah Y., Kim S. W., Otwinowski Z., and Bezprozvanny I. (2009) Secondary structure of Huntingtin amino-terminal region. Structure 17, 1205–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Darnell G., Orgel J. P, Pahl R., and Meredith S. C. (2007) Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 374, 688–704 [DOI] [PubMed] [Google Scholar]
- 58. Fodale V., Kegulian N. C., Verani M., Cariulo C., Azzollini L., Petricca L., Daldin M., Boggio R., Padova A., Kuhn R., Pacifici R., Macdonald D., Schoenfeld R. C., Park H., Isas J. M., Langen R., Weiss A., and Caricasole A. (2014) Polyglutamine- and temperature-dependent conformational rigidity in mutant huntingtin revealed by immunoassays and circular dichroism spectroscopy. PLoS ONE 9, e112262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hollenbach B., Scherzinger E., Schweiger K., Lurz R., Lehrach H., and Wanker E. E. (1999) Aggregation of truncated GST-HD exon 1 fusion proteins containing normal range and expanded glutamine repeats. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Walters R. H., and Murphy R. M. (2009) Examining Polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J. Mol. Biol. 393, 978–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perutz M. F., Johnson T., Suzuki M., and Finch J. T. (1994) Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 91, 5355–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Landrum E., and Wetzel R. (2014) Biophysical underpinnings of the repeat length dependence of polyglutamine amyloid formation. J. Biol. Chem. 289, 10254–10260 [DOI] [PMC free article] [PubMed] [Google Scholar]